

Abstract
Connections between neurons can undergo long-lasting changes in synaptic strength correlating with changes in structure. These events require the synthesis of new proteins, the availability of which can lead to cooperative and competitive interactions between synapses for the expression of plasticity. These processes can occur over limited spatial distances and temporal periods, defining dendritic regions over which activity may be integrated and could lead to the physical rewiring of synapses into functional groups. Such clustering of inputs may increase the computational power of neurons by allowing information to be combined in a greater than additive manner. The availability of new proteins may be a key modulatory step towards activity-dependent, long-term growth or elimination of spines necessary for remodelling of connections. Thus, the aberrant growth or shrinkage of dendritic spines could occur if protein levels are misregulated. Indeed, such perturbations can be seen in several mental retardation disorders, wherein either too much or too little protein translation exists, matching an observed increase or decrease in spine density, respectively. Cellular events which alter protein availability could relieve a constraint on synaptic competition and disturb synaptic clustering mechanisms. These changes may be detrimental to modifications in neural circuitry following activity.
Keywords: dendritic spines, clustered plasticity, structural plasticity, spine elimination, competition, mental retardation
1. Functional and structural plasticity
During development, the nervous system undergoes an important period of synaptogenesis in which the proper connections are established between neurons. This period is hallmarked by robust formation and pruning of synapses, a process that is shaped by activity [1]. Much less is known about the relationship between physical synaptic changes and learning in the mature brain and whether remodelling of connections can occur. The discovery of long-term potentiation (LTP) 40 years ago shed light on a unique and critical ability of neurons to change the efficacy of their connections upon learning [2]. Depending on the nature of the activity, bidirectional modifications of synaptic connections can occur. However, these changes need to be physically stored over long periods of time and much less is known about how this is accomplished; specifically, what is the relationship between the structure and function of synapses. The direct connection between these first became clear following experiments in which new spine growth upon LTP induction was demonstrated [3]. The advent of precise glutamate uncaging, in combination with two-photon imaging at single spines, allowed for the induction of synaptic plasticity at the level of single inputs that showed a direct physical enlargement of the stimulated spines [4]. A linear relationship between spine volume and the amount of current in a spine was also established [5]. Analogously, synaptic depression has been shown to lead to spine shrinkage [6,7], although the specific effects of activity on single spines have not been defined as precisely as for the case of synaptic potentiation. Taken together, these findings support the idea that bidirectional changes in efficacy correlate with bidirectional structural modifications.
2. Cooperation and competition
Notably, there are still many details to be worked out regarding how different forms of activity are stored at the synaptic level, especially with regard to changes that are long lasting. One reason for this is owing to an important mechanistic difference between short-lasting (less than 2 h) and long-lasting (3 h or more) plasticity, in that the former does not require protein synthesis, while the latter does (reviewed in [8]). These changes add an extra dimension of complexity because the availability of new proteins contributes to the facilitation of plasticity between synapses, a phenomenon that was first described by the synaptic tagging and capture hypothesis [9,10]. It describes cooperativity between differentially activated inputs, in which strong stimulation of one pathway contributes to the expression of long-lasting plasticity at the second, weakly stimulated pathway, when new proteins are synthesized and shared. The impact of protein availability on the kinetics of synaptic cooperation is highlighted by experiments probing these interactions at individual spines. When protein independent forms of plasticity are induced, cooperation between inputs, termed ‘cross-talk’, occurs for a limited time and over short distances (10 min/10 µm) [11]. By contrast, if new proteins are produced, cooperation between inputs can occur for up to 70 µm and over the course of 1.5 h [12]. The presence of proteins confers plasticity even to synapses stimulated with subthreshold activity, and can lead to potentiation and spine growth at these sites that last for hours. Indeed, earlier experiments demonstrating that dendritic stimulation-induced growth of multiple spines within a 70 µm distance hinted that such interactions could occur, although the specific location and number of stimulated spines were unknown [13]. Therefore, depending on the nature of the activity, cellular mechanisms may be activated which lead to the growth of neighbours over protracted spatial and temporal parameters. Interestingly, protein cooperation can occur even between synapses expressing different forms of plasticity. A series of experiments by Sajikumar & Frey [14] and Sajikumar et al. [15] showed that induction of LTP can be coopted to enable long-lasting long-term depression (LTD; termed cross-tagging). This implies that a similar set of proteins may be required for different types of plasticity. However, not all the proteins required were the same. The blockade of PKMζ selectively inhibited LTP while still allowing for the conversion of LTD from a short- to a long-lasting form [15]. It will be interesting to see in future studies whether and how specific proteins contribute to activity-dependent structural changes.
