

Abstract

A new strategy for enantioselective synthesis of azacyclic molecules in which dynamic kinetic equilibration of diastereomeric iminium ions precedes a stereochemistry-determining sigmatropic rearrangement is reported. The method is illustrated by the synthesis in high enantiomeric purity (generally 95–99% ee) of a variety of 1-azabicyclic molecules containing angular allyl or 3-substituted 2-propenyl side chains adjacent to nitrogen and up to three stereogenic centers. In these products, the size of the carbocyclic ring is varied widely (5–12 membered), however useful yields are obtained in forming 1-azabicyclic products containing only fused pyrrolidine and piperidine rings. Chi-rality transfer from substituents at carbons 1 and 2 of the 3-butenylamine fragment of the starting material is investigated, with methyl and phenyl substituents at the allylic position shown to provide exquisite stereocontrol (generally 98–99% chirality transfer). An attractive feature of the method is the ability to carry out the key transformation in the absence of solvent. Illustrated also is the high yielding conversion of four such products to a new family of bicyclic β-amino acids of high enantiomeric purity.
Introduction
1-Azabicyclic rings having an angular substituent adjacent to nitrogen (1, Figure 1) have a variety of potential applications. These ring systems are found in natural products exhibiting potentially useful therapeutic properties such as deoxyharringtonine,1 and in a few molecules of medicinal chemistry significance, such as 8a-phenyldecahydroquinolines (Figure 1).2 One can also readily envisage applications of conformationally constrained β-amino acids of the type depicted in Figure 1 in peptidomimetics or as organocatalysts. Nonetheless, there are remarkably few reports of applications of angularly substituted nitrogen heterocycles 1,3 particularly in light of the presence of other substituted 1-azabicyclic non-aromatic heterocycles in marketed drugs and exploratory drug candidates.4 The lack of general methods for the synthesis of heterocycles of type 1, particularly in high enantiomeric purity, could be responsible for their limited use to date.5
Figure 1. Angularly Substituted 1-Azabicyclic Ring Systems 1 and Examples of Compounds Containing this Ring System.

We reported earlier a general method for the synthesis of racemic, angularly substituted, 1-azabicylic molecules 1.6 The method is illustrated in Scheme 1 for the preparation cis-cyclopentapyrrolidine 4 from aminoketal 2. In this synthesis, a mixture of aminoketal 2, 1 equiv of tri-fluoroacetic acid (TFA) and 2.5 equiv of dimedone is heated for several hours at 120 °C. The tetrasubsti-tuted iminium ion A is formed initially and upon heating undergoes 3,3-sigmatropic equilibration with iminium ion isomer B. The inclusion of dimedone (5,5-dimethylcyclohexane-1,3-dione, 3) selectively traps the less-stable formaldiminium ion isomer B to give presumably adduct C, which upon fragmentation delivers the cis-cyclopentapyrrolidine product 4.7 To aid in purification, this product is converted to benzyloxy (Cbz) derivative 5, which was isolated in 86—96% yield. The exomethylene fragment of the iminium ion intermediate eventually emerges as the well known dimedone-formaldehyde adduct 6.
Scheme 1. Methylene Transfer-Driven Cationic 2-Aza-Cope Rearrangement.

During the development of this method, we discovered that when the synthesis of the corresponding cis-octahydroindole 8 was carried out in CD3OD containing 3 equiv of D2O, deuterium was incorporated into the angular methine (C3a) and C7 methylene positions of product d3-8 (Scheme 2).6 This deuterium incorporation signified that the initially formed iminium ion D equilibrated with enamonium ion iomers E and F more rapidly than formaldiminium ion intermediate G was trapped to yield cis-octahydroindole product 8.
Scheme 2. Deuterium Incorporation by Iminium/Enamonium Ion Tautomerization.

On the basis of the rapid pre-equilibrium that occurs between iminium ion and enamonium ion intermediates in this synthesis of angularly substituted heterocycles, we postulated that incorporating a nonracemic stereocenter on the homoallylic side chain of an aminoketal precursor such as 9 should result in the [3,3]-sigmatropic rearrangement occurring faster with one C3a epimer to deliver potentially one enantiomer of the azabicyclic product (Scheme 3). For this process to occur with high chirality transfer, the C3a epimers of iminium ion intermediate H must equilibrate rapidly, one epimer must preferentially undergo [3,3]-sigmatropic rearrangement, and dimedone trapping also must occur more rapidly than product iminium ion J equilibrates with its enantiomer.8 At the outset, we anticipated that sig-matropic rearrangement via transition state geometry I would be preferred, because bond formation from the convex face and placement of the substituent R in a quasi-equatorial orientation should be favored.
Scheme 3. Potential Synthesis of Enantiomerically Enriched cis-Octahydroindole 8 by Coupled Dynamic Kinetic Epimerization and Chirality Transfer.

In this article, we describe the development of this new strategy into a general method for enan-tioselective synthesis of angularly substituted 1-azabicyclic molecules.9 Experiments that illuminate some of the mechanistic details of the cationic 2-aza-Cope rearrangement and other steps in the sequence are discussed also.
Results and Discussion
Synthesis of Aminoketals Containing Enantiomerically Enriched 1-Substituted 3-Butenyl Fragments and Their Conversion to 1-Azabicyclic Products
Our investigations began by preparing five aminoketal precursors 9 that varied in the nature of the homoallylic substituent. The synthesis begins with benzyl 2-oxocyclohexaneacetate (11),10 which we found most convenient to prepare by fluoride-mediated alkylation of 1-(trimethylsiloxy)cyclohexene (10) with benzyl 2-bromoacetate (Scheme 4).11 Ketalization of 11 catalyzed by scandium triflate yielded dioxolane derivative 12 in 68% overall yield from 10. Cleavage of the benzyl ester by hydrogenolysis, followed by carbodiimide-promoted coupling with (R)-1-substituted-3-butenylamines 13a–e provided amides 14a–e. The enantiomerically enriched (R)-1-substituted-3-butenylamines 13a–e were available in three steps from the corresponding aldehyde and (R)-phenylglycinol by the method of Vilaivan and co-workers.12,13 Reduction of amide intermediates 14a–e with lithium aluminum hydride delivered aminoketals 9a–e in good overall yields from ketal ester 12.
Scheme 4. Synthesis of Aminoketals 9.

In our initial study, the reaction temperature and the effect of the homoallylic substituent on chirality transfer was examined (Table 1). All reactions were conducted neat by heating mixtures of the aminoketal 9, TFA (1.0 equiv) and dimedone (2.5 equiv). In these small-scale reactions, 0.1 equiv of morpholine was added to ensure that excess TFA was not present, because dimedone is decomposed at elevated temperatures in the presence of this acid. At 120 °C, phenyl-substituted aminoketal 9a was converted to cis-octahydroindole ent-8 in 92% yield with 96% chirality transfer (entry 1).14 When this reaction was conducted at 100 °C, the rearrangement was significantly slower (entry 2); however, the extent of chirality transfer was not enhanced. No reaction was observed at 80 °C after 1 h (entry 3). The degree of chirality transfer was affected to only a small extent by the incorporation of p-Cl or o-Cl substituents or a p-OMe group in the aryl fragment (entries 4–6). Most notable was the significant decrease in chirality transfer observed when the homoallylic substituent was isopropyl (entry 7). In addition, carrying out the reaction in CD3OD at 110 °C in a sealed tube (1 equiv of TFA and 2.5 equiv dim-edone) provided ent-8 in 75% yield, however with unsatisfactory (68%) chirality transfer. The absolute configuration of ent-8 was secured by single crystal X-ray analysis of its hydrobromide salt.
Table 1. Enantioselective Synthesis of Octahydroindole 8 from Aminoketals 9a–e.

