

Abstract
Almost since the discovery of long-term potentiation (LTP) in the hippocampus, its locus of expression has been debated. Throughout the years, convincing evidence has accumulated to suggest that LTP can be supported either presynaptically, by an increase in transmitter release, or postsynaptically, by an increase in α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor number. However, whereas postsynaptic enhancement appears to be consistently obtained across studies following LTP induction, presynaptic enhancement is not as reliably observed. Such discrepancies, along with the failure to convincingly identify a retrograde messenger required for presynaptic change, have led to the general view that LTP is mainly supported postsynaptically, and certainly, research within the field for the past decade has been heavily focused on the postsynaptic locus. Here, we argue that LTP can be expressed at either synaptic locus, but that pre- and postsynaptic forms of LTP are dissociable phenomena mediated by distinct mechanistic processes, which are sensitive to different patterns of neuronal activity. This view of LTP helps to reconcile discrepancies across the literature and may put to rest a decades-long debate.
Keywords: long-term potentiation, presynaptic, postsynaptic, plasticity, hippocampus, Schaffer collaterals
1. Long-term potentiation expression at the pre- and postsynaptic locus is mechanistically distinct
While the locus of long-term potentiation (LTP) expression is disputed, the locus of LTP induction is widely accepted to be postsynaptic and dependent on N-methyl-d-aspartate receptors (NMDARs). Blockade of NMDARs is often reported to inhibit LTP induction [1], and Ca2+ influx from the receptor has been causally linked to the insertion of postsynaptic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) [2]. NMDARs, however, are not always required for the induction of LTP. In 1990, Grover & Teyler [3] reported that LTP could be induced in NMDAR blockade (50 µM (2R)-amino-5-phosphonovaleric acid (APV)) with 200 Hz, but not 100 Hz, tetanic stimulation; potentiation was not simply a result of residual NMDAR activity during high-frequency stimulation as it was induced with similar magnitude under a more potent receptor blockade (100 µM APV + 20 µM MK-801) ([4] but see [5]). LTP obtained in NMDAR blockade was later shown to require the activation of L-type voltage-gated calcium channels (L-VGCCs) [6–11]. Others subsequently reported that a similar form of potentiation could be obtained (i) when presynaptic stimuli (less than or equal to 0.1 Hz) were delivered in the presence of voltage-gated potassium channel blockers [6,8,9,11–13], (ii) when tetanic stimulation (25–100 Hz) occurred in the absence of gamma aminobutyric acid A (GABAA)-mediated inhibition [14,15] and (iii) when presynaptic stimuli (1–2 Hz) were paired with strong postsynaptic depolarization [7,16]; by contrast, no potentiation was induced by presynaptic stimulation in the absence of postsynaptic depolarization or by postsynaptic depolarization in the absence of presynaptic stimulation [4,17]. These findings suggest that the induction of L-VGCC-dependent LTP requires presynaptic activity to coincide with strong postsynaptic depolarization, and that given strong postsynaptic depolarization, LTP can be induced even with very low-frequency (less than or equal to 0.1 Hz) presynaptic stimulation. Although it may be thought that the stimulation paradigms used to obtain L-VGCC-dependent LTP represent artificial experimental conditions that would be unlikely to occur in vivo, several groups have also shown that L-VGCC-dependent LTP can be induced by theta-burst stimulation [10,18–21], which is thought to emulate physiological patterns of hippocampal activity. Moreover, the finding that inhibition of L-VGCCs augments the impairment to spatial memory caused by NMDAR antagonists, suggests that L-VGCCs support some aspects of learning and memory in vivo, independent of NMDARs [22–24].
The locus of expression of L-VGCC-dependent LTP appears to be presynaptic [16,20,25] (but see [4]). The most compelling evidence comes from Bayazitov et al. [20], who used synaptopHlourins to optically monitor activity-driven changes in presynaptic function [20]. SynaptopHlourin is a pH-sensitive variant of green fluorescent protein that has been fused to the luminal domain of the vesicular protein, VAMP2. The fluorophore is quenched within the acidic lumen of the vesicle and fluoresces upon vesicular exocytosis, when it is exposed to the pH-neutral extracellular environment. Bayazitov et al. [20] demonstrated that presynaptic function was enhanced following either theta-burst or 200 Hz stimulation and that such increases could only be abolished with the L-VGCC antagonist, nitrendipine, but not with the NMDAR antagonist, APV; the resilience of presynaptic enhancement to APV is also evident in several studies using FM dyes to monitor presynaptic function [18,26,27]. Moreover, in APV, a similar fold potentiation was observed both for the presynaptic pHlourin response and the recorded field potential, suggesting that LTP was exclusively expressed presynaptically under NMDAR blockade. Conversely, tetanus in nitrendipine resulted in an enhancement of the recorded field potential but not in the presynaptic pHlourin response, suggesting that under L-VGCC blockade, LTP was exclusively expressed postsynaptically. Such findings strongly suggest that pre- and postsynaptic forms of LTP are mechanistically distinct, with the former requiring L-VGCC activation and the latter requiring NMDAR activation.
The finding that presynaptic change can occur independently of NMDAR activation appears to be at odds with findings from other laboratories, including our own, that demonstrate that NMDAR blockade abolishes, or at least reduces, presynaptic enhancement [18,20,26–28]. It is, however, important to recognize that the NMDAR, in addition to acting as a Ca2+ source for the spine, is also a potent source of depolarization for the cell and dendrite. The NMDAR is far more permeable to Na+ than it is to Ca2+, and the activation of the receptor facilitates somatic and dendritic spiking [14,29–32]. Although postsynaptic enhancement depends on NMDARs as a source of Ca2+, presynaptic enhancement, given its dependence on L-VGCC activation, may only rely on NMDARs as a source of postsynaptic depolarization. This would explain why NMDAR antagonists abolish presynaptic potentiation during standard 100 Hz, but not during 200 Hz or theta-burst stimulation protocols, which are more effective at producing postsynaptic depolarization via AMPAR activation. It is important to note that presynaptic potentiation can also be obtained when single presynaptic stimuli are paired with postsynaptic depolarization, which rules out any specific requirement of high-frequency presynaptic activity for the enhancement of presynaptic strength [16,33]. Thus, pre- and postsynaptic forms of LTP may well be mechanistically dissociable and differentially depend on L-VGCCs and NMDARs for Ca2+ influx.
2. Reconciling the literature
The inconsistency with which presynaptic changes are reported across laboratories has cast doubt as to whether the presynaptic terminal is a locus of LTP expression. However, given the differential importance of L-VGCC activation in pre- and postsynaptic forms of LTP, the failure of some laboratories to report presynaptic enhancement might depend on the nature of the experimental conditions under which LTP is induced. L-VGCCs are activated by strong depolarization and are susceptible to desensitization during periods of prolonged depolarization (more than 100 ms) [34,35]. As such, we reason that the magnitude and duration of postsynaptic depolarization during LTP induction determines the extent of L-VGCC activation, and thus the likelihood that LTP has a presynaptic component of expression. To test this idea, we examined past studies to see whether a correlation exists between the stimulation protocol used to induce LTP and the likelihood of obtaining presynaptic enhancement. To circumvent bias, our literature search was guided by past reviews on the locus of LTP expression [2,36–42], including those predominantly supporting either a pre- [39] or postsynaptic view [2,36,37]. Collectively, the studies included in our analysis employed a variety of techniques to investigate the locus of LTP expression at Schaffer-collateral synapses, including the use of: the NMDAR-component of synaptic potentials, glial transport currents, use-dependent-receptor blockers to estimate glutamate release probability, paired pulse ratios or brief high-frequency bursts to monitor changes in short-term plasticity, and finally, FM dyes, Ca2+ indicators or pHlourins to optically monitor presynaptic function. We excluded studies using coefficient of variation analysis, minimal stimulation or paired recordings, principally because the unmasking of postsynaptically silent synapses can masquerade as presynaptic enhancements using these techniques. Postsynaptic unmasking contributes significantly to LTP expression, especially during the first few weeks of postnatal development, when synaptic plasticity is most commonly studied [43]. It is therefore difficult to judge whether changes in coefficient of variation analysis or in synaptic failure rate following LTP induction in young tissue are attributable to the enhancement of pre- or postsynaptic function. Moreover, results from minimal stimulation are potentially confounded by activity-dependent changes in axonal excitability for experiments conducted at room temperature ([44] but see [45]).
