

Abstract
Donald Hebb chose visual learning in primary visual cortex (V1) of the rodent to exemplify his theories of how the brain stores information through long-lasting homosynaptic plasticity. Here, we revisit V1 to consider roles for bidirectional ‘Hebbian’ plasticity in the modification of vision through experience. First, we discuss the consequences of monocular deprivation (MD) in the mouse, which have been studied by many laboratories over many years, and the evidence that synaptic depression of excitatory input from the thalamus is a primary contributor to the loss of visual cortical responsiveness to stimuli viewed through the deprived eye. Second, we describe a less studied, but no less interesting form of plasticity in the visual cortex known as stimulus-selective response potentiation (SRP). SRP results in increases in the response of V1 to a visual stimulus through repeated viewing and bears all the hallmarks of perceptual learning. We describe evidence implicating an important role for potentiation of thalamo-cortical synapses in SRP. In addition, we present new data indicating that there are some features of this form of plasticity that cannot be fully accounted for by such feed-forward Hebbian plasticity, suggesting contributions from intra-cortical circuit components.
Keywords: long-term potentiation, long-term depression, stimulus-selective response potentiation, monocular deprivation, amblyopia, perceptual learning
1. Introduction
The idea that information is stored in the brain by changing the strength of neuronal connections can be traced back as far as the nineteenth century [1]. However, the modern concept of the plastic synapse as a unit of information storage arose with the writing of Donald Hebb. In his famous book, The Organization of Behaviour, Hebb first considered the ways in which synaptic plasticity in visual cortex might account for the recognition of familiar visual cues by rodents [2]. He chose primary visual cortex (V1) as an exemplary system within which to frame his theories ‘not because vision has any unique significance but because it is in visual perception, with few exceptions, that the problem of patterning and form has been studied experimentally’ (p. 16). Although a huge amount of work has now been conducted in other systems since Hebb's writing, this deep understanding of V1 remains a major reason why it is an attractive system within which to investigate the mechanisms that support memory and other forms of experience-dependent plasticity.
Another advantage of V1, of course, is that we have known for 50 years that this is indeed a site of robust experience-dependent plasticity. To test the role of visual experience in the development of cortical connections that serve binocular vision, Wiesel & Hubel [3] examined the consequences of temporary monocular deprivation (MD) in young animals. Their dramatic findings revealed that visual experience indeed sculpts connections during early life. Hebbian principles were soon invoked to explain how correlated patterns of activity in the two eyes could be used to establish precise binocular connectivity [4–6]. To account for the effects of deprivation and other features of visual cortical plasticity, elaborations on Hebb's initial postulates were introduced that notably included assumptions about bidirectional synaptic plasticity and homosynaptic depression that have now been validated [7,8]. The experimental model of homosynaptic long-term depression (LTD) was introduced to facilitate investigation of the mechanisms that might underlie the effects of MD [9–12]. Here, we will review the evidence accumulated over the past 25 years that LTD mechanisms serve ocular dominance (OD) plasticity in visual cortex.
We also describe more recent experimental findings demonstrating that, as Hebb had surmised, discrimination of simple novel and familiar visual stimuli relies on long-lasting, input-specific plasticity within V1. Furthermore, we show that stimulus-selective visual plasticity uses the mechanisms of long-term synaptic potentiation (LTP) to modify visual responses. We argue that this form of stimulus-selective visual learning provides a platform upon which to gain a deep understanding of how Hebbian plasticity operates in tandem with other processes across visual cortical circuits to store information. Taken together, these studies of V1 make abundantly clear that the decades-old study of LTP and LTD has paid great dividends in furthering the understanding of experience-dependent cortical plasticity. It is also clear that Hebb's conjectures were prescient in terms of the form, function and locus of synaptic modification.
2. Ocular dominance plasticity
The study of cortical plasticity as a result of MD has revealed a great deal about how experience shapes receptive fields in V1 [13,14]. MD reliably causes OD plasticity in all mammals with binocular vision, manifest as a reduction of V1 responsiveness to input through the deprived eye and enhancement of responsiveness to input through the non-deprived eye. In mice (figure 1a), within the region of V1 described as the binocular zone (figure 1b), layer 4 cells receive converging synaptic input from the two eyes [17] (figure 1c). This input is innately biased in strength towards the eye contralateral to V1 in each hemisphere at a ratio of approximately 2 : 1. The contralateral bias can be detected with intrinsic imaging [18] or single unit recordings [19,20], and reflects the anatomical organization of the ascending visual pathway from the retinas through the dorsal lateral geniculate nucleus (dLGN) to the cortex in mice [21].
Figure 1.

Ocular dominance (OD) plasticity resulting from visual deprivation. (a) Head-fixed mice view phase-reversing sinusoidal grating stimuli while visual-evoked potentials (VEPs) and unit activity are recorded. An occluder is used to restrict visual input to one eye or the other. (b) Recordings are made from electrodes implanted in layer 4 of the binocular zone of V1 (green), receiving independent input from the contralateral (blue) and ipsilateral (yellow) eyes. (c) In binocular V1 of the mouse, thalamo-recipient principal cells in layers 2/3 and 4 receive independent inputs from contralateral and ipsilateral eyes. Pronounced feed-forward connections from layer 4 to layer 2/3 and horizontal connections within layers 2/3 also exist. Inhibitory cells receive thalamic and intra-cortical input and inhibit principal cells throughout cortex. (d) OD plasticity is assayed by suturing the contralateral eye for 3 days to deprive this eye of visual input. Monocular deprivation (MD) results in a significant reduction in the amplitude of VEPs driven by visual input through the contralateral eye. Example waveforms are displayed at the top of the figure. (e) Binocularly responsive units can be scored for ocular dominance by assessing the bias towards response to the contralateral or ipsilateral eye prior to MD (open circles). After 3 days of MD this OD index shifts away from the deprived eye towards the non-deprived eye (closed circles). (f) Pairing of low-frequency stimulation (LFS) of white matter and layer 4 cell depolarization in V1 slices induces thalamo-cortical LTD (open circles). MD occludes this Hebbian LTD, preventing it from being established long-term (closed circles). Data are reproduced from [15]. (g) Clathrin-dependent endocytosis can be blocked by expression of a peptide that mimics the cytoplasmic tail of the GluR2 subunit (GluR2-CT). Expression of the GluR2-CT peptide blocks LFS pairing-induced Hebbian thalamocortical (TC) LTD in layer 4 (dark grey) of V1 slices relative to GFP-only control (light grey). (h) This same treatment prevents the OD shift resulting from MD relative to the interleaved controls presented in (d). (i) The ocular dominance shift resulting from MD is also blocked by the GluR2-CT peptide. This block can be compared to controls shown in (e). (d, e, g, h and i) are reproduced from [16]. Throughout the figure asterisks denote comparisons revealing significance of p < 0.05.
Visual-evoked potentials (VEPs) provide a convenient assay to study the consequences of MD [22] and have the added benefit of stability over days [23], enabling explicit measurement of the contributions of loss and gain in responsiveness through the deprived and non-deprived eye, respectively, to the OD shift (albeit with the limitation that the VEP is more sensitive to changes in synaptic currents than to changes in spiking [24,25]). Suturing closed the contralateral eyelid during the fourth postnatal week causes a pronounced and significant loss of responsiveness through the deprived eye such that V1 becomes more-or-less equally responsive to input from the two eyes [20,26] (figure 1d,e). Longer periods of deprivation result additionally in potentiation of responsiveness to the non-deprived eye [19,26]. In the mouse, this capacity for gain through the non-deprived eye is retained into adulthood while the deprived eye depression no longer occurs reliably if MD is initiated from the second month of the animal's postnatal life onwards [23,27,28]. Two major questions have shaped this area of research: first, what are the mechanisms that mediate OD plasticity? Second, what are the mechanisms that change the quality of OD plasticity as the animal matures? Here, we will focus on the first question, with a particular emphasis on the means by which responsiveness of V1 to input through the deprived eye is reduced. This is a question of considerable practical importance, as the loss of cortical responsiveness triggered by poor quality vision during early childhood is a leading cause of human visual disability worldwide [29].
Degrading image formation by lid closure has no effect on average firing rates of dLGN neurons, but replaces temporally and spatially structured activity with spontaneous activity that can be described as ‘noise’ [30]. Building on Hebb's initial postulates about synaptic modification, Bienenstock, Cooper & Munro (BCM) [31] proposed that this synaptic noise in cortex actively triggers the loss of synaptic strength when the postsynaptic target of these synapses is weakly activated under the influence of other inputs. Thus, the BCM theory proposed that homosynaptic LTD of excitatory afferent synapses is chiefly responsible for the loss of visual cortical responsiveness after MD. The BCM theory inspired the experiments that eventually demonstrated the existence of homosynaptic LTD (reviewed by Bear [7] and Cooper & Bear [8]).