| |||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| entry | R | ee (%) 9a |
temp (°C) |
time(h) | yield (%) |
ee (%) ent-8b | chirality transfer (%) |
| 1 | Ph (9a) | 93 | 120 | 5 | 92 | 89 | 96 |
| 2 | Ph (9a) | 93 | 100 | 22 | 84 | 88 | 95 |
| 3 | Ph (9a) | 93 | 80 | 1 | 0 | — | — |
| 4 | 4-OMeC6H4(9b) | 92 | 120 | 5 | 92 | 82 | 89 |
| 5 | 4-ClC6H4(9c) | 94 | 120 | 5 | 90 | 89 | 95 |
| 6 | 2-ClC6H4(9d) | 94 | 120 | 5 | 88 | 87 | 93 |
| 7 | i-Pr (9e) | 46 | 120 | 22 | 51 | 24 | 52 |
Enantiomeric purity of the homoallylic amine fragment of 9 was determined by enantioselective HPLC.
Enantiomeric purity was determined by enantioselective HPLC analysis of the corresponding N-benzoyl derivative.
We briefly investigated the scope of this enantioselective construction of allyl-substituted 1-azabicyclic molecules by examining three additional precursors containing a (R)-1-phenyl-3-butenyl fragment. Aminoacetal substrates 15–17 were prepared by sequences identical (or analogous) to that reported in Scheme 4.15 As summarized in Scheme 5, cis-hexahydrocyclopenta[b]pyrrole carbamate 18 and cis-octahydrocyclopenta[b]pyridine carbamate 19 were formed in 95% and 80% yield from precursors 15 and 16, respectively, and with 96–97% chirality transfer. In each case, only the cis steroisomer of the product was detected. In contrast, decahydroquinoline carbamate 20 was produced as a nearly 1:1 mixture of cis and trans epimers. These isomers could be separated, and each was shown to have an en-antiomeric purity of 93% ee, corresponding to 97% chirality transfer. The absolute configuration of 18 was determined by single crystal X-ray analysis of the hydrobromide salt of the parent hexahydrocyclo-penta[b]pyrrole;9 the absolute configuration for 19 and 20 was assigned in analogy to that of related products 8 and 18.
Scheme 5. Enantioselective Synthesis of Allyl-Substituted Heterocycles 18–20.

Synthesis of 2-Substituted 3-Butenylamines of High Enantiomeric Purity
Although chirality transfer was typically high with substrates having an aryl substituent at the homoallylic position, we entertained the possibility that this selectivity might be even higher with substrates having a substituent at the allylic carbon. We initiated these studies by developing an enantioselective synthesis of (R)-2-phenylbut-2-enylamine. In order to have convenient access to both enantiomers, we chose a route wherein the stereocenter is set by a catalytic enantioselective reaction using a catalyst for which both enantiomers are commercially available (Scheme 6). Using a microwave modification16 of a method first introduced by Trost,17 molybdenum-catalyzed enantioselective allylic alkylation of cinnamyl methyl carbonate (21) with dimethyl sodiomalonate gave (S)-pentenoate 23 in high yield and 99% ee. Krapcho decarboxylation of the malonate,18 followed by saponification of the carboxylic ester delivered (R)-acid 24.19 Curtius rearrangement of 24 with diphenylphosphoryl azide in t-BuOH, followed by cleavage of the Boc group with trifluoroacetic acid delivered (R)-2-phenylbut-2-enylamine (26) in 83% yield from the precursor acid. This sequence has been demonstrated on multigram scale.
Scheme 6. Synthesis of (R)-2-Phenylbut-2-en-1-amine.

To explore the effect of the 2-butenyl substituent on chirality transfer of reactions of the corresponding aminoacetals, a short synthesis of (S)-2-methylbut-3-en-1-ammonium chloride (29) was developed also (Scheme 7). Enantioselective allylic alkylation of allylbromide 27 (available in one step from (E)-1,4-dibromobut-2-ene)20 with methylmagnesium bromide, catalyzed by 1 mol % CuBr•SMe2 and 1.2 mol % Taniaphos delivered allylic carbamate 28 in 91% yield and in 95% ee.21 Subsequent removal of the tosyl and tert-butoxycarbonyl groups gave the enantiomerically enriched butenylamine hy-drochloride salt 29 in 78% overall yield from allylic bromide 27.
Scheme 7. Synthesis of (S)-2-Methylbut-3-en-1-ammonium Chloride.

Synthesis of Enantiomerically Enriched Aminoacetal Precursors Containing 2-Substituted 3-Butenyl Fragments and Their Conversion to 1-Azabicyclic Products
From homoallylic amine 26 or ammonium salt 29, a variety of aminoacetal precursors were prepared from the corresponding cyclo-alkanone acetal acids by the general method shown in Scheme 4.15 Our initial studies focused on the reaction of aminoacetal 30 and used the conditions utilized in our earlier studies with the analogous substrate 15 having the phenyl substituent at the homoallylic position. In this way, cis-hexahydrocyclopenta[b]pyrrole carbamate 31 was formed in 89% yield and a remarkable 99% ee, corresponding to complete chirality transfer (eq 1). When the morpholine buffer was omitted, the yield was depressed slightly (80%).
 |
(1) |
We next examined whether dimedone was essential to the success of the transformation shown in eq 1 (Table 2). In the absence of dimedone (and morpholine), product 31 was formed in 21% yield (entry 3). On the assumption that 1,3-propanediol was the agent trapping what would be in this case a formaldiminium ion of the 2-aza-Cope rearrangement product, the reaction was carried out in the presence of 3 equiv of this additive. However, the yield of 31 was not improved (entry 4). Methanol was considered also to be a potential trap for the formaldiminium ion intermediate; however, carrying out the reaction in methanol did not result in appreciably higher yields after 24 h at 60 °C (enries 5 and 6). Although we believe that other trapping agents as efficacious as dimedone could likely be found, we decided to explore instead the scope of the efficient, highly enantioselective, reaction shown in eq 1.
Table 2. The Reaction of Aminoacetal 30 with Trifluoroacetic Acid to Form Product 31 in the Presence of Various Additivesa.

| ||||
|---|---|---|---|---|
|
| ||||
| entry | additive | temp (°C) |
time (h) |
yield (%) |
| 1b | dimedone (2.5 equiv) | 120 | 0.5 | 80 |
| 2 | dimedone (2.5 equiv), morpholine (0.1 equiv) | 120 | 0.5 | 89 |
| 3c | none | 120 | 0.5 | 21 |
| 4 | 1,3-propanediol (3 equiv) | 120 | 0.5 | 21 |
| 5 | MeOHd | 60 | 24 | 24 |
| 6 | 5% H2O in MeOHd | 60 | 24 | 7 |
Reactions used a 1:1 molar ratio of rac-30 and trifluoroacetic acid and the indicated additive(s).
Reactant 30 was prepared from (S)-butenylamine 26 (99% ee).
When enantioenriched 30 was used, product 31 was formed with >98% chirality transfer.
Methanol was the solvent in these reaction; the starting concentration of rac-30 was 0.5 M.
A comparison of phenyl and methyl stereocontrolling groups and results of our initial exploration of this route to functionalized 1-azacyclic ring systems is summarized in Table 3. Angularly substituted cis-hexahydropenta[b]pyrroles 31, cis-octahydroindoles 32, and cis-octahydrocyclopenta[b]pyridines 33 containing a (E)-3-substituted-2-propenyl side chain were obtained in good yields (71-89% yields) and high 95–99% ee (entries 1–6). Chirality transfer was complete, within experimental uncertainty, regardless of the nature of the allylic substituent on the butenyl fragment. Carrying out the synthesis of 31a (87% yield, 99% ee) on gram scale gave the product in comparable yield with no reduction in enantioselectivity, demonstrating the practicality of this method. The high-yield formation of cis-octahydroindole 34a as exclusively a single C7 methyl epimer, indicates that both stereogenic centers of the carbocyclic aminoacetal precursor epimerized by iminium/enamonium tautomerization prior to 2-aza-Cope rearrangement/dimedone trapping. In this case, the methyl-substituted aminoketal provided cis-octahydroindole 34b in slightly diminished diastereoselectivity (9:1 dr) and chirality transfer (96%, entry 8). The absolute configuration of cis-octahydrocyclopenta[b]pyridine 33a was determined by X-ray analysis of the corresponding secondary amine hydrobromide salt,9 whereas the absolute configuration of products 31a and 32a was determined by chemical correlation with azacyclic products 18 and 8. Absolute configurations of the other azabicy-clic products reported in Table 3 were assigned by analogy.
Table 3. Enantioslective Synthesis of 1-Azabicyclic Molecules Containing an Angular 3-Phenyl or 3-Methylpropenyl Substituents.