We examined a total of 38 studies, which assess LTP expression across 53 experimental conditions (table 1). Presynaptic changes were reported in 23 of the 38 studies and in 23 of the 53 experimental conditions. LTP was generally induced either by brief, high-frequency tetanic stimulation (50–200 Hz) or by a pairing protocol, in which lower frequency stimulation (generally less than 2 Hz but ranging between 0.2 and 100 Hz) was delivered while voltage-clamping the postsynaptic neuron between −10 and 10 mV, often for tens of seconds. From our meta-analysis, we find that LTP is significantly more likely to have a presynaptic component of expression when induced by tetanic stimulation (20 of 35 conditions) rather than by pairing (3 of 18 conditions) (X2 = 7.92; p = 0.005). LTP induced by pairing, rather than tetanic stimulation, also appeared to be insensitive to L-VGCC blockers [7,10,18–20]. Perhaps, one reason for these findings is that prolonged periods of depolarization that are involved in pairing protocols, although effective at relieving the Mg2+ block of NMDARs, may desensitize L-VGCCs; the resulting LTP is therefore insensitive to L-VGCC antagonists and lacks a presynaptic component of expression. That said, pairing protocols can elicit L-VGCC-dependent LTP when postsynaptic depolarization consists of several brief, rather than one long, voltage step; this protocol may more effectively activate L-VGCCs without triggering channel desensitization [7].
Table 1.
Studies examining the presynaptic expression of LTP. NMDAR, NMDA-receptor-mediated component of synaptic response; SRP, synaptic refractory period; STP, short-term plasticity; GTC, glial transport current; PPR, paired pulse ratio; DNXQ, 6,7-dinitroquinoxaline-2,3-dione; CNQX, 6-cyano-7-nitroquinoxaline-2,3-dione.
| citationa | method | protocol | %LTPb | Δprec | additional notes |
|---|---|---|---|---|---|
| tetanus-induced LTP | |||||
| Muller & Lynch [46] | NMDAR | (4 at 100 Hz) × 10 at 5 Hz | 163 | yes (119%) | LTP induced in low Mg2+ |
| Muller et al. [47,48] | NMDAR | (4 at 100 Hz) × 10 at 5 Hz | 130 | no | LTP induced in low Mg2+ |
| NMDAR | (4 at 100 Hz) × 10 at 5 Hz | 120 | yes (120%) | LTP induced in low Mg2+ and DNQX | |
| Muller et al. [49] | NMDAR | (4 at 100 Hz) × 10 at 5 Hz | 148 | no | NMDAR assessed using bursts (4 at 100 Hz) |
| Bashir et al. [50] | NMDAR | 25 at 100 Hz | 143 | yes (143%) | LTP induced in CNQX |
| Asztely et al. [51] | NMDAR | 10 at 50 Hz | 114 | yes (114%) | LTP induced in low Mg2+ and CNQX |
| Clark & Collingridge [52] | NMDAR | 100 at 100 Hz | 160 | yes (160%) | |
| Kullmann et al. [53] | NMDAR | (5 at 100 Hz) × 50 at 5 Hz | 150 | yes (120%) | |
| Mainen et al. [54] | NMDAR | (50–100 at 100 Hz) × 1–3 | 265 | no | GluR2 knockout mouse |
| polyamine use-dependent block | (50–100 at 100 Hz) × 1–3 | 254 | no | GluR2 knockout mouse | |
| Muller & Lynch [55] | PPR | (4 at 100 Hz) × 10 at 5 Hz | 150 | no | |
| Zalutsky & Nicoll [56] | PPR | (100 at 100 Hz) × 4 at 0.1 Hz | 163 | no | |
| Foster & McNaughton [57] | PPR | (8 at 400 Hz) × 4 | 125 | no | |
| Schulz et al. [58] | PPR | (50 at 100 Hz) × 10 at 5 Hz × 1–6 | 179 | yes | |
| Schulz [59] | PPR + other occlusion/rescue experiments | (50 at 100 Hz) × 10 at 5 Hz × 1–6 | 250 | yes | see reference for more details on occlusion/rescue exps. |
| Kleschevnikov et al. [60] | PPR | 100 at 100 Hz (strong) | 280 | yes | |
| PPR | 100 at 100 Hz (weak) | 150 | no | ||
| Volianskis & Jensen [61] | PPR | (4 at 100 Hz) × 10 at 5 Hz | 160 | yes | |
| Pananceau et al. [62] | STP and PPR | (25 at 200 Hz) × 5 | 214 | no | STP assessed w/5 at 20 and 50 Hz |
| Yasui et al. [63] | STP | 50–100 at 100 Hz | 144 | yes | STP assessed w/10 at 10 Hz |
| Volianskis et al. [64] | STP | (4 at 100 Hz) × 10 at 5 Hz | 180 | yes | STP assessed w/7 at 12.5 Hz; decayed 2 h post-tetanus |
| Luscher et al. [65] | GTCs | (50 at 50 Hz) × 4 at 0.05 Hz OR (100 at 50 Hz) × 4 at 0.05 Hz | 159 | no | LTP induced in CNQX |
| GTC | (100 at 50 Hz) × 4 at 0.05 Hz | 179 | no | ||
| Diamond et al. [66] | GTC | (100 at 100 Hz) × 3 | 170 | no | |
| Johnstone & Raymond [67,68] | FM-dye | ((5 at 100 Hz) × 10 at 5 Hz) × 1 | 130 | no | |
| FM-dye and PPR | ((5 at 100 Hz) × 10 at 5 Hz) × 4 at 0.0033 Hz | 180 | yes | ||
| FM-dye and PPR | ((5 at 100 Hz) × 10 at 5 Hz) × 8 at 0.0033 Hz | 184 | yes | ||
| Zakharenko et al. [18,19] | FM-dye | 50 at 50 Hz | 129 | no | |
| FM-dye | (100 at 100 Hz) × 4 at 0.05 Hz | 154 | no | ||
| FM-dye | (40 at 200 Hz) × 10 at 0.2 Hz | 185 | yes | ||
| FM-dye | ((4 at 100 Hz) × 5 at 5 Hz) × 3 at 0.0033 Hz | 210 | yes | ||
| Bayazitov et al. [20] | pHlourin | (40 at 200 Hz) × 10 at 0.2 Hz | 200 | yes | pHlourin response assessed w/50 at 10 Hz |
| pHlourin | ((4 at 100 Hz) × 5 at 5 Hz) × 3 at 0.0033 Hz | 220 | yes | pHlourin response assessed w/50 at 10 Hz | |
| Emptage et al. [28], Ward et al. [69] | Ca2+ imaging and PPR | (20 at 100 Hz) × 3 at 0.75 Hz | 265 | yes | |
| Enoki et al. [33] | Ca2+ imaging | (20 at 100 Hz) × 3 at 0.75 Hz OR 100 at 0.003 Hz, each paired with three postsynaptic spikes | 199 | yes | |
| pairing-induced LTP | |||||
| Perkel & Nicoll [70] | NMDAR | (100 at 100 Hz) × 2 at 0.1 Hz | 175 | no | LTP induced in CNQX |
| NMDAR | 40 at 2 Hz; 0 mV | 190 | no | LTP induced in CNQX | |
| NMDAR | 50 at 0.5–0.7 Hz; 30 mV | 165 | no | ||
| Kauer et al. [71] | NMDAR | 19 at 2 Hz; 0 mV | 150 | no | LTP induced in CNQX |
| NMDAR | (100 at 100 Hz) × 2 at 0.05 Hz; 0 mV | 180 | no | LTP induced in CNQX | |
| NMDAR | (100 at 100 Hz) × 2 at 0.05 Hz; 0 mV | 152 | no | ||
| Plant et al. [72] | NMDAR | 50–100 at 0.5–2 Hz; −10 to 0 mV | 155 | no | |
| Kullmann et al. [53] | NMDAR | 120 at 2 Hz; 0 mV | 162 | yes (110%) | |
| MK801 | 100 at 100 Hz × 2; 0 mV | 162 | yes | use-dependent block | |
| Manabe & Nicoll [73] | MK801 | 40 at 2 Hz; 0 mV | 158 | no | use-dependent block |
| Manabe et al. [74] | PPR | 80 at 2 Hz; 0 mV | 178 | no | |
| PPR | 100 at 100 Hz; 0 mV | 178 | no | ||
| Palmer et al. [45] | PPR | 40 at 0.5 Hz; 0 mV | 234 | yes | P6 rodent slices |
| PPR | 40 at 0.5 Hz; 0 mV | 223 | no | P12 rodent slices | |
| Hjelmstad et al. [75] | SRP | 100 at 1 Hz; 0 mV | 200 | no | SRP probed w/2 at 250 Hz |
| SRP | (100 at 100 Hz) × 2 at 0.67 Hz; 0 mV | 200 | no | SRP period probed w/2 at 250 Hz | |
| 4-AP occlusion | ((100 at 100 Hz) × 2 at 0.67 Hz) × 5; 0 mV | 260 | no | ||
| Selig et al. [76] | STP | 120 at 1 Hz; −10 mV | 286 | no | STP assessed w/7 at 25 Hz |
aBlank spaces represent additional experiments conducted by same study cited in the row above. Studies with very similar experimental conditions have been combined and are represented in one row.
b% LTP is expressed as % of baseline measured 30 min post-tetanus. %LTP was estimated from graphs in studies where LTP magnitude was not mentioned in the text. In instances where a study conducted multiple experiments under similar conditions, %LTP was taken as the average across experiments.
cQuantitative changes in presynaptic efficacy were reported in some studies and are shown in brackets where appropriate.