Facilitative interactions are not the only ones influenced by the presence of newly made proteins. The dendritic availability of new proteins, or limited availability, sets up a competitive environment for the expression of plasticity if these proteins need to be shared. Indeed, field recording experiments demonstrated competition for the functional expression of LTP when multiple pathways are stimulated under protein synthesis blockade [16]. On a different scale, competition for structural plasticity is observed between neighbouring spines (approx. 20 µm apart) when protein availability is limited. After costimulation of multiple spines under these conditions, their physical growth was negatively correlated with one another [12]. Thus, the availability of proteins during long-lasting forms of plasticity may contribute to the physical organization of synapses by providing a constraint on how many coactive inputs ultimately cooperate with one another at a particular time and within a particular space. Determining the specific parameters over which competition functions will be key to understanding the learning rules that underlie structural plasticity and neuronal function.
3. Spine shrinkage and elimination with long-term depression
While protein-driven cooperative and competitive interactions during potentiation have begun to be characterized, much less is known about whether such interactions, and their resulting structural consequences, occur during protein synthesis-dependent synaptic depression. There is some evidence to support the idea that bidirectional structural changes can occur. Upon the induction of NMDA-mediated synaptic depression, which does not require protein synthesis, shrinkage or retraction of spines can occur [6,7,17]. As the presence of proteins has been shown to facilitate spine growth, it was unclear how they would impact structural changes following synaptic depression. A recent study by Ramiro-Cortés and Israely examines this question by studying the structural correlates of a well-known protein synthesis-dependent form of synaptic depression mediated by metabotropic glutamate receptors (mGluRs) [18,19]. Inducing depression with the group I mGluR agonist DHPG led to the robust shrinkage and elimination of spines in a protein synthesis-dependent manner, which importantly, could be observed for up to 24 h [19]. An interesting question still to be addressed is whether structural correlates of synaptic depression exist at the single spine level, and if so, whether proteins can facilitate functional and structural plasticity between multiple synapses. Thus far, the induction of structural LTD through glutamate uncaging at single spines has been biased towards occurring through NMDA-dependent mechanisms [17] and does not necessarily lead to a reduction in spine volumes [17]. It remains to be determined whether protein synthesis-dependent depression can be induced structurally at the level of single spines or whether it requires the activation of several inputs.
It is worth mentioning that the bidirectional protein synthesis-dependent structural plasticity discussed thus far, appears to be more universal in its ability to induce structural changes at spines of all sizes, compared with its shorter lived counterpart. Specifically, it has been reported that protein synthesis-independent synaptic potentiation is easier to induce at smaller spines [4], and likewise these smaller spines are more likely to shrink following NMDA-mediated synaptic depression [17] (figure 1). Conversely, strong potentiation leads to growth of spines of different sizes when new proteins are made [12]. Similarly to the case of potentiation, protein synthesis-dependent LTD leads to spine shrinkage and elimination that is independent of spine size [19] (figure 1). Although spine shrinkage or elimination induced by mGluR-LTD requires protein synthesis [19] and the pool of proteins required seems to be similar to those needed for late-LTP [14], it is unknown which or how new proteins can lead to these structural changes. One candidate is Arc/Arg3.1, which is translated upon mGluR-LTD induction, leading to the endocytosis of AMPA receptors on postsynapses [20,21]. Endocytosis may be one way by which to reduce spine surface area, and this could provide a mechanism by which to effect concurrent spine shrinkage and depression of synaptic conductances following activity [22]. It remains to be determined how specific molecular pathways are involved in implementing the functional consequences of LTD. These findings highlight protein synthesis-dependent synaptic plasticity as a potentially robust mechanism by which to remodel spines of various sizes, a mechanism that may be useful in order to rewire neuronal connections, as discussed below.
Figure 1.
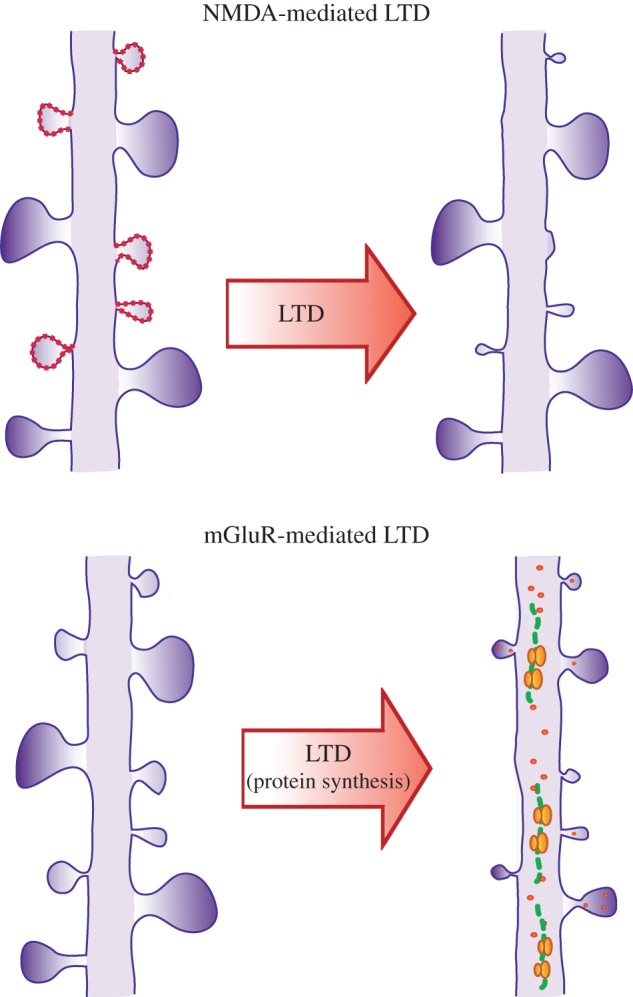
Structural plasticity mediated by LTD. LTD mediated by NMDAR induces shrinkage preferentially of small spines (depicted with dotted lines) and does not necessarily require protein synthesis. By contrast, LTD mediated by mGluRs, specifically group I, induces spine shrinkage and elimination that requires protein synthesis and is independent of spine size; both small and large spines can potentially be reduced in size in response to synaptic depression. (Online version in colour.)
