

Abstract
The endocrine hormone leptin plays a key role in regulating food intake and body weight via its actions in the hypothalamus. However, leptin receptors are highly expressed in many extra-hypothalamic brain regions and evidence is growing that leptin influences many central processes including cognition. Indeed, recent studies indicate that leptin is a potential cognitive enhancer as it markedly facilitates the cellular events underlying hippocampal-dependent learning and memory, including effects on glutamate receptor trafficking, neuronal morphology and activity-dependent synaptic plasticity. However, the ability of leptin to regulate hippocampal synaptic function markedly declines with age and aberrant leptin function has been linked to neurodegenerative disorders such as Alzheimer's disease (AD). Here, we review the evidence supporting a cognitive enhancing role for the hormone leptin and discuss the therapeutic potential of using leptin-based agents to treat AD.
Keywords: leptin, hippocampus, synaptic plasticity, long-term potentiation, AMPA receptor trafficking, Alzheimer's disease
1. Introduction
Leptin is a peptide hormone that is principally made and secreted by white adipose tissue and circulates in the plasma at levels closely correlated with body fat [1]. Leptin readily enters the brain via regulated and saturable transport across the blood–brain barrier [2]. It is well established that the ability of leptin to regulate specific hypothalamic neurons is pivotal for controlling feeding behaviour and body weight. In a fed state, leptin serves as a potent signal for satiety; however, withdrawal of the leptin signal occurs very rapidly following food restriction or fasting [3]. The central actions of leptin are not restricted to the neural control of feeding behaviour, as leptin can also influence various developmental processes in the immature brain. Indeed, in support of extra-hypothalamic targets, leptin receptors are widely distributed throughout the central nervous system (CNS), with high levels of expression detected in the hippocampus and cerebellum in particular [4–6]. Leptin receptor expression in the hypothalamus is altered by changes in the circulating levels of leptin [7]. In hippocampal neurons, the expression of leptin receptors is also reportedly influenced by fasting [8]. Several studies have also demonstrated expression of leptin mRNA and protein throughout the CNS, suggesting that leptin may be released locally from specific neuronal populations [9].
The diabetes (db) gene encodes the leptin receptor (ObR; [10]), a class I cytokine receptor, that signals by associating with and activating Janus tyrosine kinases (JAKs). The main pathways activated downstream of JAKs in neurons are PI 3-kinase (phosphoinositide 3-kinase), ERK MAPK (mitogen-activated protein kinase) and STAT3 (signal transducer and activator of transcription). Six splice variants of ObR (a–f) have been identified, with the long form, ObRb, being the main signalling competent isoform. The short isoforms (ObRa,c,d,f) are thought to control the internalization and degradation of leptin, whereas ObRe that lacks a trans-membrane region buffers the plasma levels of leptin.
In accordance with the high levels of leptin receptor expression detected at hippocampal synapses [6], evidence is growing that leptin is a potent modulator of hippocampal excitatory synaptic function [11–15]. Indeed, studies in obese leptin-insensitive rodents (Zucker fa/fa rats; db/db mice) have identified deficits in hippocampal long-term potentiation (LTP) and long-term depression (LTD) as well as spatial memory [16,17]. Furthermore, direct administration of leptin into rodent hippocampus results in enhanced performance in various memory tasks [18]. In cellular studies performed in juvenile hippocampal slices (P14–21), exposure to leptin facilitates the induction of hippocampal LTP [14,16]. Leptin also reverses LTP (depotentiation) evoked at CA1 synapses when applied within a specific time window after LTP induction [13]. Furthermore, under conditions of enhanced excitability leptin induces a novel form of NMDA receptor-dependent LTD in juvenile hippocampal slices [11]. Thus, it is clear that the hormone leptin has the capacity to potently modify excitatory synaptic transmission and synaptic plasticity at early stages of postnatal development. However, there is limited knowledge of how leptin's ability to modulate various CNS functions is altered with age or indeed if the leptin system is altered in age-related CNS-driven disease.
Evidence is growing that metabolic systems functionally decline with age, and impairments in energy metabolism are correlated with faster rates of ageing and a greater risk of developing neurodegenerative disease. However, our understanding of how leptin influences hippocampal synaptic function during the ageing process is limited. Recent evidence indicates that age-related changes in leptin receptor-dependent signalling cascades occur. Thus, in aged rats a decline in STAT3 activation is observed that is linked to a decrease in leptin responsiveness [19]. Conversely, elevations in SOCS-3 and PTP1B (protein tyrosine phosphatase 1B) levels, which limit leptin receptor signal transduction, are evident in aged animals [20,21]. Recent studies have identified links between age-related alterations in leptin levels and cognitive performance [22]; however, the cellular basis for the age-dependent alterations in the cognitive enhancing effects of leptin are unclear.
Here, we summarize recent studies showing that neuronal sensitivity to leptin declines with age and in turn how this impacts on the efficacy of hippocampal excitatory synaptic transmission. We also discuss recent evidence that not only implicates dysfunctions in the leptin system in age-related disorders such as Alzheimer's disease (AD), but also the potential benefits of using leptin-based therapies to treat AD.
2. Leptin-induced long-term potentiation at adult hippocampal CA1 synapses
Most central synapses that exhibit synaptic plasticity are glutamatergic in nature. Four main types of glutamate receptors exist (α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptors, kainate receptors, N-methyl-d-aspartate (NMDA) receptors and metabotropic glutamate receptors (mGluRs), and these play key roles in various aspects of synaptic plasticity. NMDA receptors contribute little to excitatory synaptic transmission under basal conditions [23,24]. However, it is well documented that the synaptic activation of NMDA receptors is pivotal for activity-dependent LTP and LTD at hippocampal CA1 synapses [25–27].
Several lines of evidence indicate that leptin is a potent regulator of excitatory synaptic transmission at hippocampal CA1 synapses. Indeed, our laboratory was the first to report that application of leptin to juvenile hippocampal slices (P11–18) induces a rapid depression of excitatory synaptic transmission that readily reverses on leptin washout [14]. In accordance with this, transient synaptic depressions have been reported in response to leptin in both mouse and rat hippocampus at similar stages of postnatal development [28,29]. However, the leptin-driven synaptic depression observed during early postnatal development is in marked contrast to the effects of this hormone in adult tissue. Thus, leptin results in a persistent increase in excitatory synaptic transmission (leptin-induced LTP) in adult (12–16 week old) hippocampal slices [12,29], an effect requiring leptin receptor activation as robust leptin-induced LTP was observed in Zucker lean, but not leptin-insensitive, Zucker fa/fa, rats [12]. In hippocampal neurons, leptin receptors are expressed at both presynaptic and postsynaptic sites [6], and consequently leptin-induced LTP could potentially be expressed at either locus. However, no significant changes in paired pulse facilitation ratio (PPR) and coefficient of variation (CV) accompany the leptin-driven increase in synaptic efficacy indicating a postsynaptic expression mechanism. Leptin had no effect on excitatory synaptic transmission in slices treated with the competitive NMDA receptor antagonist D-AP5, indicating involvement of an NMDA receptor-dependent process. Synaptic activation of NMDA receptors was also pivotal for leptin-induced LTP as leptin had no effect when synaptic stimulation was stopped in two-input experiments [12].
