

Abstract
Despite decades of study, the mechanisms by which synapses express the increase in strength during long-term potentiation (LTP) remain an area of intense interest. Here, we have studied how AMPA receptor subunit composition changes during the early phases of hippocampal LTP in CA1 pyramidal neurons. We studied LTP at silent synapses that initially lack AMPA receptors, but contain NMDA receptors. We show that strongly inwardly rectifying AMPA receptors are initially incorporated at silent synapses during LTP and are then subsequently replaced by non-rectifying AMPA receptors. These findings suggest that silent synapses initially incorporate GluA2-lacking, calcium-permeable AMPA receptors during LTP that are then replaced by GluA2-containing calcium-impermeable receptors. We also show that LTP consolidation at CA1 synapses requires a rise in intracellular calcium concentration during the early phase of expression, indicating that calcium influx through the GluA2-lacking AMPA receptors drives their replacement by GluA2-containing receptors during LTP consolidation. Taken together with previous studies in hippocampus and in other brain regions, these findings suggest that a common mechanism for the expression of activity-dependent glutamatergic synaptic plasticity involves the regulation of GluA2-subunit composition and highlights a critical role for silent synapses in this process.
Keywords: glutamate, synaptic plasticity, hippocampus
1. Introduction
A major body of evidence supports the idea that long-term potentiation (LTP) is an important synaptic mechanism underlying diverse forms of learning and memory in the mammalian brain [1,2]. Considerable effort has been made to define the molecular mechanisms for the induction and expression of LTP. This work shows that LTP is triggered by the activation of NMDA receptors, leading to an influx of calcium that causes an increase in the strength of synapses mediated primarily by an increase in the number and responsiveness of AMPA receptors expressed at the postsynaptic membrane [3]. AMPA receptor number is increased owing to the trafficking of receptors to the synapse and involves a number of interacting proteins [3,4]. LTP at silent synapses exemplifies the regulation of AMPA receptor number. Silent synapses are a subset of synapses in hippocampus and neocortex that lack postsynaptic AMPA receptors, yet contain NMDA receptors. Hence, at resting membrane potentials, these synapses are functionally silent owing to the voltage-dependent block of NMDA receptors by magnesium ions. However, upon depolarization of the postsynaptic neuron, the NMDA receptor voltage-dependent block is relieved, and when combined with synaptic glutamate release, this causes the induction of LTP [5–9]. Silent synapses are found in greater number in early development [10–15]. One hypothesis is that they represent conditional synaptic connections requiring activity-dependent strengthening to become functional. Such a mechanism is proposed to be important, for example, in experience-dependent synaptic plasticity underlying development of topographical maps in primary sensory cortex [12–16].
One area of recent controversy, concerning the recruitment of AMPA receptors to synapses during LTP, is the subunit composition of these receptors. The core AMPA receptor complex is tetrameric, made up of combinations of the GluA1–4 subunits [17]. In cortical pyramidal neurons, the vast majority of AMPA receptors are thought to contain the GluA2 subunit. Indeed, the presence of the GluA2 subunit, in its typical postnatally edited form, renders the receptor ion channel impermeable to calcium and insensitive to voltage-dependent blockade by intracellular polyamines, for example spermine [17,18]. Consistent with this view, the great majority of AMPA receptors on cortical pyramidal neurons exhibit no inward rectification, which reflects their lack of voltage-dependent blockade by intracellular polyamines. Moreover, calcium influx through GluA2-lacking AMPA receptors is believed to be excitotoxic to pyramidal neurons [19], suggesting an important functional role of GluA2 incorporation in the AMPA complex in these neurons. Nevertheless, an emerging body of evidence suggests that GluA2-lacking AMPA receptors are involved in several forms of synaptic plasticity. Early studies showed that GluA2-lacking AMPA receptors are present at synapses on cerebellar stellate neurons and calcium influx through these receptors causes a form of synaptic plasticity in which GluA2-lacking are replaced by GluA2-containing receptors [20–23]. We subsequently showed that a similar process may occur at hippocampal CA1 synapses during LTP. We found that immediately after LTP, inwardly rectifying GluA2-lacking AMPA receptors are transiently expressed for approximately 15 min before being replaced by non-rectifying GluA2-containing receptors in an activity-dependent process that consolidates LTP [24].