| |||||
|---|---|---|---|---|---|
|
| |||||
| entry | product | R2 | yield (%) |
ee (%)a |
chirality transfer (%) |
| 1 |

|
Ph, 31a |
89 | 99 | >98 |
| 2 | Me, 31b |
73 | 95 | >98 | |
| 3 |

|
Ph, 32a |
82 | 99 | >98 |
| 4 | Me, 32b |
74 | 95 | >98 | |
| 5 |

|
Ph, 33a |
89 | 99 | >98 |
| 6 | Me, 33b |
71 | 95 | >98 | |
| 7 |

|
Ph, 34a |
81 | 99 | >98 |
| 8 | Me, 34b |
70 | 91b | 96 | |
Enantiomeric excess was determined by enantioselective HPLC.
This product was a 9:1 mixture of epimers at the angular methine carbon.
Results of our further investigations of the scope of this chemistry using precursors containing a (R)-2-phenylbutenylamine fragment are summarized in Table 4. At 120 °C, decahydroquinoline 35 was formed as a 1.7:1 mixture of cis and trans stereoisomers (entry 1). Stereoselectivity was increased slightly when the reaction was conducted in methanol at 60 °C (entry 2).22 Both the cis- and trans-decahydroquinoline products were formed in 99% ee. 1-Azabicyclic products containing an (E)-3-phenyl-2-propenyl side chain adjacent to nitrogen can be prepared with various sized rings carbocyclic rings (entries 3–5). The cis steroisomer of the product was strongly favored when the azacyclic ring is five-membered. Extension of this chemistry to the synthesis of 1-azabicyclic molecules in which the azacyclic unit is a medium ring appears problematic. For example, decahydrocyclopenta[b]azocine 38 was formed in only 16% yield after an extended reaction time of 22 h at 120 °C (entry 6). We attribute this low conversion to low efficiency in forming the initial 8-membered-ring iminium ion.
Table 4. Further Scope of the Enantioselective Synthesis of 1-Azabicyclic Molecules Containing an Angular 3-Phenylpropenyl Substituent.

| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| entry | product | temp (°C) | time (h) | dr (cis:trans)a |
yield (%) | ee (%)b |
| 1 |

|
120 | 0.5 | 1.7:1 | 86 | 99c |
| 2 | 60c | 20 | 2:1 | 72 | 99d | |
| 3 |

|
120 | 0.5 | 9:1 | 74 | 99e |
| 4 | 60c | 20 | 15.2:1 | 70 | 99e | |
| 5 |

|
120 | 0.5 | >10:1 | 79 | 99 |
| 6 |

|
120 | 22 | >10:1 | 16f | nd |
Diastereoselectivities were determined by analysis of the 1H NMR spectra.
Enantiomeric excesswas determined by enantioselective HPLC.
Reaction carried out in methanol (0.5 M).
Both isomerswere obtained in 99% ee.
Enantiomeric excess of the major isomer; the enantiomeric excess of the minor isomer was not determined.
Enantiomeric excess was not determined.
As the stereocontrolling step in this construction of 1-azacyclic molecules is an iminium ion [3,3]-sigmatropic rearrangement, the organized six-membered chair transition structure should allow the geometry of a double bond in the starting material to be transformed to a new stereocenter at C1 of the allylic side chain of the product. To examine this possibility, three unsaturated aminoacetal substrates, 39a–c, were prepared from (R)-2-phenyl-3E-alkenylamines having Me, Ph, or cyclohexyl substituents at C4. Exposure of aminoketal 39a, prepared from (R)-2-phenyl-3E-pentenylamine of 87% ee, to trifluoro-acetic acid at 120 °C for 30 min, followed by Cbz protection gave cis-hexahydrocyclopenta[b]pyrrole 40 as a single stereoisomer in 75% yield. The enantiomeric purity (87% ee) of 40 indicated again that chirality transfer was essentially complete. Similar results were obtained in the formation of products 41 and 42, with the exception of their higher enantiomeric purity (99% ee) resulting from the greater dia-stereomeric purity of precursors 39b and 39c.23 However, the yields of 41 and 42 were diminished, presumably because the larger size of the phenyl and cyclohexyl substituents results in destabilizing steric interactions in the iminium ion [3,3]-sigmatropic rearrangement.
Synthesis of Uncommon β-Amino Acids in High Enantiomeric Purity
β-Amino acids are valuable building blocks in peptide-based drug design, as proteolytic degradation is greatly reduced in vivo, increasing their bioavailability.24 For example, replacing proline with the more rigid analog, oc-tahydroindole-2-carboxylic acid, has been exploited in several bradykinin B2 antagonists to improve both enzymatic stability and potency.25 Moreover, proline-based catalysts have been shown to be powerful catalysts for a wide variety of transformations26 with recent examples exemplifying 3-pyrrolidinecarboxylic acid catalysts.27 Oxidative cleavage of the angular-allyl side chain of the 1-azabicyclic molecules prepared in the manner reported herein would provide a variety of new, potentially useful, α-amino acids. To demonstrate this potential, we optimized a high-yielding, two-step sequence to achieve this objective. Application of this sequence to prepare four Cbz-protected α-amino acids is reported in Scheme 9.
Scheme 9. Synthesis of Four Representative Cbz-Protected β-Aminoacids.

Mechanistic Discussion
Our current understanding of the high transfer of chirality observed in the azacyclic construction reported in this article is derived in part from our studies of the transformation of aminoketal 30 to cis-hexahydrocyclopenta[b]pyrrole 47. The success of this dynamic kinetic epimerization rearrangement sequence was predicated on iminium/enamonium equilibration occurring faster than the [3,3]-sigmatropic rearrangement step. The rapid tautomerization of iminium ion and enamonium ion intermediates K–M was confirmed to occur more rapidly than the 2-aza-Cope rearrangement when the reaction of aminoketal 30 was carried out in CD3OD (Scheme 10). As expected, azabicyclic carbamate 31 was isolated with deuterium incorporated in the angular C3a and C6 meth-ylene positions; analysis of this product by electrospray mass spectrometry indicated an 8:4:0:1 ratio of d3:d2:d1:d0 deuterium incorporation.28
Scheme 10. Deuterium Incorporation in cis-Hexahydrocyclopenta[b]pyrrole 47 via Rapid Iminium/Enamonium Ion Equilibratium.

We next probed the reversibility of the aza-Cope rearrangement and methylene transfer steps (Scheme 11). The irreversibility of the methylene transfer step was demonstrated by heating secondary amine 47 with dimedone-formaldehyde adduct 6 (1.25 equiv), trifluoroacetic acid (1 equiv), and mor-pholine (0.1 equiv) at 120 °C in CD3OD for 48 h. Benzyloxycarbonyl protection of recovered 47 and mass spectrometric analysis showed that deuterium had not been incorporated. In contrast, heating cis-hexahydrocyclopenta[b]pyrrole 47 in CD3OD at 120 °C for 24 h with paraformaldehyde (3 equiv), tri-fluoroacetic acid (1 equiv) and morpholine (0.1 equiv), followed by the addition of excess dimedone at 120 °C provided, after Cbz protection, carbamate 31 containing extensive deuterium incorporation (d3:d2:d1:d0 = 36:15:2:1). This latter result establishes that in the absence of dimedone, iminium ion isomers K and N (see Scheme 10) equilibrate under the reaction conditions.
Scheme 11. Probing the Reversibility of the Aza-Cope Rearrangement and Dimedone-Trapping Steps.