Just as with LTP, Hebbian LTD was first identified in the hippocampus [9,32], but it has since been observed at many synapses throughout the brain, including those impinging on excitatory neurons within layer 4 of V1 that receive direct dLGN input [15,16,33,34]. There is evidence that LTD in V1 is developmentally constrained [33–35], perhaps contributing to the loss in adulthood of deprived-eye depression following MD. LTD induction in V1 requires activation of NMDA (N-methyl-d-aspartate) receptors (NMDAR), but the downstream mechanisms vary according to the layer [15]. In layer 4, which receives most input from the thalamus, LTD is caused by NMDAR-mediated endocytosis of postsynaptic AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptors (AMPAR) and is sensitive to (i) specific peptides that interfere with the recognition of the GluA2 subunit C-terminus by the endocytosis machinery [36,37], (ii) the levels of the immediate early gene product Arc, which regulates AMPAR endocytosis rate [38,39] and (iii) the NR2A/NR2B subunit ratio of cortical NMDAR which regulates the intracellular signalling that triggers endocytosis [40–42]. A distinct form of LTD is expressed outside of layer 4 that depends on endocannabinoid signalling via the CB1 receptor and is likely to be expressed presynaptically [15,43], but there is evidence that some synapses in layer 3 also express LTD via AMPAR endocytosis [35]. It is well established that OD plasticity in vivo requires activation of cortical NMDAR [23,44–46], and the LTD model suggests how NMDAR might trigger the loss of visual responsiveness [47]. This ‘LTD hypothesis’ is now very well supported by experimental findings. Over the past decade extensive research has shown (i) that MD in visual cortex triggers LTD-like synaptic modifications and (ii) that the molecular mechanisms of LTD are required for the effects of MD.
Mimicry and occlusion are the two main criteria used to assess whether two different triggers of synaptic plasticity converge onto a common set of mechanisms [7]. This approach has been taken to establish, for example, that one-trial learning in the hippocampus [48,49] induces plasticity akin to LTP. A biochemical signature of LTD is the loss of surface expressed AMPAR and concomitant changes in AMPAR subunit phosphorylation at specific residues, and these same changes have been observed in visual cortex following brief MD [50]. Furthermore, the induction of LTD using electrical stimulation of the dLGN causes depression of VEP amplitude similar to that observed after MD. Thus, deprivation-induced depression and LTD mimic one another. In addition, the induction of synaptic depression by MD in vivo reduces the amount of LTD that can be achieved ex vivo [15,50] (figure 1f), suggesting that deprivation-induced depression also occludes LTD. Together, these findings indicate that MD induces LTD in visual cortex.
The common requirement of LTD and OD plasticity for NMDAR activation supports the hypothesis that LTD mechanisms are required for the effects of MD; however, it is known that NMDAR contribute to multiple forms of synaptic plasticity, e.g. LTP, as well as to polysynaptic information processing in the cortex. Thus, additional evidence has been required to establish that the molecular mechanisms of LTD actually contribute importantly to the functional consequences of MD. One approach has been to take advantage of highly specific manipulations of NMDAR-dependent AMPAR endocytosis. Two independent studies have now shown that intracellular delivery of peptides that interfere specifically with AMPAR endocytosis (G2CT and GluR23Y) prevent the OD shift in mouse visual cortex after 3–4 days of MD [16,35] (figure 1g,i). In addition, OD plasticity is completely absent in layer 4 of V1 in the Arc knockout mouse, which has impaired LTD and AMPAR trafficking [18]. In layer 3, where LTD has been shown to depend at least partly on cannabinoid signalling, the cannabinoid CB1 receptor blocker AM251 prevents the OD shift [51]. Finally, mice deficient in the NR2A subunit of NMDAR, which have impaired LTD and facilitated LTP in layer 4, exhibit a qualitatively different OD shift after MD that is expressed solely by potentiation of responses to the non-deprived eye; deprived eye depression is completely absent [42]. Thus, the mechanisms revealed by the study of homosynaptic LTD appear to be essential for the loss of cortical response after MD.
A requirement for LTD of excitatory synapses in triggering deprived-eye depression after MD does not rule out important contributions for modifications of GABA (gamma-aminobutyric acid)-mediated cortical inhibition in the expression of the OD shift [52–55], and successive generations of scientists have examined this issue with various methods in kittens and mice (reviewed by Smith & Bear [56]). To investigate whether inhibition plays an important role in the expression of OD plasticity in the mouse, experiments were performed recently using local infusion of inhibitors of GABAA receptors, bicuculline methiodide (BMI) [57] and gabazine (LA Khibnik, KK Cho, MF Bear 2010, unpublished data) (figure 2a,b), before and after induction of the OD shift. Removal of inhibition by these drugs has no effect on the innate 2 : 1 contralateral : ipsilateral eye OD ratio in layer 4 [57], indicating that this property is generated by the degree of feed-forward excitatory input to V1 [21]. Moreover, the drop in this OD ratio to 1 : 1 following MD is also maintained in the presence of BMI [57] (figure 2c). Thus, although modifications of inhibition clearly can occur as a result of MD [55], they are not required for expression of OD plasticity in principal cells (figure 1f–h). Much further work will be required to elucidate the functional importance of plasticity in inhibitory interneurons in V1.
Figure 2.
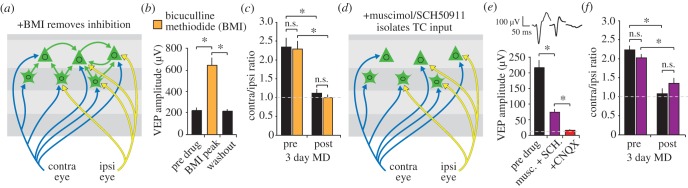
OD plasticity does not require inhibition for expression. (a) An in vivo pharmacological approach applies drugs locally around the VEP recording site in V1 in awake animals. Phasic inhibition can be blocked by applying the GABAA receptor antagonist bicuculline methiodide (BMI). (b) VEP amplitude is significantly enhanced as a result of BMI application (orange) but recovers after washout. (c) Measurement of VEPs driven independently through the two eyes reveals the approximately 2 : 1 ratio of the amplitude of contra- and ipsi-laterally driven VEPs, respectively. Despite the increase in VEP amplitude with BMI application this ratio is unchanged. After 3 days of MD, the contra : ipsi ratio falls significantly to approximately 1 : 1. Again this new ratio is unchanged in the presence of BMI, despite the overall increase in VEP amplitude, indicating a lack of requirement for modulated inhibition in the maintenance of the initial ratio and its shift as a result of MD. (d) Unit activity and, hence, all intra-cortical synaptic activity can be blocked through application of the GABAA receptor agonist muscimol. Thalamo-cortical input can be maintained also by locally infusing the GABAB receptor blocker SCH50911 to prevent non-specific muscimol binding pre-synaptically. (e) A TC VEP of significantly reduced amplitude can be isolated in the presence of the muscimol–SCH50911 cocktail (purple). This remaining VEP is completely abolished by the AMPAR antagonist CNQX (red), indicating that it is post-synaptically mediated. (f) The approximate 2 contra : 1 ipsi OD VEP ratio is maintained in the presence of this cocktail, as is expression of the shift as a result of 3 days of MD. (b, c, e and f) are reproduced from [57]. Asterisks denote significance of p < 0.05 while n.s. denotes higher p-values.
Even if the cause of the OD shift is a modification of excitatory synaptic transmission, the question remains as to which excitatory synapses are primarily responsible. It is known from the pioneering work of Hubel and Wiesel that long-term MD, lasting weeks or months, can shrink dLGN axon arbours, but it has been less clear the extent to which modification of thalamocortical (TC) synaptic transmission is responsible for the rapid loss of visual responsiveness during MD [58]. This question was addressed recently in the mouse using an in vivo pharmacological strategy to isolate purely TC synaptic VEPs [57]. By co-applying a cocktail of the GABAA receptor agonist muscimol with the GABAB receptor blocker SCH50911, it was possible to prevent all unit firing in an area of cortex while preserving TC input [59]. Under these circumstances, purely synaptic TC VEPs can be recorded in awake animals before or after deprived eye depression induced by three days of MD. Prior to any OD shift, the TC VEP shows a normal 2 : 1 contralateral:ipsilateral eye response ratio, indicating that TC input from the contralateral eye is twice as strong overall as input from the ipsilateral eye [21]. Deprived eye depression pushes the OD ratio to 1 : 1 and this ratio is retained in the presence of the inhibitory cocktail, indicating that OD shifts can be fully accounted for through TC plasticity (figure 2d–f). This conclusion receives additional support from an ultrastructural study showing shrinkage and loss of TC synapses in layer 4 after only 3 days of MD [60]. Although these experiments do not provide any insights into the degree to which MD-induced plasticity is distributed across visual cortical excitatory and inhibitory circuit elements, they do demonstrate that there is no requirement for further plasticity to account for the full OD shift.
Longer periods of MD (more than 5 days) cause potentiation of the cortical response to input through the non-deprived (ipsilateral) eye. Consensus has not yet been achieved on the mechanism for this homeostatic compensation, and it is reasonable to think that different mechanisms might predominate in different layers (thalamorecipient layer 4 versus layers 2/3), and at different ages (juvenile versus adult). With respect to visually evoked responses through the non-deprived eye, several observations have been made in binocular V1 that we summarize below.