Since these early studies on the involvement of GluA2-lacking, calcium-permeable AMPA receptors in cerebellar and hippocampal synaptic plasticity, a number of studies have shown that similar mechanisms may underlie plasticity in other brain areas, including primary sensory cortex, ventral tegmental area (VTA), amygdala and hypothalamus [18,25–28]. Despite this, considerable controversy remains for hippocampal CA1 LTP on the role of GluA2-lacking AMPA receptors. Some studies have failed to observe any change in the GluA2 content of AMPA receptors during LTP expression [29,30], whereas others provide evidence in favour of the hypothesis [31,32]. One possibility to explain these discrepant results is that different subsets of CA1 synapses differentially incorporate GluA2-lacking or GluA2-containing AMPA receptors immediately after LTP induction, depending on their previous history of plasticity or stage of maturity [32]. In particular, one possibility is that silent synapses, which are thought to represent an immature form, may selectively express GluA2-lacking AMPA receptors immediately after LTP induction. In this study, we directly tested this idea by studying the rectification properties of AMPA receptors incorporated at silent synapses during LTP. By pharmacologically isolating the AMPA receptor responses after LTP, we measured the rectification properties of these newly incorporated AMPA receptors. We show that in the majority of cases, silent synapses incorporate strongly inwardly rectifying AMPA receptors immediately after LTP and these are subsequently replaced by non- or outwardly rectifying receptors. In a separate set of experiments, we tested the role for intracellular calcium in the consolidation of LTP. Using UV light to uncage an intracellular calcium chelator immediately following LTP induction, we show that calcium chelation causes reversal of LTP. Taken together with previous work on the requirement for synaptic activation of GluA2-lacking AMPA receptors for the consolidation of LTP [24], this work provides evidence that silent synapses preferentially incorporate GluA2-lacking receptors during LTP and the calcium influx through these receptors is required for LTP consolidation by driving their switch to GluA2-containing receptors.
2. Results
(a). Inwardly rectifying AMPA receptors are transiently incorporated at silent synapses during long-term potentiation
We tested the hypothesis that GluA2-lacking, calcium-permeable AMPA receptors are inserted into silent synapses as a mechanism for LTP expression in hippocampal CA1 pyramidal neurons in acute slices prepared from 5- to 8-day-old rats. To detect GluA2-lacking AMPA receptors, we measured the rectification properties of the newly incorporated receptors. To isolate a putative silent synapse, we used minimal stimulation and reduced the intensity to that just subthreshold for eliciting AMPA receptor-mediated excitatory postsynaptic currents (EPSCs) at a holding potential of −70 mV and collected a baseline of 100 stimuli at 0.5 Hz. To induce LTP, we then switched the membrane potential to 0 mV for the next 100 trials, without interrupting the ongoing 0.5 Hz stimulation. At the end of the LTP induction period, the membrane potential was once again returned to −70 mV, without interrupting the ongoing stimulation, and 100 μM D-AP5 was bath applied for the remaining duration of the experiment. At this point, in a subset of cells, AMPA EPSCs were now observed showing that LTP had been successfully induced at the originally silent synapse (figure 1a). In the remainder of the experiments in which no AMPA EPSCs were detected, presumably no silent synapse was being activated during baseline. This protocol is very similar to that used previously for the induction of LTP at silent synapses [5].
Figure 1.

Inwardly rectifying AMPA receptors are incorporated at silent synapses during LTP: (a) example silent synapse experiment showing EPSC amplitude versus time (i) and series resistance (ii) during a whole-cell voltage-clamp recording from a CA1 pyramidal neuron in a hippocampal slice. Holding potential is indicated for different parts of the recording. (b) Example averaged traces from the experiment in (a) taken at the times indicated (average of all traces in the indicated time period). (c) Averaged traces from another example experiment exhibiting rectifying AMPA EPSCs during LTP1. (d) Averaged traces from a further example experiment: in this case rectifying AMPA EPSCs were not observed during LTP1.