To gain additional insight into this chemistry, the reaction of amino acetal ent-48 with 1 equiv of TFA was studied by NMR (Scheme 12). Within 3 min at room temperature, ent-48 was converted to a 1:1 mixture of diastereomeric bicyclic isomers 49 and 50. This mixture was essentially unchanged after 12 h at room temperature. At temperatures above 40 °C, loss of 1,3-propanediol occurred to generate a mixture of products. The major component is iminium ion O (diagnostic 13C NMR signal at 221 ppm), with additional products assigned as enamonium ions P and Q (diagnostic vinylic 13C NMR signals between 132–123 ppm). This mixture was largely unchanged after heating at 60 °C for 12 h or at 120 °C for 5 min. Addition of excess dimedone to the latter sample and further heating at 130 °C generated ent-51 as the major azacyclic product.
Scheme 12. Intermediates Formed During the Formation of cis-hexahydrocyclopenta[b]pyrrole ent-51.

Based upon the studies reported in Schemes 10–12, we propose the following mechanism (Scheme 13). In the presence of 1 equiv of TFA, an aminoketal such as 30 is transformed at elevated temperatures, by way of enamonium tautomer L, into an equilibrium mixture composed largely of tetrasubstituted iminium ions R and S. In the slow step of the sequence, iminium ion diastereomer S preferentially undergoes [3,3]-sigmatropic rearrangement via favored chair-transition structure T having the phenyl substituent pseudoequatorial to give cis-hexahydrocyclopenta[b]pyrrole formaldiminium ion U. The exquisite chirality transfer observed in the transformations reported herein requires that intermediate U, once formed, does not equilibrate with isomers S and R, because [3,3]-sigmatropic rearrangement of the latter (vide infra) would generate ent-U and erode enantioselectivity. Thus, trapping of intermediate U by dimedone and fragmentation of the dimedone adduct must be irreversible in order to form the 3aS,6aR product 47 in high enantioselectivity.29
Scheme 13. Proposed Mechanism for the Formation of (3aS,6aR)-cis-Hexahydrocyclopenta[b]pyrrole 47 from Amino Acetal 30.
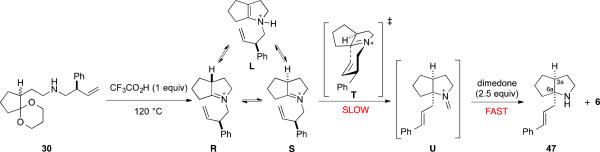
The mechanism proffered in Scheme 13 ascribes the high chirality transfer (>98%) observed in the transformation of 30 to 47 to preferential [3,3]-sigmatropic rearrangement of iminium ion diastere-omers S via transition structure T. As depicted in Figure 2, ent-47 would be the result of [3,3]-sigmatropic rearrangement of diastereomer R from the convex face via boat geometry V.30 The near perfect chirality transfer observed in forming 47 is consistent with transition structure V being at least 3 kcal/mol (more likely 3.5 kcal/mol) higher in energy than chair transition structure T.31 In addition to the high enantioselectivity observed in forming cis-hexahydrocyclopenta[b]pyrrole 47, the side chain of 47 is introduced with high E stereoselectivity. Quantitative HPLC analysis calibrated with an authentic sample of the (Z)-3-phenyl-2-propenyl isomer of the Cbz derivative 3032 demonstrated that stereoselec-tivity in forming product 47 having a (E)-3-phenyl-2-propenyl side chain was 150:1. This isomer ratio establishes that chair-transition structure W for [3,3]-sigmatropic rearrangement of iminium ion dia-stereomer R is similarly disfavored (by 3.9 kcal/mol relative to the favored transition structure T).
Figure 2. Higher Energy Pathways Leading to Azacyclic Products ent-47 and 52.

