

Abstract
Synaptic plasticity is fundamental to the neural processes underlying learning and memory. Interestingly, synaptic plasticity itself can be dynamically regulated by prior activity, in a process termed ‘metaplasticity’, which can be expressed both homosynaptically and heterosynaptically. Here, we focus on heterosynaptic metaplasticity, particularly long-range interactions between synapses spread across dendritic compartments, and review evidence for intracellular versus intercellular signalling pathways leading to this effect. Of particular interest is our previously reported finding that priming stimulation in stratum oriens of area CA1 in the hippocampal slice heterosynaptically inhibits subsequent long-term potentiation and facilitates long-term depression in stratum radiatum. As we have excluded the most likely intracellular signalling pathways that might mediate this long-range heterosynaptic effect, we consider the hypothesis that intercellular communication may be critically involved. This hypothesis is supported by the finding that extracellular ATP hydrolysis, and activation of adenosine A2 receptors are required to induce the metaplastic state. Moreover, delivery of the priming stimulation in stratum oriens elicited astrocytic calcium responses in stratum radiatum. Both the astrocytic responses and the metaplasticity were blocked by gap junction inhibitors. Taken together, these findings support a novel intercellular communication system, possibly involving astrocytes, being required for this type of heterosynaptic metaplasticity.
Keywords: metaplasticity, intercellular signalling, long-term potentiation, long-term depression, gap junctions, astrocytes
1. Introduction
It is now commonly accepted that synaptic plasticity in the form of long-term potentiation (LTP) and long-term depression (LTD) is a fundamental component of the neural processes underlying learning and memory. As memory mechanisms, however, LTP and LTD present a number of challenges to the neuronal network. In particular, unregulated synaptic plasticity has the potential to produce runaway effects that lead to functional maladaption and impaired cognitive function. For example, if the strength of connections reaches saturated floor or ceiling levels, synaptic plasticity becomes unavailable for further change in that same direction, and previously stored memories may become impaired [1]. Moreover, removing restrictions on synaptic potentiation also impairs learning and memory [2]. At more extreme levels, there could be dramatic pathological effects such as synapses being potentiated to a level that causes excitoxicity, or depressed to non-function. A key issue then is to understand how synaptic plasticity is regulated in order to optimise encoding and storage of information.
One class of plasticity regulatory mechanisms is metaplasticity, which refers to activity-dependent and persistent changes in the state of synapses or neurons that alter the magnitude or duration of subsequent synaptic plasticity [3]. Metaplasticity is a particularly attractive regulatory mechanism, because it dynamically links the history of neuronal activity with the current response. While the term metaplasticity was first formally coined in 1996 [4], the preceding Bienenstock, Cooper and Munro (BCM) theory of synaptic plasticity incorporated a metaplasticity-type feature that adjusts (‘slides’) the threshold for LTP according to the cell's previous history of activity [5] (figure 1). The purpose of this feature is to address the instability problem noted above from unregulated plasticity.
Figure 1.

BCM-like heterosynaptic metaplasticity. (a) The BCM model [5] posits a synaptic plasticity function whereby a high level of coincident pre- and postsynaptic activity induces LTP, whereas a low level of coincident activity induces LTD. Prior high levels of postsynaptic activity shifts the modification threshold (θM) of the synaptic plasticity function to the right. The converse effect occurs following low levels of postsynaptic activity. Critically, this change in synaptic plasticity induction occurs across all of a cell's synapses regardless of whether they participated in the activity inducing the metaplastic state. (b) Data from the visual cortex support the predictions of the BCM model insofar as dark rearing animals, by reducing the level of activity, causes a leftward metaplastic shift in the synaptic plasticity function. (Adapted and with permission from [6].) (c) A similar effect has been demonstrated in hippocampus slices where, as compared to control conditions (black squares), priming stimulation of one pathway (S1, inset) produces a BCM-like metaplastic shift in subsequent synaptic plasticity both for the stimulated pathway (white squares) and for an independent heterosynaptic pathway (S2, inset) in stratum radiatum (grey squares). (Adapted and with permission from [7].) (d) We have shown that BCM-like heterosynaptic inhibition of stratum radiatum LTP (high-frequency stimulation, Rad. HFS; pathway S2/R2, inset) can also be induced when priming stimulation is delivered in stratum oriens (Or.prime; S1, inset) [8]. (e) Consistent with a BCM-like effect, stratum radiatum LTD (induced by low-frequency stimulation, Rad. LFS) is heterosynaptically facilitated by priming stimulation in stratum oriens. fEPSP, field excitatory postsynaptic potential.
A particular feature of the BCM sliding threshold is that it applies simultaneously to all synapses spread across the cell. That is, for a given postsynaptic neuron, both the synapses participating in the activity that induces the metaplastic state (the homosynaptic pathway) and those that do not participate (heterosynaptic pathways) are proposed to undergo the same changes in subsequent plasticity. We refer to such regulation as heterosynaptic metaplasticity. Overall, the BCM theory has been hugely influential in the plasticity field by formalizing testable predictions not only about the direction and degree of plasticity arising from conjunctive pre- and postsynaptic activity, but also about the activity-dependent regulation of the LTP threshold. These predictions have been confirmed, at least in part, by studies in the visual cortex and hippocampus [6–10].
Not all experimentally described heterosynaptic metaplasticity effects conform to BCM principles however [8], raising questions about the mechanisms that underlie these various phenomena. For example, how do they vary with respect to their mechanisms of induction, their expression mechanisms that alter subsequent plasticity, and very importantly, their mechanisms that coordinate the metaplasticity effect between synapses scattered across the dendritic arbour? Here, we review and discuss these issues, with a particular focus on metaplasticity that is cell-wide, or at least spread broadly across dendritic compartments.
2. Intracellular pathways
Most studies of heterosynaptic metaplasticity have focused naturally on intracellular mechanisms. A key question is the cellular location that governs the setting of plasticity thresholds. In other words, is synaptic/cellular activity integrated by a central mechanism to direct a cell-wide state change, or is metaplasticity triggered directly at the over- or under active synapses and then transmitted to heterosynaptic locations? Equally, is the expression of the metaplasticity at synapses or at other neuronal elements such as dendrites?