In the cells in which LTP was induced, we could then study the rectification properties of the newly incorporated AMPA receptors as follows: a 5-min period of trials were recorded at −70 mV immediately after LTP induction followed by a 5-min period at +40 mV to investigate whether the newly inserted AMPA receptors exhibited inward rectification. In a subset of cells, a period at 0 mV was also collected. We called this early phase of LTP, ‘LTP1’. Following this, the cell was returned to −70 mV and data collected for another 15 min before the cell was once again depolarized to 0 and +40 mV to measure rectification properties of the AMPA receptors (‘LTP2’). Note that the presence of the NMDA receptor antagonist, D-APV, after LTP induction, as well as the GABAA receptor blocker picrotoxin throughout, enabled the rectification properties of the newly inserted AMPA receptors to be measured in pharmacological isolation.
As shown in figure 1a–c, we found that in the majority of experiments, the newly inserted AMPA receptors at silent synapses were completely or very strongly inwardly rectifying in the early LTP phase (LTP1). However, in one experiment we observed non-rectifying AMPA receptors newly incorporated (figure 1d). In the later LTP2 phase, we found that the AMPA receptors had switched to being non-rectifying or outwardly rectifying in the majority of experiments (figure 1a–d). When all experiments were taken together (n = 6), the appearance of AMPA receptor-mediated EPSCs at a holding potential of −70 mV during LTP1 was not associated with the appearance of AMPA receptor-mediated EPSCs at +40 mV; however, during LTP2 AMPA receptor-mediated EPSCs were observed at both potentials (figure 2a). Thus, during LTP1, AMPA receptor-mediated EPSCs were strongly inwardly rectifying AMPA receptors on average, whereas during LTP2 AMPA receptor-mediated EPSCs no longer exhibited inward rectification and were now typically non- or outwardly rectifying (figure 2b–d). Therefore, we show that inwardly rectifying AMPA receptors are initially incorporated at silent synapses during LTP and are subsequently replaced by non-rectifying receptors.
Figure 2.

Summary data showing that inwardly rectifying AMPA receptors are typically incorporated at silent synapses during LTP: (a) mean EPSC amplitude versus time for all experiments (data average of 10 responses). (b) Mean rectification index values for AMPA EPSCs during LTP1 and LTP2. Asterisk (*) indicates p < 0.05. (c) Current-voltage plots for AMPA EPSCs during LTP1 and (d) for LTP2. Line and filled diamonds represent mean data; values for individual experiments are shown by other symbols.
(b). An increase in intracellular calcium is required for consolidation of long-term potentiation
Our data support the model [18,24] in which GluA2-lacking, calcium-permeable inwardly rectifying AMPA receptors are initially incorporated into CA1 pyramidal cell synapses during LTP and are subsequently switched for GluA2-containing calcium-impermeable non-rectifying AMPA receptors. As GluA2-lacking AMPA receptors are calcium permeable, one hypothesis is that this calcium influx is required for their activity-dependent replacement during LTP stabilization. Indeed, previous work suggests that activation of the newly incorporated GluA2-lacking AMPA receptors is required for their replacement by GluA2-containing receptors [21,22,24]. At CA1 synapses, we have previously shown that blockade of GluA2-lacking AMPA receptors by philanthotoxin reverses LTP when applied within the first 15 min following induction and that synaptic activity is also required for LTP consolidation during this early LTP period [24]. One prediction from these findings is that an increase in intracellular calcium concentration owing to calcium influx through the GluA2-lacking AMPA receptors is necessary for LTP consolidation. To test this idea, we used the caged calcium chelator, diazo-2. This caged version of the high-affinity calcium chelator, BAPTA, can be used in neurons to prevent a rise in intracellular calcium concentration with little effect on resting calcium levels [33,34]. We included 10 mM diazo-2 in the intracellular solution during whole-cell patch-clamp recordings from CA1 pyramidal neurons in acute hippocampal slices prepared from two week old rats, and then used UV light to uncage the chelator following LTP induction. We interleaved these experiments with controls in which no diazo-2 was included but in which UV light was still applied, or in which diazo-2 was included in the whole-cell solution but no UV light was applied. We found that inclusion of diazo-2 did not prevent the induction of LTP; however, uncaging of diazo-2 2 min after LTP induction did cause the potentiation to reverse back to baseline after 20–25 min. In the interleaved controls, UV light alone or diazo-2 without UV light did not prevent establishment of stable LTP (figure 3a,d). These findings suggest that an increase in intracellular calcium concentration during the early phase of LTP is required for its maintenance and support the idea that calcium influx through GluA2-lacking AMPA receptors is required for LTP consolidation.