Overall, the data summarized above indicate that the initiation and maintenance of long-lasting, bidirectional structural changes requires protein synthesis, and that within a dendritic domain, spines can compete or cooperate in order to express the different forms of plasticity. These interactions may comprise a mechanism by which to remodel synaptic connectivity depending on the nature of the activity. In order for such remodelling to occur, certain inputs would be potentiated while others would be reduced or lost, through growth or elimination of spines, respectively. Thus, plasticity mechanisms which extend beyond the level of a single synapse may have significant implications for how a neuron's structure is shaped in the long term.
4. Synapse clustering
One such outcome could be the clustering of synapses, which has been the subject of growing interest over recent years owing to its implications for the storage capacity of a neuron and a network. Synapses do not necessarily act as independent, linearly summed units on the dendritic arbour; instead, if multiple nearby synapses are activated in concert, supralinear processing can occur (for review, see [23]). This supralinear processing is caused by a local depolarization giving rise to a dendritic spike, facilitated by voltage-gated channels in the cell membrane. This spike will travel efficiently to the soma and has a high probability of making the cell fire. Thus, synapses which are activated concurrently and within a small spatial range will have a disproportionately large effect on the cell's output, because only a small number need to be activated to cause firing [24–26]. If a neuron's inputs could be rewired to achieve a clustered organization, the cell would be able to distinguish a larger number of patterns and have a higher processing power [24,25,27]. Furthermore, the potential to disconnect and reconnect the cells can change the overall wiring diagram of the network, allowing it to store more information [28].
Recently, there have been various findings which have shown functional clustering of synapses—i.e. the concurrent activation of inputs closely together on a dendritic branch—both in vitro and in vivo [29–31]. These studies used either calcium imaging to directly observe synapse activation [29,31] or the presence of tagged GluA1 receptors in the synapse as a marker of recent LTP [30], to conclude that in pyramidal neurons of the hippocampus or neocortex, synapses which are close together have an increased likelihood of being activated within a short time frame of one another. In these studies, such clustering was observed at different stages of development, from just after birth to young adult, and over distances of 8 –20 µm. Interestingly, these spatial parameters fit well within the bounds of cooperative and competitive plasticity mechanisms that occur during synaptic potentiation. It will be interesting to determine whether the spatial organization of synapses influences how specific proteins act, depending on the type of plasticity and the type of cooperativity that is induced [32].
If the functional clustering discussed above could drive the physical creation of anatomically distinct groups of spines, then it should be possible to find evidence of these structural clusters in vivo. Indeed, Yadav et al. [33] found that anatomical spine clusters (groups of three or more) occur significantly more frequently than chance within apical dendrites of layer III cells of the monkey prefrontal cortex. This study reveals the capability of neurons to spatially organize spines, but it does not prove a causal relationship between functional and structural changes. This latter point requires that the formation of clusters be driven by activity or learning. To begin to address this issue, two studies have looked for cluster formation in neural regions that are involved in encoding a specific task; they find that learning increases the prevalence of clustered synapses in those particular areas. In one case, rearing owls wearing prism goggles lead to an increase in clustered spines specifically in fields of the barn owl auditory system that correspond to the newly shifted topographic map [34]. Subsequently, learning a forelimb reaching task in mice was shown to correlate with the appearance of clustered spines in the motor cortex [35]. These studies suggest that activity in a given region could lead to structural clustering. It will be important to determine which activity patterns lead to branch specific clustering, and if concurrent activation of spines during learning precedes their structural organization. This question will require the monitoring of both the activity and the structure of dendritic regions throughout the learning process.