(a). Central integration of neuronal activity
A central metaplasticity integrator proposed by the BCM theory is the time-averaged history of cell firing, although whether the expression of the metaplastic state is via synaptic or non-synaptic mechanisms was not defined. In support of the theory, neuronal discharge generated by either antidromic stimulation or intra-somatic current injections can modify subsequent synaptic plasticity in a BCM-like manner [9,11]. However, under other conditions, cell firing is neither necessary nor sufficient for BCM-like heterosynaptic metaplasticity [8,12]. In fact, cell firing in these cases facilitated rather than inhibited subsequent LTP. The more recent calcium-dependent plasticity (CaDP) model, derived from the BCM model, also suggests a central mediator of metaplastic induction, i.e. time-integrated membrane voltage [13]. Moreover, the CaDP model specifically proposes that this integrator regulates a distributed synaptic expression mechanism, namely the modification of synaptic N-methyl-d-aspartate (NMDA) receptor conductances. Indeed, during visual cortex development, BCM-like metaplasticity is governed by activity-dependent regulation of the NMDA receptor subunit composition [14,15], an expression mechanism that also underlies certain metaplasticity effects in the hippocampus [16]. However, others have proposed that heterosynaptic metaplasticity can be mediated by a widespread modification of cellular excitability through changes in ion channels that control the discharge properties of neurons such as Ih or the slow after-hyperpolarization, for which there is experimental support [17–21]. It should be noted that these various potential expression mechanisms are not mutually exclusive.
One well-studied type of heterosynaptic metaplasticity that does not conform to the BCM theory is encapsulated by the synaptic tag and capture (STC) model of plasticity. Here, the duration of LTD/LTP is centrally regulated by protein synthesis such that prior neural activity facilitates rather than impairs LTP heterosynaptically [22]. In this model, a pool of proteins required for consolidation of LTD and LTP can be made available by suitably strong afferent stimulation, or even by simply postsynaptic spiking [23,24]. Crucially, the capture and usage of these proteins can occur at heterosynaptic locations where there is only relatively weak synaptic activity that is normally insufficient to induce the late phase of synaptic plasticity. Interestingly, proteins that are produced by strong stimulation of one pathway can contribute to ‘cross-capture’, allowing LTP-inducing stimulation to reinforce LTD at heterosynaptic locations and vice versa [25,26].
On the surface this mechanism appears to suggest a simple, uniform cell-wide regulation of synaptic plasticity duration; however, the nature of these interactions is complicated. Although capture between basal and apical dendritic compartments can occur, it happens under limited conditions [27–30]. Instead STC interactions are more usually confined to specific dendritic compartments. Moreover, differences even exist in the effectiveness of STC mechanisms between proximal and distal synapses on the apical dendrite [31]. An interesting aspect of the STC model is that the mechanism requires additional neuromodulatory input [25,32], highlighting an interesting route to heterosynaptic metaplasticity; that is, while the postsynaptic induction pathway is exclusively intracellular, more than one coincident presynaptic input plus non-glutamatergic transmission can be involved. In line with this, prior stimulation of other brain areas, particularly the basolateral amygdala, can also transform decremental LTP to a more stable form through a protein synthesis-dependent mechanism [33]. Interestingly, negative interactions that impair plasticity duration can also arise, particularly through competition for newly synthesized proteins. This occurs either when protein synthesis is limited or when strong induction protocols are used [30,34]. This competitive mechanism may represent an important brake on the extent to which STC mechanisms can enhance LTP, helping to avoid some of the positive-feedback instability on synaptic efficacy that the sliding threshold feature of the BCM theory was also designed to avoid.
(b). Metaplasticity triggered at and spreading between synapses
Can a metaplastic state be triggered directly at synapses and then spread heterosynaptically? There is little experimental evidence to indicate that this can occur to any significant degree. While metaplasticity consistent with most BCM principles has been demonstrated or modelled, this is typically confined to the active (or inactive) synapses [35,36]. However under some conditions, it is clear that synaptic plasticity at adjacent synapses can be metaplasticity regulated through the diffusion of intracellular factors. For example, inducing LTP at a single synapse can spread an LTP-permissive metaplastic state across the dendrite to other spines approximately 10 µm away [37]. While this effect is in the opposite direction to BCM predictions, it represents proof of principle for the intracellular spread of a metaplasticity state, lending credence to the possibility that similar molecules could spread other metaplasticity effects heterosynaptically, at least to immediately adjacent synapses. Inositol trisphosphate (IP3) and cyclic adenosine monophosphate are two diffusible messengers that could act in such a way, as G-protein signalling culminating in the formation of these molecules triggers metaplastic states favouring LTD and LTP, respectively [38].
3. Intercellular pathways
The extensive spatial distribution of change that characterizes heterosynaptic metaplasticity in some experimental models raises the possibility that such long-range communication employs intercellular mechanisms beyond just the postsynaptic neurons (figure 2). For example, we recently described a heterosynaptic metaplasticity that spread from basal to apical dendrites in CA1 pyramidal neurons, without requirement for action potential generation or even somatic depolarization [8]. We identified activation of M1-acetylcholine receptors (M1-AChRs) and IP3-mediated release of Ca2+ from intracellular stores as contributing mechanisms. However, the fact that extracellularly released molecules contribute to the induction of this form of metaplasticity suggest it is not mediated purely by an intracellular signalling pathway within CA1 pyramidal cells (see below). Thus, we have considered the possibility that intercellular signalling may play a crucial role in inducing this metaplastic state [40].
Figure 2.

Intercellular pathways for heterosynaptic metaplasticity. (a) GABAergic interneurons in the hippocampus can mediate local heterosynaptic facilitation of LTP. Following presynaptic activity (red axon), glutamate (Glu) release and activation of postsynaptic glutamate receptors (GluR), retrograde endocannabinoid (eCB) signalling from the postsynaptic neuron to presynaptic type 1 cannabinoid receptors (CB1R) on GABAergic interneurons persistently reduces GABA release and thus activation of GABAA receptors (GABAAR), thereby facilitating LTP at nearby synapses (green halo, [39]). (b) Another potential intercellular pathway for heterosynaptic metaplasticity involves astrocytes. Long-range signalling through the astrocytic network may alter subsequent synaptic plasticity at distant synapses, including those in different dendritic compartments (left-hand side of figure). Here, for example, activation of inputs to basal dendrites (the red presynaptic axon) results in heterosynaptic metaplasticity (orange halos) in the apical dendrites. Activation of a single astrocyte may also produce heterosynaptic metaplasticity, albeit likely over a more limited spatial extent (right-hand side of figure). The illustrated pathways for intercellular mediation of heterosynaptic metaplasticity are almost certainly not exhaustive. (a) GABAergic interneurons in the hippocampus can mediate local heterosynaptic facilitation of LTP. Following presynaptic activity (red axon), glutamate (Glu) release and activation of postsynaptic glutamate receptors (GluR), retrograde endocannabinoid (eCB) signalling from the postsynaptic neuron to presynaptic type 1 cannabinoid receptors (CB1R) on GABAergic interneurons persistently reduces GABA release and thus activation of GABAA receptors (GABAAR), thereby facilitating LTP at nearby synapses (green halo, [39]).