It is well documented that NMDA receptor activation promotes AMPA receptor trafficking to synapses during hippocampal LTP [30]. Recent evidence indicates that the molecular composition of synaptic AMPA receptors is altered following activity-dependent changes in synaptic strength ([31–33]; but also note [34,35]). Similarly, alterations in AMPA receptor trafficking are implicated in leptin-induced LTP as an increase in AMPA receptor rectification accompanied the leptin-driven increase in synaptic efficacy. Application of philanthotoxin, a selective inhibitor of GluA2-lacking AMPA receptors, also resulted in reversal of leptin-induced LTP [12], consistent with an increase in the synaptic density of GluA2-lacking AMPA receptors underlying this effect of leptin.
In accordance with electrophysiological studies, leptin increased the surface expression of GluA1, but not GluA2, in biotinylation assays performed in adult hippocampal slices [12]. In immunocytochemical studies, leptin readily increased GluA1 surface expression in cultured hippocampal neurons. The surface expression of GluA2 is also enhanced by leptin, but much higher concentrations of leptin are required for this effect [12]. Temporal differences also exist in the regulation of GluA1 versus GluA2 subunits by leptin. Thus, in dual immunolabelling studies, exposure to leptin (50 nM) for 30 or 60 min resulted in comparable increases in the surface expression of both GluA1 and GluA2 (figure 1; data is available on Dryad; http://dx.doi.org/10.5061/dryad.jj17h Data files: Fig. 1 data). However, exposure of hippocampal neurons to leptin for longer periods of time (up to 180 min) caused significant reductions in GluA2 surface expression (86 ± 0.05% of control at 180 min; n = 27; p < 0.05; statistical analyses were performed using ANOVA (analysis of variance). In contrast, a sustained increase in GluA1 surface expression was observed after longer duration exposure to leptin (90–180 min) such that GluA1 surface expression was increased to 156 ± 0.08% of control (n = 27; p < 0.01; after 180 min leptin treatment; figure 1). Thus, there are clear temporal and potency differences in the regulation of different AMPA receptor subunits by the hormone leptin.
Figure 1.
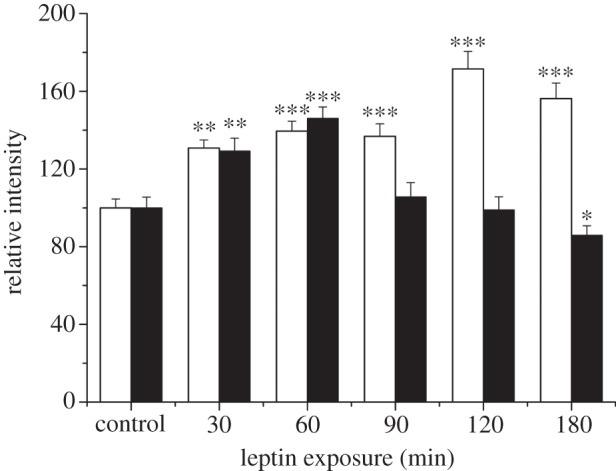
Differential regulation of GluA1 and GluA2 surface expression by leptin. Histogram of pooled data illustrating the effects of leptin on the surface expression of GluA1 (open bars) and GluA2 (filled bars) on hippocampal neurons (7–11 DIC). Leptin (50 nM) evoked a significant increase in GluA1 surface immunostaining after all exposure times (30–180 min). By contrast, exposure to leptin for up to 60 min increased GluA2 surface staining, whereas a significant reduction in GluA2 surface staining was observed after treatment with leptin for between 90 and 180 min.
3. A key role for phosphatase and tensin homologue in the regulation of GluA1 trafficking by leptin
Under physiological conditions, the circulating leptin levels lie within the low nanomolar range [36]. Consequently, the GluA1 subunit is likely to be the predominant target for leptin. Recent studies have probed the cellular mechanisms underlying leptin regulation of GluA1 trafficking to hippocampal synapses [12]. The density of surface receptors is tightly regulated by both exocytotic and endocytotic mechanisms. However, the increase in GluA1 surface expression induced by leptin involves increased delivery of GluA1 to synapses, as specific inhibitors of exocytosis, but not endocytosis, prevented the effects of leptin. Whole cell dialysis with inhibitors of exocytosis also blocked leptin-induced LTP in adult hippocampal slices, thereby supporting a role for increased delivery of AMPA receptors to synapses in this process.
Previous studies indicate that PI 3-kinase, which converts PtdIns(4,5)P2 into PtdIns(3,4,5,)P3, is pivotal for NMDA receptor-driven trafficking of AMPA receptors to hippocampal synapses during LTP [30]. PI 3-kinase is also implicated in the leptin-dependent increase in GluA1 surface expression in hippocampal neurons, as elevations in PtdIns(3,4,5,)P3 staining accompany this process [12]. Furthermore, the effects of leptin on excitatory synaptic strength and GluA1 surface expression were blocked by PI 3-kinase inhibitors. A recent report also supports a role for PI 3-kinase in trafficking AMPA receptors to synapses, as increased synthesis of PtdIns(3,4,5,)P3 results in enhanced AMPA-mediated synaptic transmission [37]. However, in addition to PI 3-kinase, the cellular levels of PtdIns(3,4,5,)P3 are also tightly controlled by phosphatase and tensin homologue (PTEN), the phosphatase that antagonises PI 3-kinase activity by dephosphorylating PtdIns(3,4,5,)P3 to PtdIns(4,5)P2. Consequently, leptin-driven inhibition of PTEN would also result in elevated PtdIns(3,4,5,)P3 levels. In support of a possible role for PTEN, leptin activation of hypothalamic KATP channels reportedly involves inhibition of PTEN [38]. Similarly in hippocampal neurons, expression of dominant-negative PTEN mutants (C124S or G129E) not only mirrored but also occluded the leptin-driven increase in GluA1 surface expression, suggesting involvement of PTEN inhibition in this process. The ability of leptin to insert GluA2-lacking AMPA receptors into synapses and increase miniature excitatory postsynaptic current (mEPSC) amplitude was also absent in neurons transfected with the PTEN mutants [12]. Moreover, pharmacological inhibition of PTEN with bisperoxovanadium (bpV) evoked a persistent increase in excitatory synaptic strength and it blocked the effects of leptin on synaptic efficacy in adult hippocampal slices. Thus, these findings are consistent with inhibition of PTEN and subsequent PtdIns(3,4,5,)P3-dependent delivery of AMPA receptors to synapses underlying the leptin-induced increase in synaptic efficacy in adult hippocampus.