Conclusions and Outlook
In summary, a method of some generality for the enantioselective synthesis of angularly substituted 1-azabicyclic structures having up to three stereocenters is reported. This synthesis illustrates a new strategy for enantioselective synthesis of azacyclic molecules in which dynamic kinetic equilibration of dia-stereomeric iminium ions precedes a stereochemistry determining sigmatropic rearrangement. Critical to the success of this method is the facility of iminium ion/enamonium ion interconversions, and identifying an irreversible step—in this case trapping of the less-stable iminium isomer of a 2-aza-Cope equilibration by reaction with dimedone—to dictate stereoselection.
The method was illustrated by the enantioselective synthesis of a variety of 1-azabicyclic molecules containing angular allyl or 3-substituted 2-propenyl side chains. In these structures, the size of the car-bocyclic ring was varied widely (5–12 membered), however useful yields were obtained in forming bi-cyclic products containing only fused pyrrolidine and piperidine rings. A notable aspect of the method is formation of the structurally rare 1-azacyclic products in high enantiomeric purity (95–99% ee). Also demonstrated is the high yielding conversion of four such products to a new family of bicyclic β-amino acids of high enantiomeric purity, molecules of potential utility for the synthesis of peptidomimetics and scaffolds for medicinal chemistry (Scheme 9). An attractive feature of the method is the ability to carry out the key transformation in the absence of solvent. An unappealing feature of the method as currently practiced is the stoichiometric formation of the dimedone-formaldehyde adduct. The potential to mitigate this drawback by identifying alternate, more attractive, iminium ion trapping steps is suggested by preliminary studies, but to date has not been realized in a high-yielding manner.
Experimental Section
Preparation of Aminoketals Containing a Homoallylic Stereogenic Center
General Procedure A for Oxidative Cleavage of the Chiral Auxiliary
(R)-1-Phenylbut-3-en-1-amine (13a)12
Lead acetate (4.50 g, 10.1 mmol) was added to a solution of (2R)-2-phenyl-2-[(1′R)-1′-phenylbut-3′-enylamino]ethanol12 (2.26 g, 8.45 mmol), CH2Cl2 (15 mL), and MeOH (15 mL) at 0 °C. The mixture was stirred at 0 °C for 30 min. Hydroxylamine hydro-chloride (5.87 g, 84.5 mmol) was added and the mixture was stirred at 0 °C for 30 min before concentration in vacuo. The residue was washed with hexanes (50 mL) and suspended in CH2Cl2 (50 mL) followed by filtration of the lead precipitate. The filtrate was extracted with 1 N HCl (3 × 30 mL). The combined aqueous layers were washed with Et2O (30 mL), treated with 10% NaOH until pH 14, and extracted with Et2O (30 mL). The organic layer was washed with brine (20 mL), dried (MgSO4), and concentrated in vacuo to afford 13a as a pale yellow oil (890 mg, 6.08 mmol, 72%). Characterization data were consistent with previously reported values.12 HPLC analysis indicated an enantiomeric excess of 93% [CHIRALCEL OD-H column; flow: 0.5 mL/min; hexanes:i-PrOH:Et2NH = 900:100:1; λ = 254 nm; major enantiomer tR = 10.5 min; minor enantiomer tR = 14.3 min. .
(R)-1-(4-Methoxyphenyl)but-3-en-1-amine (13b)
Following general procedure A, 13b (1.08 g, 6.11 mmol, 74% yield) was obtained as a yellow oil. Characterization data were consistent with previously reported values.12 HPLC analysis indicated an enantiomeric excess of 92% [CHIRALCEL OD-H column; flow: 0.5 mL/min; hexanes:i-PrOH:Et2NH = 900:100:1; λ = 254 nm; major enantiomer tR = 13.1 min; minor enantiomer tR = 16.8 min]. .
(R)-1-Butyl-3-butenyl-1-(4-chlorophenyl)amine (13c)
Following general procedure A, 13c (1.21 g, 6.70 mmol, 72% yield) was obtained as a colorless oil. Characterization data were consistent previously reported values.12 HPLC analysis indicated an enantiomeric excess of 94% [CHIRALCEL AD column; flow: 0.5 mL/min; hexanes:i-PrOH:Et2NH = 950:50:1; λ = 254 nm; major enantiomer tR = 14.6 min; minor enantiomer tR = 14.0 min]. .
(R)-1-Butyl-3-butenyl-1-(2-chlorophenyl)amine (13d)
Following general procedure A, 13d (1.10 g, 6.11 mmol, 73% yield) was obtained as a colorless oil. Characterization data were consistent with previously reported values.12 HPLC analysis indicated an enantiomeric excess of 94% [CHIRALCEL AD column; flow: 0.5 mL/min; hexanes:i-PrOH:Et2NH = 950:50:1; λ = 254 nm; major enantiomer tR = 11.9 min; minor enantiomer tR = 11.4 min]. .
General Procedure B for the Fluoride-Mediated Alkylation of Enoxysilanes
Benzyl 2-(2-oxocyclohexyl)acetate (11)
To a stirred suspension of TASF (Me) (330 mg, 1.20 mmol) in THF (1.2 mL) at −78 °C was added a solution of 1-cyclohexenyloxytrimethylsilane (200 mg, 1.20 mmol) and benzyl 2-bromoacetate (0.22 mL, 1.4 mmol) in THF (1.8 mL) dropwise via syringe over 5 min. The resulting mixture was stirred at room temperature for 24 h, then diluted with hexanes (25 mL), washed with H2O (10 mL), dried (MgSO4), filtered and concentrated in vacuo. Purification of the crude residue by flash chromatography on silica gel (9:1 hexanes:EtOAc) provided 11 as a colorless oil (0.22 g, 0.91 mmol, 77%). 1H NMR (500 MHz, CDCl3) δ 7.37–7.30 (m, 5H), 5.18–5.08 (m, 2H), 2.94–2.80 (m, 2H), 2.48–2.32 (m, 2H), 2.26–2.07 (m, 3H), 1.92–1.85 (m, 1H), 1.79–1.58 (m, 2H), 1.44 (qd, J = 12.6, 3.7 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 210.8, 172.4, 136.0, 128.5, 128.09, 128.05, 66.2, 47.1, 41.8, 34.4, 33.8, 27.7, 25.1; IR (film, cm-1) 3066, 3035, 2937, 2861, 1733, 1710; Anal. Calcd for C15H18O3: C, 73.15; H, 7.37. Found: C, 72.89; H, 7.47.
General Procedure C for Ketalization of Cycloalkanones
(1,5-Dioxaspiro[5.5]undec-7-yl)acetic acid benzyl ester (12)
To a solution of ketoester 11 (3.9 g, 16 mmol), 1,3-propanediol (22 mL, 39 mmol), and trimethylorthoformate (8.6 mL, 78 mmol) in ace-tonitrile (160 mL) at 0 °C was added scandium triflate (77 mg, 0.16 mmol). The reaction was maintained at 0 °C for 30 min, then quenched with saturated aqueous NaHCO3 (∼5 mL) and extracted with Et2O (3 × 75 mL). The combined organic layers were washed with water (3 × 50 mL) and brine (25 mL), dried (MgSO4), filtered, concentrated in vacuo and purified on silica gel by flash chromatography (1:9 Et2O:pentane) to provide 12 (4.2 g, 14 mmol, 88%) as a colorless oil. Characterization data were consistent with previously reported values.6