Already there is good reason to consider intercellular signalling as a mechanism for heterosynaptic metaplasticity. Under normal circumstances, extrinsic input such as from inhibitory interneurons plays a key role in gating the induction of synaptic plasticity. If the efficacy of such inputs could be regulated in an activity-dependent manner, this would be a locus of expression for a metaplastic state. Indeed, retrograde endocannabinoid signalling from excitatory synapses induces both transient and persistent suppressions of gamma-aminobutyric acid (GABA) release that can locally facilitate LTP [39,41]. Extrinsic input from other brain areas can also mediate heterosynaptic metaplasticity as prior stimulation of the basolateral amygdala can persistently modify subsequent plasticity in the hippocampus [42]. Moreover, the prior history of amygdala activity influences the induction of such effects in both the hippocampus [43] and prefrontal cortex [44], albeit thus far only when the timing between amygdala stimulation and plasticity induction is short.
Interestingly, regulation of GABAergic transmission was not a mediator of the cell-wide heterosynaptic metaplasticity we have recently described [8]. Instead, we identified two other signalling mechanisms, both of which are commonly implicated in intercellular communication. First, the effect requires a purinergic signalling cascade involving the extracellular hydrolysis of adenosine triphosphate (ATP) to adenosine by ectonucleotidases [40], and activation of adenosine A2 receptors (figure 3). ATP exerts diverse and diffuse effects through its numerous sites of release, whether from presynaptic terminals, nodes of Ranvier or gap junction hemichannels [45–47]. Following hydrolysis, adenosine acts on several receptor subtypes both homo- and heterosynaptically to modulate network activity (for a review, see [48]). In reduced preparations like the hippocampal slice, such intercellular contributions to metaplasticity could be mediated by release of neurotransmitter from adjacent neurons activated through ephaptic coupling, by inhibitory interneurons or by non-neuronal cells. But, since blocking action potentials or GABAergic transmission in our preparation did not prevent the induction of heterosynaptic metaplasticity [8], non-neuronal intercellular communication is likely to play the critical role [40]. This conclusion is supported by the finding that metaplastic regulation of stratum radiatum LTP by stratum oriens priming is abolished by the gap junction inhibitors carbenoxolone (figure 4a) and meclofenamic acid, plus a peptide inhibitor of astrocytic hemichannels and gap junctions containing connexin-43 [40]. Gap junctions formed by tightly apposed connexin hemichannels allow for a dynamic and highly plastic mode of fast intercellular communication [49], especially for non-neural cells such as astrocytes. For example, diffusible messengers such as Ca2+ and IP3 travel between cells via gap junctions [50]. These channels are therefore capable of mediating widespread modulatory signalling within neural networks. There is a growing body of literature demonstrating that astrocytes play an important and active role in neural function and cognition (for a review, see [51]). Astrocytic processes are intimately associated spatially with synapses [52–54] and can be activated through a number of signalling pathways inherently coupled to both pre- and postsynaptic neuronal activity, most notably through neurotransmitters binding to astrocytic G-protein-coupled receptors (e.g. [55–60]). Astrocytes can also influence neuronal function and synaptic plasticity through active control of extracellular ion concentrations, re-uptake of neurotransmitters from the synaptic cleft and release of gliotransmitters ([56,61–64], but see also [65,66]). This notion of a synapse containing not only pre- and postsynaptic elements but also astrocytic elements is referred to as the ‘tripartite synapse’ [67–69]. It is the focus of considerable recent research activity, in part due to the potentially dramatic increase in computational power this may afford to neural networks [51,70,71].
Figure 3.
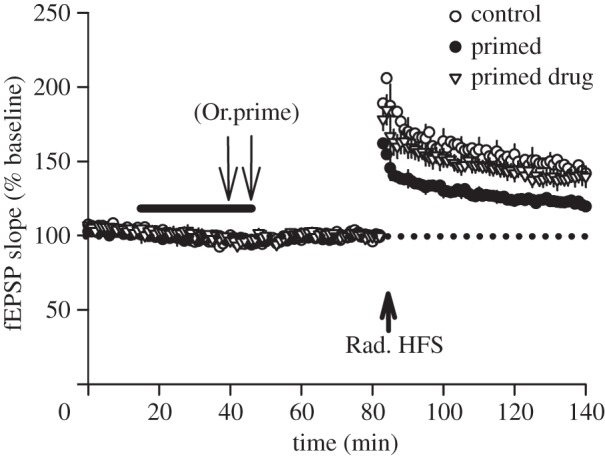
Heterosynaptic metaplasticity in the hippocampal slice is dependent on activation of adenosine A2 receptors. In field potential recordings from CA1 of acute hippocampal slices (refer for methods [40]), LTP (2 × 100 Hz) in stratum radiatum of CA1 is inhibited in slices which first receive priming stimulation (3 × 100 Hz, repeated after 5 min) delivered to stratum oriens afferents (Or.prime). This effect is inhibited by co-administration of the A2AR antagonist ZM241385 and the A2BR antagonist MRS1754 (50 nM each, bar), bath applied prior to and during priming (control: n = 5, 144 ± 4%; primed: n = 8, 122 ± 2%; drug: n = 5, 139 ± 6%, F2,15 = 10.12, p = 0.002). Data are expressed as a percentage of the averaged baseline responses. Arrows denote time-points of oriens priming or radiatum HFS.
Figure 4.