Tetanic stimulation did not always produce presynaptic changes. However, given the high voltage-threshold property of L-VGCCs, the likelihood of generating presynaptic potentiation would depend on the ability for tetanus to produce sufficiently strong postsynaptic depolarization. Consistent with this, Zakharenko et al. [18,19] and Bayazitov et al. [20] demonstrated, using optical techniques, that theta-burst or 200 Hz stimulation generated an L-VGCC-sensitive form of LTP involving robust presynaptic enhancements, whereas no such L-VGCC-sensitive enhancements were induced by 50 or 100 Hz stimulation [18–20,25]. As stated previously, the enhanced probability of obtaining presynaptic changes under high-frequency stimulation probably reflects the requirement for strong postsynaptic depolarization rather than for high-frequency presynaptic activity per se [6–11,16,33]. Other experimental conditions may also influence the level of postsynaptic depolarization achieved during tetanus including the temperature of the preparation, the divalent cation concentration, GABAA-receptor antagonists, as well as the intensity and duration of presynaptic stimulation used during tetanus, all of which vary considerably across studies. As such, tetanic stimulation might preferentially generate presynaptic enhancement under some experimental conditions, but not others.
We further examined whether the magnitude of LTP generated by tetanic stimulation reflects the likelihood that LTP is associated with presynaptic enhancement, regardless of the actual pattern of stimulation and the experimental conditions under which it is induced. We reason that stimulation achieving sufficiently strong depolarization would recruit both pre- and postsynaptic components of LTP, and therefore generate larger enhancements in synaptic activity. Consistent with this notion, we find that the average amplitude of LTP was 194.59 ± 9.62% (n = 17) when it was associated with presynaptic enhancement, but only 153.50 ± 7.77% (n = 12) when it was not (U = 34; p = 0.003) (figure 1). Moreover, presynaptic enhancement was reported in 91.67% of experiments (n = 11/12) that produced LTP with a magnitude greater than or equal to 180% (figure 1; dashed line), but only 35.3% of experiments (n = 6/17) produced LTP with a lower magnitude (X2 = 9.21; p = 0.002). Only experiments that induced LTP using tetanic stimulation under standard experimental conditions were included in our analysis (29 of 35 conditions); as such, experiments in which LTP was induced in AMPAR blockade or in GluR2 knockout animals were excluded (6 of 35 conditions). Collectively, these findings demonstrate that LTP at the presynaptic terminal is not some enigmatic and sporadic process, but a predictable form of plasticity whose induction is likely to depend on the levels of postsynaptic depolarization achieved during tetanus.
Figure 1.
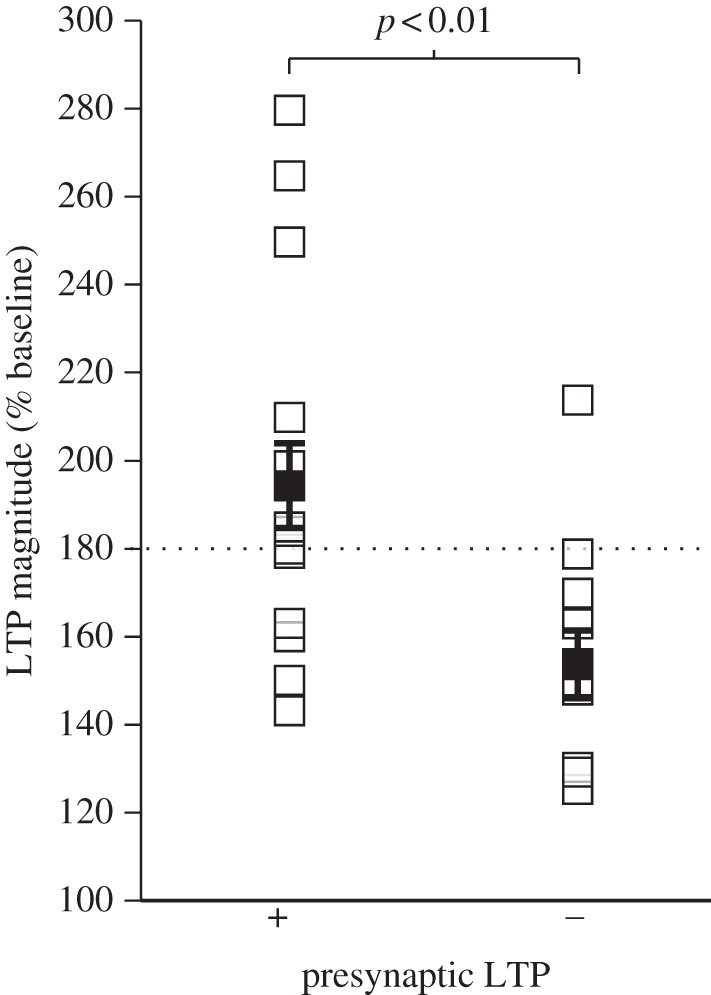
LTP magnitude predicts a presynaptic component of expression. LTP magnitude following tetanic stimulation is shown for 29 experimental conditions, 17 of which report a presynaptic component of expression (+). LTP with a magnitude greater than or equal to 180% (dashed line) had a higher probability of being associated with a presynaptic component of expression (91.67%) than LTP with a lower magnitude (35.3%).
3. Nitric oxide as a retrograde messenger
LTP at the presynaptic locus is dependent on postsynaptic depolarization. How this event is signalled is not known, but it is thought to depend on a postsynaptically generated retrograde signal. Unfortunately, the failure to identify a convincing messenger has cast doubt on a presynaptic locus of LTP. Although several putative messengers have been proposed [25,77,78], the most commonly investigated candidate has been, and continues to be, nitric oxide (NO). NO was first suggested as a retrograde signal in plasticity by Schuman & Madison [79] and O'Dell et al. [80], who demonstrated that inhibition of NO signalling impaired the induction of LTP, a finding that had been previously reported by Bohme et al. [81]. Similar impairments in LTP could be achieved by scavenging extracellular NO using haemoglobin, suggesting that NO was required to act across the synapse to potentiate synaptic responses [79]. The inherently diffuse nature of NO signalling would appear to contradict the site-specificity of LTP. Zhuo et al. [82,83], however, demonstrated that NO application had no effect on synaptic responses until paired with a weak tetanus, which alone failed to generate LTP, suggesting that NO was only effective at potentiating responses at active synapses [82,83]. Subsequent studies demonstrated that NO synthesis is activity dependent and that both neuronal and endothelial variant of nitric oxide synthase (NOS) are expressed postsynaptically in CA1 pyramidal neurons [84], and that genetic deletion of NOS [85–87], or pharmacological inhibition of NOS in vivo [88], impairs LTP at Schaffer-collateral synapses.
Perhaps, the most compelling evidence for NO as a retrograde messenger came in 1996, from Arancio et al. [89]. In their study, the authors demonstrated that LTP induction was blocked by (i) extracellular NO scavengers, (ii) intracellular NO scavengers applied to either pre- or postsynaptic neurons and (iii) injection of NOS inhibitors in the post-, but not pre-, synaptic neuron. They further showed (i) that photolytic release of NO could generate LTP when paired with presynaptic stimulation (ii) and that potentiation could be blocked by extracellular NO scavengers when NO was photoreleased in the post-, but not presynaptic compartment. Their findings strongly suggest that extracellular diffusion of postsynaptically synthesized NO into active presynaptic terminals is both necessary and sufficient for the induction of LTP.
Although the study by Arancio et al. [89] demonstrates that NO acts at the presynaptic terminal, evidence for its role in the actual enhancement of presynaptic strength has come more recently. In 2003, Nikonenko et al. [90] found that tetanic stimulation induced structural changes within the axon, including outgrowth of filopodia and the restructuring of presynaptic boutons. These changes could be abolished with NO inhibitors and could be elicited with bath application of NO donors. Stanton et al. [91] later demonstrated that activity-dependent potentiation of presynaptic function, as assessed with FM dyes, was also dependent on NO signalling [91]; these findings have since been confirmed by two additional studies using FM dyes and paired pulse ratio to monitor presynaptic enhancements [27,67].