Figure 3.

UV uncaging of a calcium buffer after LTP induction prevents stabilization of LTP: (a) mean data for EPSC amplitude versus time for whole-cell patch-clamp experiments from CA1 pyramidal neurons with diazo-2 included in the intracellular solution (open circles); UV light applied at the time point is indicated by the arrow. For controls (filled circles), either UV light was applied to neurons with no diazo-2 in the intracellular solution (n = 6), or neurons contained diazo-2 and no UV light was applied (n = 2). Bar and asterisk (*) indicate significant difference (p < 0.05) between diazo-2 and control datasets at the time indicated. (b) Example averaged EPSC traces from a diazo-2 and a control experiment taken at the times indicted in (a).
3. Discussion
Here, we have studied the properties of AMPA receptors that are newly inserted at silent synapses during LTP. We have also investigated a role for an increase in intracellular calcium concentration in the consolidation of LTP. We show that fully or strongly inwardly rectifying AMPA receptors are transiently incorporated at silent synapses during LTP. These unique rectification properties strongly indicate that GluA2-lacking, calcium-permeable AMPA receptors are preferentially incorporated at silent synapses and mediate the earliest phase of LTP expression. We further show that the rectifying receptors are subsequently replaced by non-rectifying receptors, indicating that the newly incorporated GluA2-lacking receptors are replaced by GluA2-containing receptors. Finally, we show that an increase in intracellular calcium is required for the consolidation of LTP, suggesting that the calcium influx through the newly incorporated GluA2-lacking AMPA receptors drives the switch to GluA2-containing receptors during LTP consolidation. These findings are summarized in figure 4.
Figure 4.
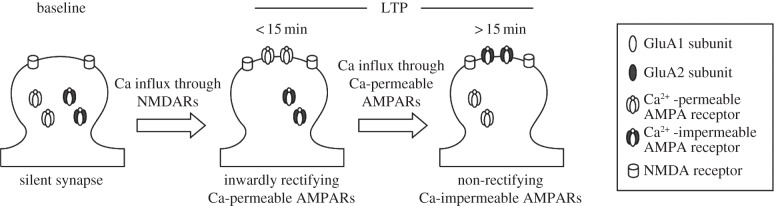
Schematic for the mechanism of LTP expression at silent synapses: during baseline, silent synapses lack surface-expressed AMPA receptors but contain NMDA receptors. Immediately after LTP induction (less than 15 min), GluA2-lacking AMPA receptors are selectively incorporated at silent synapses. Calcium influx through GluA2-lacking AMPA receptors drives their subsequent replacement by GluA2-containing receptors (more than 15 min) during LTP consolidation.
This work characterizing newly incorporated AMPA receptors at silent synapses represents the most direct analysis of the rectification properties of synaptic AMPA receptors during LTP, to date. The design of the experiment allows the newly incorporated receptors to be analysed in isolation because they are added to synapses that originally lack AMPA receptors and the rectification properties are measured in the presence of NMDA receptor blockade. Thus, a pure population of newly incorporated receptors are assayed. Our work shows that the new AMPA receptors exhibit very strong inward rectification consistent in magnitude with that observed at CA1 synapses from neurons in which GluA2 has been knocked out [35]. This suggests that silent synapses preferentially incorporate AMPA receptors that lack the GluA2 subunit. This work confirms the previous finding [24] that the inwardly rectifying population of AMPA receptors expressed during LTP is transient. In agreement with that study, the rectifying population of receptors is only present during the early phase of LTP and is replaced by non-rectifying receptors after approximately 20–30 min.