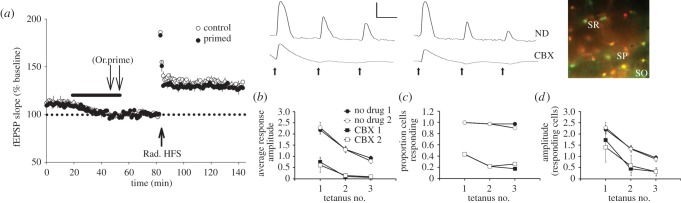
Heterosynaptic metaplasticity and long-range astrocyte responses are blocked by a gap junction inhibitor. (a) LTP in stratum radiatum is not attenuated by stratum oriens priming stimulation if it occurs in the presence of carbenoxolone (30 µM, black bar: control: n = 6, 130 ± 5%; primed: n = 6, 127 ± 3%, t = 0.53, p = 0.61, n.s.). (b) Following delivery of the same oriens priming stimulation, the average amplitude of astrocytic calcium responses (ΔF/F0) in stratum radiatum was significantly greater in the no drug condition (collapsed across bursts, p < 0.0001; n = 40 cells from seven slices) when compared with the carbenoxolone condition (CBX; n = 23 cells from three slices). For both the no drug and CBX conditions, there was a decline in average amplitude over successive tetani in a burst (p < 0.0001) but no interaction between the condition and tetanus number (p = 0.33). (c) Almost all identifiable astrocytes in stratum radiatum responded (greater than 0.1 increase) to each of the tetani in the no drug condition as compared to only a few cells responding in the presence of carbenoxolone. (d) Excluding astrocytes which did not respond to each of the tetani, the amplitude of responses was still significantly greater in the no drug condition (p < 0.001). Error bars represent s.e.m. Waveforms are from a representative cell from both the no drug (ND) and carbenoxolone (CBX) conditions showing responses to burst 1 and burst 2. Arrows indicate tetanus delivery. Scale bar: amplitude 1, time 10 s. Fluorescence overlay showing SR101 (red) and Fluo-4 (green) (stratum oriens: SO; stratum pyramidale: SP; stratum radiatum: SR). Additional methods for b–d and group numbers for d can be found in the electronic supplementary material.
Astrocytes are ideally placed to mediate both local and widespread heterosynaptic effects on synaptic plasticity as a single astrocyte likely ensheaths multiple neuronal somata, hundreds of dendrites [53] and thousands of individual synapses [52]. Additionally, signalling can occur across the astrocytic network through gap junctions and ATP-mediated Ca2+ waves [72], indicating that activation of astrocytes could provide for very long-range communication and associated influence over synaptic plasticity. IP3-mediated release of calcium from intracellular stores is critical for the generation of astrocytic calcium responses [73–75] and is potentially involved in communication between astrocytes [63] and release of gliotransmitters [76]. These Ca2+ elevations are triggered by a number of G-protein-coupled receptors, including mAChRs [62,77]. Furthermore, brief adenosine 2B receptor (A2BR) activation triggers spontaneous Ca2+ elevations throughout astrocytic networks that persist for at least 20 min [78]. Such spatially and temporally widespread signalling is in keeping with the requirements for generating heterosynaptic metaplasticity.
The above considerations raise the possibility that hippocampal astrocytes can in fact communicate widely enough across the CA1 layers to be able in principle to mediate long-range heterosynaptic metaplasticity that spreads from basilar to apical dendritic compartments. To address this, we have undertaken calcium imaging of CA1 astrocytes filled with Fluo-4 and labelled with sulforhodamine-101 by injection of these compounds directly into the hippocampus prior to slice preparation (see electronic supplementary material). Using the same stratum oriens stimulation parameters that inhibit LTP and promote LTD in stratum radiatum (6 × 100 Hz, 1 s trains [8]), we observed that each high-frequency train of priming stimulation reliably induced a calcium elevation not only in stratum oriens astrocytes (not shown), but also in stratum radiatum astrocytes as far from the cell body layers as was imaged (297 µm; mean 87 ± 10 µm; figure 4b,d). The greatest increase occurred in response to the first high-frequency stimulation (HFS) in a set of three trains (20 s apart). A second set of trains 5 min later gave a nearly identical set of responses. To test the hypothesis that this communication between CA1 layers was mediated by gap junctional communication, we repeated the experiments in the presence of carbenoxolone. This drug, at the same concentration used to inhibit the metaplasticity, greatly inhibited the calcium response in radiatum astrocytes, often completely eliminating the responses altogether, particularly to the second and third trains in a burst of stimulation (figure 4c).
If astrocytes do mediate the heterosynaptic metaplasticity, which gliotransmitter(s) might be signalling back to neurons to modulate plasticity? Astrocytic ATP, converted to adenosine and acting on A1Rs, is one candidate as it is implicated in heterosynaptic depression [73–75] (but also see [66]). Astrocytic glutamate is another candidate, as cholinergic activation of astrocytes induces mGluR or NMDAR-dependent LTP in neurons in vivo [60,79,80]. Moreover, astrocytic activation of neuronal A1Rs and NMDARs regulates plasticity thresholds [81,82]. However, these receptors do not contribute to heterosynaptic metaplasticity in our model [8,40]. Interestingly, A2Rs are already implicated in an inhibitory form of metaplasticity [83], although the precise mechanism of action remains unknown. HFS and activation of A2BRs can trigger the release of cytokines from astrocytes [84,85], and this has been proposed as a metaplastic mechanism for inhibiting LTP both homo- and heterosynaptically [86], and indeed, we have recently shown that A2BR activation can generate a cell-wide inhibition of LTP [40]. It is therefore possible that priming stimulation in stratum oriens modulates plasticity in stratum radiatum by eliciting widespread cytokine release from astrocytes, a hypothesis we consider worthy of future investigation.
Taken together, the pattern of results we have obtained so far is strongly suggestive of an intercellular signalling pathway mediating BCM-like long-range heterosynaptic metaplasticity in the hippocampus. We have also established proof of principle that the participating intercellular network may include astrocytes, which are activated extensively by afferent stimulation and which are capable of regulating LTP and LTD induction. However, to fully test this hypothesis, experiments are needed that directly manipulate astrocyte function both during and after priming stimulation.