4. Reconciling the literature
Although NO appears to be a promising candidate for a retrograde signal, its role in plasticity remains controversial, principally because some studies fail to find LTP impairments following the inhibition of NO signalling. Much like the presynaptic expression of LTP, the importance of NO looks to be dependent on the stimulus paradigm used to induce LTP. For example, Johnston & Raymond [68] demonstrated that NO inhibitors only affected LTP induced by multiple trains of theta-burst stimulation, as opposed to a single train, which in their hands failed to enhance presynaptic strength [68]. We therefore reason that NO inhibition is most likely to impair LTP when it has a presynaptic component of expression. To examine this idea, we looked at studies investigating the effects of NO inhibitors on LTP at Schaffer-collateral synapses; all relevant studies searched on PubMed (search terms: LTP and NO) were included. Although, these studies did not specifically monitor presynaptic strength, we looked to see whether, across studies, the sensitivity of LTP to NO inhibitors was correlated with the magnitude of LTP, which we have already shown reflects the likelihood that an enhancement in presynaptic function has occurred post-tetanus (figure 1).
We examined a total of 36 experiments across 21 studies (table 2); experiments were divided into NO-sensitive and NO-insensitive, depending on whether NO blockade reduced the expression of LTP. We find that the magnitude of control LTP is 162 ± 5.5% in NO-sensitive experiments (25/36), but only 136 ± 8.0% in NO-insensitive experiments (11/36) (U = 84.5; p = 0.02). We also divided experiments based on those reporting (i) strong LTP, as defined as having a magnitude greater than or equal to 180%, which has a high probability (91.67%) of being associated with presynaptic changes (figure 1) and (ii) those reporting weak LTP (less than 180%), which is less likely (35.3%) to be associated with presynaptic changes. Although the age and temperature of the preparation, as well as the type and concentration of NO inhibitors varied greatly across experiments (table 2), we find that NO inhibition reduced LTP in 10 of 10 experiments that yielded strong LTP but in only 16 of 26 experiments that yielded weak LTP (X2 = 11.08; p = 0.0009). Such findings suggest that the degree to which plasticity is dependent on NO signalling depends on the magnitude, and potentially the locus, of LTP. It should be mentioned, however, that independent of its role as a retrograde signal, NO has effects on postsynaptic signalling; as a result, inhibition of NO synthesis may have additionally affected postsynaptic plasticity under certain experimental conditions [99,103,106,108,109].
Table 2.
Studies examining the involvement of nitric oxide in LTP. SD, Sprague-Dawley.
| citationa | protocol | %LTPb | ΔLTP in NO blockadec | age/animal | temp. (°C) | NO inhibitorsd |
|---|---|---|---|---|---|---|
| Bohme et al. [81] | (100 at 100 Hz × 2) at 0.02 Hz | 146 | decreased (9%) | 5–6 weeks SD | 32°C | l-NoArg (0.1 μM) |
| Schuman & Madison [79] | (100 at 100 Hz) × 4–5 at 0.033–0.066 Hz | 143 | decreased (0–10%) | 2–3 weeks SD | 22°C | l-NoArg (100 μM), l-MeArg (100 um), Hg (100 μM) |
| O'Dell et al. [80] | (100 at 100 Hz) × 2 at 0.05 Hz | 205 | decreased (20%) | Age? Guinea pig | 24°C | l-NoArg (50 μM), l-MeArg (1000 um intracellular), Hg (20 μM) |
| Bon et al. [92] | (100 at 100 Hz × 2) at 0.02 Hz | 200 | decreased (43%) | 5–6 weeks SD | 32°C | l-NoArg (0.1–100 nM), Hg (10–100 nM) |
| Gribkoff & Lum-Ragan [93] | (100 at 100 Hz); 50% max intensity | 135 | no change (40%) | 4–12 weeks F-344 male rat | 32°C | l-NoArg (50–200 μM), NMMA (100 μM) |
| 100 at 100 Hz × 2) at 0.017 Hz; max intensity | 190 | decreased (25%) | 4–12 weeks F-344 male rat | 32°C | l-NoArg (100 μM), NMMA (100 μM) | |
| Haley et al. [94] | (4 at 100 Hz) × 10 at 5 Hz | 137 | decreased (2%) | 4–6 weeks SD | 31°C | l-NoArg (10 nM–10 μM), Hg (100 μM) |
| Haley et al. [95] | (25 at 100 Hz) × 2 at 0.2 Hz | 119 | decreased (6%) | 4–6 weeks SD | 31°C | l-NoArg (10–1000 μM) |
| (50 at 100 Hz) × 2 at 0.1 Hz | 115 | no change (17%) | 4–6 weeks SD | 31°C | l-NoArg (10–1000 μM) | |
| (25 at 100 Hz) × 2 at 0.2 Hz; 2× intensity | 118 | no change (15%) | 4–6 weeks SD | 31°C | l-NoArg (10–100 μM) | |
| Kato & Zorumski [96] | 30 at 100 Hz | 109 | increased (33%) | 3–4 weeks male albino rat | 30°C | l-NoArg (5–100 μM); Hg (10 μM) |
| Chetkovich et al. [97] | 100 at 100 Hz × 3 at l0.02 Hz; 50% max intensity | 150 | decreased (13%) | approximately 4 weeks SD | 32°C | l-NoArg (100 μM) |
| 100 at 100 Hz × 3 at l0.02 Hz; max intensity | 175 | no change (75%) | approximately 4 weeks SD | 32°C | l-NoArg (100 μM) | |
| Musleh et al. [98] | (4 at 100 Hz) × 10 at 5 Hz | 142 | decreased (−8%) | 4–6 weeks SD | 35°C | l-NoArg (20 μM), l-MeArg (100 μM), Hg (50 μM) |
| Williams et al. [99] | (20 at 100 Hz) × 6 at 0.33 Hz | 130 | decreased (0%) | 5–7 weeks SD | 24°C | l-NoArg (100 μM) l-NAME (0.1 mM), Hg (20 μM) |
| (20 at 100 Hz) × 6 at 0.33 Hz | 180 | decreased (22%) | 5–7 weeks SD | 24°C | l-NoArg (100 μM) | |
| (20 at 100 Hz) × 6 at 0.33 Hz+Bicuculline | 156 | no change (60%) | 5–7 weeks SD | 29°C | l-NoArg (100 μM) | |
| (20 at 100 Hz) × 6 at 0.33 Hz | 159 | no change (56%) | 16–24 weeks SD | 29°C | l-NoArg (0.1–1 mM), l-NAME (0.1 mM), Hg (20 μM) | |
| Nicolarakis & Bennett [100] | (50 at 100 Hz) × 2 at 0.1 Hz | 192 | decreased (32%) | 3–5 weeks Wistar | 21–23°C | l-NAME (100–300 μM) |
| Cummings et al. [101] | (100 at 100 Hz) × 4 at 0.033 Hz | 150 | decreased (56%) | 2–3 weeks SD | 25–29°C | l-NoArg (100 μM) |
| O'Dell et al. [86] | (4 at 100 Hz) × 25 at 5 Hz; baseline intensity | 125 | decreased (5%) | Age? mouse | 30°C | l-NoArg (50 μM) |
| (4 at 100 Hz) × 25 at 5 Hz; 50% max intensity | 190 | decreased (60%) | 30°C | l-NoArg (50 μM), | ||
| Boulton et al. [102] | 100 at 100 Hz | 168 | decreased (32%) | 4–6 weeks? Wistar | 24°C | l-NoArg (100 μM) |
| 100 at 100 Hz | 180 | decreased (34%) | 4–6 weeks? Wistar | 30°C | l-NoArg (100 μM) | |
| Malen & Chapman [103] | 900 at 30 Hz | 123 | decreased (3%) | 2–20 weeks SD | 32°C | l-NAME (100 μM) |
| 50 at 100 Hz | 115 | no change (17%) | 2–20 weeks SD | 32°C | l-NAME (100 μM) | |
| Zhou et al. [104,105] | 100 at 100 Hz | 163 | decreased (−6%) | 4–6 weeks SD | 28–30°C | l-NoArg (100 μM) |
| 100 at 100 Hz ×2; 2 × baseline duration | 210 | decreased (81%) | 4–6 weeks SD | 28–30°C | l-NoArg (100 μM) | |
| Wilson et al. [87] | 10 at 100 Hz × 3 | 138 | decreased (8%) | 8–12 weeks mouse | 29–31°C | l-NoArg (200 μM) |
| 10 at 100 Hz × 3; 2 × baseline duration | 150 | decreased (40%) | 8–12 weeks mouse | 29–31°C | l-NoArg (200 μM) | |
| Ko & Kelly [106] | (25 at 100 Hz) × 5 at 0.2 Hz | 180 | decreased (25%) | 5–8 weeks SD | 32°C | l-NAME (100 μM), C-PTIO (30 μM), MGD-Fe (75/150 μM) |
| Bon & Garthwaite [107] | 100 at 100 Hz | 150 | decreased (25%) | 6–8 weeks SD | 30°C | l-NoArg (100 μM), l-NIO (100 μM) |
| Johnstone & Raymond [67] | ((4 at 100 Hz) × 10 at 5 Hz) × 1 | 130 | no change (35%) | 6–8 weeks Wistar | approximately 22°C | l-NAME (100 μM), cPTIO (40 μM) |
| ((4 at 100 Hz) × 10 at 5 Hz) × 4 at 0.003 Hz | 150 | no change (60%) | 6–8 weeks Wistar | approximately 22°C | ||
| ((4 at 100 Hz) × 10 at 5 Hz) × 8 at 0.0033 Hz | 180 | decreased (40%) | 6–8 weeks Wistar | approximately 22°C |
aBlank spaces represent additional experiments conducted by same study cited in the row above. Studies with very similar experimental conditions have been combined and are represented in one row.