4. Age-dependent modulation of hippocampal excitatory synaptic transmission by leptin
Several studies indicate that leptin transiently depresses excitatory synaptic transmission in juvenile hippocampus [14,28,29]. By contrast, application of leptin to hippocampal slices from younger animals (P5–8) results in a persistent synaptic depression (leptin-induced LTD) that is sustained following leptin washout [29]. Conversely, in adult (12–16 week old) hippocampal slices leptin induces a long-lasting enhancement of excitatory synaptic transmission (leptin-induced LTP; [12,29]). Leptin also readily induces LTP at hippocampal CA1 synapses in slices from older (12–14 month) animals, but the magnitude of leptin-induced LTP is markedly less at this age [29]. Thus, there are clear age-dependent differences in the direction and magnitude of synaptic modulation by leptin in the hippocampal CA1 region.
The cellular mechanisms underlying the divergent age-dependent effects of leptin on hippocampal synaptic function have been examined. In particular, the locus of leptin's effects was verified by analysis of two parameters linked to presynaptic release probability: PPR and CV [12,29]. Both the transient and persistent synaptic depressions induced by leptin were not associated with alterations in PPR or CV, indicating the involvement of a postsynaptic expression mechanism. Similarly, and in accordance with earlier studies [12], no significant changes in PPR and CV accompanied leptin-induced LTP in adult and aged hippocampus, thereby also indicating involvement of a postsynaptically expressed process.
5. The age-dependent effects of leptin involve distinct NMDA receptor subunits
It is well established that NMDA receptor activation is pivotal for various forms of activity-dependent synaptic plasticity in the mammalian CNS. NMDA receptor activation is also necessary for leptin modulation of hippocampal excitatory synaptic plasticity, including its ability to facilitate LTP [14], induce a novel form of LTD [11] and reverse established LTP (depotentiation; [13]). Similarly, leptin-driven regulation of excitatory synaptic transmission in the developing and adult hippocampus is NMDA receptor-dependent, as exposure of hippocampal slices to the competitive NMDA receptor antagonist D-AP5 blocked the effects of leptin at all ages [29]. It is known that the subunit composition and synaptic localization of NMDA receptors varies during development [39] and there is functional diversity in the roles played by different NMDA receptor subunits. Indeed, molecularly distinct NMDA receptors are implicated in hippocampal and cortical synaptic plasticity at different developmental stages [40–42]. In a similar manner, distinct NMDA receptor subunits are required for the bi-directional effects of leptin on hippocampal synaptic function. Thus, the synaptic depressions evoked by leptin at P5–8 and P11–18 involve GluN2B-containing NMDA receptors, whereas GluN2A subunits are pivotal for leptin-induced LTP in the adult and ageing hippocampus [29].
The role of different NMDA receptor subunits at different ages correlates well with the reported contribution of GluN2 subunits to synaptic NMDA receptors as the density of synaptic GluN2B subunits is significantly higher early in postnatal development, whereas expression of GluN2A subunits increases with age. In hippocampal neurons, PI 3-kinase and mitogen-activated protein kinase (MAPK) (extracellular signal-regulated protein kinase; ERK) are the main signalling cascades activated downstream of leptin receptors [43], and activation of both signalling pathways mediates facilitation of hippocampal NMDA responses by leptin [14]. However, divergent leptin-driven signalling pathways underlie the age-dependent effects of leptin on synaptic transmission. Thus, ERK activation is crucial for the synaptic depressions induced by leptin at early postnatal stages, whereas PI 3-kinase is implicated in leptin-induced hippocampal LTP in adult [29]. Our previous studies indicate that in cerebellar granule cells, leptin selectively enhances GluN2B responses via the ERK pathway [44]. Thus, it is possible that divergent signalling cascades couple leptin receptors to molecularly distinct NMDA receptors, thereby resulting in the opposing age-dependent effects of leptin on excitatory synaptic transmission.
6. Leptin-driven changes in synaptic efficacy display parallels to activity-dependent synaptic plasticity
Several studies have demonstrated that the magnitude of NMDA receptor-dependent LTP at hippocampal CA1 synapses attenuates with age [45–47]. The ability of leptin to induce LTP in adult hippocampus is also markedly reduced in aged animals [29]. Similarities also exist in the role that different NMDA receptor subunits play in HFS-induced LTP and leptin-induced LTP, suggesting that the two processes use similar expression mechanisms (figure 2). Indeed, in two-input occlusion experiments, leptin-induced LTP occluded the ability of high-frequency stimulation (HFS) to induce LTP and vice versa [29]. In addition, increased trafficking of GluA2-lacking AMPA receptors to hippocampal CA1 synapses is reported to underlie LTP induced by both HFS and leptin [12,32]. Analogous signalling pathways are also implicated in both forms of LTP as PI 3-kinase inhibitors block delivery of AMPA receptor to synapses during leptin-induced LTP and activity-dependent LTP ([12,29,30]; figure 2).
Figure 2.

Schematic of common signalling pathways underlying activity-dependent and leptin-dependent synaptic plasticity. During LTP, activation of NMDA receptors stimulates PI 3-kinase and subsequent inhibition of GSK3β at hippocampal CA1 synapses. Activation of this pathway promotes delivery of AMPA receptors to synapses which in turn results in a persistent increase in the efficacy of excitatory synaptic transmission (LTP). In a similar manner, following leptin binding to leptin receptors, the activity of PI 3-kinase is increased resulting in AMPA receptor exocytosis and a sustained increased in synaptic efficacy (leptin-induced LTP). Although neuronal leptin receptors are capable of inhibiting GSK3β, via PI 3-kinase, it is unclear if GSK3β plays a role in leptin-induced LTP. During LTD, stimulation of NMDA receptors activates PP1 leading to increased GSK3β activity and subsequent AMPA receptor endocytosis and LTD. Activation of the JAK2-STAT3 pathway has also recently been implicated in NMDA receptor-dependent LTD. Although the JAK2-STAT3 pathway is a key downstream target of neuronal leptin receptors, it is not known if this pathway plays a role in leptin-dependent synaptic plasticity. (Online version in colour.)