General Procedure D for Sequential Debenzylation and Amide Bond Formation
2-(1,5-Dioxaspiro[5.5]undec-7-yl)-N-[(1R)-1-phenylbut-3-en-1-yl]acetamide (14a)
Ketal ester 12 (1.82 g, 5.98 mmol), NaHCO3 (1.82 g, 21.7 mmol), and Pd (OH)2/C (182 mg, 20 % on carbon, wet) in EtOAc (60 mL) was evacuated and backfilled three times with N2, then three times with H2. The mixture was stirred at room temperature under a balloon atmosphere of H2 for 2.5 h before filtering the solution over Celite, eluting with EtOAc (30 mL). The filtrate was concentrated to give the resulting acid, which was used without further purification.
N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDCI, 1.9 g, 7.2 mmol) was added to a solution of the crude acid, amine 13a (0.88 g, 5.9 mmol), and DMAP (73 mg, 0.60 mmol) in CH2Cl2 (13 mL). The solution was maintained at room temperature for 21 h and concentrated in vacuo. The crude residue was purified by flash chromatography (1:3 EtOAc:hexanes) to afford 14a (1.8 g, 5.1 mmol, 86%) as a colorless solid. mp 128–130 °C; 1H NMR (500 MHz, CDCl3) δ 7.34–7.22 (m, 5H), 6.30 (app dd, J = 31.7, 8.3 Hz, 1H), 5.70–5.65 (m, 1H), 5.12–5.05 (m, 3H), 4.07–4.05 (m, 1H), 3.92–3.89 (m, 1H), 3.80–3.66 (m, 2 H), 2.81–2.77 (m, 1H), 2.60–2.54 (m, 3H), 2.10–2.00 (m, 1H), 2.00–1.80 (m, 2H), 1.65–1.55 (m, 3H), 1.40–1.05 (m, 5H); 13C NMR (125 MHz, CDCl3) δ 173.1, 142.4, 142.3, 134.44, 134.41, 128.7, 127.3, 126.70, 126.67, 118.2, 118.1, 98.94, 98.92, 59.3, 59.18, 59.17, 52.5, 52.4, 41.0, 40.9, 37.1, 29.2, 28.2, 25.89, 25.85, 25.0, 22.4; IR (thin film, cm-1) 3290, 1644, 1536, 1447, 1337; HRMS (ESI) m/z: [M + H]+ Calcd for C21H30NO3 344.2226; Found 344.2237.
2-(1,5-Dioxaspiro[5.5]undec-7-yl)-N-[(1R)-1-(4-methoxyphenyl)but-3-en-1-yl]acetamide (14b)
Following general procedure D, 14b (1.75 g, 4.71 mmol, 88% yield) was obtained as a colorless solid. mp 79–80 °C; 1H NMR (500 MHz, CDCl3) δ 7.33–7.23 (m, 2H), 6.81 (app d, J = 7.0 Hz, 2H), 6.30 (app dd, J = 35.0, 7.8 Hz, 1H), 5.77–5.70 (m, 1H), 5.15–5.06 (m, 3H), 3.99 (app ds, J = 11.7, 2.9 Hz, 1H), 3.84 (app dq, J = 15.6, 5.2 Hz, 1H), 3.84 (s, 3H), 3.73-3.71 (m, 1H), 2.74 (app dt, J = 15.1, 5.0 Hz, 1H), 2.51–2.48 (m, 3H), 2.01–1.80 (m, 3H), 1.59–1.52 (m, 3H), 1.47–1.23 (m, 5H); 13C NMR (125 MHz, CDCl3) δ 173.0, 172.9, 158.8, 134.57, 134.55, 134.5, 134.4, 127.81, 127.79, 118.0, 117.9, 114.1, 114.04, 114.02, 98.90, 98.89, 98.88, 98.87, 59.2, 59.1, 55.45, 52.0, 51.88, 40.9, 40.8, 37.01, 36.99, 29.10, 28.14, 5.9, 25.8, 25.0, 22.4; IR (thin film, cm-1) 3296, 2935, 1638, 1513, 1246; HRMS (ESI) m/z: [M + H]+ Calcd for C22H32NO4 374.2331; Found: 374.2334.
N-[(1R)-1-(4-Chlorophenyl)but-3-en-1-yl]-2-(1,5-dioxaspiro[5.5]undec-7-yl)acetamide (14c)
Following general procedure D, 14c (1.80 g, 4.76 mmol, 89% yield) was obtained as a colorless solid. mp 135–138 °C; 1H NMR (500 MHz, CDCl3) δ 7.27 (app t, J = 8.4 Hz), 7.20 (app t, J = 6.8 Hz, 2H), 6.38 (dd, J = 21.4, 7.6 Hz, 1H), 5.68–5.61 (m, 1H), 5.10–5.02 (m, 3H), 4.04 (app q, J = 11.8, 2.8 Hz, 1H), 3.90 (app q, J = 10.3, 2.5 Hz, 1H), 3.80–3.75 (m, 2H), 2.80 (ddd, J = 19.5, 9.4, 6.8 Hz, 1H), 2.63 (broad s, 1H), 2.50 (t, J = 6.8 Hz, 2H), 2.03–1.84 (m, 3H), 1.72–1.56 (m, 3H), 1.43–1.15 (m, 5H); 13C NMR (125 MHz, CDCl3) δ 173.2, 173.1, 141.1, 141.0, 133.99, 133.96, 133.0, 128.82, 128.80, 128.2, 128.0, 118.6, 118.5, 99.0, 98.9, 59.3, 59.21, 59.19, 52.0, 51.8, 40.8, 37.0, 28.1, 25.9, 22.4; IR (thin film, cm-1) 3288, 2935, 1638, 1542, 1493; HRMS (ESI) m/z: [M + H]+ Calcd for C21H29ClNO3 378.1836; Found: 378.1834.
N-[(1R)-1-(2-Chlorophenyl)but-3-en-1-yl]-2-(1,5-dioxaspiro[5.5]undec-7-yl)acetamide (14d)
Following general procedure D, 14d (1.81 g, 4.78 mmol, 91% yield) was obtained as a colorless solid. mp 86–88 °C; 1H NMR (500 MHz, CDCl3) δ 7.34 (dt, J = 7.7, 1.5 Hz, 1H), 7.27–7.16 (m, 3H), 6.50 (t, J = 6.5 Hz, 1H), 5.73–5.65 (m, 1H), 5.39 (dtd, J = 13.5, 7.9, 5.5 Hz, 1H), 5.13–5.07 (m, 2H), 4.05 (dtd, J = 18.5, 12.0, 3.0 Hz, 1H), 3.92 (qd, J = 11.5, 2.5 Hz, 1H), 3.83–3.68 (m, 2H), 2.82 (ddd, J = 15.0, 10.0, 5.0 Hz, 1H), 2.63–2.49 (m, 3H), 2.10–2.02 (m, 1H), 2.00–1.84 (m, 2H), 1.66–1.54 (m, 3H), 1.44–1.15 (m, 5H);13C NMR (125 MHz, CDCl3) δ 173.1, 173.0, 139.70, 139.65, 134.21, 134.17, 133.0, 132.9, 130.22, 130.21, 128.4, 128.0, 127.9, 127.0, 126.96, 126.95, 118.4, 118.3, 98.93, 98.92, 98.91, 98.90, 59.23, 59.18, 59.17, 50.6, 50.5, 39.3, 39.2, 36.83, 36.82, 29.14, 29.08, 28.2, 25.9, 25.8, 25.0, 22.4; IR (thin film, cm-1) 3319, 3070, 1642, 1538, 1478; HRMS (ESI) m/z: [M + Na]+ Calcd for C21H28ClNO3Na 400.1655; Found: 400.1659.
2-(1,5-Dioxaspiro[5.5]undec-7-yl)-N-[(1R)-1-isopropylbut-3-en-1-yl]-acetamide (14e)
Following general procedure D, 14e (747 mg, 2.40 mmol, 68% yield) was obtained as a colorless solid from (R)-2-methylhex-5-en-3-amine (51% ee).33 mp 88–92 °C; 1H NMR (500 MHz, CDCl3) δ 5.80–5.73 (m, 2H), 5.07–5.03 (m, 2H), 4.06 (tt, J = 12.0, 3.0 Hz, 1H), 3.95–3.84 (m, 2H), 3.83–3.77 (m, 2H), 2.80 (dt, J = 14.5, 3.0 Hz, 1H), 2.61 (br s, 1H), 2.26 (dt, J = 15.0, 5.0 Hz, 1H), 2.15–2.10 (m, 2H), 1.93 (ddd, J = 14.6, 7.5, 2.8 Hz, 2H), 1.78–1.71 (m, 2H), 1.69–1.63 (m, 1H), 1.62–1.55 (m, 2H), 1.43–1.18 (m, 5H), 0.90 (ddd, J = 14.5, 6.8, 2.2 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 173.41, 173.38, 135.4, 117.24, 117.21, 99.0, 98.9, 59.3, 59.22, 59.19, 53.4, 53.3, 37.12, 37.09, 37.0, 31.5, 31.4, 29.1, 29.0, 28.3, 28.2, 25.9, 25.0, 22.5, 19.48, 19.46, 18.1; IR (thin film, cm-1) 3294, 2941, 1654, 1538, 1108; HRMS (ESI) m/z: [M + Na]+ Calcd for C18H31NO3Na 332.2202; found: 332.2210.
General Procedure E for Reduction of an Amide with Lithium Aluminum Hydride
(1R)-N-[2-(1,5-Dioxaspiro[5.5]undec-7-yl)ethyl]-1-(4-methoxyphenyl)but-3-en-1-amine (9b)