5. Synaptic interactions leading to spine clustering
The findings detailed above make a plausible argument for the clustering of dendritic spines in response to activity. Such remodelling could benefit the system by increasing the computational capacity of a neuron and circuit. For example, clustered synapses could more efficiently summate weak inputs and lead to neuronal firing. Remaining to be determined is what type of activity could lead to cluster formation; over what time scale would the formation of clusters be advantageous as a learning mechanism and what are the exact events by which activity could promote clusters. As outlined in figure 2, we propose that the following steps, which combine synaptic tagging and capture with spine remodelling and turnover, may be an avenue by which synaptic clustering can occur on the dendrite. Some of these have experimental support, whereas others will require further investigation in order to ascertain their biological relevance.
Figure 2.

Competition and cooperation in structural remodelling. (i) A dendritic region receives a pattern of activity. Some spines receive strong late-LTP inputs, in this example, spine a. Others (b,c,d,e) receive weaker early-LTP inputs, which set a synaptic tag. (ii) Signalling mechanisms including protein synthesis are initiated around spine a, which travel outwards along the dendrite. Tagged spines in the surrounding regions (b,c,d) compete for the resources. Spine e is outside the range of the proteins so will not enter the competition. (iii) The winning spines (a,b,c) are selectively strengthened. The losing spine (d) does not express long-lasting plasticity. At longer time scales: (iv) new spine growth can be initiated around the site of plasticity (dashed spines), while previously existing spines that were not strengthened, are removed (faint spines). (iv) A structural cluster is formed, reflecting the summation of activity which came into the dendrite. (Online version in colour.)
6. Activity-dependent structural correlates in vivo
A crucial mechanistic step to achieving activity-dependent structural clustering would be the ability to physically remodel connections, potentially through spine loss and gain. Chronic two-photon imaging in mice has allowed for the examination of spine dynamics in vivo over the course of many months (for reviews, see [35,36]). These data indicate that in the adult brain, different pools of spines exist—some of which turn over and some of which appear stable over the lifetime of the animal. These two elements provide essential components of a system which retains the ability to learn into adulthood, while being able to store long-lasting memories. Further studies have investigated the link between learning and spine remodelling in vivo. Some of the observations include: (i) spine formation—following a successful reaching task [37]; (ii) spine loss—associated with Pavlovian fear conditioning [38] and (iii) increased spine turnover-related to songbird learning [39]. All of these demonstrate structural flexibility of neurons in the respective brain areas involved in the encoding of behaviours. They also show that not all learning necessarily results in the same types of structural modifications. These results add weight to the idea that spine dynamics, and hence structural plasticity, may be the substrates for the storage of information.
7. Behavioural implications
The availability of proteins for cooperative and competitive interactions may have interesting outcomes for how information is processed by neurons during the encoding of behaviours. Indeed, the necessity for new protein synthesis has previously been established to be important for long-lasting memory formation [40]. Therefore, the coincidence of a salient event that leads to the production of new proteins may facilitate the subsequent encoding of otherwise weakly relevant information, potentially within the same engram. Indeed, experiments in which such ‘behavioural tagging’ was tested recently demonstrated the facilitation of memory storage [41]. When animals were trained in an inhibitory avoidance paradigm, which normally produces short-term learning, they were able to perform the task for up to an hour. However, when they were exposed to a novel open field prior to the training, the inhibitory avoidance learning lasted for over 24 h. This effect, which required dopamine receptors, was protein synthesis dependent because the infusion of the inhibitor anisomycin into the hippocampus prevented this facilitation. Thus, the exposure to novelty, which probably releases dopamine-induced protein synthesis allowing the facilitation of subsequent learning [41]. An additional study similarly found that novelty-induced behavioural tagging could occur during the learning of a non-fear inducing spatial learning task in which animals learned to locate a food reward within an arena [42]. Although it is not known which neural circuits encode this information, it is intriguing to consider that the various aspects of such events may be encoded within the same synaptic clusters. In this way, the availability of proteins during information processing would facilitate cognitive function by enhancing the ability to bind divergent, yet relevant, information together. Importantly, our brain is continuously presented with new information, yet not all of this is incorporated into any given memory. Perhaps a limited window of protein availability could serve to include timely experiences into a common learning trace, while avoiding the random incorporation of irrelevant information into any active circuit.
8. What is the relevance of synaptic competition for brain function?
We have discussed the fact that synaptic and structural plasticity can occur over a defined region when multiple inputs are coactive, by competing for limiting proteins that are necessary for the expression of plasticity. The physical boundaries over which these processes function could delineate a region for cooperative and competitive interactions, whereby spines could either grow or shrink. In addition to these potentiation mechanisms, competition for long-lasting synaptic depression (requiring protein availability) may also be involved in establishing which spines are lost or maintained, although this has yet to be explored experimentally. In this way, protein availability could select a subset of synapses to be incorporated into a physical cluster through either synaptic potentiation or depression. Therefore, normal levels of protein synthesis could provide an optimal functional range for: (i) the integration of information over long time scales and (ii) the refinement of neuronal connections through competitive interactions. An imbalance of protein translation could therefore affect the optimal range of protein concentration and result in altered spine densities (figure 3).
Figure 3.