(a). Functional implications of heterosynaptic metaplasticity
Looking across studies, there is a clearly a wide range of mechanisms mediating heterosynaptic metaplasticity, perhaps reflecting the range of preparations used (in vivo versus in vitro, specific brain regions, etc.), but also perhaps reflecting the diverse capability of neural networks for this class of metaplasticity. Regardless, heterosynaptic metaplasticity has the potential to powerfully modulate local network function, particularly when mediated by third-party cells such as astrocytes. It is possible that such network metaplasticity acts to enhance the signal-to-noise ratio between active and quiescent inputs, thus maximizing the distinction between salient and non-salient information. Such a distinction may be augmented by the synaptic release of agents that also regulate plasticity. For example, the facilitation of LTP by A2AR activation may be balanced by heterosynaptic depression mediated by astrocytic activation of A1Rs [48]. Furthermore, whereas endocannabinoids can induce homosynaptic depression they can also activate astrocytes which mediate heterosynaptic facilitation via glutamate release [63]. Another function of BCM-like metaplasticity may be to prolong the duration of synaptic plasticity by rendering newly established synaptic weightings resistant to change for a period of time. Alternatively, where dramatic perturbations of normal neural activity occur, such metaplasticity may serve a neuroprotective role by braking subsequent plasticity, and facilitating homeostatic synaptic scaling in the opposite direction to normalize overall synaptic weights. In this scenario, metaplasticity could promote functionally adaptive responses such as preventing potentiation of synaptic inputs to the point of excitoxicity. It is interesting to note in this context the considerable mechanistic overlap between neuroprotective preconditioning effects which prevent excitoxicity and metaplasticity [3,87,88].
Funding statement
We thank the Royal Society of New Zealand Marsden fund, the Neurological Foundation of New Zealand, the National Health and Medical Research Council of Australia and the Australian Research Council for grant support.
References
- 1.Brun VH, Ytterbo K, Morris RG, Moser MB, Moser EI. 2001. Retrograde amnesia for spatial memory induced by NMDA receptor-mediated long-term potentiation. J. Neurosci. 21, 356–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Migaud M, et al. 1998. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature 396, 433–439 (doi:10.1038/24790) [DOI] [PubMed] [Google Scholar]
- 3.Abraham WC. 2008. Metaplasticity: tuning synapses and networks for plasticity. Nat. Rev. Neurosci. 9, 387–399 (doi:10.1038/nrn2356) [DOI] [PubMed] [Google Scholar]
- 4.Abraham WC, Bear MF. 1996. Metaplasticity: the plasticity of synaptic plasticity. Trends Neurosci. 19, 126–130 (doi:10.1016/S0166-2236(96)80018-X) [DOI] [PubMed] [Google Scholar]
- 5.Bienenstock EL, Cooper LN, Munro PW. 1982. Theory for the development of neuron selectivity: orientation specificity and binocular interaction in visual cortex. J. Neurosci. 2, 32–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Philpot BD, Espinosa JS, Bear MF. 2003. Evidence for altered NMDA receptor function as a basis for metaplasticity in visual cortex. J. Neurosci. 23, 5583–5588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang H, Wagner JJ. 1999. Priming-induced shift in synaptic plasticity in the rat hippocampus. J. Neurophysiol. 82, 2024–2028 [DOI] [PubMed] [Google Scholar]
- 8.Hulme SR, Jones OD, Ireland DR, Abraham WC. 2012. Calcium-dependent but action potential-independent BCM-like metaplasticity in the hippocampus. J. Neurosci. 32, 6785–6794 (doi:10.1523/JNEUROSCI.0634-12.2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abraham WC, Mason-Parker SE, Bear MF, Webb S, Tate WP. 2001. Heterosynaptic metaplasticity in the hippocampus in vivo: a BCM-like modifiable threshold for LTP. Proc. Natl Acad. Sci. USA 98, 10 924–10 929 (doi:10.1073/pnas.181342098) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cooper LN, Bear MF. 2012. The BCM theory of synapse modification at 30: interaction of theory with experiment. Nat. Rev. Neurosci. 13, 798–810 (doi:10.1038/nrn3353) [DOI] [PubMed] [Google Scholar]
- 11.Yasuda R, Sabatini BL, Svoboda K. 2003. Plasticity of calcium channels in dendritic spines. Nat. Neurosci. 6, 948–955 (doi:10.1038/nn1112) [DOI] [PubMed] [Google Scholar]
- 12.Bukalo O, Campanac E, Hoffman DA, Fields RD. 2013. Synaptic plasticity by antidromic firing during hippocampal network oscillations. Proc. Natl Acad. Sci. USA 110, 5175–5180 (doi:10.1073/pnas.1210735110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yeung LC, Shouval HZ, Blais BS, Cooper LN. 2004. Synaptic homeostasis and input selectivity follow from a calcium-dependent plasticity model. Proc. Natl Acad. Sci. USA 101, 14 943–14 948 (doi:10.1073/pnas.0405555101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cho KK, Khibnik L, Philpot BD, Bear MF. 2009. The ratio of NR2A/B NMDA receptor subunits determines the qualities of ocular dominance plasticity in visual cortex. Proc. Natl Acad. Sci. USA 106, 5377–5382 (doi:10.1073/pnas.0808104106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Philpot BD, Cho KK, Bear MF. 2007. Obligatory role of NR2A for metaplasticity in visual cortex. Neuron 53, 495–502 (doi:10.1016/j.neuron.2007.01.027) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu Z, Chen RQ, Gu QH, Yan JZ, Wang SH, Liu SY, Lu W. 2009. Metaplastic regulation of long-term potentiation/long-term depression threshold by activity-dependent changes of NR2A/NR2B ratio. J. Neurosci. 29, 8764–8773 (doi:10.1523/JNEUROSCI.1014-09.2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Narayanan R, Johnston D. 2007. Long-term potentiation in rat hippocampal neurons is accompanied by spatially widespread changes in intrinsic oscillatory dynamics and excitability. Neuron 56, 1061–1075 (doi:10.1016/j.neuron.2007.10.033) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brager DH, Johnston D. 2007. Plasticity of intrinsic excitability during long-term depression is mediated through mGluR-dependent changes in Ih in hippocampal CA1 pyramidal neurons. J. Neurosci. 27, 13 926–13 937 (doi:10.1523/JNEUROSCI.3520-07.2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fan Y, Fricker D, Brager DH, Chen X, Lu HC, Chitwood RA, Johnston D. 2005. Activity-dependent decrease of excitability in rat hippocampal neurons through increases in Ih. Nat. Neurosci. 8, 1542–1551 (doi:10.1038/nn1568) [DOI] [PubMed] [Google Scholar]