b%LTP is expressed as % of baseline measured 30 min post-tetanus. %LTP was estimated from graphs in studies where LTP magnitude was not mentioned in the text. In instances where a study conducted multiple experiments under similar conditions, %LTP was taken as the average across experiments.
c%LTP obtained with NO inhibition is included in brackets.
dl-NoArg, N omega-nitro-l-arginine; l-MeArg, NG-methyl-l-arginine; Hg, haemoglobin; NMMA, l-NG-monomethylarginine; l-NAME, l-NG-nitroarginine methyl ester; C-PTIO, 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide; MGD-Fe, iron-N-methyl-d-glucamine dithiocarbamate complex; l-NIO, N5-(1-iminoethyl)-l-ornithine.
There have also been disagreements regarding the effect of exogenous NO on synaptic function. Bohme et al. [81] first demonstrated that NO donors persistently potentiated synaptic responses; similar effects were later confirmed using NO donors, free NO, and photoactivated NO [80–83,89,90,92,103,104]. By contrast, two groups have failed to elicit LTP with NO application [110–112]. Exogenous NO, therefore, appears to have varied effects on synaptic responses across studies. However, it is important to recognize that, like any transmitter in the nervous system, NO has a diverse repertoire of effects on neuronal function [113]. As with glutamate, the specific effect of NO at a synapse will very likely depend on (i) the spatio-temporal dynamics and concentration of signalling, (ii) the current pattern of neuronal activity and (iii) the state of the synapse. For NO, the parameters required for the induction of LTP remain largely unknown and may not always be emulated by the application of exogenous NO, in whatever form [113]. The fact that the vast majority of studies manage to potentiate synaptic responses using exogenous NO, while having little knowledge of the dynamics of endogenous NO signalling, is remarkable in and of itself, and certainly a compelling demonstration that NO signalling has the potential to induce LTP; though, as with glutamate, this potential is likely to be realized only under certain conditions.
5. Concluding remarks
Discrepancies in the literature have raised doubts over a presynaptic locus of LTP. We have argued that these discrepancies actually reflect the presence of two mechanistically distinct forms of LTP: one, which is expressed postsynaptically and dependent on Ca2+ influx from NMDARs and the other, which is expressed presynaptically and dependent on Ca2+ influx from L-VGCCs. Experimental protocols that successfully activate L-VGCCs are most likely to recruit a presynaptic component of LTP expression and are also most likely to involve a retrograde signal, such as NO. As research continues to elucidate the mechanistic basis of presynaptic plasticity, one thing is becoming clear: the current, postsynaptic-centric dogma of LTP needs to change in order to reflect the more comprehensive understanding of synaptic plasticity that is supported by a growing body of literature. There are two sides to the synapse, and both can change.
References
- 1.Collingridge GL, Kehl SJ, McLennan H. 1983. Excitatory amino acids in synaptic transmission in the Schaffer collateral-commissural pathway of the rat hippocampus. J. Physiol. 334, 33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Luscher C, Malenka RC. 2012. NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harb. Perspect Biol. 4, a005710 ( 10.1101/cshperspect.a005710) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grover LM, Teyler TJ. 1990. Two components of long-term potentiation induced by different patterns of afferent activation. Nature 347, 477–479. ( 10.1038/347477a0) [DOI] [PubMed] [Google Scholar]
- 4.Grover LM. 1998. Evidence for postsynaptic induction and expression of NMDA receptor independent LTP. J. Neurophysiol. 79, 1167–1182. [DOI] [PubMed] [Google Scholar]
- 5.Pananceau M, Gustafsson B. 1997. NMDA receptor dependence of the input specific NMDA receptor-independent LTP in the hippocampal CA1 region. Brain Res. 752, 255–260. ( 10.1016/S0006-8993(96)01471-0) [DOI] [PubMed] [Google Scholar]
- 6.Aniksztejn L, Ben-Ari Y. 1991. Novel form of long-term potentiation produced by a K+ channel blocker in the hippocampus. Nature 349, 67–69. ( 10.1038/349067a0) [DOI] [PubMed] [Google Scholar]
- 7.Kullmann DM, Perkel DJ, Manabe T, Nicoll RA. 1992. Ca2+ entry via postsynaptic voltage-sensitive Ca2+ channels can transiently potentiate excitatory synaptic transmission in the hippocampus. Neuron 9, 1175–1183. ( 10.1016/0896-6273(92)90075-O) [DOI] [PubMed] [Google Scholar]
- 8.Huang YY, Malenka RC. 1993. Examination of TEA-induced synaptic enhancement in area CA1 of the hippocampus: the role of voltage-dependent Ca2+ channels in the induction of LTP. J. Neurosci. 13, 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanse E, Gustafsson B. 1994. TEA elicits two distinct potentiations of synaptic transmission in the CA1 region of the hippocampal slice. J. Neurosci. 14, 5028–5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morgan SL, Teyler TJ. 2001. Electrical stimuli patterned after the theta-rhythm induce multiple forms of LTP. J. Neurophysiol. 86, 1289–1296. [DOI] [PubMed] [Google Scholar]
- 11.Huber KM, Mauk MD, Kelly PT. 1995. Distinct LTP induction mechanisms: contribution of NMDA receptors and voltage-dependent calcium channels. J. Neurophysiol. 73, 270–279. [DOI] [PubMed] [Google Scholar]
- 12.Petrozzino JJ, Connor JA. 1994. Dendritic Ca2+ accumulations and metabotropic glutamate receptor activation associated with an N-methyl-D-aspartate receptor-independent long-term potentiation in hippocampal CA1 neurons. Hippocampus 4, 546–558. ( 10.1002/hipo.450040504) [DOI] [PubMed] [Google Scholar]
- 13.Platt B, Behnisch T, Reymann KG. 1995. Metabotropic glutamate receptors are involved in TEA-induced long-term potentiation in area CA1 of the hippocampus. Neuropharmacology 34, 1339–1341. ( 10.1016/0028-3908(95)00123-N) [DOI] [PubMed] [Google Scholar]
- 14.Grover LM, Yan C. 1999. Blockade of GABAA receptors facilitates induction of NMDA receptor-independent long-term potentiation. J. Neurophysiol. 81, 2814–2822. [DOI] [PubMed] [Google Scholar]
- 15.Hsu KS, Ho WC, Huang CC, Tsai JJ. 1999. Prior short-term synaptic disinhibition facilitates long-term potentiation and suppresses long-term depression at CA1 hippocampal synapses. Eur. J. Neurosci. 11, 4059–4069. ( 10.1046/j.1460-9568.1999.00819.x) [DOI] [PubMed] [Google Scholar]
- 16.Stricker C, Cowan AI, Field AC, Redman SJ. 1999. Analysis of NMDA-independent long-term potentiation induced at CA3-CA1 synapses in rat hippocampus in vitro. J. Physiol. 520, 513–525. ( 10.1111/j.1469-7793.1999.00513.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grover LM, Yan C. 1999. Evidence for involvement of group II/III metabotropic glutamate receptors in NMDA receptor-independent long-term potentiation in area CA1 of rat hippocampus. J. Neurophysiol. 82, 2956–2969. [DOI] [PubMed] [Google Scholar]
- 18.Zakharenko SS, Zablow L, Siegelbaum SA. 2001. Visualization of changes in presynaptic function during long-term synaptic plasticity. Nat. Neurosci. 4, 711–717. ( 10.1038/89498) [DOI] [PubMed] [Google Scholar]
- 19.Zakharenko SS, Patterson SL, Dragatsis I, Zeitlin SO, Siegelbaum SA, Kandel ER, Morozov A. 2003. Presynaptic BDNF required for a presynaptic but not postsynaptic component of LTP at hippocampal CA1-CA3 synapses. Neuron 39, 975–990. ( 10.1016/S0896-6273(03)00543-9) [DOI] [PubMed] [Google Scholar]