There are also parallels in the cellular mechanisms underlying leptin-induced LTD and NMDA receptor-dependent LTD. Thus, GluN2B subunits are implicated in low-frequency stimulation (LFS)-induced LTD [40,48] and the LTD induced by leptin at P5–8 [29]. The involvement of similar expression mechanisms is supported by findings from two input experiments as leptin-induced LTD occludes LFS-induced LTD and vice versa [29]. It is well documented that removal of AMPA receptors from synapses is crucial for NMDA receptor-dependent LTD [49]. Thus, as leptin-induced LTD involves a postsynaptic expression mechanism, it is feasible that LTD induced by leptin at P5–8 also involves internalization of AMPA receptors. It is also known that AMPA receptor endocytosis during NMDA receptor-dependent LTD is triggered by activation of protein phosphatases [50]. By contrast, however, an ERK-dependent cascade is implicated in leptin-induced LTD, as selective inhibitors of ERK activation block leptin action at P5–8 [29]. The role of ERK in leptin-induced LTD displays similarities to mGluR-dependent LTD, as activation of ERK is necessary for endocytosis of AMPA receptors and LTD [51]. Thus, although AMPA receptor internalization may be common to both leptin-induced LTD at P5–8 and LFS-induced LTD, it is likely that divergent signalling pathways promote AMPA receptor removal from synapses.
The ability of leptin to induce LTD under conditions of enhanced excitability (at P14–18) also displays parallels to NMDA receptor-dependent LTD [11]. Indeed, recent studies indicate that the serine/threonine kinase, GSK3β plays a pivotal role in NMDA receptor-dependent LTD, as activation of PP1 is reported to dephosphorylate and activate GSK3β, which in turn promotes AMPA receptor endocytosis and LTD [52]. In accordance with this, the magnitude of leptin-induced LTD (at P14–18) is significantly enhanced following inhibition of PI 3-kinase, suggesting that leptin-induced LTD is negatively regulated by PI 3-kinase. As inhibition of PI 3-kinase would relieve Akt-driven inhibition of GSK3β, the possibility that stimulation of GSK3β plays a role in leptin-induced LTD at P14–18 cannot be excluded. Moreover, the JAK2/STAT3 pathway, a key component of neuronal leptin receptor signal transduction, has also been implicated in NMDA receptor-dependent LTD [53]. Thus, it is feasible that leptin-dependent JAK2/STAT3 signalling also contributes to persistent reduction in synaptic efficacy induced by leptin, although this remains to be established.
7. Parallels between leptin and insulin action in regulating hippocampal synaptic function
It is well known that the hormone insulin is secreted by pancreatic beta cells in response to food intake. Insulin levels also correlate with energy balance, as levels of insulin fall with starvation and rise with obesity. Like leptin, central administration of insulin results in suppression of food intake [54]. Evidence is also growing that like leptin, peripherally derived insulin is readily transported into the brain and has the capacity to regulate synaptic plasticity at hippocampal synapses. Indeed, application of insulin to acute hippocampal slices results in the induction of a novel form of NMDA receptor-dependent LTD [55,56], a process involving tyrosine phosphorylation and endocytosis of GluA2 [57]. In a manner similar to leptin, insulin facilitates the induction of LTP [58], enhances NMDA receptor function and promotes delivery of NMDA receptors to the cell surface [59,60].
It is well documented that type II diabetes is associated with dementia and cognitive deficits [61]. Diabetic rodent models with either insulin deficiency or insulin resistance commonly display impairments in spatial learning and hippocampal synaptic plasticity [62,63]. Deficits in NMDA receptor-driven signalling have been observed in streptozotocin-induced diabetic rodents [64]. It is known that obesity, owing to leptin resistance, is a common feature of type II diabetes. Thus, it is likely that a combination of resistance to insulin and leptin, and the resultant impairments in hippocampal synaptic plasticity, contribute to the cognitive deficits observed in type II diabetics.
8. Leptin and neurodegenerative disorders
It is known that age is one of the major risks for developing neurodegenerative disorders such as AD. As life expectancy rises, it is not surprising that the incidence of AD is rapidly increasing. In addition to age, lifestyle and diet are important factors in determining the risk of developing AD. In particular, evidence from clinical studies indicates that mid-life obesity significantly increases the risk of AD. As obesity is mainly due to leptin resistance, it is likely that resistance to leptin and/or leptin dysfunction contribute to AD. Indeed, weight loss is a common feature of AD, and clinical studies indicate that the circulating levels of leptin are significantly attenuated in AD patients [65]. A recent prospective study found that the incidence of AD was much lower in non-obese individuals with high circulating leptin levels [66], which further supports a link between leptin levels and the incidence of this disease. Studies in rodent AD models have also detected correlations between leptin and neurodegeneration as leptin levels are significantly reduced in APPSwe and CRND8 murine models of AD [67,68].
9. Leptin prevents synaptic disruption and neuronal cell death in Alzheimer's disease models
Recent evidence indicates that leptin protects neurons from a variety of toxic insults, including apoptotic stimuli and ischaemic conditions [69,70]. In AD, accumulation of β-amyloid (Aβ) and formation of amyloid plaques are critically involved in hippocampal and cortical neuron degeneration. Indeed, exposure of neurons to toxic levels of Aβ significantly reduces neuronal viability. However, a recent study has shown that leptin inhibits Aβ-induced toxicity, as exposure to this hormone increases the viability of cortical neurons treated with Aβ [69]. Leptin also directly interferes with the accumulation of Aβ, as leptin is reported to inhibit β-secretase activity, thereby reducing production of Aβ [71]. In addition, cytoplasmic Aβ levels are lowered by leptin, as neuronal uptake of Aβ is increased in the presence of leptin [71]. Another key pathological hallmark of AD is neurofibrillary tangles comprising hyper-phosphorylated tau. Recent studies indicate that leptin regulates the levels of phosphorylated tau, as leptin not only reduces neuronal accumulation of tau but also limits tau phosphorylation via inhibition of GSK3β [68]. In cortical neurons, leptin markedly reduces Aβ-stimulated increases in phosphorylated tau (p-tau; [69]). In the same study, Doherty et al. [69] detected elevated levels of p-tau in cortical tissue from Zucker fa/fa rats, suggesting that dysfunctions in the leptin system increases the expression of proteins linked to AD pathogenesis. In behavioural paradigms, improvements in cognitive function have been reported following leptin treatment. Thus, leptin enhances performance in memory tasks in SAMP8 mice which display Aβ-induced neuronal toxicity [67]. Improvements in novel object recognition, contextual and cued fear conditioning tests have also been reported following leptin treatment in CRND8 mice [68] that overexpress mutant forms of the human APP gene [72]. Thus, in murine models of AD, treatment with leptin not only lowers neuronal levels of toxic Aβ and p-tau but it also alleviates the cognitive deficits associated with this disease.