— Non-deprived eye response potentiation can occur independently from deprived-eye depression. These phenomena are dissociated in adults [23,27] and in juvenile mice lacking the NR2A subunit of NMDAR [42] and tumour necrosis factor alpha (TNFα) [61], and on the C57BL/6JOlaHsd (6JOla) genetic background [62].
— Potentiation of VEPs requires visual experience through the non-deprived eye. Comparable changes are not observed following binocular deprivation or in the monocular segment after contralateral eye deprivation [26,63,64]. However, exceptions are worth noting. In some experiments [26,61,65], but not all [18], the deprived-eye response can drift up concurrently with open-eye potentiation. When it occurs, however, this effect is less pronounced than the potentiation of the open eye input [66].
— Non-deprived eye response potentiation is behaviourally significant. Long-term MD leads to a gain in visual grating acuity through the non-deprived eye [67]. Of relevance to the upward drift in the deprived-eye response mentioned above, there is no evidence to our knowledge of any ‘spontaneous’ recovery of visual acuity through the deprived eye with prolonged deprivation. However, it is well known that substantial visual acuity is maintained despite deprivation in the monocular segment of the visual field [68].
— Non-deprived eye response potentiation requires activation of cortical NMDAR. In juvenile mice, initiation of treatment with an NMDAR blocker following 3 days of MD, when deprived-eye depression is asymptotic, completely blocks potentiation of open-eye VEPs (and prevents upward drift of the deprived eye responses) [42]. Similarly, open eye response potentiation of units and VEPs is prevented in adults by treatment with an NMDAR blocker [27] or by adult-onset, cortex-specific genetic deletion of NMDARs [23].
Two models have dominated the discussion of potential mechanism. According to a ‘metaplasticity’ model, deprivation of the contralateral eye enables LTP-like strengthening of synapses serving the non-deprived ipsilateral eye via a metaplastic shift in the LTP threshold [7,8,69]. According to a ‘synaptic scaling’ model, the reduction in cortical activity wrought by deprivation and depression of the contralateral eye inputs causes all excitatory cortical synapses to undergo a general process of upward scaling of strength [61,65,70].
Observations that (i) open-eye potentiation requires visual experience and can occur in an input-specific manner [18,66], (ii) the LTP threshold in visual cortex is reduced by brief periods of visual deprivation, in part by a change in the NR2A/B ratio of synaptic NMDAR [40–42,71] and (iii) open-eye potentiation is abolished by NMDAR blockade [23,27,42], all support the metaplasticity model. Findings that (i) increases in the average miniature excitatory postsynaptic current (EPSC) amplitude and intrinsic excitability in layer 2/3 pyramidal neurons correlate with open-eye potentiation [70], (ii) deprived-eye responses can also rebound slightly at the same time as open-eye potentiation [26,65] and (iii) open-eye potentiation in vivo and synaptic scaling (but not LTP) ex vivo are both absent in juvenile mice lacking TNFα [61], all support the scaling model. We note that evidence supporting scaling is not inconsistent with metaplasticity. Because interocular correlations are still possible during MD for visual stimuli with low spatial frequencies, upward drift of deprived-eye responses could reflect an associative LTP-like process during open-eye potentiation [63]. And, as discussed at length elsewhere [72], failures to observe correlated changes in electrically induced synaptic plasticity ex vivo (e.g. LTP) and experience-dependent changes in vivo (e.g. non-deprived eye potentiation) do not constitute strong evidence that they are unrelated. On the other hand, it is more difficult to reconcile the input specificity and NMDAR-dependence of open-eye potentiation with scaling (see [8] for review).
Two very recent studies add a new twist to this issue. In one study, it was reported that two genetically defined mouse strains (TNFα−/− and 6JOla) fail to display synaptic scaling after MD, but exhibit normal open-eye potentiation in adult mice (>P90) [62]. These findings clearly indicate that scaling is not a mechanism for adult open-eye potentiation. However, as reported previously for the TNFα−/− mutants, juvenile mice on the 6JOla background failed to show both scaling and open-eye potentiation. These data suggest that the requirements for this form of plasticity vary according to age.
Even more provocative are data from an elegant recent study that employed intracellular recordings of eye-specific synaptic responses in vivo [25]. The authors confirmed that depression of VEPs and unit responses to deprived-eye stimulation is accounted for by rapid and long-lasting depression of visually evoked EPSCs. Unexpectedly, however, they found that inhibitory postsynaptic currents (IPSCs) driven by the non-deprived eye were markedly depressed after long-, but not short-term MD. Consequently, the ratio of excitation to inhibition (E/I) driven by the non-deprived eye is greatly increased after MD. Thus, input-specific modulation of inhibition offers a new mechanism to consider for non-deprived eye potentiation. How this modulation occurs remains to be determined, but the NMDAR dependence and input-specificity of non-deprived eye potentiation favours a model in which inhibition driven by the non-deprived eye is selectively reduced via LTD of excitatory TC synapses onto inhibitory interneurons.
Understanding these forms of plasticity has clinical as well as theoretical implications. MD is a model for some forms of amblyopia, a condition that affects a large number of people in the developed world and an even greater proportion of those that live in the developing world [29]. Amblyopia results from a wide variety of fairly common ocular dysfunctions, such as cataracts, strabismus or anisometropia. If these ocular dysfunctions are not treated until late in childhood, beyond the closure of the so-called critical period (as happens most often in the developing world), then permanent visual impairment results. This functional deficit is largely unrecoverable even if the eyes are returned to perfect working order.
Three major approaches are currently being pursued to improve the prognosis for the recovery of vision in adult patients, all exploiting knowledge of synaptic plasticity: first, finding ways to re-establish the conditions required for juvenile plasticity in the adult cortex [73]; second, exploiting the principles of metaplasticity to encourage the potentiation of synaptic inputs that have withered as a consequence of long-term deprivation [69,74,75]; and third, providing appropriate sensory experience through deprived, and perhaps non-deprived eyes, to induce plasticity that will maximally drive recovery of function [76]. This third approach attempts to exploit a pervasive and fascinating category of sensory plasticity known as perceptual learning [77]. As we now discuss, recent work in our laboratory has uncovered mechanisms for this type of learning in V1.
3. Stimulus selective response potentiation
Perceptual learning, resulting in improved perception through repeated sensory experience, occurs in adults as well as children [78,79]. This broad category of learning is characterized by an exquisite selectivity for the experienced stimulus. In the visual domain, gains in perception can be limited to the orientation or spatial frequency of the stimulus [80] and, in some cases, restricted to the eye through which the stimulus has been viewed [81,82]. These features are commonly invoked as psychophysical evidence that contributing plasticity resides prior to integration of sensory primitives into complex and generalized representations [83]. Indeed, evidence exists in humans and animals that stimulus-selective changes in the responsiveness of V1 occur during visual perceptual learning [84,85]. However, until recently, there have been few experimental findings to establish the exact location and mechanisms of plasticity supporting it.
Unlike many other species, orientation selectivity within V1 of rodents observes a confetti-like interleaved arrangement, meaning that VEPs recorded at any one site within V1 have approximately equal amplitudes across all orientations (figure 3a). This arrangement enables relatively easy internally controlled assessment of changes in the response to oriented stimuli through experience. Stimulus-selective response potentiation (SRP) is a form of plasticity observed in thalamo-recipient layer 4 of binocular V1 that shares features with visual perceptual learning [86]. SRP occurs in awake, head-fixed mice (figure 1a), in the absence of explicit reward or punishment, as they view a phase-reversing sinusoidal grating stimulus, resulting in an increase in the responsiveness of V1 (figure 3b). This increase in responsiveness is measured as a potentiation of the amplitude of VEPs one day after initial stimulus presentation, approaching saturation by around 5 days. SRP is highly selective for the orientation (figure 2c) and the spatial frequency of the stimulus that has been viewed (figure 3d) [87] and occurs repeatedly within an individual animal over a similar time-course for each new stimulus that is presented. SRP also lasts for at least a week and possibly many months (SF Cooke, MF Bear 2010, unpublished data) even in the absence of intervening stimulus presentation (figure 3b). Importantly, if the stimulus is viewed through just one eye (figure 1a), SRP does not transfer to the other eye (figure 3e). This observation is consistent not only with plasticity occurring prior to, or at the point of binocular integration but also suggests that plasticity is input-specific: in rodents, ocular inputs remain segregated until thalamo-recipient layers in binocular V1, at which point many cells are binocularly responsive as they receive independent synaptic input carrying information from the ipsilateral and contralateral eyes (figure 1c) [19,20]. Interestingly, SRP occurs at high but not low stimulus contrast (figure 3f), suggesting a requirement for cooperation between multiple strongly excited afferents in its induction [88]. Longevity, input specificity and this potential requirement for cooperativity are all consistent with SRP being mediated by LTP-like Hebbian synaptic plasticity. Given that it occurs in the mouse, SRP offers an opportunity for interventional experimental approaches to test this hypothesis. Although the mouse LGN has neurons with the property of orientation-selectivity, conferred by convergent feed-forward retinogeniculate connections [89], the evidence to be reviewed below places the mechanism for SRP in V1.