- 20.Le RD, Fernandez De Sevilla D, Belen PA, Fuenzalida M, Buno W. 2004. Heterosynaptic metaplastic regulation of synaptic efficacy in CA1 pyramidal neurons of rat hippocampus. Hippocampus 14, 1011–1025 (doi:10.1002/hipo.20021) [DOI] [PubMed] [Google Scholar]
- 21.Narayanan R, Johnston D. 2010. The h current is a candidate mechanism for regulating the sliding modification threshold in a BCM-like synaptic learning rule. J. Neurophysiol. 104, 1020–1033 (doi:10.1152/jn.01129.2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frey U, Morris RG. 1997. Synaptic tagging and long-term potentiation. Nature 385, 533–536 (doi:10.1038/385533a0) [DOI] [PubMed] [Google Scholar]
- 23.Dudek SM, Fields RD. 2002. Somatic action potentials are sufficient for late-phase LTP-related cell signaling. Proc. Natl Acad. Sci. USA 99, 3962–3967 (doi:10.1073/pnas.062510599) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Raymond CR. 2008. Different requirements for action potentials in the induction of different forms of long-term potentiation. J. Physiol. 586, 1859–1865 (doi:10.1113/jphysiol.2008.151035) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sajikumar S, Frey JU. 2004. Late-associativity, synaptic tagging, and the role of dopamine during LTP and LTD. Neurobiol. Learn. Mem. 82, 12–25 (doi:10.1016/j.nlm.2004.03.003) [DOI] [PubMed] [Google Scholar]
- 26.Sajikumar S, Navakkode S, Sacktor TC, Frey JU. 2005. Synaptic tagging and cross-tagging: the role of protein kinase Mzeta in maintaining long-term potentiation but not long-term depression. J. Neurosci. 25, 5750–5756 (doi:10.1523/JNEUROSCI.1104-05.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alarcon JM, Barco A, Kandel ER. 2006. Capture of the late phase of long-term potentiation within and across the apical and basilar dendritic compartments of CA1 pyramidal neurons: synaptic tagging is compartment restricted. J. Neurosci. 26, 256–264 (doi:10.1523/JNEUROSCI.3196-05.2006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sajikumar S, Navakkode S, Korz V, Frey JU. 2007. Cognitive and emotional information processing: protein synthesis and gene expression. J. Physiol. 584, 389–400 (doi:10.1113/jphysiol.2007.140087) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sajikumar S, Navakkode S, Frey JU. 2007. Identification of compartment- and process-specific molecules required for ‘synaptic tagging’ during long-term potentiation and long-term depression in hippocampal CA1. J. Neurosci. 27, 5068–5080 (doi:10.1523/JNEUROSCI.4940-06.2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pavlowsky A, Alarcon JM. 2012. Interaction between long-term potentiation and depression in CA1 synapses: temporal constrains, functional compartmentalization and protein synthesis. PLoS ONE 7, e29865 (doi:10.1371/journal.pone.0029865) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parvez S, Ramachandran B, Frey JU. 2010. Functional differences between and across different regions of the apical branch of hippocampal CA1 dendrites with respect to long-term depression induction and synaptic cross-tagging. J. Neurosci. 30, 5118–5123 (doi:10.1523/JNEUROSCI.5808-09.2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frey U, Morris RG. 1998. Synaptic tagging: implications for late maintenance of hippocampal long-term potentiation. Trends Neurosci. 21, 181–188 (doi:10.1016/S0166-2236(97)01189-2) [DOI] [PubMed] [Google Scholar]
- 33.Frey S, Bergado-Rosado J, Seidenbecher T, Pape HC, Frey JU. 2001. Reinforcement of early long-term potentiation (early-LTP) in dentate gyrus by stimulation of the basolateral amygdala: heterosynaptic induction mechanisms of late-LTP. J. Neurosci. 21, 3697–3703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fonseca R, Nagerl UV, Morris RG, Bonhoeffer T. 2004. Competing for memory: hippocampal LTP under regimes of reduced protein synthesis. Neuron 44, 1011–1020 (doi:10.1016/j.neuron.2004.10.033) [DOI] [PubMed] [Google Scholar]
- 35.Lee MC, Yasuda R, Ehlers MD. 2010. Metaplasticity at single glutamatergic synapses. Neuron 66, 859–870 (doi:10.1016/j.neuron.2010.05.015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kalantzis G, Shouval HZ. 2009. Structural plasticity can produce metaplasticity. PLoS ONE 4, e8062 (doi:10.1371/journal.pone.0008062) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harvey CD, Svoboda K. 2007. Locally dynamic synaptic learning rules in pyramidal neuron dendrites. Nature 450, 1195–1200 (doi:10.1038/nature06416) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang S, Trevino M, He K, Ardiles A, Pasquale R, Guo Y, Palacios A, Huganir R, Kirkwood A. 2012. Pull-push neuromodulation of LTP and LTD enables bidirectional experience-induced synaptic scaling in visual cortex. Neuron 73, 497–510 (doi:10.1016/j.neuron.2011.11.023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chevaleyre V, Castillo PE. 2004. Endocannabinoid-mediated metaplasticity in the hippocampus. Neuron 43, 871–881 (doi:10.1016/j.neuron.2004.08.036) [DOI] [PubMed] [Google Scholar]