10. Leptin and synaptic function in Alzheimer's disease
Several studies indicate that acute exposure to Aβ elicits detrimental effects on synaptic function, events thought to mirror the aberrant synaptic changes occurring in the early stages of AD. Indeed, Aβ prevents the induction of LTP and it facilitates LTD at hippocampal CA1 synapses [73,74]. In addition, exposure to Aβ promotes removal of glutamate receptors from synapses that is likely to contribute to synaptic disruption in AD [75–77]. A recent study has shown that prior treatment of hippocampal slices with leptin prevents Aβ inhibition of hippocampal LTP, as HFS failed to induce LTP in Aβ-treated slices whereas robust LTP was evident in slices exposed to leptin and Aβ [69]. Leptin treatment also inhibits the ability of Aβ to facilitate the induction of hippocampal LTD. Moreover, Aβ-driven removal of AMPA receptors from hippocampal synapses is significantly attenuated in leptin-treated neurons [69]. A PI 3-kinase dependent process is implicated in the protective effects of leptin on synaptic function, as leptin failed to prevent both Aβ-driven facilitation of hippocampal LTD and the decrease in GluA1 surface expression following PI 3-kinase inhibition [69]. The crucial role of PI 3-kinase in preventing the aberrant effects of Aβ on synaptic function correlates well with recent studies. Indeed, inhibition of GSK3β, a downstream target of PI 3-kinase, prevents inhibition of hippocampal LTP by Aβ [74]. Furthermore, GSK3 inhibitors are also reported to rescue LTP in a murine model of AD [78]. Thus, it is feasible that activation of PI 3-kinase and subsequent inhibition of GSK3β play a key role in leptin-dependent reversal of Aβ inhibition of LTP.
Recent molecular studies also support the notion that alterations in the leptin system contribute to synaptic disruption early in AD. Indeed, levels of endophilin I, a protein that regulates synaptic vesicle endocytosis and increases the probability of glutamate release [79] are elevated in post-mortem AD tissue. Similarly, recent studies indicate that cortical levels of endophilin I are also significantly elevated in leptin-insensitive Zucker fa/fa rats [69]. Moreover, exposure of cortical neurons to leptin significantly reduces the increase in endophilin I levels induced by Aβ [69]. Thus, dysfunctions in the leptin system may indirectly result in hippocampal synaptic disruption by promoting alterations in endophilin I levels.
11. Conclusion
It is well documented that the endocrine hormone leptin regulates many hypothalamic-driven functions, including energy balance, reproduction and bone formation. However, recent reports indicate that leptin has cognitive enhancing properties as it markedly influences the cellular events underlying hippocampal-dependent learning and memory. Indeed, leptin promotes rapid alterations in glutamate receptor trafficking and excitatory synaptic strength at hippocampal synapses. However, in accordance with other metabolic systems, the ability of leptin to regulate hippocampal synaptic function significantly attenuates with age. In addition, cognitive impairments in age-related neurodegenerative disorders, for instance AD, have recently been linked to aberrant leptin function. However, recent studies have revealed that treatment with leptin counteracts some of pathological events in AD, including disruption of hippocampal synaptic function and neuronal degeneration. Thus, developing novel strategies that boost the cognitive enhancing and neuroprotective actions of leptin may be a beneficial therapeutic approach in AD.
Data accessibility
Data is available on Dryad at http://dx.doi.org/10.5061/dryad.jj17h Data files: Fig. 1 data.
Funding statement
This work was supported by The Cunningham Trust and Medical Research Scotland.
References
- 1.Maffei M, et al. 1995. Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat. Med. 1, 1155–1161. ( 10.1038/nm1195-1155) [DOI] [PubMed] [Google Scholar]
- 2.Banks WA, Kastin AJ, Huang W, Jaspan JB, Maness LM. 1996. Leptin enters the brain by a saturable system independent of insulin. Peptides 17, 305–311. ( 10.1016/0196-9781(96)00025-3) [DOI] [PubMed] [Google Scholar]
- 3.Spiegelman BM, Flier JS. 2001. Obesity and the regulation of energy balance. Cell 104, 531–543. ( 10.1016/S0092-8674(01)00240-9) [DOI] [PubMed] [Google Scholar]
- 4.Mercer JG, Hoggard N, Williams LM, Lawrence CB, Hannah LT, Trayhurn P. 1996. Localization of leptin receptor mRNA and the long form splice variant (Ob-Rb) in mouse hypothalamus and adjacent brain regions by in situ hybridization. FEBS Lett. 387, 113–116. ( 10.1016/0014-5793(96)00473-5) [DOI] [PubMed] [Google Scholar]
- 5.Savioz A, Charnay Y, Huguenin C, Graviou C, Greggio B, Bouras C. 1997. Expression of leptin receptor mRNA (long form splice variant) in the human cerebellum. Neuroreport 8, 3123–3126. ( 10.1097/00001756-199709290-00023) [DOI] [PubMed] [Google Scholar]
- 6.Shanley LJ, O'Malley D, Irving AJ, Ashford ML, Harvey J. 2002. Leptin inhibits epileptiform-like activity in rat hippocampal neurones via PI 3-kinase-driven activation of BK channels. J. Physiol. 545, 933–944. ( 10.1113/jphysiol.2002.029488) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baskin DG, Seeley RJ, Kuijper JL, Lok S, Weigle DS, Erickson JC, Palmiter RD, Schwartz MW. 1998. Increased expression of mRNA for the long form of the leptin receptor in the hypothalamus is associated with leptin hypersensitivity and fasting. Diabetes 47, 538–543. ( 10.2337/diabetes.47.4.538) [DOI] [PubMed] [Google Scholar]
- 8.Lin S, Huang XF. 1997. Fasting increases leptin receptor mRNA expression in lean but not obese (ob/ob) mouse brain. Neuroreport 8, 3625–3629. ( 10.1097/00001756-199711100-00040) [DOI] [PubMed] [Google Scholar]