Despite the accumulating evidence for a role of GluA2-lacking AMPA receptors in the expression of LTP in several brain regions [18,25–28], there is controversy as to the role GluA2-lacking AMPA receptors play in the expression of hippocampal LTP. Two studies failed to find evidence for such a role forGluA2-lacking AMPA receptors [29,30]; however, two other studies do provide evidence for incorporation of GluA2-lacking AMPA receptors in LTP [31,32]. Based on the present results, one possibility to explain the inconsistent data is that GluA2-lacking AMPA receptors are selectively incorporated at silent synapses in CA1 pyramidal neurons. Silent synapses are developmentally downregulated and may also be regulated in response to experience. Thus, a possibility is that there was a lack of silent synapses in studies that failed to observe changes in GluA2 content during LTP.
One important proposed role of the GluA2-lacking AMPA receptors initially incorporated during LTP is that they drive consolidation of LTP in a mechanism requiring calcium influx through these receptors. This has been proposed as a mechanism to tag newly potentiated synapses that then require subsequent activity to stabilize the potentiation at these inputs [18,24]. Indeed, it has been shown that such newly potentiated synapses require synaptic activity for LTP consolidation and that blockade of GluA2-lacking AMPA receptors by philanthotoxin reverses LTP [24]. The current data showing reversal of LTP with diazo-2 uncaging after induction, now provide further evidence for such a model by showing the requirement for a rise in intracellular calcium concentration for the consolidation of the LTP. It is formally possible that intracellular diazo-2 uncaging is chelating a calcium rise produced by mechanisms other than calcium permeation through GluA2-lacking AMPA receptors. However, this seems unlikely as other calcium sources, such as NMDA receptor-dependent calcium influx or release of calcium from intracellular stores, have previously been shown not to be required for the stability of LTP after its induction [36]. For example, as shown in figures 1 and 2, LTP is stable in the presence NMDA receptor blockade. Moreover, although calcium-permeable AMPA receptors have a lower calcium-permeability compared with, for example, NMDA receptors, calcium transients in neurons resulting from the synaptic activation of calcium-permeable AMPA receptors are observed, supporting the idea that such calcium influx is functionally important. Previous work using intracellular uncaging of a calcium chelator showed that diazo-4 uncaging more than 2 s after LTP induction had no effect on subsequent LTP stability [37]. However, slices from animals 2–5 weeks of age were used in this study and silent synapses are developmentally downregulated in CA1 hippocampus by this age [10]; therefore, the lack of effect of calcium chelation on LTP consolidation shows that this study is consistent with the hypothesis that GluA2-lacking AMPA receptors are selectively incorporated at silent synapses.
In summary, we have shown that strongly inwardly rectifying AMPA receptors are incorporated at CA1 silent synapses during LTP, but are subsequently replaced by non-rectifying AMPA receptors. Further, we show that uncaging of an intracellular calcium chelator prevents consolidation of LTP. Our data are consistent with a mechanism in which GluA2-lacking, calcium-permeable AMPA receptors are selectively expressed at silent synapse during the early phase of LTP and replaced by GluA2-containing receptors during LTP consolidation in a mechanism requiring calcium influx through the calcium-permeable AMPA receptors. The requirement for calcium influx through GluA2-lacking AMPA receptors to drive the switch to GluA2-containing receptors during CA1 LTP consolidation is very similar to the mechanism described for plasticity at cerebellar stellate cell synapses [21]. This suggests that such a mechanism may be a common process in the brain underlying the GluA2-subunit composition switch during similar forms of plasticity described at inputs in primary sensory cortex, VTA, amygdala and hypothalamus [18,25–28].