- 40.Jones OD, Hulme SR, Abraham WC. 2013. Purinergic receptor- and gap junction-mediated intercellular signalling as a mechanism of heterosynaptic metaplasticity. Neurobiol. Learn. Mem. 105, 31–39 (doi:10.1016/j.nlm.2013.05.010) [DOI] [PubMed] [Google Scholar]
- 41.Diana MA, Marty A. 2004. Endocannabinoid-mediated short-term synaptic plasticity: depolarization-induced suppression of inhibition (DSI) and depolarization-induced suppression of excitation (DSE). Br. J. Pharmacol. 142, 9–19 (doi:10.1038/sj.bjp.0705726) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li Z, Richter-Levin G. 2012. Stimulus intensity-dependent modulations of hippocampal long-term potentiation by basolateral amygdala priming. Front. Cell Neurosci. 6, 21 (doi:10.3389/fncel.2012.00021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nakao K, Matsuyama K, Matsuki N, Ikegaya Y. 2004. Amygdala stimulation modulates hippocampal synaptic plasticity. Proc. Natl Acad. Sci. USA 101, 14 270–14 275 (doi:10.1073/pnas.0405709101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Richter-Levin G, Maroun M. 2010. Stress and amygdala suppression of metaplasticity in the medial prefrontal cortex. Cereb. Cortex 20, 2433–2441 (doi:10.1093/cercor/bhp311) [DOI] [PubMed] [Google Scholar]
- 45.Jo YH, Schlichter R. 1999. Synaptic corelease of ATP and GABA in cultured spinal neurons. Nat. Neurosci. 2, 241–245 (doi:10.1038/6344) [DOI] [PubMed] [Google Scholar]
- 46.Fields RD, Ni Y. 2010. Nonsynaptic communication through ATP release from volume-activated anion channels in axons. Sci. Signal. 3, ra73 (doi:10.1126/scisignal.2001128) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pearson RA, Dale N, Llaudet E, Mobbs P. 2005. ATP released via gap junction hemichannels from the pigment epithelium regulates neural retinal progenitor proliferation. Neuron 46, 731–744 (doi:10.1016/j.neuron.2005.04.024) [DOI] [PubMed] [Google Scholar]
- 48.Cunha RA. 2008. Different cellular sources and different roles of adenosine: A1 receptor-mediated inhibition through astrocytic-driven volume transmission and synapse-restricted A2A receptor-mediated facilitation of plasticity. Neurochem. Int. 52, 65–72 (doi:10.1016/j.neuint.2007.06.026) [DOI] [PubMed] [Google Scholar]
- 49.Pereda AE, Curti S, Hoge G, Cachope R, Flores CE, Rash JE. 2013. Gap junction-mediated electrical transmission: regulatory mechanisms and plasticity. Biochim. Biophys. Acta 1828, 134–146 (doi:10.1016/j.bbamem.2012.05.026) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saez JC, Connor JA, Spray DC, Bennett MV. 1989. Hepatocyte gap junctions are permeable to the second messenger, inositol 1,4,5-trisphosphate, and to calcium ions. Proc. Natl Acad. Sci. USA 86, 2708–2712 (doi:10.1073/pnas.86.8.2708) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Min R, Santello M, Nevian T. 2012. The computational power of astrocyte mediated synaptic plasticity. Front. Comput. Neurosci. 6, 93 (doi:10.3389/fncom.2012.00093) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bushong EA, Martone ME, Jones YZ, Ellisman MH. 2002. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J. Neurosci. 22, 183–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Halassa MM, Fellin T, Takano H, Dong JH, Haydon PG. 2007. Synaptic islands defined by the territory of a single astrocyte. J. Neurosci. 27, 6473–6477 (doi:10.1523/JNEUROSCI.1419-07.2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ventura R, Harris KM. 1999. Three-dimensional relationships between hippocampal synapses and astrocytes. J. Neurosci. 19, 6897–6906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Navarrete M, Araque A. 2008. Endocannabinoids mediate neuron-astrocyte communication. Neuron 57, 883–893 (doi:10.1016/j.neuron.2008.01.029) [DOI] [PubMed] [Google Scholar]
- 56.Min R, Nevian T. 2012. Astrocyte signaling controls spike timing-dependent depression at neocortical synapses. Nat. Neurosci. 15, 746–753 (doi:10.1038/nn.3075) [DOI] [PubMed] [Google Scholar]
- 57.Pasti L, Volterra A, Pozzan T, Carmignoto G. 1997. Intracellular calcium oscillations in astrocytes: a highly plastic, bidirectional form of communication between neurons and astrocytes in situ. J. Neurosci. 17, 7817–7830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kang J, Jiang L, Goldman SA, Nedergaard M. 1998. Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat. Neurosci. 1, 683–692 (doi:10.1038/3684) [DOI] [PubMed] [Google Scholar]
- 59.Jourdain P, et al. 2007. Glutamate exocytosis from astrocytes controls synaptic strength. Nat. Neurosci. 10, 331–339 (doi:10.1038/nn1849) [DOI] [PubMed] [Google Scholar]
- 60.Navarrete M, Perea G, Fernandez de Sevilla D, Gomez-Gonzalo M, Nunez A, Martin ED, Araque A, Scheiffele P. 2012. Astrocytes mediate in vivo cholinergic-induced synaptic plasticity. PLoS Biol. 10, e1001259 (doi:10.1371/journal.pbio.1001259) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang F, Smith NA, Xu Q, Fujita T, Baba A, Matsuda T, Takano T, Bekar L, Nedergaard M. 2012. Astrocytes modulate neural network activity by Ca2+ dependent uptake of extracellular K+. Sci. Signal. 5, ra26 (doi:10.1126/scisignal.2002334) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pita-Almenar JD, Zou S, Colbert CM, Eskin A. 2012. Relationship between increase in astrocytic GLT-1 glutamate transport and late-LTP. Learn. Mem. 19, 615–626 (doi:10.1101/lm.023259.111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Navarrete M, Araque A. 2010. Endocannabinoids potentiate synaptic transmission through stimulation of astrocytes. Neuron 68, 113–126 (doi:10.1016/j.neuron.2010.08.043) [DOI] [PubMed] [Google Scholar]
- 64.Henneberger C, Papouin T, Oliet SH, Rusakov DA. 2010. Long-term potentiation depends on release of D-serine from astrocytes. Nature 463, 232–236 (doi:10.1038/nature08673s) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Agulhon C, Fiacco TA, McCarthy KD. 2010. Hippocampal short- and long-term plasticity are not modulated by astrocyte Ca2+ signaling. Science 327, 1250–1254 (doi:10.1126/science.1184821) [DOI] [PubMed] [Google Scholar]