- 20.Bayazitov IT, Richardson RJ, Fricke RG, Zakharenko SS. 2007. Slow presynaptic and fast postsynaptic components of compound long-term potentiation. J. Neurosci. 27, 11 510–11 521. ( 10.1523/JNEUROSCI.3077-07.2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grover LM, Kim E, Cooke JD, Holmes WR. 2009. LTP in hippocampal area CA1 is induced by burst stimulation over a broad frequency range centered around delta. Learn. Mem. 16, 69–81. ( 10.1101/lm.1179109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moosmang S, et al. 2005. Role of hippocampal Cav1.2 Ca2+ channels in NMDA receptor-independent synaptic plasticity and spatial memory. J. Neurosci. 25, 9883–9892. ( 10.1523/JNEUROSCI.1531-05.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woodside BL, Borroni AM, Hammonds MD, Teyler TJ. 2004. NMDA receptors and voltage-dependent calcium channels mediate different aspects of acquisition and retention of a spatial memory task. Neurobiol. Learn. Mem. 81, 105–114. ( 10.1016/j.nlm.2003.10.003) [DOI] [PubMed] [Google Scholar]
- 24.Borroni AM, Fichtenholtz H, Woodside BL, Teyler TJ. 2000. Role of voltage-dependent calcium channel long-term potentiation (LTP) and NMDA LTP in spatial memory. J. Neurosci. 20, 9272–9276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blundon JA, Zakharenko SS. 2008. Dissecting the components of long-term potentiation. Neuroscientist 14, 598–608. ( 10.1177/1073858408320643) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ryan TA, Ziv NE, Smith SJ. 1996. Potentiation of evoked vesicle turnover at individually resolved synaptic boutons. Neuron 17, 125–134. ( 10.1016/S0896-6273(00)80286-X) [DOI] [PubMed] [Google Scholar]
- 27.Ratnayaka A, Marra V, Bush D, Burden JJ, Branco T, Staras K. 2012. Recruitment of resting vesicles into recycling pools supports NMDA receptor-dependent synaptic potentiation in cultured hippocampal neurons. J. Physiol. 590, 1585–1597. ( 10.1113/jphysiol.2011.226688) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Emptage NJ, Reid CA, Fine A, Bliss TV. 2003. Optical quantal analysis reveals a presynaptic component of LTP at hippocampal Schaffer-associational synapses. Neuron 38, 797–804. ( 10.1016/S0896-6273(03)00325-8) [DOI] [PubMed] [Google Scholar]
- 29.Schiller J, Schiller Y. 2001. NMDA receptor-mediated dendritic spikes and coincident signal amplification. Curr. Opin. Neurobiol. 11, 343–348. ( 10.1016/S0959-4388(00)00217-8) [DOI] [PubMed] [Google Scholar]
- 30.Schiller J, Major G, Koester HJ, Schiller Y. 2000. NMDA spikes in basal dendrites of cortical pyramidal neurons. Nature 404, 285–289. ( 10.1038/35005094) [DOI] [PubMed] [Google Scholar]
- 31.Mayer ML, Westbrook GL. 1987. Permeation and block of N-methyl-D-aspartic acid receptor channels by divalent cations in mouse cultured central neurones. J. Physiol. 394, 501–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Herron CE, Lester RA, Coan EJ, Collingridge GL. 1986. Frequency-dependent involvement of NMDA receptors in the hippocampus: a novel synaptic mechanism. Nature 322, 265–268. ( 10.1038/322265a0) [DOI] [PubMed] [Google Scholar]
- 33.Enoki R, Hu YL, Hamilton D, Fine A. 2009. Expression of long-term plasticity at individual synapses in hippocampus is graded, bidirectional, and mainly presynaptic: optical quantal analysis. Neuron 62, 242–253. ( 10.1016/j.neuron.2009.02.026) [DOI] [PubMed] [Google Scholar]
- 34.Hofmann F, Lacinova L, Klugbauer N. 1999. Voltage-dependent calcium channels: from structure to function. Rev. Physiol. Biochem. Pharmacol. 139, 33–87. ( 10.1007/BFb0033648) [DOI] [PubMed] [Google Scholar]
- 35.Lacinova L, Hofmann F. 2005. Ca2+- and voltage-dependent inactivation of the expressed L-type Ca(v)1.2 calcium channel. Arch. Biochem. Biophys. 437, 42–50. ( 10.1016/j.abb.2005.02.025) [DOI] [PubMed] [Google Scholar]
- 36.Nicoll RA. 2003. Expression mechanisms underlying long-term potentiation: a postsynaptic view. Phil. Trans. R. Soc. Lond. B 358, 721–726. ( 10.1098/rstb.2002.1228) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nicoll RA, Malenka RC. 1999. Expression mechanisms underlying NMDA receptor-dependent long-term potentiation. Ann. NY Acad. Sci. 868, 515–525. ( 10.1111/j.1749-6632.1999.tb11320.x) [DOI] [PubMed] [Google Scholar]
- 38.Bliss TV, Collingridge GL. 2013. Expression of NMDA receptor-dependent LTP in the hippocampus: bridging the divide. Mol. Brain 6, 5 ( 10.1186/1756-6606-6-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kullmann DM, Siegelbaum SA. 1995. The site of expression of NMDA receptor-dependent LTP: new fuel for an old fire. Neuron 15, 997–1002. ( 10.1016/0896-6273(95)90089-6) [DOI] [PubMed] [Google Scholar]
- 40.Lisman J. 2003. Long-term potentiation: outstanding questions and attempted synthesis. Phil. Trans. R. Soc. Lond. B 358, 829–842. ( 10.1098/rstb.2002.1242) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lisman J, Raghavachari S. 2006. A unified model of the presynaptic and postsynaptic changes during LTP at CA1 synapses. Sci. STKE 2006, re11 ( 10.1126/stke.3562006re11) [DOI] [PubMed] [Google Scholar]
- 42.Larkman AU, Jack JJ. 1995. Synaptic plasticity: hippocampal LTP. Curr. Opin. Neurobiol. 5, 324–334. ( 10.1016/0959-4388(95)80045-X) [DOI] [PubMed] [Google Scholar]
- 43.Abrahamsson T, Gustafsson B, Hanse E. 2008. AMPA silencing is a prerequisite for developmental long-term potentiation in the hippocampal CA1 region. J. Neurophysiol. 100, 2605–2614. ( 10.1152/jn.90476.2008) [DOI] [PubMed] [Google Scholar]
- 44.McNaughton BL, Shen J, Rao G, Foster TC, Barnes CA. 1994. Persistent increase of hippocampal presynaptic axon excitability after repetitive electrical stimulation: dependence on N-methyl-D-aspartate receptor activity, nitric-oxide synthase, and temperature. Proc. Natl Acad. Sci. USA 91, 4830–4834. ( 10.1073/pnas.91.11.4830) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Palmer MJ, Isaac JT, Collingridge GL. 2004. Multiple, developmentally regulated expression mechanisms of long-term potentiation at CA1 synapses. J. Neurosci. 24, 4903–4911. ( 10.1523/JNEUROSCI.0170-04.2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Muller D, Lynch G. 1988. Long-term potentiation differentially affects two components of synaptic responses in hippocampus. Proc. Natl Acad. Sci. USA 85, 9346–9350. ( 10.1073/pnas.85.23.9346) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Muller D, Joly M, Lynch G. 1988. Contributions of quisqualate and NMDA receptors to the induction and expression of LTP. Science 242, 1694–1697. ( 10.1126/science.2904701) [DOI] [PubMed] [Google Scholar]
- 48.Muller D, Arai A, Lynch G. 1992. Factors governing the potentiation of NMDA receptor-mediated responses in hippocampus. Hippocampus 2, 29–38. ( 10.1002/hipo.450020105) [DOI] [PubMed] [Google Scholar]
- 49.Muller D, Larson J, Lynch G. 1989. The NMDA receptor-mediated components of responses evoked by patterned stimulation are not increased by long-term potentiation. Brain Res. 477, 396–399. ( 10.1016/0006-8993(89)91435-2) [DOI] [PubMed] [Google Scholar]
- 50.Bashir ZI, Alford S, Davies SN, Randall AD, Collingridge GL. 1991. Long-term potentiation of NMDA receptor-mediated synaptic transmission in the hippocampus. Nature 349, 156–158. ( 10.1038/349156a0) [DOI] [PubMed] [Google Scholar]
- 51.Asztely F, Wigstrom H, Gustafsson B. 1992. The relative contribution of NMDA receptor channels in the expression of long-term potentiation in the hippocampal CA1 region. Eur. J. Neurosci. 4, 681–690. ( 10.1111/j.1460-9568.1992.tb00177.x) [DOI] [PubMed] [Google Scholar]