4. Material and methods
Whole-cell patch-clamp recordings were made from CA1 pyramidal neurons in acute hippocampal slices using standard techniques as previously described [24,38]. Acute slices prepared from rats aged 5–8 days old (for silent synapse experiments) or two weeks old (for diazo-2 experiments) were immersed in an extracellular solution comprising (mM): 125 NaCl, 3.25 KCl, 1.25 NaHPO4, 25 NaHCO3, 2.5 CaCl2, 1.3 MgSO4, 10 glucose, 0.1 picrotoxin, saturated with 95% O2/5% CO2. The intracellular solution was as follows (mM): 135 CsMeSO4, 8 NaCl, 10 HEPES, 0.5 EGTA, 4 Mg-ATP, 0.3 Na-GTP, 5 QX314, 0.1 spermine and pH 7.2, 285 mOsm. All recordings were performed at room temperature. For the diazo-2 experiments, 10 mM diazo-2 was added to the intracellular solution and CsMeSO4 was reduced to 130 mM. EPSCs, recorded at a holding potential of −70 mV, were elicited by extracellular stimulation of CA3 axons using a stimulating electrode placed in stratum radiatum, at a frequency of 0.5 Hz for silent synapse experiments, and at 0.2 Hz for diazo-2 experiments. Signals were amplified using an Axopatch 700B patch-clamp amplifier and filtered at 5 Hz. Data were digitized at 10 kHz and acquired on PC using Signal software. EPSC amplitude, direct current, series resistance and input resistance were measured and displayed online. Recordings were terminated if series resistance changed by more than 20%.
For silent synapse experiments, minimal stimulation was used whereby initially a stimulus intensity sufficient to evoke EPSCs was used, and then gradually reduced in intensity until just subthreshold for eliciting AMPA receptor-mediated EPSCs. One hundred trials were then collected at 0.5 Hz (at −70 mV holding potential) as a baseline followed by an LTP induction protocol in which holding potential was changed to 0 mV and stimulation continued uninterrupted for 100 trials at 0.5 Hz. The cell was returned to −70 mV and monitored for a further 100 trials to determine whether LTP had been induced, characterized by the appearance of EPSCs. We did not attempt to detect ESPCs mediated only by NMDA receptors during baseline, because the unblocking of NMDA receptors to measure such EPSCs would itself induce plasticity.
For diazo-2 experiments, LTP was induced by pairing 100 stimuli at 1 Hz with a holding potential of 0 mV. Rectification index is calculated as (EPSC+40/EPSC−70) × (7/4). For the diazo-2 uncaging experiments, a 1 s UV light pulse was applied to the recorded cell through the objective (×40). The calcium buffering properties of diazo-2 before and following uncaging has been well described in the literature [33,34]. This shows that diazo-2 at mM intracellular concentrations in whole-cell recordings from CA1 pyramidal neurons exhibits a small amount of calcium buffering, which is then greatly increased upon uncaging. This reflects the change in KD from 2.2 μM to 70 nM upon uncaging. Although we do not measure the degree of calcium buffering in this work, the ability to induce LTP in the presence of diazo-2 and the block of LTP following diazo-2 uncaging indicates that a significant increase in calcium buffering was achieved under our experimental conditions. Drugs were purchased from Sigma, Invitrogen or Tocris. For statistical comparisons, a two-tailed Student's t-test was used; p < 0.05 was considered significant.
Funding statement
This research is supported by the NINDS Intramural programme.
References
- 1.Malenka RC, Bear MF. 2004. LTP and LTD: an embarrassment of riches. Neuron 44, 5–21. ( 10.1016/j.neuron.2004.09.012) [DOI] [PubMed] [Google Scholar]
- 2.Bliss TV, Collingridge GL. 1993. A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361, 31–39. ( 10.1038/361031a0) [DOI] [PubMed] [Google Scholar]
- 3.Malinow R, Malenka RC. 2002. AMPA receptor trafficking and synaptic plasticity. Annu. Rev. Neurosci. 25, 103–126. ( 10.1146/annurev.neuro.25.112701.142758) [DOI] [PubMed] [Google Scholar]