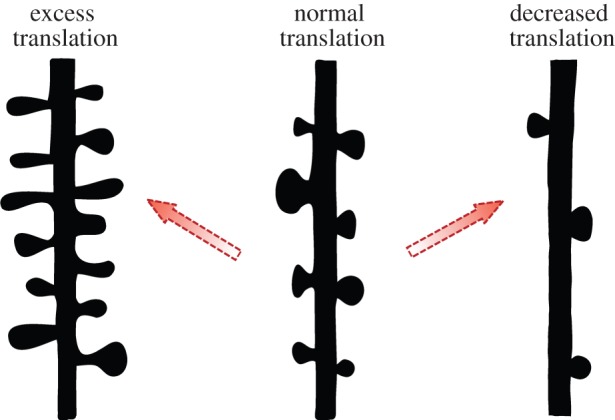
Hypothetical structural outcomes depending on protein availability. The distribution of spines on a dendritic arbour may be shaped by the balance between cooperation and competition among synapses for the expression of plasticity. The overall level of activity which a neuron experiences may contribute to maintaining an optimal balance of protein synthesis. In cases where mutations drive an increase or decrease in the general amount of proteins made, a reciprocal increase or decrease in spine density may result in a scenario which could change synaptic connectivity and neural circuits. (Online version in colour.)
Is there evidence to suggest that competitive mechanisms are involved in information storage? One clue may come from systems in which the balance of protein availability is biased towards one direction or another, shifting the ideal level necessary for neuronal function. Indeed, several mental retardation disorders have in common mutations that lead to an upregulation of protein synthesis [43,44]. A well-known example of this can be seen in the case of Fragile X, the most prevalent genetic disorder leading to cognitive deficits, when loss of the FMR1 gene product leads to an increase in protein synthesis through overactive mGluR signalling [45]. A key route to initiating translation is through the protein mammalian target of Rapamycin (mTOR) [46]. Mutations in several proteins, which regulate this pathway through PTEN/PI3K/AKT signalling, have also been linked to autism spectrum disorders, and show cognitive impairments. Specifically, these include mutations of TSC1 and TSC2 in tuberous sclerosis, NF1 in neurofibromatosis type 1, and PTEN hamartoma tumour syndrome [47]. For each of these cases, the proteins involved negatively regulate the mTOR pathway and their loss of function results in an increase in protein synthesis. Importantly, these mutations also result in structural alterations of increased spine density [48–51]. Thus, the resulting increase in protein levels in these disorders may be responsible for both the functional and structural abnormalities observed, perhaps by relieving a constraint on competitive processes that occur in neurons during the encoding of information.
The above described disorders contain mutations which lead to increased protein synthesis, and hence protein availability. How would a reciprocal decrease in protein translation affect cognitive function and dendritic structure? Some clues can be gleaned from the following two examples of Down syndrome and Rett syndrome, in which indeed cognitive dysfunction and reduced dendritic spine density are observed. In both of these disorders, perhaps owing to the many genes affected in each, the underlying mechanisms are not fully understood; however, there is evidence to suggest that the downregulation of protein translation could be partially responsible. In Down syndrome, chromosomal triplication of 21q results in the increased gene dosage of over 300 genes. However, a critical region was identified in rare cases with truncated duplications, from which the DSCR1 gene was identified (DSCR1) [52]. This protein was recently shown to interact with Fragile X mental retardation 1 protein (FMRP) and enhance its function in translational repression [53]. Thus, a mutation which leads to the opposite cellular effect of Fragile X, also demonstrates the opposite structural changes. In the case of Rett syndrome, mutations in the DNA methyltransferase MeCP2, which regulates the transcriptional silencing of genes, were found to be responsible for the disorder [54]. Recent evidence indicates that MeCP2 regulation correlates with the activity-dependent induction of BDNF transcription, and results in reduced dendritic growth and spine maturation [55]. As BDNF is known to trigger long-lasting synaptic plasticity and protein synthesis [56], chronic reductions in its transcription could lead to a state of decreased protein availability in the neuron, particularly in regions undergoing synaptic plasticity. Reductions in protein translation may affect the strengthening of inputs and predispose neurons to undergo dendritic spine pruning. Thus, assessing competitive plasticity mechanisms in these backgrounds will provide important insights regarding the role of protein availability during plasticity.
9. Concluding remarks
Since the original discovery of LTP, we have learned much about the complexity involved in modifying synaptic weights. We have discussed above how varied plasticity mechanisms requiring the synthesis of new proteins can effect long-lasting changes in synaptic and structural plasticity. These include events that lead to the strengthening of some synapses or that induce the shrinkage and elimination of others. In both cases, the presence of newly made proteins is critical for long-lasting structural changes. These processes may specifically delineate dendritic regions over which inputs can become integrated, and therefore may drive the physical creation of spine clusters. Such physical rewiring into clusters has implications for short-term processing of information, enhancing the subsequent efficacy of activity by optimizing dendritic integration. Importantly, such interactions may require an optimal balance of protein availability to allow for synaptic competition to occur between the inputs. In fact, interfering with this balance, either through blockade of protein synthesis or blockade of protein degradation, is detrimental to synaptic function [57,58]. In the absence of such plasticity constraints, the physical organization of spines may become compromised. Indeed, when too many proteins are made, for example in the case of several mental retardation disorders, spine density is aberrantly increased, while the counterpart is also true (figure 3). The production of too few proteins leads to reduced dendritic spine density in certain examples of cognitive dysfunction. Thus, in the long term, the connectivity of neural circuits may be impacted if spines cannot properly cooperate or compete. Further defining the learning rules for structural plasticity will be important for understanding how activity can shape connectivity in the brain.