- 9.Morash B, Li A, Murphy PR, Wilkinson M, Ur E. 1999. Leptin gene expression in the brain and pituitary gland. Endocrinology 140, 5995–5998. ( 10.1210/en.140.12.5995) [DOI] [PubMed] [Google Scholar]
- 10.Tartaglia LA, et al. 1995. Identification and expression cloning of a leptin receptor, OB-R. Cell 83, 1263–1271. ( 10.1016/0092-8674(95)90151-5) [DOI] [PubMed] [Google Scholar]
- 11.Durakoglugil M, Irving AJ, Harvey J. 2005. Leptin induces a novel form of NMDA receptor-dependent long-term depression. J. Neurochem. 95, 396–405. ( 10.1111/j.1471-4159.2005.03375.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moult PR, Cross A, Santos SD, Carvalho AL, Lindsay Y, Connolly CN, Irving AJ, Leslie NR, Harvey J. 2010. Leptin regulates AMPA receptor trafficking via PTEN inhibition. J. Neurosci. 30, 4088–4101. ( 10.1523/JNEUROSCI.3614-09.2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moult PR, Milojkovic B, Harvey J. 2009. Leptin reverses long-term potentiation at hippocampal CA1 synapses. J. Neurochem. 108, 685–696. ( 10.1111/j.1471-4159.2008.05810.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shanley LJ, Irving AJ, Harvey J. 2001. Leptin enhances NMDA receptor function and modulates hippocampal synaptic plasticity. J. Neurosci. 21, RC186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oomura Y, et al. 2006. Leptin facilitates learning and memory performance and enhances hippocampal CA1 long-term potentiation and CaMK II phosphorylation in rats. Peptides 27, 2738–2749. ( 10.1016/j.peptides.2006.07.001) [DOI] [PubMed] [Google Scholar]
- 16.Li XL, Aou S, Oomura Y, Hori N, Fukunaga K, Hori T. 2002. Impairment of long-term potentiation and spatial memory in leptin receptor-deficient rodents. Neuroscience 113, 607–615. ( 10.1016/S0306-4522(02)00162-8) [DOI] [PubMed] [Google Scholar]
- 17.Winocur G, Greenwood CE, Piroli GG, Grillo CA, Reznikov LR, Reagan LP, McEwen BS. 2005. Memory impairment in obese Zucker rats: an investigation of cognitive function in an animal model of insulin resistance and obesity. Behav. Neurosci. 119, 1389–1395. ( 10.1037/0735-7044.119.5.1389) [DOI] [PubMed] [Google Scholar]
- 18.Wayner MJ, Armstrong DL, Phelix CF, Oomura Y. 2004. Orexin-A (Hypocretin-1) and leptin enhance LTP in the dentate gyrus of rats in vivo. Peptides 25, 991–996. ( 10.1016/j.peptides.2004.03.018) [DOI] [PubMed] [Google Scholar]
- 19.Scarpace PJ, Matheny M, Shek EW. 2000. Impaired leptin signal transduction with age-related obesity. Neuropharmacology 39, 1872–1879. ( 10.1016/S0028-3908(00)00014-9) [DOI] [PubMed] [Google Scholar]
- 20.Morrison CD, White CL, Wang Z, Lee SY, Lawrence DS, Cefalu WT, Zhang ZY, Gettys TW. 2007. Increased hypothalamic protein tyrosine phosphatase 1B contributes to leptin resistance with age. Endocrinology 148, 433–440. ( 10.1210/en.2006-0672) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peralta S, Carrascosa JM, Gallardo N, Ros M, Arribas C. 2002. Ageing increases SOCS-3 expression in rat hypothalamus: effects of food restriction. Biochem. Biophys. Res. Commun. 296, 425–428. ( 10.1016/S0006-291X(02)00906-3) [DOI] [PubMed] [Google Scholar]
- 22.Holden KF, Lindquist K, Tylavsky FA, Rosano C, Harris TB, Yaffe K, Health ABC study. 2009. Serum leptin level and cognition in the elderly: findings from the health ABC study. Neurobiol. Aging 30, 1483–1489. ( 10.1016/j.neurobiolaging.2007.11.024) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andreasen M, Lambert JD, Jensen MS. 1989. Effects of new non-N-methyl-d-aspartate antagonists on synaptic transmission in the in vitro rat hippocampus. J. Physiol. 414, 317–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davies SN, Collingridge GL. 1989. Role of excitatory amino acid receptors in synaptic transmission in area CA1 of rat hippocampus. Proc. R. Soc. Lond. B 236, 373–384. [DOI] [PubMed] [Google Scholar]
- 25.Collingridge GL, Kehl SJ, McLennan H. 1983. Excitatory amino acids in synaptic transmission in the Schaffer collateral-commissural pathway of the rat hippocampus. J. Physiol. 334, 33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dudek SM, Bear MF. 1992. Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-d-aspartate receptor blockade. Proc. Natl Acad. Sci. USA 89, 4363–4367. ( 10.1073/pnas.89.10.4363) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mulkey RM, Malenka RC. 1992. Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of the hippocampus. Neuron 9, 967–975. ( 10.1016/0896-6273(92)90248-C) [DOI] [PubMed] [Google Scholar]
- 28.Xu L, Rensing N, Yang XF, Zhang HX, Thio LL, Rothman SM, Weisenfeld AE, Wong M, Yamada KA. 2008. Leptin inhibits 4-aminopyridine- and pentylenetetrazole-induced seizures and AMPAR-mediated synaptic transmission in rodents. J. Clin. Invest. 118, 272–280. ( 10.1172/JCI33009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moult PR, Harvey J. 2011. NMDA receptor subunit composition determines the polarity of leptin-induced synaptic plasticity. Neuropharmacology 61, 924–936. ( 10.1016/j.neuropharm.2011.06.021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Man HY, et al. 2003. Activation of PI3-kinase is required for AMPA receptor insertion during LTP of mEPSCs in cultured hippocampal neurons. Neuron 38, 611–624. ( 10.1016/S0896-6273(03)00228-9) [DOI] [PubMed] [Google Scholar]
- 31.Ho MT, Pelkey KA, Topolnik L, Petralia RS, Takamiya K, Xia J, Huganir RL, Lacaille JC, McBain CJ. 2007. Developmental expression of Ca2+-permeable AMPA receptors underlies depolarization-induced long-term depression at mossy fiber CA3 pyramid synapses. J. Neurosci. 27, 11 651–11 662. ( 10.1523/JNEUROSCI.2671-07.2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Plant K, Pelkey KA, Bortolotto ZA, Morita D, Terashima A, McBain CJ, Collingridge GL, Isaac JT. 2006. Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nat. Neurosci. 9, 602–604. ( 10.1038/nn1678) [DOI] [PubMed] [Google Scholar]