Figure 3.

Stimulus-selective response potentiation (SRP). (a) In the rodent, unlike in cats, ferrets and primates, a confetti-like, interleaved arrangement of orientation selectivity exists in primary visual cortex (V1). As a consequence, visual evoked potentials (VEPs) of approximately equal trough-peak amplitude are driven by visual stimuli of any orientation at a single recording site within binocular V1 without prior modification through experience. (b) SRP occurs over days in awake mice as they view full-field sinusoidal grating stimuli. VEP amplitude undergoes potentiation through SRP that is selective for the orientation that has been experienced, as revealed by presenting a new orientation (in this case X + 45°). SRP then independently occurs to this new orientation at an equivalent rate and magnitude to that observed for X°. SRP can occur sequentially over multiple stimulus orientations. Example waveforms are shown at the top. (c) SRP is exquisitely orientation-selective so that the familiar orientation (blue) evokes significantly larger VEPs than a novel (red) orientation that is shifted by as little as 5°. (d) SRP is selective for the spatial frequency as well as the orientation of the stimulus. An oriented grating presented only at 0.05 cycles per degree (CPD) induces SRP (blue) that does not transfer across presentations of the same orientation at other spatial frequencies. This is revealed by comparisons to a novel orientation (red) presented across the same range of spatial frequencies because VEPs vary in amplitude depending on spatial frequency regardless of prior experience. (e) SRP can be induced through one eye only (in this case the ipsi-lateral eye in yellow) and this plasticity fails to transfer to the opposite eye (in this case the contra-lateral eye in blue). (f) SRP does not occur at low stimulus contrasts (grey) as compared to high contrasts within the same animals (black). (b, e and f) are reproduced from [86]. (c,d) are reproduced from [87]. Asterisks denote significance of p < 0.05.
A number of molecular features of SRP indicate that it is supported by similar mechanisms to LTP. First, NMDARs are required for SRP induction. SRP is blocked by the systemic application of the selective NMDAR antagonist CPP (3-(2-carboxypiperazin-4-yl)propyl-1-phosphonic acid [86] and SRP is also blocked by local application of another antagonist, AP5, within V1 (SF Cooke, RW Komorowski, JP Gavornik, MF Bear 2013, unpublished data). The NMDAR is a central induction mechanism in canonical forms of LTP [90] and, more specifically, the induction of LTP in layer 4 of V1 depends upon this receptor [15,42]. Thus, it is likely that the NMDAR, through its combined ligand and voltage gating, serves as the detector of coincident pre and post-synaptic activity required for the induction of Hebbian synaptic plasticity that supports SRP in V1. Second, prevention of activity-dependent insertion of GluR1-containing AMPAR into the post-synaptic membrane prevents SRP expression. This block is accomplished by locally expressing a dominant-negative peptide in V1 that mimics the cytoplasmic tail of the GluR1 subunit (GluR1-CT), thereby sequestering an expression mechanism of many forms of LTP [91]. Third, SRP is completely absent in a mouse that lacks the immediate early gene Arc [18], which is required for the long-term maintenance of some forms of hippocampal LTP and many forms of memory [92,93]. As yet, it is unclear how Arc contributes to LTP maintenance, although as discussed above it plays an important role in the opposing process of LTD where it has a defined role in activity-dependent AMPAR endocytosis [94]. Finally, SRP is also susceptible to erasure by V1 infusion of the myristolated peptide ZIP (Z-pseudosubstrate inhibitory peptide) [87]. This peptide has the remarkable property of reversing established LTP in the hippocampus [95] and erasing long-standing associative memories when applied locally in the cortex [96] and elsewhere [95], albeit by an unresolved mechanism [97,98]. Thus, there are multiple molecular mechanisms at play locally within V1 that are common to LTP and SRP.
Recently, we directly tested the involvement of LTP-like plasticity in the induction and maintenance of SRP using an approach of mimicry and mutual occlusion similar to those that have been applied in the hippocampus [48,49,99–103] and motor cortex [104]. We first demonstrated that LTP of TC synapses within mouse V1, induced through tetanic stimulation of the dLGN (figure 4a–c), increases the amplitude of VEPs (figure 4d). This observation is consistent with previous findings in rodents [105–107] and indicates that experimentally induced LTP can mimic the effects of SRP on visually driven synaptic activity in V1. Having saturated LTP at these synapses with multiple bursts of theta frequency stimulation, we then went on to test the impact of prior LTP on the subsequent induction of SRP. In all of these experiments, one hemisphere underwent LTP and the other was used as a control (figure 4c). Within each animal, a greater degree of SRP occurred in the control hemisphere than the hemisphere that had undergone LTP (figure 4e), indicating that saturation of synaptic plasticity through tetanic stimulation occluded subsequent potentiation through visual experience, and implying shared mechanisms. We then conducted the reverse experiment, in which SRP was saturated to a single orientation prior to the induction of LTP. In this case, the impact of LTP on VEP amplitude was limited to oriented stimuli that were novel to the animal (figure 4f), indicating that, just as with the effects of visual experience in inducing SRP, the occluding impact of SRP on LTP is also highly stimulus-selective [87].
Figure 4.

SRP and LTP mutually occlude one another: (a) Geniculo-cortical LTP can be induced in vivo by acutely placing a stimulating electrode into the dorsal lateral geniculate nucleus (dLGN) of isoflurane anaesthetised mice and recording electrically-evoked potentials in layer 4 through chronically implanted recording electrodes that can also record VEPs in the same animals when awake. (b) LTP induced with theta burst stimulation (TBS) applied repeatedly to the dLGN lasts for at least the hour of recording time prior to recovery of animals from anaesthesia. (c) Interactions between LTP and SRP can be tested by inducing LTP in just one hemisphere (black) and comparing VEP amplitudes with those recorded in the other, control hemisphere (open) before or after SRP. (d) VEP amplitude was significantly greater in the LTP hemisphere (black) than the control hemisphere (open) whether VEPs were sampled 1 day later or 5 days later, indicating the impact of LTP on responsiveness to visual stimuli. (e) Although VEPs are significantly greater in amplitude in the TBS hemisphere than in the control hemisphere there is less SRP in this hemisphere as a consequence, so that VEP amplitude is no longer significantly different after 5 days of repeated presentation of the same grating stimulus. (f) In the reverse experiment, after SRP has been saturated to one familiar orientation (blue), TBS-induced LTP significantly impacts only those VEPs driven by a novel orientation (red), indicating that SRP occludes the effects of LTP on the VEP. (b–f) are reproduced from [87]. (g) A schematic describing the simple interpretation of these results shows that feed-forward TC plasticity forms templates of strengthened synapses for familiar stimuli (blue) in thalamo-recipient layer 4 that overlap minimally with patterns driven by other, novel oriented stimuli (red). LTP indiscriminately potentiates large numbers of synapses impacting response to all stimuli (black), whether familiar or novel. Asterisks denote significance of p < 0.05.
The conclusion from this work is that sparse subsets of synapses conveying very selective information undergo Hebbian potentiation as a result of experience. The occlusion of subsequent plasticity by any other means, whether experimental or natural, is limited to that large remaining population of synapses that have not previously been altered by experience (figure 4g). The selectivity of the occlusion effect to familiar stimuli in SRP is notable as it rules out potential contributions of meta-plasticity to the block that prior learning imposes on LTP, or that LTP imposes on subsequent learning. The stimulus-selectivity of the occlusion effect in our SRP–LTP study contrasts somewhat with other studies that have taken this approach [48,99,104], including a recent study describing the occluding effects of visual perceptual learning on LTP in ex vivo visual cortical slices [108], because these studies demonstrate an apparently robust and general occluding impact on LTP by single instances of very specific learning. On the other hand, in contrast to our previous work on SRP, these same studies have demonstrated that learning of obvious ethological importance to the animal occludes LTP. If SRP does indeed represent a form of perceptual learning it will be fascinating to determine what behavioural consequences arise from this form of plasticity.
4. Stimulus-selective response potentiation is more than thalamo-cortical long-term potentiation
We have so far described several lines of evidence that indicate an important role in SRP for feed-forward Hebbian plasticity similar to LTP. However, there are several interesting observations suggesting that a great deal of further work is required to fully understand SRP. In fact, SRP may provide an opportunity to determine how Hebbian plasticity cooperates with other critical processes in the encoding, consolidation and retrieval of memory. One notable feature of SRP is that it is not expressed within a recording session but takes several hours to emerge [86] (figure 5a). This feature contrasts with most forms of LTP, which are typically immediately established by the appropriate pattern of stimulation. It will be important to determine the events that unfold during this lag prior to the emergence of SRP, during which the animal is no longer viewing the stimulus and is returned to its home cage. Previous studies of the Arc null mutant mouse indicate that short-term memory and synaptic plasticity remain intact but that after an hour or two memories are lost and synaptic change returns to baseline [92]. Given the pronounced SRP deficit observed in the absence of Arc expression [18] it seems reasonable to suppose that post-learning consolidation is required if SRP is to be expressed. Further investigation is required to determine if this is the case and, more importantly, whether critical off-line processes involve cellular or systems-level consolidation processes [109].