Funding statement
This work was supported by the Champalimaud Foundation, Gulbenkian Foundation, Bial Foundation, Fundação para Ciência e Tecnologia and CONACyT.
References
- 1.Waites CL, Craig AM, Garner CC. 2005. Mechanisms of vertebrate synaptogenesis. Annu. Rev. Neurosci. 28, 251–274. ( 10.1146/annurev.neuro.27.070203.144336) [DOI] [PubMed] [Google Scholar]
- 2.Bliss TV, Lomo T. 1973. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J. Physiol. 232, 331–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Engert F, Bonhoeffer T. 1999. Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature 399, 66–70. ( 10.1038/19978) [DOI] [PubMed] [Google Scholar]
- 4.Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. 2004. Structural basis of long-term potentiation in single dendritic spines. Nature 429, 761–766. ( 10.1038/nature02617) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith MA, Ellis-Davies GC, Magee JC. 2003. Mechanism of the distance-dependent scaling of Schaffer collateral synapses in rat CA1 pyramidal neurons. J. Physiol. 548, 245–258. ( 10.1113/jphysiol.2002.036376) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou Q, Homma KJ, Poo MM. 2004. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron 44, 749–757. ( 10.1016/j.neuron.2004.11.011) [DOI] [PubMed] [Google Scholar]
- 7.Nagerl UV, Eberhorn N, Cambridge SB, Bonhoeffer T. 2004. Bidirectional activity-dependent morphological plasticity in hippocampal neurons. Neuron 44, 759–767. ( 10.1016/j.neuron.2004.11.016) [DOI] [PubMed] [Google Scholar]
- 8.Kelleher RJ, III, Govindarajan A, Tonegawa S. 2004. Translational regulatory mechanisms in persistent forms of synaptic plasticity. Neuron 44, 59–73. ( 10.1016/j.neuron.2004.09.013) [DOI] [PubMed] [Google Scholar]
- 9.Frey U, Morris RG. 1997. Synaptic tagging and long-term potentiation. Nature 385, 533–536. ( 10.1038/385533a0) [DOI] [PubMed] [Google Scholar]
- 10.Frey U, Morris RG. 1998. Weak before strong: dissociating synaptic tagging and plasticity-factor accounts of late-LTP. Neuropharmacology 37, 545–552. ( 10.1016/S0028-3908(98)00040-9) [DOI] [PubMed] [Google Scholar]
- 11.Harvey CD, Svoboda K. 2007. Locally dynamic synaptic learning rules in pyramidal neuron dendrites. Nature 450, 1195–1200. ( 10.1038/nature06416) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Govindarajan A, Israely I, Huang SY, Tonegawa S. 2011. The dendritic branch is the preferred integrative unit for protein synthesis-dependent LTP. Neuron 69, 132–146. ( 10.1016/j.neuron.2010.12.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Engert F, Bonhoeffer T. 1997. Synapse specificity of long-term potentiation breaks down at short distances. Nature 388, 279–284. ( 10.1038/41815) [DOI] [PubMed] [Google Scholar]
- 14.Sajikumar S, Frey JU. 2004. Late-associativity, synaptic tagging, and the role of dopamine during LTP and LTD. Neurobiol. Learn. Mem. 82, 12–25. ( 10.1016/j.nlm.2004.03.003) [DOI] [PubMed] [Google Scholar]
- 15.Sajikumar S, Navakkode S, Sacktor TC, Frey JU. 2005. Synaptic tagging and cross-tagging: the role of protein kinase Mzeta in maintaining long-term potentiation but not long-term depression. J. Neurosci. 25, 5750–5756. ( 10.1523/JNEUROSCI.1104-05.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fonseca R, Nagerl UV, Morris RG, Bonhoeffer T. 2004. Competing for memory: hippocampal LTP under regimes of reduced protein synthesis. Neuron 44, 1011–1020. [DOI] [PubMed] [Google Scholar]
- 17.Oh WC, Hill TC, Zito K. 2013. Synapse-specific and size-dependent mechanisms of spine structural plasticity accompanying synaptic weakening. Proc. Natl Acad. Sci. USA 110, E305–E312. ( 10.1073/pnas.1214705110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huber KM, Kayser MS, Bear MF. 2000. Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science 288, 1254–1257. ( 10.1126/science.288.5469.1254) [DOI] [PubMed] [Google Scholar]