- 52.Clark KA, Collingridge GL. 1995. Synaptic potentiation of dual-component excitatory postsynaptic currents in the rat hippocampus. J. Physiol. 482, 39–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kullmann DM, Erdemli G, Asztely F. 1996. LTP of AMPA and NMDA receptor-mediated signals: evidence for presynaptic expression and extrasynaptic glutamate spill-over. Neuron 17, 461–474. ( 10.1016/S0896-6273(00)80178-6) [DOI] [PubMed] [Google Scholar]
- 54.Mainen ZF, Jia Z, Roder J, Malinow R. 1998. Use-dependent AMPA receptor block in mice lacking GluR2 suggests postsynaptic site for LTP expression. Nat. Neurosci. 1, 579–586. ( 10.1038/2812) [DOI] [PubMed] [Google Scholar]
- 55.Muller D, Lynch G. 1989. Evidence that changes in presynaptic calcium currents are not responsible for long-term potentiation in hippocampus. Brain Res. 479, 290–299. ( 10.1016/0006-8993(89)91631-4) [DOI] [PubMed] [Google Scholar]
- 56.Zalutsky RA, Nicoll RA. 1990. Comparison of two forms of long-term potentiation in single hippocampal neurons. Science 248, 1619–1624. ( 10.1126/science.2114039) [DOI] [PubMed] [Google Scholar]
- 57.Foster TC, McNaughton BL. 1991. Long-term enhancement of CA1 synaptic transmission is due to increased quantal size, not quantal content. Hippocampus 1, 79–91. ( 10.1002/hipo.450010108) [DOI] [PubMed] [Google Scholar]
- 58.Schulz PE, Cook EP, Johnston D. 1994. Changes in paired-pulse facilitation suggest presynaptic involvement in long-term potentiation. J. Neurosci. 14, 5325–5337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schulz PE. 1997. Long-term potentiation involves increases in the probability of neurotransmitter release. Proc. Natl Acad. Sci. USA 94, 5888–5893. ( 10.1073/pnas.94.11.5888) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kleschevnikov AM, Sokolov MV, Kuhnt U, Dawe GS, Stephenson JD, Voronin LL. 1997. Changes in paired-pulse facilitation correlate with induction of long-term potentiation in area CA1 of rat hippocampal slices. Neuroscience 76, 829–843. ( 10.1016/S0306-4522(96)00342-9) [DOI] [PubMed] [Google Scholar]
- 61.Volianskis A, Jensen MS. 2003. Transient and sustained types of long-term potentiation in the CA1 area of the rat hippocampus. J. Physiol. 550, 459–492. ( 10.1113/jphysiol.2003.044214) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pananceau M, Chen H, Gustafsson B. 1998. Short-term facilitation evoked during brief afferent tetani is not altered by long-term potentiation in the guinea-pig hippocampal CA1 region. J. Physiol. 508, 503–514. ( 10.1111/j.1469-7793.1998.503bq.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yasui T, Fujisawa S, Tsukamoto M, Matsuki N, Ikegaya Y. 2005. Dynamic synapses as archives of synaptic history: state-dependent redistribution of synaptic efficacy in the rat hippocampal CA1. J. Physiol. 566, 143–160. ( 10.1113/jphysiol.2005.086595) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Volianskis A, Collingridge GL, Jensen MS. 2013. The roles of STP and LTP in synaptic encoding. PeerJ 1, e3 ( 10.7717/peerj.3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Luscher C, Malenka RC, Nicoll RA. 1998. Monitoring glutamate release during LTP with glial transporter currents. Neuron 21, 435–441. ( 10.1016/S0896-6273(00)80552-8) [DOI] [PubMed] [Google Scholar]
- 66.Diamond JS, Bergles DE, Jahr CE. 1998. Glutamate release monitored with astrocyte transporter currents during LTP. Neuron 21, 425–433. ( 10.1016/S0896-6273(00)80551-6) [DOI] [PubMed] [Google Scholar]
- 67.Johnstone VP, Raymond CR. 2011. A protein synthesis and nitric oxide-dependent presynaptic enhancement in persistent forms of long-term potentiation. Learn. Mem. 18, 625–633. ( 10.1101/lm.2245911) [DOI] [PubMed] [Google Scholar]
- 68.Johnstone VP, Raymond CR. 2013. Postsynaptic protein synthesis is required for presynaptic enhancement in persistent forms of long-term potentiation. Front. Synaptic Neurosci. 5, 1 ( 10.3389/fnsyn.2013.00001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ward B, McGuinness L, Akerman CJ, Fine A, Bliss TV, Emptage NJ. 2006. State-dependent mechanisms of LTP expression revealed by optical quantal analysis. Neuron 52, 649–661. ( 10.1016/j.neuron.2006.10.007) [DOI] [PubMed] [Google Scholar]
- 70.Perkel DJ, Nicoll RA. 1993. Evidence for all-or-none regulation of neurotransmitter release: implications for long-term potentiation. J. Physiol. 471, 481–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kauer JA, Malenka RC, Nicoll RA. 1998. A persistent postsynaptic modification mediates long-term potentiation in the hippocampus. Neuron 1, 911–917. ( 10.1016/0896-6273(88)90148-1) [DOI] [PubMed] [Google Scholar]
- 72.Plant K, Pelkey KA, Bortolotto ZA, Morita D, Terashima A, McBain CJ, Collingridge GL, Isaac JT. 2006. Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nat. Neurosci. 9, 602–604. ( 10.1038/nn1678) [DOI] [PubMed] [Google Scholar]
- 73.Manabe T, Nicoll RA. 1994. Long-term potentiation: evidence against an increase in transmitter release probability in the CA1 region of the hippocampus. Science 265, 1888–1892. ( 10.1126/science.7916483) [DOI] [PubMed] [Google Scholar]
- 74.Manabe T, Wyllie DJ, Perkel DJ, Nicoll RA. 1993. Modulation of synaptic transmission and long-term potentiation: effects on paired pulse facilitation and EPSC variance in the CA1 region of the hippocampus. J. Neurophysiol. 70, 1451–1459. [DOI] [PubMed] [Google Scholar]
- 75.Hjelmstad GO, Nicoll RA, Malenka RC. 1997. Synaptic refractory period provides a measure of probability of release in the hippocampus. Neuron 19, 1309–1318. ( 10.1016/S0896-6273(00)80421-3) [DOI] [PubMed] [Google Scholar]
- 76.Selig DK, Nicoll RA, Malenka RC. 1999. Hippocampal long-term potentiation preserves the fidelity of postsynaptic responses to presynaptic bursts. J. Neurosci. 19, 1236–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Williams JH, Errington ML, Lynch MA, Bliss TV. 1989. Arachidonic acid induces a long-term activity-dependent enhancement of synaptic transmission in the hippocampus. Nature 341, 739–742. ( 10.1038/341739a0) [DOI] [PubMed] [Google Scholar]
- 78.Fitzsimonds RM, Poo MM. 1998. Retrograde signaling in the development and modification of synapses. Physiol. Rev. 78, 143–170. [DOI] [PubMed] [Google Scholar]
- 79.Schuman EM, Madison DV. 1991. A requirement for the intercellular messenger nitric oxide in long-term potentiation. Science 254, 1503–1506. ( 10.1126/science.1720572) [DOI] [PubMed] [Google Scholar]
- 80.O'Dell TJ, Hawkins RD, Kandel ER, Arancio O. 1991. Tests of the roles of two diffusible substances in long-term potentiation: evidence for nitric oxide as a possible early retrograde messenger. Proc. Natl Acad. Sci. USA 88, 11 285–11 289. ( 10.1073/pnas.88.24.11285) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bohme GA, Bon C, Stutzmann JM, Doble A, Blanchard JC. 1991. Possible involvement of nitric oxide in long-term potentiation. Eur. J. Pharmacol. 199, 379–381. ( 10.1016/0014-2999(91)90505-K) [DOI] [PubMed] [Google Scholar]
- 82.Zhuo M, Kandel ER, Hawkins RD. 1994. Nitric oxide and cGMP can produce either synaptic depression or potentiation depending on the frequency of presynaptic stimulation in the hippocampus. Neuroreport 5, 1033–1036. ( 10.1097/00001756-199405000-00004) [DOI] [PubMed] [Google Scholar]
- 83.Zhuo M, Small SA, Kandel ER, Hawkins RD. 1993. Nitric oxide and carbon monoxide produce activity-dependent long-term synaptic enhancement in hippocampus. Science 260, 1946–1950. ( 10.1126/science.8100368) [DOI] [PubMed] [Google Scholar]