- 66.Lovatt D, Xu Q, Liu W, Takano T, Smith NA, Schnermann J, Tieu K, Nedergaard M. 2012. Neuronal adenosine release, and not astrocytic ATP release, mediates feedback inhibition of excitatory activity. Proc. Natl Acad. Sci. USA 109, 6265–6270 (doi:10.1073/pnas.1120997109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Araque A, Parpura V, Sanzgiri RP, Haydon PG. 1999. Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 22, 208–215 (doi:10.1016/S0166-2236(98)01349-6) [DOI] [PubMed] [Google Scholar]
- 68.Perea G, Navarrete M, Araque A. 2009. Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci. 32, 421–431 (doi:10.1016/j.tins.2009.05.001) [DOI] [PubMed] [Google Scholar]
- 69.Santello M, Cali C, Bezzi P. 2012. Gliotransmission and the tripartite synapse. Adv. Exp. Med. Biol. 970, 307–331 (doi:10.1007/978-3-7091-0932-8_14) [DOI] [PubMed] [Google Scholar]
- 70.De Pitta M, Volman V, Berry H, Parpura V, Volterra A, Ben-Jacob E. 2012. Computational quest for understanding the role of astrocyte signaling in synaptic transmission and plasticity. Front. Comput. Neurosci. 6, 98 (doi:10.3389/fncom.2012.00098) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tewari S, Majumdar K. 2011. A mathematical model for astrocytes mediated LTP at single hippocampal synapses. J. Comput. Neurosci. 33, 341–370 (doi:10.1007/s10827-012-0389-5) [DOI] [PubMed] [Google Scholar]
- 72.Giaume C, Koulakoff A, Roux L, Holcman D, Rouach N. 2010. Astroglial networks: a step further in neuroglial and gliovascular interactions. Nat. Rev. Neurosci. 11, 87–99 (doi:10.1038/nrn2757) [DOI] [PubMed] [Google Scholar]
- 73.Zhang JM, Wang HK, Ye CQ, Ge W, Chen Y, Jiang ZL, Wu CP, Poo MM, Duan S. 2003. ATP released by astrocytes mediates glutamatergic activity-dependent heterosynaptic suppression. Neuron 40, 971–982 (doi:10.1016/S0896-6273(03)00717-7) [DOI] [PubMed] [Google Scholar]
- 74.Serrano A, Haddjeri N, Lacaille JC, Robitaille R. 2006. GABAergic network activation of glial cells underlies hippocampal heterosynaptic depression. J. Neurosci. 26, 5370–5382 (doi:10.1523/JNEUROSCI.5255-05.2006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen J, et al. 2013. Heterosynaptic long-term depression mediated by ATP released from astrocytes. Glia 61, 178–191 (doi:10.1002/glia.22425) [DOI] [PubMed] [Google Scholar]
- 76.Ben AS, Pont-Lezica L, Bechade C, Pascual O. 2010. Is astrocyte calcium signaling relevant for synaptic plasticity? Neuron Glia Biol. 6, 147–155 (doi:10.1017/S1740925X10000207) [DOI] [PubMed] [Google Scholar]
- 77.Panatier A, Theodosis DT, Mothet JP, Touquet B, Pollegioni L, Poulain DA, Oliet SH. 2006. Glia-derived D-serine controls NMDA receptor activity and synaptic memory. Cell 125, 775–784 (doi:10.1016/j.cell.2006.02.051) [DOI] [PubMed] [Google Scholar]
- 78.Kawamura M, Jr, Kawamura M. 2011. Long-term facilitation of spontaneous calcium oscillations in astrocytes with endogenous adenosine in hippocampal slice cultures. Cell Calcium 49, 249–258 (doi:10.1016/j.ceca.2011.02.009) [DOI] [PubMed] [Google Scholar]
- 79.Takata N, Mishima T, Hisatsune C, Nagai T, Ebisui E, Mikoshiba K, Hirase H. 2011. Astrocyte calcium signaling transforms cholinergic modulation to cortical plasticity in vivo. J. Neurosci. 31, 18 155–18 165 (doi:10.1523/JNEUROSCI.5289-11.2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen N, Sugihara H, Sharma J, Perea G, Petravicz J, Le C, Sur M. 2012. Nucleus basalis-enabled stimulus-specific plasticity in the visual cortex is mediated by astrocytes. Proc. Natl Acad. Sci. USA 109, E2832–E2841 (doi:10.1073/pnas.1206557109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pascual O, et al. 2005. Astrocytic purinergic signaling coordinates synaptic networks. Science 310, 113–116 (doi:10.1126/science.1116916) [DOI] [PubMed] [Google Scholar]
- 82.Bonansco C, Couve A, Perea G, Ferradas CA, Roncagliolo M, Fuenzalida M. 2011. Glutamate released spontaneously from astrocytes sets the threshold for synaptic plasticity. Eur. J. Neurosci. 33, 1483–1492 (doi:10.1111/j.1460-9568.2011.07631.x) [DOI] [PubMed] [Google Scholar]
- 83.Fujii S, Kuroda Y, Ito KL, Yoshioka M, Kaneko K, Yamazaki Y, Sasaki H, Kato H. 2000. Endogenous adenosine regulates the effects of low-frequency stimulation on the induction of long-term potentiation in CA1 neurons of guinea pig hippocampal slices. Neurosci. Lett. 279, 121–124 (doi:10.1016/S0304-3940(99)00980-5) [DOI] [PubMed] [Google Scholar]
- 84.Moidunny S, Vinet J, Wesseling E, Bijzet J, Shieh CH, van Ijzendoorn SC, Bezzi P, Boddeke HW, Biber K. 2012. Adenosine A(2B) receptor-mediated leukemia inhibitory factor release from astrocytes protects cortical neurons against excitotoxicity. J. Neuroinflammation 9, 198 (doi:10.1186/1742-2094-9-198) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vazquez JF, Clement HW, Sommer O, Schulz E, van Calker D. 2008. Local stimulation of the adenosine A2B receptors induces an increased release of IL-6 in mouse striatum: an in vivo microdialysis study. J. Neurochem. 105, 904–909 (doi:10.1111/j.1471-4159.2007.05191.x) [DOI] [PubMed] [Google Scholar]
- 86.Jankowsky JL, Derrick BE, Patterson PH. 2000. Cytokine responses to LTP induction in the rat hippocampus: a comparison of in vitro and in vivo techniques. Learn. Mem. 7, 400–412 (doi:10.1101/lm.32600) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gidday JM. 2006. Cerebral preconditioning and ischaemic tolerance. Nat. Rev. Neurosci. 7, 437–448 (doi:10.1038/nrn1927) [DOI] [PubMed] [Google Scholar]
- 88.Hedou GF, Koshibu K, Farinelli M, Kilic E, Gee CE, Kilic U, Baumgärtel K, Hermann DM, Mansuy IM. 2008. Protein phosphatase 1-dependent bidirectional synaptic plasticity controls ischemic recovery in the adult brain. J. Neurosci. 28, 154–162 (doi:10.1523/JNEUROSCI.4109-07.2008) [DOI] [PMC free article] [PubMed] [Google Scholar]