- 33.Lu Y, Allen M, Halt AR, Weisenhaus M, Dallapiazza RF, Hall DD, Usachev YM, McKnight GS, Hell JW. 2007. Age-dependent requirement of AKAP150-anchored PKA and GluR2-lacking AMPA receptors in LTP. EMBO J. 26, 4879–4890. ( 10.1038/sj.emboj.7601884) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Adesnik H, Nicoll RA. 2007. Conservation of glutamate receptor 2-containing AMPA receptors during long-term potentiation. J. Neurosci. 27, 4598–4602. ( 10.1523/JNEUROSCI.0325-07.2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gray EE, Fink AE, Sariñana J, Vissel B, O'Dell TJ. 2007. Long-term potentiation in the hippocampal CA1 region does not require insertion and activation of GluR2-lacking AMPA receptors. J. Neurophysiol. 98, 2488–2492. ( 10.1152/jn.00473.2007) [DOI] [PubMed] [Google Scholar]
- 36.Caro JF, Sinha MK, Kolaczynski JW, Zhang PL, Considine RV. 1996. Leptin: the tale of an obesity gene. Diabetes 45, 1455–1462. ( 10.2337/diab.45.11.1455) [DOI] [PubMed] [Google Scholar]
- 37.Arendt KL, Royo M, Fernández-Monreal M, Knafo S, Petrok CN, Martens JR, Esteban JA. 2010. PIP3 controls synaptic function by maintaining AMPA receptor clustering at the postsynaptic membrane. Nat. Neurosci. 13, 36–44. ( 10.1038/nn.2462) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ning K, Miller LC, Laidlaw HA, Watterson KR, Gallagher J, Sutherland C, Ashford ML. 2009. Leptin-dependent phosphorylation of PTEN mediates actin restructuring and activation of ATP-sensitive K+ channels. J. Biol. Chem. 284, 9331–9340. ( 10.1074/jbc.M806774200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Monyer H, Sprengel R, Schoepfer R, Herb A, Higuchi M, Lomeli H, Burnashev N, Sakmann B, Seeburg PH. 1992. Heteromeric NMDA receptors: molecular and functional distinction of subtypes. Science 256, 1217–1221. [DOI] [PubMed] [Google Scholar]
- 40.Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M, Auberson YP, Wang YT. 2004. Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science 304, 1021–1024. [DOI] [PubMed] [Google Scholar]
- 41.Bartlett TE, Bannister NJ, Collett VJ, Dargan SL, Massey PV, Bortolotto ZA, Fitzjohn SM, Bashir Zl, Collingridge GL, Lodge D. 2007. Differential roles of NR2A and NR2B-containing NMDA receptors in LTP and LTD in the CA1 region of two-week old rat hippocampus. Neuropharmacology 52, 60–70. [DOI] [PubMed] [Google Scholar]
- 42.Massey PV, Johnson BE, Moult PR, Auberson YP, Brown MW, Molnar E, Collingridge GL, Bashir Zl. 2004. Differential roles of NR2A and NR2B-containing NMDA receptors in cortical long-term potentiation and long-term depression. J. Neurosci. 24, 7821–7828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harvey J. 2007. Leptin regulation of neuronal excitability and cognitive function. Curr. Opin. Pharmacol. 7, 3–7. ( 10.1016/j.coph.2006.11.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Irving AJ, Wallace L, Durakoglugil D, Harvey J. 2006. Leptin enhances NR2B-mediated N-methyl-d-aspartate responses via a mitogen-activated protein kinase-dependent process in cerebellar granule cells. Neuroscience 138, 1137–1148. ( 10.1016/j.neuroscience.2005.11.042) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deupree DL, Turner DA, Watters CL. 1991. Spatial performance correlates with in vitro potentiation in young and aged Fischer 344 rats. Brain Res. 554, 1–9. ( 10.1016/0006-8993(91)90164-Q) [DOI] [PubMed] [Google Scholar]
- 46.Tombaugh GC, Rowe WB, Chow AR, Michael TH, Rose GM. 2002. Theta-frequency synaptic potentiation in CA1 in vitro distinguishes cognitively impaired from unimpaired aged Fischer 344 rats . J. Neurosci. 22, 9932–9940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rosenzweig ES, Rao G, McNaughton BL, Barnes CA. 1997. Role of temporal summation in age-related long-term potentiation-induction deficits. Hippocampus 7, 549–558. () [DOI] [PubMed] [Google Scholar]
- 48.Kutsuwada T, et al. 1996. Impairment of suckling response, trigeminal neuronal pattern formation, and hippocampal LTD in NMDA receptor epsilon 2 subunit mutant mice. Neuron 16, 333–344. ( 10.1016/S0896-6273(00)80051-3) [DOI] [PubMed] [Google Scholar]
- 49.Collingridge GL, Isaac JT, Wang YT. 2004. Receptor trafficking and synaptic plasticity. Nat. Rev. Neurosci. 5, 952–962. [DOI] [PubMed] [Google Scholar]
- 50.Massey PV, Bashir ZI. 2007. Long-term depression: multiple forms and implications for brain function. Trends Neurosci. 30, 176–184. ( 10.1016/j.tins.2007.02.005) [DOI] [PubMed] [Google Scholar]
- 51.Gallagher SM, Daly CA, Bear MF, Huber KM. 2004. Extracellular signal-regulated protein kinase activation is required for metabotropic glutamate receptor-dependent long-term depression in hippocampal area CA1. J. Neurosci. 24, 4859–4864. ( 10.1523/JNEUROSCI.5407-03.2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Peineau S, et al. 2007. LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron 53, 703–717. ( 10.1016/j.neuron.2007.01.029) [DOI] [PubMed] [Google Scholar]
- 53.Nicolas CS, et al. 2012. The Jak/STAT pathway is involved in synaptic plasticity. Neuron 73, 374–390. ( 10.1016/j.neuron.2011.11.024) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schwartz MW, Figlewicz DP, Baskin DG, Woods SC, Porte D., Jr 1992. Insulin in the brain: a hormonal regulator of energy balance. Endocr. Rev. 13, 387–414. [DOI] [PubMed] [Google Scholar]
- 55.Man HY, Lin JW, Ju WH, Ahmadian G, Liu L, Becker LE, Sheng M, Wang YT. 2000. Regulation of AMPA receptor-mediated synaptic transmission by clathrin-dependent receptor internalization. Neuron 25, 649–662. [DOI] [PubMed] [Google Scholar]