Under an atmosphere of dry N2, LiAlH4 (1.64 g, 43.1 mmol) was added to a solution of 14b (1.61 g, 4.31 mmol) and Et2O (110 mL) at 0 °C. The suspension was stirred at room temperature for 22 h. After cooling to 0 °C, water (3 mL), 10% NaOH (3 mL), and water (3 mL) were slowly added sequentially and the mixture was stirred at room temperature for 0.5 h. After filtration, the filtrate was dried (MgSO4) and concentrated in vacuo. The crude residue was purified by flash chromatography (100:1 EtOAc:Et3N) to afford 9b as a colorless oil (1.31 g, 3.66 mmol, 85%). 1H NMR (500 MHz, CDCl3) δ 7.23 (dd, J = 8.7, 3.9 Hz, 2H), 6.86 (d, J = 8.7 Hz, 2H), 5.71 (ddt, J = 17.4, 10.0, 7.2 Hz, 1H), 5.09–5.01 (m, 2H), 4.01 (td, J = 11.5, 2.5 Hz, 1H), 3.88 (td, J = 11.5, 3.0 Hz, 1H), 3.81–3.73 (m, 5H), 3.62 (q, J = 6.6 Hz, 1H), 2.53–2.42 (m, 2H), 2.40–2.34 (m, 3H), 1.99–1.84 (m, 2H), 1.54–1.47 (m, 5H), 1.43–1.15 (m, 7H); 13C NMR (125 MHz, CDCl3) δ 158.68, 158.67, 136.5, 136.0, 128.41, 128.38, 117.40, 117.36, 113.80, 113.79, 99.17, 99.16, 62.4, 62.2, 59.13, 59.12, 59.07, 59.06, 55.42, 55.41, 46.9, 46.4, 43.29, 43.25, 28.8, 28.44, 28.35, 28.0, 27.8, 25.9, 22.48, 22.46; IR (thin film, cm-1) 2933, 2860, 1511, 1245, 1106; HRMS (ESI) m/z: [M + H]+ Calcd for C22H34NO3 360.2539; Found: 360.2536.
(1R)-N-[2-(1,5-Dioxaspiro[5.5]undec-7-yl)ethyl]-1-phenylbut-3-en-1-amine (9a)
Following general procedure E, 9a (1.56 g, 1.62 mmol, 97% yield) was obtained as a pale yellow oil; changes from the standard procedure include the use of THF (70 mL) and Et2O (30 mL) as the solvent mixture (to assist in the solubility of the amide). 1H NMR (500 MHz, CDCl3) δ 7.32–7.31 (m, 4H), 7.26–7.21 (m, 1H),5.76–5.608 (m, 1H), 5.10–5.02 (m, 2H), 4.01 (td, J = 11.5, 3.0 Hz, 1H), 3.88 (td, J = 11.0, 2.5 Hz, 1H), 3.80–3.74 (m, 2H), 3.66 (q, J = 6.2 Hz, 1H), 2.54–2.45 (m, 2H), 2.44–2.36 (m, 3H), 2.00–1.84 (m, 2H), 1.63–1.47 (m, 7H), 1.44–1.16 (m, 7H); 13C NMR (125 MHz, CDCl3) δ 144.4, 135.9, 128.42, 128.41, 128.39, 128.38, 127.44, 127.43, 127.42, 127.39, 127.38, 127.01, 126.99, 117.53, 117.49, 99.14, 99.13, 63.0, 62.8, 59.12, 59.11, 59.05, 59.0, 47.0, 46.4, 43.3, 43.2, 28.8, 28.5, 28.4, 28.3, 28.0, 27.8, 25.8, 22.5, 22.4; IR (thin film, cm-1) 2931, 2860, 1453, 1245, 1108; HRMS (ESI) m/z: [M + H]+ Calcd for C21H32NO2 330.2433; Found: 330.2426.
(1R)-1-(4-Chlorophenyl)-N-[2-(1,5-dioxaspiro[5.5]undec-7-yl)ethyl]but-3-en-1-amine (9c)
Following general procedure E, 9c (1.50 g, 4.13 mmol, 98% yield) was obtained as a colorless oil; changes from the standard procedure include the use of THF (16 mL) as the solvent mixture (to assist in the solubility of the amide). 1H NMR (500 MHz, CDCl3) δ 7.32–7.24 (m, 4H), 5.69 (ddt, J = 10.5, 7.5 Hz, 1H), 5.08–5.02 (m, 2H), 4.01 (tt, J = 11.0, 2.0 Hz, 1H), 3.88 (td, J = 11.5, 3.0 Hz, 1H), 3.77–3.75 (m, 2H), 3.64 (q, J = 6.5 Hz, 1H), 2.50–2.45 (m, 2H), 2.38–2.33 (m, 3H), 1.99–1.84 (m, 2H), 1.55–1.46 (m, 5H), 1.42–1.15 (m, 7H); 13C NMR (125 MHz, CDCl3) δ 143.0, 142.9, 135.34, 135.33, 132.47, 132.45, 128.81, 128.80, 128.79, 128.78, 128.77, 128.75, 128.74, 128.73, 128.72, 128.52, 128.50, 117.9, 117.8, 99.08, 99.07, 99.06, 62.36, 62.35, 62.19, 62.18, 62.17, 59.10, 59.09, 59.03, 59.01, 47.0, 46.4, 43.21, 43.19, 30.5, 28.8, 28.4, 28.3, 28.2, 28.1, 27.8, 25.8, 24.5, 22.42, 22.40; IR (thin film, cm-1) 2933, 2860, 1490, 1245, 1108; HRMS (ESI) m/z: [M + Na]+ Calcd for C21H30ClNO2Na 386.1863; Found: 386.1860.
(1R)-1-(2-Chlorophenyl)-N-[2-(1,5-dioxaspiro[5.5]undec-7-yl)ethyl]but-3-en-1-amine (9d)
Following general procedure E, 9d (1.35 g, 3.69 mmol, 83% yield) was obtained as a colorless oil; changes from the standard procedure include the use of THF (16 mL) as a solvent (to assist in the solubility of the amide) and the purification of 9d on silica gel by flash chromatography (100:100:1 hex-anes:EtOAc:Et3N). 1H NMR (500 MHz, CDCl3) δ 7.55 (ddd, J = 7.8, 4.2, 1.7 Hz, 1H), 7.34 (dt, J = 7.9, 0.9 Hz, 1H), 7.27 (tt, J = 7.5, 1.5 Hz, 1H), 7.17 (td, J = 7.4, 1.7 Hz, 1H), 5.83–5.75 (m, 1H), 5.14–5.06 (m, 2H), 4.25 (ddd, J = 12.5, 8.0, 5.0 Hz, 1H), 4.06–4.00 (m, 1H), 3.90 (tt, J = 11.0, 3.0 Hz, 1H), 3.82–3.76 (m, 2H), 2.55–2.48 (m, 3H), 2.44–2.36 (m, 1H), 2.30 (dt, J = 14.0, 8.0 Hz, 1H), 2.03–1.95 (m, 1H), 1.94–1.86 (m, 1H), 1.57–1.53 (m, 4H), 1.42 (dq, J = 13.0, 2.5 Hz, 1H), 1.38–1.28 (m, 2H), 1.27–1.17 (m, 4H); 13C NMR (125 MHz, CDCl3) δ 141.39, 141.36, 135.43, 135.42, 133.8, 133.7, 129.58, 129.56, 128.32, 128.25, 127.86, 127.85, 127.1, 127.0, 117.9, 117.8, 99.13, 99.12, 59.13, 59.11, 59.05, 59.0, 58.4, 58.2, 46.8, 46.4, 41.5, 41.4, 31.8, 31.1, 28.8, 28.5, 28.4, 28.3, 28.0, 27.8, 25.83, 25.82, 24.5, 22.8, 22.5, 22.4, 14.3; IR (thin film, cm-1) 2933, 2860, 1461, 1441, 1108; HRMS (ESI) m/z: [M + H]+ Calcd for C21H31ClNO2 364.2043; Found: 364.2043.
(1R)-N-[2-(1,5-Dioxaspiro[5.5]undec-7-yl)ethyl]-1-isopropylbut-3-en-1-amine (9e)
Following general procedure E, 9e (488 mg, 1.65 mmol, 85% yield) was obtained as a pale yellow oi. 1H NMR (500 MHz, CDCl3) δ 5.80 (ddt, J = 17.0, 10.0, 7.5 Hz, 1H), 5.10–04 (m, 2H), 4.03 (td, J = 11.0, 3.0 Hz, 1H), 3.90 (td, J = 11.5, 3.0 Hz, 1H), 3.82–3.79 (m, 2H), 2.68–2.62 (m, 1H), 2.55–2.45 (m, 2H), 2.37–2.33 (m, 1H), 2.23–2.18 (m, 1H), 2.08–2.01 (m, 1H), 1.99–1.87 (m, 2H), 1.84–1.77 (m, 1H), 1.66–1.53 (m, 6H), 1.45–1.21 (m, 8H), 0.91–0.88 (m, 6H); 13C NMR (125 MHz, CDCl3) δ 137.2, 116.7, 99.19, 99.18, 62.92, 62.91, 62.89, 59.10, 59.09, 59.03, 47.1, 47.0, 42.8, 35.34, 35.31, 30.2, 30.1, 28.9, 28.8, 28.42, 28.36, 27.9, 25.9, 24.4, 22.47, 22.46, 19.0, 18.9, 18.19, 18.17; IR (thin film, cm-1) 2935, 2863, 1465, 1445, 1109; HRMS (ESI) m/z: [M + H]+ Calcd for C18H34NO2 296.2590; Found: 296.2585.
Representative Procedure for the Synthesis of Octahydroindole ent-8 from Aminoacetals 9
(3aR,7aS)-7a-Allyloctahydro-1H-indole (ent-8)
A stirring mixture of 9a (0.20 g, 0.62 mmol), TFA (47 μ L, 0.62 mmol), and morpholine (5.4 μL, 0.062 mmol) was heated at 120 °C. Dimedone (0.22 g, 1.5 mmol) was added and the solution was maintained at 120 °C for 5 h. After cooling to room temperature, the reaction mixture was dissolved in Et2O (10 mL). The mixture was extracted with 1 N HCl (2 × 10 mL), and the combined aqueous layers were washed with Et2O (30 mL), treated with 10% NaOH until pH 14, and extracted with Et2O (30 mL). The organic layer was washed with brine (20 mL), dried (MgSO4), filtered, and concentrated in vacuo to afford ent-8 as yellow oil (94 mg, 0.57 mmol, 92%). ; HPLC analysis of the derived benzoyl protected amine S1 indicated an enantiomeric excess of 88% (see below). 1H NMR (500 MHz, CDCl3) δ 5.87-5.79 (m, 1H), 5.09-5.02 (m, 2H), 3.03-2.91 (m, 2H), 2.25 (ddt, J = 13.9, 8.0, 1.2 Hz, 1H), 2.11 (ddt, J = 15.1, 7.0, 6.3 Hz, 1H), 1.89-1.82 (m, 1H), 1.76-1.63 (m, 3H), 1.57-1.50 (m, 1H), 1.48-1.27 (m, 7H); 13C NMR (125 MHz, CDCl3) δ 135.0, 117.8, 61.9, 42.8, 42.5, 42.3, 31.4, 29.8, 26.8, 22.5, 22.1; IR (thin film, cm-1) 3300, 2923, 1667, 1407; HRMS (ESI) m/z: [M + H]+ Calcd for C11H20N 166.1596; Found: 166.1594.