Figure 5.
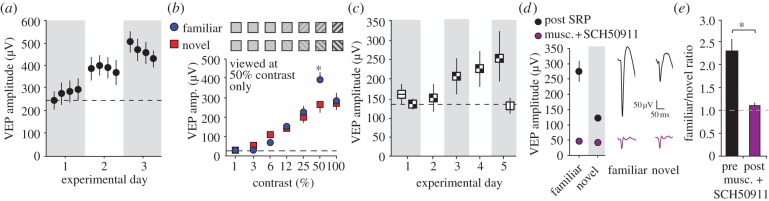
The complexity of SRP. (a) SRP does not emerge within the recording session as multiple samples within a half hour recording session yield VEPs of similar amplitude. Gains in amplitude are only apparent the following day. (b) SRP is selective for the contrast at which the stimulus is viewed. An oriented grating presented only at 50% contrast induces SRP (blue) that does not transfer across presentations of the same orientation at other contrasts, including the higher contrast of 100%. This is revealed by comparisons to a novel orientation (red) presented across the same range of contrasts because VEPs vary in amplitude depending on contrast regardless of prior experience. (c) SRP can occur to a compound chessboard stimulus. VEPs driven by the chessboard stimulus are of equivalent amplitude to those driven by a component orientation on day 1. However, SRP to the chessboard stimulus does not transfer to a second, novel component stimulus within an interleaved test session on day 5. (a–c) are reproduced from [86]. (d) Application of a cocktail of muscimol and SCH50911 (purple) isolates a purely TC VEP within layer 4 (as described in figure 2d). This treatment prevents expression of SRP such that VEPs driven by either familiar or novel stimuli are not significantly different in amplitude. Example VEPs are shown on the right side. (e) The ratio of VEP amplitude driven by familiar and novel stimuli falls significantly from approximately 2 : 1 prior to cocktail application to 1 : 1 after cocktail application, indicating that SRP is not simply expressed through potentiation of layer 4 TC synapses. (d) and (e) display new and previously unpublished data. Asterisks denote significance of p < 0.05.
SRP also has a number of other features that suggest that it is a complex network phenomenon. First, SRP is so stimulus-selective that it is hard to envisage how it could be accomplished purely through thalamo-cortical potentiation. Shifts from the familiar orientation of as little as five degrees result in a significant reduction in VEP amplitude [87] (figure 3c), demonstrating a greater degree of tuning in the mouse than any one individual simple cell appears to construct from its untuned TC input [110]. This observation strongly suggests a network representation in which fine-grained orientation tuning is integrated from neuronal populations with differing but overlapping selectivity. Second, SRP is specific not just for the spatial frequency and orientation of the stimulus but also for the contrast of the stimulus (figure 5b). This is difficult to explain from the perspective of simple potentiation of TC synapses because stimuli that are identical in all aspects other than contrast should share a large subset of fibres originating in the dLGN and projecting to V1. We would therefore anticipate that SRP at 50% contrast would at least be partly inherited by a stimulus that is the same size, orientation and spatial frequency but presented at 100% contrast. This is not the case. Finally, presentation of a relatively complex chessboard stimulus, which comprises both vertical and horizontal lines, can drive SRP. However, there is no transfer of SRP from this compound stimulus to its component horizontal and vertical lines when they are isolated from each other (figure 5c). The same is true for the reverse experiment, in which SRP occurs to a range of stimuli, including both cardinal and oblique orientations, without there being any transfer to a compound chessboard stimulus that effectively contains all four of these orientations [86]. It will be a great challenge to understand exactly how the network can segregate representations of compound from component stimuli and, perhaps most puzzlingly, how the same stimulus presented at different contrasts can be recognized as two different stimuli given the typical contrast invariance that is a feature of the visual system [13].
These considerations inspired us to ask whether SRP is apparent at layer 4 TC synapses using the same in vivo pharmacological technique as we have already described in the context of OD plasticity. Here, we present the findings of this experiment, which are new and previously unpublished. In contrast to the normal OD ratio and its shift as a result of MD, both of which continue to be expressed in the presence of a GABA receptor agonist cocktail [57], the ratio of the VEP amplitude evoked by familiar and novel stimuli (familiar/novel) dropped significantly from 2.068 ± 0.264 prior to application of the cocktail to 1.156 ± 0.063 when the drug was present at the recording site in V1 (n = 6 mice; Mann–Whitney U rank sum t-test, T = 57.000, p = 0.002) (figure 5d,e). Thus, SRP does not appear to be the inverse of the consequences of MD, despite the fact that, in terms of scale, the impact on VEPs that are unperturbed by pharmacological intervention is approximately equivalent for deprived eye depression over 3 days (approx. halved from baseline) and potentiation as a result of SRP (approx. doubled from baseline). It is plausible, however, that synaptic plasticity that supports SRP does occur at TC synapses in layer 4 but is so much sparser than that resulting from the dramatic deprivation incurred through eye closure that a further intra-cortical amplification process is required to noticeably impact visual cortical responsiveness as assayed with VEPs. A second alternative is that potentiation may occur at thalamo-cortical input to layer 2/3 (figure 1c), which is well documented in the mouse [64], and is therefore not detectable when recording in layer 4. Again, this interpretation requires some intra-cortical feedback to explain the observation that, under normal conditions, SRP is readily apparent in VEPs recorded in layer 4. Finally, it is important to consider the possibility that SRP is supported exclusively by intra-cortical plasticity, in contrast to the effects of MD, and that, just as is observed in visual cortical slices under the right degree of inhibition, appropriate patterns of stimulation of thalamic inputs can di-synaptically induce lasting potentiation at layer 4–2/3 intra-cortical synapses [11,111]. Distinguishing between these possibilities will require a great deal of further experimental work.
5. Conclusion
The discovery of LTP in the hippocampus [112] provided the first experimental demonstration of lasting Hebbian synaptic plasticity. LTP generated a platform upon which to experimentally dissect the ubiquitous but varied molecular implementation of Hebbian plasticity throughout the nervous system. This discovery also introduced an experimental approach that enabled the discovery of other forms of plasticity including the opposing process of synaptic LTD [9,32].
The hippocampus has been a popular model for studying LTP and LTD because it has a simple structure, an implicated role in episodic memory [113] and robust ex vivo transverse slices [114]. It is important to remember, however, that Donald Hebb considered primary visual cortex (V1) to be the most intuitive framework within which to present his influential theories of learning, principally because the functional organization was better understood than the hippocampus, a fact that is still true today. There are also other reasons to study experience-dependent plasticity within V1: owing to its proximity to sensory input, incoming information remains relatively unprocessed, enabling strict experimental constraint. Additionally, it is a surface structure and can therefore be accessed with relative ease in vivo, allowing for the application of a wide variety of recording and imaging techniques. A dogma exists that primary sensory cortices are immutable feature detectors in the adult animal and do not store memory. However, a growing body of evidence suggests that this is not the case. In this article, we have discussed not only Hebbian contributions to modification of V1 in the juvenile animal, in the form of OD plasticity, but also clear evidence that Hebbian plasticity may underlie adult perceptual learning in V1. We believe that the study of perceptual learning may facilitate an understanding of how Hebbian plasticity, which is theoretically compelling as a mnemonic mechanism, participates with other circuit elements to store and retrieve information.
6. Methods
All animals were male C57BL/6 mice (Charles River Laboratory International, Wilmington, MA). Mice were group-housed with food and water available ad libitum and maintained on a 12 L : 12 D cycle. All SRP experiments were conducted as described in [87] but used 400 phase reversals of 0.05 cycles per degree, 100% contrast, full field, sinusoidal grating stimuli averaged for each VEP recording session. Cannulation, preparation of the 4 mM muscimol/6 mM SCH50911 cocktail and drug infusions were conducted exactly as described in [57]. Statistical comparisons were made on the ratio of VEP amplitude driven by the familiar and novel stimuli (familiar VEP amplitude/novel VEP amplitude) before and during drug application. As data did not show normality of distribution or equality of variance, Mann–Whitney U rank sum t-tests were used for statistical comparisons.
Acknowledgements
The authors would like to thank colleagues Arnold Heynen and Jeffrey Gavornik for helpful discussions and Erik Sklar and Suzanne Meagher for their assistance in preparing this manuscript. They also acknowledge all of the former students and post-docs of the Bear lab that contributed to the work that has been reproduced in this manuscript.
Funding statement
The research described in this article was supported by the Howard Hughes Medical Institute (HHMI), the National Eye Institute (NEI) and the Picower Institute Innovations Fund (PIIF).