- 4.Collingridge GL, Isaac JT, Wang YT. 2004. Receptor trafficking and synaptic plasticity. Nat. Rev. Neurosci. 5, 952–962. ( 10.1038/nrn1556) [DOI] [PubMed] [Google Scholar]
- 5.Isaac JT, Nicoll RA, Malenka RC. 1995. Evidence for silent synapses: implications for the expression of LTP. Neuron 15, 427–434. ( 10.1016/0896-6273(95)90046-2) [DOI] [PubMed] [Google Scholar]
- 6.Isaac JT. 2003. Postsynaptic silent synapses: evidence and mechanisms. Neuropharmacology 45, 450–460. ( 10.1016/S0028-3908(03)00229-6) [DOI] [PubMed] [Google Scholar]
- 7.Kerchner GA, Nicoll RA. 2008. Silent synapses and the emergence of a postsynaptic mechanism for LTP. Nat. Rev. Neurosci. 9, 813–825. ( 10.1038/nrn2501) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liao D, Hessler NA, Malinow R. 1995. Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature 375, 400–404. ( 10.1038/375400a0) [DOI] [PubMed] [Google Scholar]
- 9.Kullmann DM. 1994. Amplitude fluctuations of dual-component EPSCs in hippocampal pyramidal cells: implications for long-term potentiation. Neuron 12, 1111–1120. ( 10.1016/0896-6273(94)90318-2) [DOI] [PubMed] [Google Scholar]
- 10.Durand GM, Kovalchuk Y, Konnerth A. 1996. Long-term potentiation and functional synapse induction in developing hippocampus. Nature 381, 71–75. ( 10.1038/381071a0) [DOI] [PubMed] [Google Scholar]
- 11.Hanse E, Durand GM, Garaschuk O, Konnerth A. 1997. Activity-dependent wiring of the developing hippocampal neuronal circuit. Semin. Cell Dev. Biol. 8, 35–42. ( 10.1006/scdb.1996.0119) [DOI] [PubMed] [Google Scholar]
- 12.Wu G, Malinow R, Cline HT. 1996. Maturation of a central glutamatergic synapse. Science 274, 972–976. ( 10.1126/science.274.5289.972) [DOI] [PubMed] [Google Scholar]
- 13.Ashby MC, Isaac JT. 2011. Maturation of a recurrent excitatory neocortical circuit by experience-dependent unsilencing of newly formed dendritic spines. Neuron 70, 510–521. ( 10.1016/j.neuron.2011.02.057) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Isaac JT, Crair MC, Nicoll RA, Malenka RC. 1997. Silent synapses during development of thalamocortical inputs. Neuron 18, 269–280. ( 10.1016/S0896-6273(00)80267-6) [DOI] [PubMed] [Google Scholar]
- 15.Busetto G, Higley MJ, Sabatini BL. 2008. Developmental presence and disappearance of postsynaptically silent synapses on dendritic spines of rat layer 2/3 pyramidal neurons. J. Physiol. 586, 1519–1527. ( 10.1113/jphysiol.2007.149336) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Daw MI, Scott HL, Isaac JT. 2007. Developmental synaptic plasticity at the thalamocortical input to barrel cortex: mechanisms and roles. Mol. Cell Neurosci. 34, 493–502. ( 10.1016/j.mcn.2007.01.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dingledine R, Borges K, Bowie D, Traynelis SF. 1999. The glutamate receptor ion channels. Pharmacol. Rev. 51, 7–61. [PubMed] [Google Scholar]
- 18.Isaac JT, Ashby MC, McBain CJ. 2007. The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron 54, 859–871. ( 10.1016/j.neuron.2007.06.001) [DOI] [PubMed] [Google Scholar]
- 19.Liu SJ, Zukin RS. 2007. Ca2+-permeable AMPA receptors in synaptic plasticity and neuronal death. Trends Neurosci. 30, 126–134. ( 10.1016/j.tins.2007.01.006) [DOI] [PubMed] [Google Scholar]
- 20.Liu SJ, Cull-Candy SG. 2002. Activity-dependent change in AMPA receptor properties in cerebellar stellate cells. J. Neurosci. 22, 3881–3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu SQ, Cull-Candy SG. 2000. Synaptic activity at calcium-permeable AMPA receptors induces a switch in receptor subtype. Nature 405, 454–458. ( 10.1038/35013064) [DOI] [PubMed] [Google Scholar]
- 22.Gardner SM, Takamiya K, Xia J, Suh JG, Johnson R, Yu S, Huganir RL. 2005. Calcium-permeable AMPA receptor plasticity is mediated by subunit-specific interactions with PICK1 and NSF. Neuron 45, 903–915. ( 10.1016/j.neuron.2005.02.026) [DOI] [PubMed] [Google Scholar]