- 56.Huang CC, Lee CC, Hsu KS. 2004. An investigation into signal transduction mechanisms involved in insulin-induced long-term depression in the CA1 region of the hippocampus. J. Neurochem. 89, 217–231. [DOI] [PubMed] [Google Scholar]
- 57.Ahmadian G, et al. 2004. Tyrosine phosphorylation of GluR2 is required for insulin-stimulated AMPA receptor endocytosis and LTD. EMBO J. 23, 1040–1050. ( 10.1038/sj.emboj.7600126) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van der Heide LP, Kamal A, Artola A, Gispen WH, Ramakers GM. 2005. Insulin modulates hippocampal activity-dependent synaptic plasticity in a N-methyl-d-aspartate receptor and phosphatidyl-inositol-3-kinase-dependent manner. J. Neurochem. 94, 1158–1166. ( 10.1111/j.1471-4159.2005.03269.x) [DOI] [PubMed] [Google Scholar]
- 59.Skeberdis VA, Lan J, Zheng X, Zukin RS, Bennett MV. 2001. Insulin promotes rapid delivery of N-methyl-d-aspartate receptors to the cell surface by exocytosis. Proc. Natl Acad. Sci. USA 98, 3561–3566. ( 10.1073/pnas.051634698) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu L, Brown JC, 3rd, Webster WW, Morrisett RA, Monaghan DT. 1995. Insulin potentiates N-methyl-d-aspartate receptor activity in Xenopus oocytes and rat hippocampus. Neurosci. Lett. 192, 5–8. ( 10.1016/0304-3940(95)11593-L) [DOI] [PubMed] [Google Scholar]
- 61.Gispen WH, Biessels GJ. 2000. Cognition and synaptic plasticity in diabetes mellitus. Trends Neurosci. 23, 542–549. [DOI] [PubMed] [Google Scholar]
- 62.Biessels GJ, Kamal A, Ramakers GM, Urban IJ, Spruijt BM, Erkelens DW, Gispen WH. 1996. Place learning and hippocampal synaptic plasticity in streptozotocin-induced diabetic rats. Diabetes 45, 1259–1266. [DOI] [PubMed] [Google Scholar]
- 63.Kamal A, Biessels GJ, Urban IJ, Gispen WH. 1999. Hippocampal synaptic plasticity in streptozotocin-diabetic rats: impairment of long-term potentiation and facilitation of long-term depression. Neuroscience 90, 737–745. [DOI] [PubMed] [Google Scholar]
- 64.Di Luca M, Ruts L, Gardoni F, Cattabeni F, Biessels GJ, Gispen WH. 1999. NMDA receptor subunits are modified transcriptionally and post-translationally in the brain of streptozotocin-diabetic rats. Diabetologia 42, 693–701. [DOI] [PubMed] [Google Scholar]
- 65.Power DA, Noel J, Collins R, O'Neill D. 2001. Circulating leptin levels and weight loss in Alzheimer's disease patients. Dement. Geriatr. Cogn. Disord. 12, 167–170. ( 10.1159/000051252) [DOI] [PubMed] [Google Scholar]
- 66.Lieb W, et al. 2009. Association of plasma leptin levels with incident Alzheimer disease and MRI measures of brain aging. JAMA 302, 2565–2572. ( 10.1001/jama.2009.1836) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Farr SA, Banks WA, Morley JE. 2006. Effects of leptin on memory processing. Peptides 27, 1420–1425. ( 10.1016/j.peptides.2005.10.006) [DOI] [PubMed] [Google Scholar]
- 68.Greco SJ, Bryan KJ, Sarkar S, Zhu X, Smith MA, Ashford JW, Johnston JM, Tezapsidis N, Casadesus G. 2010. Leptin reduces pathology and improves memory in a transgenic mouse model of Alzheimer's disease. J. Alzheimers Dis. 19, 1155–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Doherty GH, Beccano-Kelly D, Yan SD, Gunn-Moore FJ, Harvey J. 2013. Leptin prevents hippocampal synaptic disruption and neuronal cell death induced by amyloid β. Neurobiol. Aging 34, 226–237. ( 10.1016/j.neurobiolaging.2012.08.003) [DOI] [PubMed] [Google Scholar]
- 70.Zhang F, Wang S, Signore AP, Chen J. 2007. Neuroprotective effects of leptin against ischemic injury induced by oxygen-glucose deprivation and transient cerebral ischemia. Stroke 38, 2329–2336. ( 10.1161/STROKEAHA.107.482786) [DOI] [PubMed] [Google Scholar]
- 71.Fewlass DC, Noboa K, Pi-Sunyer FX, Johnston JM, Yan SD, Tezapsidis N. 2004. Obesity-related leptin regulates Alzheimer's Abeta. FASEB J. 18, 1870–1878. ( 10.1096/fj.04-2572com) [DOI] [PubMed] [Google Scholar]
- 72.Chishti MA, et al. 2001. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J. Biol. Chem. 276, 21562–21570. ( 10.1074/jbc.M100710200) [DOI] [PubMed] [Google Scholar]
- 73.Shankar GM, et al. 2008. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842. ( 10.1038/nm1782) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jo J, et al. 2011. Aβ(1–42) inhibition of LTP is mediated by a signaling pathway involving caspase-3, Akt1 and GSK-3β. Nat. Neurosci. 14, 545–547. ( 10.1038/nn.2785) [DOI] [PubMed] [Google Scholar]
- 75.Liu SJ, Gasperini R, Foa L, Small DH. 2010. Amyloid-beta decreases cell-surface AMPA receptors by increasing intracellular calcium and phosphorylation of GluR2. J. Alzheimers Dis. 21, 655–666. [DOI] [PubMed] [Google Scholar]
- 76.Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R. 2006. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron 52, 831–843. ( 10.1016/j.neuron.2006.10.035) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Snyder EM, et al. 2005. Regulation of NMDA receptor trafficking by amyloid-beta . Nat. Neurosci. 8, 1051–1058. ( 10.1038/nn1503) [DOI] [PubMed] [Google Scholar]
- 78.Ma T, et al. 2010. Dysregulation of the mTOR pathway mediates impairment of synaptic plasticity in a mouse model of Alzheimer's disease. PLoS ONE 5, e12845 ( 10.1371/journal.pone.0012845) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Weston MC, Nehring RB, Wojcik SM, Rosenmund C. 2011. Interplay between VGLUT isoforms and endophilin A1 regulates neurotransmitter release and short-term plasticity. Neuron 69, 1147–1145. ( 10.1016/j.neuron.2011.02.002) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data is available on Dryad at http://dx.doi.org/10.5061/dryad.jj17h Data files: Fig. 1 data.