References
- 1.Bain A. 1873. Mind and body. The theories of their relation. New York, NY: D. Appleton and Company. [Google Scholar]
- 2.Hebb DO. 1949. The organization of behavior. New York, NY: Wiley. [Google Scholar]
- 3.Wiesel TN, Hubel DH. 1963. Single-cell responses in striate cortex of kittens deprived of vision in one eye. J. Neurophysiol. 26, 1003–1017. [DOI] [PubMed] [Google Scholar]
- 4.Stent GS. 1973. A physiological mechanism for Hebb's postulate of learning. Proc. Natl Acad. Sci. USA 70, 997–1001. ( 10.1073/pnas.70.4.997) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Katz LC, Shatz CJ. 1996. Synaptic activity and the construction of cortical circuits. Science 274, 1133–1138. ( 10.1126/science.274.5290.1133) [DOI] [PubMed] [Google Scholar]
- 6.Rauschecker JP, Singer W. 1981. The effects of early visual experience on the cat's visual cortex and their possible explanation by Hebb synapses. J. Physiol. 310, 215–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bear MF. 2003. Bidirectional synaptic plasticity: from theory to reality. Phil. Trans. R. Soc. Lond. B 358, 649–655. ( 10.1098/rstb.2002.1255) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cooper LN, Bear MF. 2012. The BCM theory of synapse modification at 30: interaction of theory with experiment. Nat. Rev. Neurosci. 13, 798–810. ( 10.1038/nrn3353) [DOI] [PubMed] [Google Scholar]
- 9.Dudek SM, Bear MF. 1992. Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-d-aspartate receptor blockade. Proc. Natl Acad. Sci. USA 89, 4363–4367. ( 10.1073/pnas.89.10.4363) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dudek SM, Bear MF. 1993. Bidirectional long-term modification of synaptic effectiveness in the adult and immature hippocampus. J. Neurosci. 13, 2910–2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kirkwood A, Bear MF. 1994. Homosynaptic long-term depression in the visual cortex. J. Neurosci. 14, 3404–3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kirkwood A, Dudek SM, Gold JT, Aizenman CD, Bear MF. 1993. Common forms of synaptic plasticity in the hippocampus and neocortex in vitro. Science 260, 1518–1521. ( 10.1126/science.8502997) [DOI] [PubMed] [Google Scholar]
- 13.Hubel DH. 1988. Eye, brain and vision. New York, NY: Scientific American Library. [Google Scholar]
- 14.Wiesel TN. 1982. Postnatal development of the visual cortex and the influence of environment. Nature 299, 583–591. ( 10.1038/299583a0) [DOI] [PubMed] [Google Scholar]
- 15.Crozier RA, Wang Y, Liu CH, Bear MF. 2007. Deprivation-induced synaptic depression by distinct mechanisms in different layers of mouse visual cortex. Proc. Natl Acad. Sci. USA 104, 1383–1388. ( 10.1073/pnas.0609596104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoon BJ, Smith GB, Heynen AJ, Neve RL, Bear MF. 2009. Essential role for a long-term depression mechanism in ocular dominance plasticity. Proc. Natl Acad. Sci. USA 106, 9860–9865. ( 10.1073/pnas.0901305106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Drager UC. 1974. Autoradiography of tritiated proline and fucose transported transneuronally from the eye to the visual cortex in pigmented and albino mice. Brain Res. 82, 284–292. ( 10.1016/0006-8993(74)90607-6) [DOI] [PubMed] [Google Scholar]
- 18.McCurry CL, Shepherd JD, Tropea D, Wang KH, Bear MF, Sur M. 2010. Loss of Arc renders the visual cortex impervious to the effects of sensory experience or deprivation. Nat. Neurosci. 13, 450–457. ( 10.1038/nn.2508) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Drager UC. 1978. Observations on monocular deprivation in mice. J. Neurophysiol. 41, 28–42. [DOI] [PubMed] [Google Scholar]
- 20.Gordon JA, Stryker MP. 1996. Experience-dependent plasticity of binocular responses in the primary visual cortex of the mouse. J. Neurosci. 16, 3274–3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coleman JE, Law K, Bear MF. 2009. Anatomical origins of ocular dominance in mouse primary visual cortex. Neuroscience 161, 561–571. ( 10.1016/j.neuroscience.2009.03.045) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Porciatti V, Pizzorusso T, Maffei L. 1999. The visual physiology of the wild type mouse determined with pattern VEPs. Vision Res. 39, 3071–3081. ( 10.1016/S0042-6989(99)00022-X) [DOI] [PubMed] [Google Scholar]
- 23.Sawtell NB, Frenkel MY, Philpot BD, Nakazawa K, Tonegawa S, Bear MF. 2003. NMDA receptor-dependent ocular dominance plasticity in adult visual cortex. Neuron 38, 977–985. ( 10.1016/S0896-6273(03)00323-4) [DOI] [PubMed] [Google Scholar]
- 24.Mitzdorf U. 1985. Current source-density method and application in cat cerebral cortex: investigation of evoked potentials and EEG phenomena. Physiol. Rev. 65, 37–100. [DOI] [PubMed] [Google Scholar]
- 25.Ma W-P, Li Y-T, Tao HW. 2013. Downregulation of cortical inhibition mediates ocular dominance plasticity during the critical period. J. Neurosci. 33, 11 276–11 280. ( 10.1523/JNEUROSCI.5598-12.2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frenkel MY, Bear MF. 2004. How monocular deprivation shifts ocular dominance in visual cortex of young mice. Neuron 44, 917–923. ( 10.1016/j.neuron.2004.12.003) [DOI] [PubMed] [Google Scholar]
- 27.Sato M, Stryker MP. 2008. Distinctive features of adult ocular dominance plasticity. J. Neurosci. 28, 10 278–10 286. ( 10.1523/JNEUROSCI.2451-08.2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hofer SB, Mrsic-Flogel TD, Bonhoeffer T, Hubener M. 2006. Lifelong learning: ocular dominance plasticity in mouse visual cortex. Curr. Opin. Neurobiol. 16, 451–459. ( 10.1016/j.conb.2006.06.007) [DOI] [PubMed] [Google Scholar]
- 29.Doshi NR, Rodriguez ML. 2007. Amblyopia. Am. Fam. Physician 75, 361–367. [PubMed] [Google Scholar]
- 30.Linden ML, Heynen AJ, Haslinger RH, Bear MF. 2009. Thalamic activity that drives visual cortical plasticity. Nat. Neurosci. 12, 390–392. ( 10.1038/nn.2284) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bienenstock EL, Cooper LN, Munro PW. 1982. Theory for the development of neuron selectivity: orientation specificity and binocular interaction in visual cortex. J. Neurosci. 2, 32–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mulkey RM, Malenka RC. 1992. Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of the hippocampus. Neuron 9, 967–975. ( 10.1016/0896-6273(92)90248-C) [DOI] [PubMed] [Google Scholar]
- 33.Dudek SM, Friedlander MJ. 1996. Developmental down-regulation of LTD in cortical layer IV and its independence of modulation by inhibition. Neuron 16, 1097–1106. ( 10.1016/S0896-6273(00)80136-1) [DOI] [PubMed] [Google Scholar]
- 34.Jiang B, Trevino M, Kirkwood A. 2007. Sequential development of long-term potentiation and depression in different layers of the mouse visual cortex. J. Neurosci. 27, 9648–9652. ( 10.1523/JNEUROSCI.2655-07.2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang K, Xiong W, Yang G, Kojic L, Wang YT, Cynader M. 2011. The regulatory role of long-term depression in juvenile and adult mouse ocular dominance plasticity. Sci. Rep. 1, 203 ( 10.1038/srep00203) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee SH, Liu L, Wang YT, Sheng M. 2002. Clathrin adaptor AP2 and NSF interact with overlapping sites of GluR2 and play distinct roles in AMPA receptor trafficking and hippocampal LTD. Neuron 36, 661–674. ( 10.1016/S0896-6273(02)01024-3) [DOI] [PubMed] [Google Scholar]
- 37.Ahmadian G, et al. 2004. Tyrosine phosphorylation of GluR2 is required for insulin-stimulated AMPA receptor endocytosis and LTD. EMBO J. 23, 1040–1050. ( 10.1038/sj.emboj.7600126) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Waung MW, Pfeiffer BE, Nosyreva ED, Ronesi JA, Huber KM. 2008. Rapid translation of Arc/Arg3.1 selectively mediates mGluR-dependent LTD through persistent increases in AMPAR endocytosis rate. Neuron 59, 84–97. ( 10.1016/j.neuron.2008.05.014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shepherd JD, Bear MF. 2011. New views of Arc, a master regulator of synaptic plasticity. Nat. Neurosci. 14, 279–284. ( 10.1038/nn.2708) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Philpot BD, Espinosa JS, Bear MF. 2003. Evidence for altered NMDA receptor function as a basis for metaplasticity in visual cortex. J. Neurosci. 23, 5583–5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Philpot BD, Cho KK, Bear MF. 2007. Obligatory role of NR2A for metaplasticity in visual cortex. Neuron 53, 495–502. ( 10.1016/j.neuron.2007.01.027) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cho KK, Khibnik L, Philpot BD, Bear MF. 2009. The ratio of NR2A/B NMDA receptor subunits determines the qualities of ocular dominance plasticity in visual cortex. Proc. Natl Acad. Sci. USA 106, 5377–5382. ( 10.1073/pnas.0808104106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Feldman DE. 2009. Synaptic mechanisms for plasticity in neocortex. Annu. Rev. Neurosci. 32, 33–55. ( 10.1146/annurev.neuro.051508.135516) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bear MF, Kleinschmidt A, Gu QA, Singer W. 1990. Disruption of experience-dependent synaptic modifications in striate cortex by infusion of an NMDA receptor antagonist. J. Neurosci. 10, 909–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Daw NW, Gordon B, Fox KD, Flavin HJ, Kirsch JD, Beaver CJ, Ji Q, Reid SN, Czepita D. 1999. Injection of MK-801 affects ocular dominance shifts more than visual activity. J. Neurophysiol. 81, 204–215. [DOI] [PubMed] [Google Scholar]