- 19.Ramiro-Cortés Y, Israely I. 2013. Long lasting protein synthesis- and activity-dependent spine shrinkage and elimination after synaptic depression. PLoS ONE 8, e71155 ( 10.1371/journal.pone.0071155) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Waung MW, Pfeiffer BE, Nosyreva ED, Ronesi JA, Huber KM. 2008. Rapid translation of Arc/Arg3.1 selectively mediates mGluR-dependent LTD through persistent increases in AMPAR endocytosis rate. Neuron 59, 84–97. ( 10.1016/j.neuron.2008.05.014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Snyder EM, Philpot BD, Huber KM, Dong X, Fallon JR, Bear MF. 2001. Internalization of ionotropic glutamate receptors in response to mGluR activation. Nat. Neurosci. 4, 1079–1085. ( 10.1038/nn746) [DOI] [PubMed] [Google Scholar]
- 22.Collingridge GL, Isaac JT, Wang YT. 2004. Receptor trafficking and synaptic plasticity. Nat. Rev. Neurosci. 5, 952–962. ( 10.1038/nrn1556) [DOI] [PubMed] [Google Scholar]
- 23.Johnston D, Narayanan R. 2008. Active dendrites: colorful wings of the mysterious butterflies. Trends Neurosci. 31, 309–316. ( 10.1016/j.tins.2008.03.004) [DOI] [PubMed] [Google Scholar]
- 24.Poirazi P, Mel BW. 2001. Impact of active dendrites and structural plasticity on the memory capacity of neural tissue. Neuron 29, 779–796. ( 10.1016/S0896-6273(01)00252-5) [DOI] [PubMed] [Google Scholar]
- 25.Polsky A, Mel BW, Schiller J. 2004. Computational subunits in thin dendrites of pyramidal cells. Nat. Neurosci. 7, 621–627. ( 10.1038/nn1253) [DOI] [PubMed] [Google Scholar]
- 26.Branco T, Hausser M. 2011. Synaptic integration gradients in single cortical pyramidal cell dendrites. Neuron 69, 885–892. ( 10.1016/j.neuron.2011.02.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Govindarajan A, Kelleher RJ, Tonegawa S. 2006. A clustered plasticity model of long-term memory engrams. Nat. Rev. Neurosci. 7, 575–583. ( 10.1038/nrn1937) [DOI] [PubMed] [Google Scholar]
- 28.Chklovskii DB, Mel BW, Svoboda K. 2004. Cortical rewiring and information storage. Nature 431, 782–788. ( 10.1038/nature03012) [DOI] [PubMed] [Google Scholar]
- 29.Kleindienst T, Winnubst J, Roth-Alpermann C, Bonhoeffer T, Lohmann C. 2011. Activity-dependent clustering of functional synaptic inputs on developing hippocampal dendrites. Neuron 72, 1012–1024. ( 10.1016/j.neuron.2011.10.015) [DOI] [PubMed] [Google Scholar]
- 30.Makino H, Malinow R. 2011. Compartmentalized versus global synaptic plasticity on dendrites controlled by experience. Neuron 72, 1001–1011. ( 10.1016/j.neuron.2011.09.036) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takahashi N, Kitamura K, Matsuo N, Mayford M, Kano M, Matsuki N, Ikegaya Y. 2012. Locally synchronized synaptic inputs. Science 335, 353–356. ( 10.1126/science.1210362) [DOI] [PubMed] [Google Scholar]
- 32.Sajikumar S, Korte M. 2011. Metaplasticity governs compartmentalization of synaptic tagging and capture through brain-derived neurotrophic factor (BDNF) and protein kinase Mzeta (PKMzeta). Proc. Natl Acad. Sci. USA 108, 2551–2556. ( 10.1073/pnas.1016849108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yadav A, Gao YZ, Rodriguez A, Dickstein DL, Wearne SL, Luebke JI, Hof PR, Weaver CM. 2012. Morphologic evidence for spatially clustered spines in apical dendrites of monkey neocortical pyramidal cells. J. Comp. Neurol. 520, 2888–2902. ( 10.1002/cne.23070) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McBride TJ, Rodriguez-Contreras A, Trinh A, Bailey R, Debello WM. 2008. Learning drives differential clustering of axodendritic contacts in the barn owl auditory system. J. Neurosci. 28, 6960–6973. ( 10.1523/JNEUROSCI.1352-08.2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fu M, Zuo Y. 2011. Experience-dependent structural plasticity in the cortex. Trends Neurosci. 34, 177–187. ( 10.1016/j.tins.2011.02.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holtmaat A, Svoboda K. 2009. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat. Rev. Neurosci. 10, 647–658. ( 10.1038/nrn2699) [DOI] [PubMed] [Google Scholar]
- 37.Xu T, et al. 2009. Rapid formation and selective stabilization of synapses for enduring motor memories. Nature 462, 915–919. ( 10.1038/nature08389) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lai CS, Franke TF, Gan WB. 2012. Opposite effects of fear conditioning and extinction on dendritic spine remodelling. Nature 483, 87–91. ( 10.1038/nature10792) [DOI] [PubMed] [Google Scholar]