- 23.Liu SJ, Cull-Candy SG. 2005. Subunit interaction with PICK and GRIP controls Ca2+ permeability of AMPARs at cerebellar synapses. Nat. Neurosci. 8, 768–775. ( 10.1038/nn1468) [DOI] [PubMed] [Google Scholar]
- 24.Plant K, Pelkey KA, Bortolotto ZA, Morita D, Terashima A, McBain CJ, Collingridge GL, Isaac JT. 2006. Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nat. Neurosci. 9, 602–604. ( 10.1038/nn1678) [DOI] [PubMed] [Google Scholar]
- 25.Shepherd JD. 2012. Memory, plasticity and sleep: a role for calcium permeable AMPA receptors? Front. Mol. Neurosci. 5, 49 ( 10.3389/fnmol.2012.00049) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Man HY. 2011. GluA2-lacking, calcium-permeable AMPA receptors—inducers of plasticity? Curr. Opin. Neurobiol. 21, 291–298. ( 10.1016/j.conb.2011.01.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tukey DS, et al. 2013. Sucrose ingestion induces rapid AMPA receptor trafficking. J. Neurosci. 33, 6123–6132. ( 10.1523/JNEUROSCI.4806-12.2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hong I, Kim J, Kim J, Lee S, Ko HG, Nader K, Kaang BK, Tsien RW, Choi S. 2013. AMPA receptor exchange underlies transient memory destabilization on retrieval. Proc. Natl Acad. Sci. USA 110, 8218–8223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adesnik H, Nicoll RA. 2007. Conservation of glutamate receptor 2-containing AMPA receptors during long-term potentiation. J. Neurosci. 27, 4598–4602. ( 10.1523/JNEUROSCI.0325-07.2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gray EE, Fink AE, Sarinana J, Vissel B, O'Dell TJ. 2007. Long-term potentiation in the hippocampal CA1 region does not require insertion and activation of GluR2-lacking AMPA receptors. J. Neurophysiol. 98, 2488–2492. ( 10.1152/jn.00473.2007) [DOI] [PubMed] [Google Scholar]
- 31.Guire ES, Oh MC, Soderling TR, Derkach VA. 2008. Recruitment of calcium-permeable AMPA receptors during synaptic potentiation is regulated by CaM-kinase I. J. Neurosci. 28, 6000–6009. ( 10.1523/JNEUROSCI.0384-08.2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu Y, Allen M, Halt AR, Weisenhaus M, Dallapiazza RF, Hall DD, Usachev YM, McKnight GS, Hell JW. 2007. Age-dependent requirement of AKAP150-anchored PKA and GluR2-lacking AMPA receptors in LTP. EMBO J. 26, 4879–4890. ( 10.1038/sj.emboj.7601884) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sah P, Clements JD. 1999. Photolytic manipulation of [Ca2+]i reveals slow kinetics of potassium channels underlying the afterhyperpolarization in hippocampal pyramidal neurons. J. Neurosci. 19, 3657–3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kamiya H, Zucker RS. 1994. Residual Ca2+ and short-term synaptic plasticity. Nature 371, 603–606. ( 10.1038/371603a0) [DOI] [PubMed] [Google Scholar]
- 35.Lu W, Shi Y, Jackson AC, Bjorgan K, During MJ, Sprengel R, Seeburg PH, Nicoll RA. 2009. Subunit composition of synaptic AMPA receptors revealed by a single-cell genetic approach. Neuron 62, 254–268. ( 10.1016/j.neuron.2009.02.027) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reyes M, Stanton PK. 1996. Induction of hippocampal long-term depression requires release of Ca2+ from separate presynaptic and postsynaptic intracellular stores. J. Neurosci. 16, 5951–5960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Malenka RC, Lancaster B, Zucker RS. 1992. Temporal limits on the rise in postsynaptic calcium required for the induction of long-term potentiation. Neuron 9, 121–128. ( 10.1016/0896-6273(92)90227-5) [DOI] [PubMed] [Google Scholar]
- 38.Terashima A, Pelkey KA, Rah JC, Suh YH, Roche KW, Collingridge GL, McBain CJ, Isaac JT. 2008. An essential role for PICK1 in NMDA receptor-dependent bidirectional synaptic plasticity. Neuron 57, 872–882. ( 10.1016/j.neuron.2008.01.028) [DOI] [PMC free article] [PubMed] [Google Scholar]