- 46.Roberts EB, Meredith MA, Ramoa AS. 1998. Suppression of NMDA receptor function using antisense DNA block ocular dominance plasticity while preserving visual responses. J. Neurophysiol. 80, 1021–1032. [DOI] [PubMed] [Google Scholar]
- 47.Bear MF, Rittenhouse CD. 1999. Molecular basis for induction of ocular dominance plasticity. J. Neurobiol. 41, 83–91. () [DOI] [PubMed] [Google Scholar]
- 48.Whitlock JR, Heynen AJ, Shuler MG, Bear MF. 2006. Learning induces long-term potentiation in the hippocampus. Science 313, 1093–1097. ( 10.1126/science.1128134) [DOI] [PubMed] [Google Scholar]
- 49.Sacchetti B, Lorenzini CA, Baldi E, Bucherelli C, Roberto M, Tassoni G, Brunelli M. 2002. Time-dependent inhibition of hippocampal LTP in vitro following contextual fear conditioning in the rat. Eur. J. Neurosci. 15, 143–150. ( 10.1046/j.0953-816x.2001.01844.x) [DOI] [PubMed] [Google Scholar]
- 50.Heynen AJ, Yoon BJ, Liu CH, Chung HJ, Huganir RL, Bear MF. 2003. Molecular mechanism for loss of visual cortical responsiveness following brief monocular deprivation. Nat. Neurosci. 6, 854–862. ( 10.1038/nn1100) [DOI] [PubMed] [Google Scholar]
- 51.Liu CH, Heynen AJ, Shuler MG, Bear MF. 2008. Cannabinoid receptor blockade reveals parallel plasticity mechanisms in different layers of mouse visual cortex. Neuron 58, 340–345. ( 10.1016/j.neuron.2008.02.020) [DOI] [PubMed] [Google Scholar]
- 52.Duffy FH, Burchfiel JL, Conway JL. 1976. Bicuculline reversal of deprivation amblyopia in the cat. Nature 260, 256–257. ( 10.1038/260256a0) [DOI] [PubMed] [Google Scholar]
- 53.Bear MF, Schmechel DE, Ebner FF. 1985. Glutamic acid decarboxylase in the striate cortex of normal and monocularly deprived kittens. J. Neurosci. 5, 1262–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yazaki-Sugiyama Y, Kang S, Cateau H, Fukai T, Hensch TK. 2009. Bidirectional plasticity in fast-spiking GABA circuits by visual experience. Nature 462, 218–221. ( 10.1038/nature08485) [DOI] [PubMed] [Google Scholar]
- 55.Maffei A, Nataraj K, Nelson SB, Turrigiano GG. 2006. Potentiation of cortical inhibition by visual deprivation. Nature 443, 81–84. ( 10.1038/nature05079) [DOI] [PubMed] [Google Scholar]
- 56.Smith GB, Bear MF. 2010. Bidirectional ocular dominance plasticity of inhibitory networks: recent advances and unresolved questions. Front. Cell Neurosci. 4, 21 ( 10.3389/fncel.2010.00021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Khibnik LA, Cho KK, Bear MF. 2010. Relative contribution of feedforward excitatory connections to expression of ocular dominance plasticity in layer 4 of visual cortex. Neuron 66, 493–500. ( 10.1016/j.neuron.2010.04.012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Trachtenberg JT, Trepel C, Stryker MP. 2000. Rapid extragranular plasticity in the absence of thalamocortical plasticity in the developing primary visual cortex. Science 287, 2029–2032. ( 10.1126/science.287.5460.2029) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu BH, Wu GK, Arbuckle R, Tao HW, Zhang LI. 2007. Defining cortical frequency tuning with recurrent excitatory circuitry. Nat. Neurosci. 10, 1594–1600. ( 10.1038/nn2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Coleman JE, Nahmani M, Gavornik JP, Haslinger R, Heynen AJ, Erisir A, Bear MF. 2010. Rapid structural remodeling of thalamocortical synapses parallels experience-dependent functional plasticity in mouse primary visual cortex. J. Neurosci. 30, 9670–9682. ( 10.1523/JNEUROSCI.1248-10.2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kaneko M, Stellwagen D, Malenka RC, Stryker MP. 2008. Tumor necrosis factor-alpha mediates one component of competitive, experience-dependent plasticity in developing visual cortex. Neuron 58, 673–680. ( 10.1016/j.neuron.2008.04.023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ranson A, Cheetham CE, Fox K, Sengpiel F. 2012. Homeostatic plasticity mechanisms are required for juvenile, but not adult, ocular dominance plasticity. Proc. Natl Acad. Sci. USA 109, 1311–1316. ( 10.1073/pnas.1112204109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Blais BS, Frenkel MY, Kuindersma SR, Muhammad R, Shouval HZ, Cooper LN, Bear MF. 2008. Recovery from monocular deprivation using binocular deprivation. J. Neurophysiol. 100, 2217–2224. ( 10.1152/jn.90411.2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smith GB, Heynen AJ, Bear MF. 2009. Bidirectional synaptic mechanisms of ocular dominance plasticity in visual cortex. Phil. Trans. R. Soc. B 364, 357–367. ( 10.1098/rstb.2008.0198) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mrsic-Flogel TD, Hofer SB, Ohki K, Reid RC, Bonhoeffer T, Hubener M. 2007. Homeostatic regulation of eye-specific responses in visual cortex during ocular dominance plasticity. Neuron 54, 961–972. ( 10.1016/j.neuron.2007.05.028) [DOI] [PubMed] [Google Scholar]
- 66.Aizenman CD, Pratt KG. 2008. There's more than one way to scale a synapse. Neuron 58, 651–653. ( 10.1016/j.neuron.2008.05.017) [DOI] [PubMed] [Google Scholar]
- 67.Iny K, Heynen AJ, Sklar E, Bear MF. 2006. Bidirectional modifications of visual acuity induced by monocular deprivation in juvenile and adult rats. J. Neurosci. 26, 7368–7374. ( 10.1523/JNEUROSCI.0124-06.2006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wilson JR, Webb SV, Sherman SM. 1977. Conditions for dominance of one eye during competitive development of central connections in visually deprived cats. Brain Res. 136, 277–287. ( 10.1016/0006-8993(77)90803-4) [DOI] [PubMed] [Google Scholar]
- 69.Cho KK, Bear MF. 2010. Promoting neurological recovery of function via metaplasticity. Future Neurol. 5, 21–26. ( 10.2217/fnl.09.62) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lambo ME, Turrigiano GG. 2013. Synaptic and intrinsic homeostatic mechanisms cooperate to increase L2/3 pyramidal neuron excitability during a late phase of critical period plasticity. J. Neurosci. 33, 8810–8819. ( 10.1523/JNEUROSCI.4502-12.2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen WS, Bear MF. 2007. Activity-dependent regulation of NR2B translation contributes to metaplasticity in mouse visual cortex. Neuropharmacology 52, 200–214. ( 10.1016/j.neuropharm.2006.07.003) [DOI] [PubMed] [Google Scholar]
- 72.Bear MF. 1998. The role of LTD and LTP in development and learning. In Mechanistic relationships between development and learning (eds Carew TJ, Menzel R, Shatz CJ.), pp. 205–225. Chichester, UK: Wiley. [Google Scholar]
- 73.Baroncelli L, Maffei L, Sale A. 2011. New perspectives in amblyopia therapy on adults: a critical role for the excitatory/inhibitory balance. Front. Cell Neurosci. 5, 25 ( 10.3389/fncel.2011.00025) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Montey KL, Quinlan EM. 2011. Recovery from chronic monocular deprivation following reactivation of thalamocortical plasticity by dark exposure. Nat. Commun. 2, 317 ( 10.1038/ncomms1312) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Duffy KR, Mitchell DE. 2013. Darkness alters maturation of visual cortex and promotes fast recovery from monocular deprivation. Curr. Biol. 23, 382–386. ( 10.1016/j.cub.2013.01.017) [DOI] [PubMed] [Google Scholar]