- 39.Roberts TF, Tschida KA, Klein ME, Mooney R. 2010. Rapid spine stabilization and synaptic enhancement at the onset of behavioural learning. Nature 463, 948–952. ( 10.1038/nature08759) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kandel ER. 2001. The molecular biology of memory storage: a dialogue between genes and synapses. Science 294, 1030–1038. ( 10.1126/science.1067020) [DOI] [PubMed] [Google Scholar]
- 41.Moncada D, Viola H. 2007. Induction of long-term memory by exposure to novelty requires protein synthesis: evidence for a behavioral tagging. J. Neurosci. 27, 7476–7481. ( 10.1523/JNEUROSCI.1083-07.2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang SH, Redondo RL, Morris RG. 2010. Relevance of synaptic tagging and capture to the persistence of long-term potentiation and everyday spatial memory. Proc. Natl Acad. Sci. USA 107, 19 537–19 542. ( 10.1073/pnas.1008638107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bear MF, Dolen G, Osterweil E, Nagarajan N. 2008. Fragile X: translation in action. Neuropsychopharmacology 33, 84–87. ( 10.1038/sj.npp.1301610) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bourgeron T. 2009. A synaptic trek to autism. Curr. Opin. Neurobiol. 19, 231–234. ( 10.1016/j.conb.2009.06.003) [DOI] [PubMed] [Google Scholar]
- 45.Bear MF, Huber KM, Warren ST. 2004. The mGluR theory of fragile X mental retardation. Trends Neurosci. 27, 370–377. ( 10.1016/j.tins.2004.04.009) [DOI] [PubMed] [Google Scholar]
- 46.Hoeffer CA, Klann E. 2010. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 33, 67–75. ( 10.1016/j.tins.2009.11.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kelleher RJ, III, Bear MF. 2008. The autistic neuron: troubled translation? Cell 135, 401–416. ( 10.1016/j.cell.2008.10.017) [DOI] [PubMed] [Google Scholar]
- 48.Huang W, et al. 2013. mTORC2 controls actin polymerization required for consolidation of long-term memory. Nat. Neurosci. 16, 441–448. ( 10.1038/nn.3351) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tavazoie SF, Alvarez VA, Ridenour DA, Kwiatkowski DJ, Sabatini BL. 2005. Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat. Neurosci. 8, 1727–1734. ( 10.1038/nn1566) [DOI] [PubMed] [Google Scholar]
- 50.Lin YL, Lei YT, Hong CJ, Hsueh YP. 2007. Syndecan-2 induces filopodia and dendritic spine formation via the neurofibromin-PKA-Ena/VASP pathway. J. Cell Biol. 177, 829–841. ( 10.1083/jcb.200608121) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fraser MM, Bayazitov IT, Zakharenko SS, Baker SJ. 2008. Phosphatase and tensin homolog, deleted on chromosome 10 deficiency in brain causes defects in synaptic structure, transmission and plasticity, and myelination abnormalities. Neuroscience 151, 476–488. ( 10.1016/j.neuroscience.2007.10.048) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fuentes JJ, Pritchard MA, Planas AM, Bosch A, Ferrer I, Estivill X. 1995. A new human gene from the Down syndrome critical region encodes a proline-rich protein highly expressed in fetal brain and heart. Hum. Mol. Genet. 4, 1935–1944. ( 10.1093/hmg/4.10.1935) [DOI] [PubMed] [Google Scholar]
- 53.Wang W, Zhu JZ, Chang KT, Min KT. 2012. DSCR1 interacts with FMRP and is required for spine morphogenesis and local protein synthesis. EMBO J. 31, 3655–3666. ( 10.1038/emboj.2012.190) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. 1999. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188. ( 10.1038/13810) [DOI] [PubMed] [Google Scholar]
- 55.Zhou Z, et al. 2006. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron 52, 255–269. ( 10.1016/j.neuron.2006.09.037) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kang H, Schuman EM. 1996. A requirement for local protein synthesis in neurotrophin-induced hippocampal synaptic plasticity. Science 273, 1402–1406. ( 10.1126/science.273.5280.1402) [DOI] [PubMed] [Google Scholar]
- 57.Yi JJ, Ehlers MD. 2005. Ubiquitin and protein turnover in synapse function. Neuron 47, 629–632. ( 10.1016/j.neuron.2005.07.008) [DOI] [PubMed] [Google Scholar]
- 58.Cajigas IJ, Will T, Schuman EM. 2010. Protein homeostasis and synaptic plasticity. EMBO J. 29, 2746–2752. ( 10.1038/emboj.2010.173) [DOI] [PMC free article] [PubMed] [Google Scholar]