- 84.Dinerman JL, Dawson TM, Schell MJ, Snowman A, Snyder SH. 1994. Endothelial nitric oxide synthase localized to hippocampal pyramidal cells: implications for synaptic plasticity. Proc. Natl Acad. Sci. USA 91, 4214–4218. ( 10.1073/pnas.91.10.4214) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Son H, Hawkins RD, Martin K, Kiebler M, Huang PL, Fishman MC, Kandel ER. 1996. Long-term potentiation is reduced in mice that are doubly mutant in endothelial and neuronal nitric oxide synthase. Cell 87, 1015–1023. ( 10.1016/S0092-8674(00)81796-1) [DOI] [PubMed] [Google Scholar]
- 86.O'Dell TJ, Huang PL, Dawson TM, Dinerman JL, Snyder SH, Kandel ER, Fishman MC. 1994. Endothelial NOS and the blockade of LTP by NOS inhibitors in mice lacking neuronal NOS. Science 265, 542–546. ( 10.1126/science.7518615) [DOI] [PubMed] [Google Scholar]
- 87.Wilson RI, Godecke A, Brown RE, Schrader J, Haas HL. 1999. Mice deficient in endothelial nitric oxide synthase exhibit a selective deficit in hippocampal long-term potentiation. Neuroscience 90, 1157–1165. ( 10.1016/S0306-4522(98)00479-5) [DOI] [PubMed] [Google Scholar]
- 88.Doyle C, Holscher C, Rowan MJ, Anwyl R. 1996. The selective neuronal NO synthase inhibitor 7-nitro-indazole blocks both long-term potentiation and depotentiation of field EPSPs in rat hippocampal CA1 in vivo. J. Neurosci. 16, 418–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Arancio O, Kiebler M, Lee CJ, Lev-Ram V, Tsien RY, Kandel ER, Hawkins RD. 1996. Nitric oxide acts directly in the presynaptic neuron to produce long-term potentiation in cultured hippocampal neurons. Cell 87, 1025–1035. ( 10.1016/S0092-8674(00)81797-3) [DOI] [PubMed] [Google Scholar]
- 90.Nikonenko I, Jourdain P, Muller D. 2003. Presynaptic remodeling contributes to activity-dependent synaptogenesis. J. Neurosci. 23, 8498–8505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Stanton PK, Winterer J, Zhang XL, Muller W. 2005. Imaging LTP of presynaptic release of FM1–43 from the rapidly recycling vesicle pool of Schaffer collateral-CA1 synapses in rat hippocampal slices. Eur. J. Neurosci. 22, 2451–2461. ( 10.1111/j.1460-9568.2005.04437.x) [DOI] [PubMed] [Google Scholar]
- 92.Bon C, Bohme GA, Doble A, Stutzmann JM, Blanchard JC. 1992. A role for nitric oxide in long-term potentiation. Eur. J. Neurosci. 4, 420–424. ( 10.1111/j.1460-9568.1992.tb00891.x) [DOI] [PubMed] [Google Scholar]
- 93.Gribkoff VK, Lum-Ragan JT. 1992. Evidence for nitric oxide synthase inhibitor-sensitive and insensitive hippocampal synaptic potentiation. J. Neurophysiol. 68, 639–642. [DOI] [PubMed] [Google Scholar]
- 94.Haley JE, Wilcox GL, Chapman PF. 1992. The role of nitric oxide in hippocampal long-term potentiation. Neuron 8, 211–216. ( 10.1016/0896-6273(92)90288-O) [DOI] [PubMed] [Google Scholar]
- 95.Haley JE, Malen PL, Chapman PF. 1993. Nitric oxide synthase inhibitors block long-term potentiation induced by weak but not strong tetanic stimulation at physiological brain temperatures in rat hippocampal slices. Neurosci. Lett. 160, 85–88. ( 10.1016/0304-3940(93)90919-C) [DOI] [PubMed] [Google Scholar]
- 96.Kato K, Zorumski CF. 1993. Nitric oxide inhibitors facilitate the induction of hippocampal long-term potentiation by modulating NMDA responses. J. Neurophysiol. 70, 1260–1263. [DOI] [PubMed] [Google Scholar]
- 97.Chetkovich DM, Klann E, Sweatt JD. 1993. Nitric oxide synthase-independent long-term potentiation in area CA1 of hippocampus. Neuroreport 4, 919–922. ( 10.1097/00001756-199307000-00020) [DOI] [PubMed] [Google Scholar]
- 98.Musleh WY, Shahi K, Baudry M. 1993. Further studies concerning the role of nitric oxide in LTP induction and maintenance. Synapse 13, 370–375. ( 10.1002/syn.890130409) [DOI] [PubMed] [Google Scholar]
- 99.Williams JH, Li YG, Nayak A, Errington ML, Murphy KP, Bliss TV. 1993. The suppression of long-term potentiation in rat hippocampus by inhibitors of nitric oxide synthase is temperature and age dependent. Neuron 11, 877–884. ( 10.1016/0896-6273(93)90117-A) [DOI] [PubMed] [Google Scholar]
- 100.Nicolarakis PJ, Lin YQ, Bennett MR. 1994. Effect of nitric oxide synthase inhibition on long-term potentiation at associational-commissural and mossy fibre synapses on CA3 pyramidal neurones. Br. J. Pharmacol. 111, 521–524. ( 10.1111/j.1476-5381.1994.tb14768.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cummings JA, Nicola SM, Malenka RC. 1994. Induction in the rat hippocampus of long-term potentiation (LTP) and long-term depression (LTD) in the presence of a nitric oxide synthase inhibitor. Neurosci. Lett. 176, 110–114. ( 10.1016/0304-3940(94)90883-4) [DOI] [PubMed] [Google Scholar]
- 102.Boulton CL, Southam E, Garthwaite J. 1995. Nitric oxide-dependent long-term potentiation is blocked by a specific inhibitor of soluble guanylyl cyclase. Neuroscience 69, 699–703. ( 10.1016/0306-4522(95)00349-N) [DOI] [PubMed] [Google Scholar]
- 103.Malen PL, Chapman PF. 1997. Nitric oxide facilitates long-term potentiation, but not long-term depression. J. Neurosci. 17, 2645–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhuo M, Laitinen JT, Li XC, Hawkins RD. 1998. On the respective roles of nitric oxide and carbon monoxide in long-term potentiation in the hippocampus. Learn. Mem. 5, 467–480. [PMC free article] [PubMed] [Google Scholar]
- 105.Zhuo M, Laitinen JT, Li XC, Hawkins RD. 1999. On the respective roles of nitric oxide and carbon monoxide in long-term potentiation in the hippocampus. Learn. Mem. 6, 63–76. [PMC free article] [PubMed] [Google Scholar]
- 106.Ko GY, Kelly PT. 1999. Nitric oxide acts as a postsynaptic signaling molecule in calcium/calmodulin-induced synaptic potentiation in hippocampal CA1 pyramidal neurons. J. Neurosci. 19, 6784–6794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bon CL, Garthwaite J. 2003. On the role of nitric oxide in hippocampal long-term potentiation. J. Neurosci. 23, 1941–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang HG, et al. 2005. Presynaptic and postsynaptic roles of NO, cGK, and RhoA in long-lasting potentiation and aggregation of synaptic proteins. Neuron 45, 389–403. ( 10.1016/j.neuron.2005.01.011) [DOI] [PubMed] [Google Scholar]
- 109.Taqatqeh F, Mergia E, Neitz A, Eysel UT, Koesling D, Mittmann T. 2009. More than a retrograde messenger: nitric oxide needs two cGMP pathways to induce hippocampal long-term potentiation. J. Neurosci. 29, 9344–9350. ( 10.1523/JNEUROSCI.1902-09.2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Boulton CL, Irving AJ, Southam E, Potier B, Garthwaite J, Collingridge GL. 1994. The nitric oxide–cyclic GMP pathway and synaptic depression in rat hippocampal slices. Eur. J. Neurosci. 6, 1528–1535. ( 10.1111/j.1460-9568.1994.tb00543.x) [DOI] [PubMed] [Google Scholar]
- 111.Murphy KP, Bliss TV. 1999. Photolytically released nitric oxide produces a delayed but persistent suppression of LTP in area CA1 of the rat hippocampal slice. J. Physiol. 515, 453–462. ( 10.1111/j.1469-7793.1999.453ac.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Murphy KP, Williams JH, Bettache N, Bliss TV. 1994. Photolytic release of nitric oxide modulates NMDA receptor-mediated transmission but does not induce long-term potentiation at hippocampal synapses. Neuropharmacology 33, 1375–1385. ( 10.1016/0028-3908(94)90039-6) [DOI] [PubMed] [Google Scholar]
- 113.Garthwaite J, Boulton CL. 1995. Nitric oxide signaling in the central nervous system. Annu. Rev. Physiol. 57, 683–706. ( 10.1146/annurev.ph.57.030195.003343) [DOI] [PubMed] [Google Scholar]