- 76.Li RW, Ngo C, Nguyen J, Levi DM. 2011. Video-game play induces plasticity in the visual system of adults with amblyopia. PLoS Biol. 9, e1001135 ( 10.1371/journal.pbio.1001135) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Levi DM, Li RW. 2009. Perceptual learning as a potential treatment for amblyopia: a mini-review. Vis. Res. 49, 2535–2549. ( 10.1016/j.visres.2009.02.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gilbert CD, Sigman M, Crist RE. 2001. The neural basis of perceptual learning. Neuron 31, 681–697. ( 10.1016/S0896-6273(01)00424-X) [DOI] [PubMed] [Google Scholar]
- 79.Sasaki Y, Nanez JE, Watanabe T. 2010. Advances in visual perceptual learning and plasticity. Nat. Rev. Neurosci. 11, 53–60. ( 10.1038/nrn2737) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fiorentini A, Berardi N. 1980. Perceptual learning specific for orientation and spatial frequency. Nature 287, 43–44. ( 10.1038/287043a0) [DOI] [PubMed] [Google Scholar]
- 81.Karni A, Sagi D. 1991. Where practice makes perfect in texture discrimination: evidence for primary visual cortex plasticity. Proc. Natl Acad. Sci. USA 88, 4966–4970. ( 10.1073/pnas.88.11.4966) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fahle M, Edelman S, Poggio T. 1995. Fast perceptual learning in hyperacuity. Vis. Res. 35, 3003–3013. ( 10.1016/0042-6989(95)00044-Z) [DOI] [PubMed] [Google Scholar]
- 83.Seitz AR. 2011. Perceptual learning: stimulus-specific learning from low-level visual plasticity? Curr. Biol. 21, R814–R815. ( 10.1016/j.cub.2011.08.042) [DOI] [PubMed] [Google Scholar]
- 84.Furmanski CS, Schluppeck D, Engel SA. 2004. Learning strengthens the response of primary visual cortex to simple patterns. Curr. Biol. 14, 573–578. ( 10.1016/j.cub.2004.03.032) [DOI] [PubMed] [Google Scholar]
- 85.Schoups A, Vogels R, Qian N, Orban G. 2001. Practising orientation identification improves orientation coding in V1 neurons. Nature 412, 549–553. ( 10.1038/35087601) [DOI] [PubMed] [Google Scholar]
- 86.Frenkel MY, Sawtell NB, Diogo AC, Yoon B, Neve RL, Bear MF. 2006. Instructive effect of visual experience in mouse visual cortex. Neuron 51, 339–349. ( 10.1016/j.neuron.2006.06.026) [DOI] [PubMed] [Google Scholar]
- 87.Cooke SF, Bear MF. 2010. Visual experience induces long-term potentiation in the primary visual cortex. J. Neurosci. 30, 16 304–16 313. ( 10.1523/JNEUROSCI.4333-10.2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.McNaughton BL. 2003. Long-term potentiation, cooperativity and Hebb's cell assemblies: a personal history. Phil. Trans. R. Soc. Lond. B 358, 629–634. ( 10.1098/rstb.2002.1231) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Scholl B, Tan AY, Corey J, Priebe NJ. 2013. Emergence of orientation selectivity in the mammalian visual pathway. J. Neurosci. 33, 10 616–10 624. ( 10.1523/JNEUROSCI.0404-13.2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bliss TV, Collingridge GL. 1993. A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361, 31–39. ( 10.1038/361031a0) [DOI] [PubMed] [Google Scholar]
- 91.Shi S, Hayashi Y, Esteban JA, Malinow R. 2001. Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell 105, 331–343. ( 10.1016/S0092-8674(01)00321-X) [DOI] [PubMed] [Google Scholar]
- 92.Plath N, et al. 2006. Arc/Arg3.1 is essential for the consolidation of synaptic plasticity and memories. Neuron 52, 437–444. ( 10.1016/j.neuron.2006.08.024) [DOI] [PubMed] [Google Scholar]
- 93.Guzowski JF, Lyford GL, Stevenson GD, Houston FP, McGaugh JL, Worley PF, Barnes CA. 2000. Inhibition of activity-dependent Arc protein expression in the rat hippocampus impairs the maintenance of long-term potentiation and the consolidation of long-term memory. J. Neurosci. 20, 3993–4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chowdhury S, Shepherd JD, Okuno H, Lyford G, Petralia RS, Plath N, Kuhl D, Huganir RL, Worley PF. 2006. Arc/Arg3.1 interacts with the endocytic machinery to regulate AMPA receptor trafficking. Neuron 52, 445–459. ( 10.1016/j.neuron.2006.08.033). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pastalkova E, Serrano P, Pinkhasova D, Wallace E, Fenton AA, Sacktor TC. 2006. Storage of spatial information by the maintenance mechanism of LTP. Science 313, 1141–1144. ( 10.1126/science.1128657) [DOI] [PubMed] [Google Scholar]
- 96.Shema R, Sacktor TC, Dudai Y. 2007. Rapid erasure of long-term memory associations in the cortex by an inhibitor of PKM zeta. Science 317, 951–953. ( 10.1126/science.1144334) [DOI] [PubMed] [Google Scholar]
- 97.Volk LJ, Bachman JL, Johnson R, Yu Y, Huganir RL. 2013. PKM-zeta is not required for hippocampal synaptic plasticity, learning and memory. Nature 493, 420–423. ( 10.1038/nature11802) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lee AM, et al. 2013. Prkcz null mice show normal learning and memory. Nature 493, 416–419. ( 10.1038/nature11803) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gruart A, Munoz MD, Delgado-Garcia JM. 2006. Involvement of the CA3-CA1 synapse in the acquisition of associative learning in behaving mice. J. Neurosci. 26, 1077–1087. ( 10.1523/JNEUROSCI.2834-05.2006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Doyere V, Redini-Del Negro C, Dutrieux G, Le Floch G, Davis S, Laroche S. 1995. Potentiation or depression of synaptic efficacy in the dentate gyrus is determined by the relationship between the conditioned and unconditioned stimulus in a classical conditioning paradigm in rats. Behav. Brain Res. 70, 15–29. ( 10.1016/0166-4328(94)00179-J) [DOI] [PubMed] [Google Scholar]
- 101.Barnes CA, Jung MW, McNaughton BL, Korol DL, Andreasson K, Worley PF. 1994. LTP saturation and spatial learning disruption: effects of task variables and saturation levels. J. Neurosci. 14, 5793–5806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Castro CA, Silbert LH, McNaughton BL, Barnes CA. 1989. Recovery of spatial learning deficits after decay of electrically induced synaptic enhancement in the hippocampus. Nature 342, 545–548. ( 10.1038/342545a0) [DOI] [PubMed] [Google Scholar]
- 103.Moser EI, Krobert KA, Moser MB, Morris RG. 1998. Impaired spatial learning after saturation of long-term potentiation. Science 281, 2038–2042. ( 10.1126/science.281.5385.2038) [DOI] [PubMed] [Google Scholar]
- 104.Rioult-Pedotti MS, Friedman D, Donoghue JP. 2000. Learning-induced LTP in neocortex. Science 290, 533–536. ( 10.1126/science.290.5491.533) [DOI] [PubMed] [Google Scholar]
- 105.Heynen AJ, Bear MF. 2001. Long-term potentiation of thalamocortical transmission in the adult visual cortex in vivo. J. Neurosci. 21, 9801–9813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kuo MC, Dringenberg HC. 2012. Comparison of long-term potentiation (LTP) in the medial (monocular) and lateral (binocular) rat primary visual cortex. Brain Res. 1488, 51–59. ( 10.1016/j.brainres.2012.10.006). [DOI] [PubMed] [Google Scholar]
- 107.Clapp WC, Eckert MJ, Teyler TJ, Abraham WC. 2006. Rapid visual stimulation induces N-methyl-d-aspartate receptor-dependent sensory long-term potentiation in the rat cortex. Neuroreport 17, 511–515. ( 10.1097/01.wnr.0000209004.63352.10) [DOI] [PubMed] [Google Scholar]
- 108.Sale A, De Pasquale R, Bonaccorsi J, Pietra G, Olivieri D, Berardi N, Maffei L. 2011. Visual perceptual learning induces long-term potentiation in the visual cortex. Neuroscience 172, 219–225. ( 10.1016/j.neuroscience.2010.10.078) [DOI] [PubMed] [Google Scholar]
- 109.Morris RG, Moser EI, Riedel G, Martin SJ, Sandin J, Day M, O'Carroll C. 2003. Elements of a neurobiological theory of the hippocampus: the role of activity-dependent synaptic plasticity in memory. Phil. Trans. R. Soc. Lond. B 358, 773–786. ( 10.1098/rstb.2002.1264) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Niell CM, Stryker MP. 2008. Highly selective receptive fields in mouse visual cortex. J. Neurosci. 28, 7520–7536. ( 10.1523/JNEUROSCI.0623-08.2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kirkwood A, Bear MF. 1994. Hebbian synapses in visual cortex. J. Neurosci. 14, 1634–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bliss TV, Lomo T. 1973. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J. Physiol. 232, 331–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Scoville WB, Milner B. 1957. Loss of recent memory after bilateral hippocampal lesions. J. Neurol. Neurosurg. Psychiatr. 20, 11–21. ( 10.1136/jnnp.20.1.11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Skrede KK, Westgaard RH. 1971. The transverse hippocampal slice: a well-defined cortical structure maintained in vitro. Brain Res. 35, 589–593. ( 10.1016/0006-8993(71)90508-7) [DOI] [PubMed] [Google Scholar]