7a-Allyl-1-benzoyloctahydro-1H-indole (S1)
Benzoyl chloride (37 mg, 0.26 mmol) was added to a solution of ent-8 (31 mg, 0.19 mmol), Et3N (40 mg, 0.40 mmol) and CH2Cl2 (0.5 mL) at room temperature. The solution was maintained at room temperature for 1 h. After the addition of Et2O (10 mL), the organic layer was washed with water (5 mL), 1N HCl (5 mL), 10% NaOH (5 mL) and brine (5 mL). The organic layer was dried (MgSO4), filtered, and concentrated in vacuo. The crude residue was purified by flash chromatography (1:10 EtOAc:hexanes) to afford the title compound as a colorless solid (44 mg, 0.17 mmol, 87%). HPLC analysis indicated an enantiomeric excess of 89% [CHIRALCEL AD column; flow: 1.0 mL/min; 96.5% n-hexane/3.5% i-PrOH; λ = 220 nm; major enantiomer tR = 28.3 min; minor enantiomer tR = 25.5 min minor enantiomer tR=25.5 min]. mp 46-47 °C; . . 1H NMR (500 MHz, CDCl3) δ 7.41–7.35 (m, 5H), 5.91–5.83 (m, 1H), 5.16–5.13 (m, 2H), 3.33–3.30 (m, 2H), 3.25 (dd, J = 13.9, 6.5 Hz, 1H), 2.60–2.53 (m, 2H), 2.28–2.23 (m, 1H), 1.91–1.83 (m, 1H), 1.77 (td, J = 13.1, 4.1 Hz, 1H), 1.71–1.56 (m, 4H), 1.50–1.30 (m, 3H); 13C NMR (125 MHz, CDCl3) δ 170.0, 139.0, 134.7, 129.4, 128.4, 126.6, 118.3, 65.5, 50.7, 39.7, 37.5, 32.8, 27.3, 25.2, 22.8, 21.5; IR (thin film, cm-1) 1630, 1403, 1218, 1189, 1138; HRMS (ESI) m/z: [M + Na]+ Calcd for C18H23NONa 292.1677; Found: 292.1680.
Synthesis and Characterization of Aminoketals 15–17 and Their Precursors
Benzyl 2-(2-oxocyclopentyl)acetate (S2)
Following general procedure B, S2 (2.3 g, 9.9 mmol, 78%) was obtained as a colorless oil. This product was purified on silica gel by flash chromatography (19:1 hexanes:EtOAc to 9:1 hexanes:EtOAc). Characterization data were consistent with previously reported values.34
Benzyl 2-(6,10-dioxaspiro[4.5]decan-1-yl)acetate (S3)
Following general procedure C, S3 (5.8 g, 20 mmol, 96%) was obtained as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.37-7.28 (m, 5H), 5.14 (d, J = 12.4 Hz, 1H), 5.08 (d, J = 12.4 Hz, 1H), 3.90–3.77 (m, 4H), 2.71 (dd, J = 15.0, 3.8 Hz, 1H), 2.38–2.25 (m, 2H), 2.11 (ddd, J = 13.1, 8.7, 6.9 Hz, 1H), 1.96–1.86 (m, 2H), 1.82 (ddd, J = 13.1, 9.4, 6.1 Hz, 1H), 1.71–1.62 (m, 2H), 1.39–1.31 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 173.5, 136.3, 128.4, 128.1, 128.0, 108.1, 65.9, 62.1, 60.6, 45.1, 33.8, 29.9, 28.6, 25.8, 20.8; IR (thin film, cm-1) 2958, 2867, 1733; HRMS (ES) m/z: [M + Na]+ Calcd for C17H22O4Na 313.1416; Found: 313.1420.
2-(6,10-Dioxaspiro[4.5]dec-1-yl)-N-[(1R)-1-phenylbut-3-en-1-yl]acetamide (S4)
Following general procedure D, S4 (1.5 g, 4.5 mmol, 89%) was obtained as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.33–7.23 (m, 5H), 7.07 (br s, 0.5H), 6.90 (br s, 0.5H), 5.74–5.66 (m, 1H), 5.11–5.01 (m, 3H), 3.94–3.78 (m, 4H), 3.75–3.71 (m, 1H), 2.69 (ddd, J = 15.7, 7.8, 3.6 Hz, 1H), 2.62–2.52 (m, 2H), 2.29–2.03 (m, 3H), 1.96–1.81 (m, 3H), 1.75–1.57 (m, 3H), 1.39–1.24 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 172.5, 142.3, 142.1, 134.5, 128.64, 128.63, 127.4, 127.3, 126.9, 126.73, 126.70, 126.69, 117.90, 117.89, 108.2, 108.1, 62.4, 62.3, 60.7, 60.6, 52.9, 52.7, 46.2, 46.1, 41.0, 40.7, 36.2, 36.1, 30.7, 30.54, 30.53, 30.2, 30.0, 25.9, 25.7, 21.5, 21.3; IR (thin film, cm-1) 3292, 1638, 1542, 1152; HRMS (ESI) m/z: [M + Na]+ Calcd for C20H27NO3Na 352.1889; Found: 352.1894.
N-[2-(6,10-Dioxaspiro[4.5]dec-1-yl)-ethyl]-(1R)-1-phenylbut-3-en-1-amine (S5)
Following general procedure E, S5 (1.15 g, 3.67 mmol, 93%) was obtained as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.27–7.22 (m, 4H), 7.19–7.16 (m, 1H), 5.67 (ddt, J = 16.9, 9.4, 7.5 Hz, 1H), 5.04–4.97 (m, 2H), 3.85–3.75 (m, 4H), 3.62 (t, J = 6.9, 1H), 2.50–2.34 (m, 4H), 2.10–1.99 (m, 1H), 1.90–1.49 (m, 8H), 1.39–1.27 (m, 2H), 1.24–1.13 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 144.44, 144.38, 135.8, 128.36, 128.35, 127.41, 127.40, 127.0, 126.9, 117.4, 108.83, 108.81, 62.83, 62.75, 62.19, 62.17, 60.7, 46.9, 46.8, 46.6, 46.5, 43.21, 43.20, 30.7, 29.4, 29.22, 29.17, 29.1, 26.07, 26.05, 21.28, 21.25; IR (thin film, cm-1) 2954, 2861, 1453, 1246, 1108; HRMS (ESI) m/z: [M + H]+ Calcd for C20H30NO2 316.2277; Found: 316.2282.
Benzyl 3-(2-oxocyclopentyl)propanoate (S6)
Ketone S6 was prepared using an adaptation of the procedure of Cotarco and co-workers.35 A stirring solution of cyclopentanone (19 mL, 0.21 mol), 4-methoxyphenol (0.17 g, 1.4 mmol), cyclohexylamine (1.7 mL, 15 mmol) and acetic acid (0.15 mL, 2.5 mmol) was heated to 80 °C for 10 min, then warmed to 130 °C over 30 min. Benzyl acrylate (16 mL, 0.11 mol) was added via syringe pump at a rate of 6.8 mL/h. Upon complete addition, the reaction was cooled to 25 °C and the residue was purified directly on silica gel by flash chromatography (1:9-1:4 EtOAc:hexanes) to provide S6 (13 g, 52 mmol, 47%) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.40-7.31 (m, 5H), 5.15 (s, 2H), 2.55-2.45 (m, 2H), 2.31 (dd, J = 18.8, 8.6 Hz, 1H), 2.23-2.17 (m, 1H), 2.16-2.07 (m, 3H), 2.05-1.97 (m, 1H), 1.82-1.73 (m, 1H), 1.73-1.63 (m, 1H), 1.56-1.46 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 219.9, 172.7, 135.8, 128.3, 128.1, 128.0, 65.9, 47.9, 37.7, 31.9, 29.2, 24.6, 20.3; IR (thin film, cm-1) 2960, 2877, 1733; Anal. Calcd for C15H18O3: C, 73.15; H, 7.37. Found: C, 73.26; H, 7.40; HRMS (ES) m/z: [M + Na]+ Calcd for C15H18O3Na 269.1154; Found: 269.1144.
Benzyl 3-(6,10-dioxaspiro[4.5]decan-1-yl)propanoate (S7)
Following general procedure C, S7 (4.6 g, 16 mmol, 56%) was obtained as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.38–7.27 (m, 5H), 5.14–5.08 (m, 2H), 3.90–3.79 (m, 4H), 2.54–2.42 (m, 2H), 2.09 (dt, J = 13.3, 8.5 Hz, 1H), 1.90–2.01 (m, 2H), 1.87–1.74 (m, 3H), 1.70–1.53 (m, 3H) 1.37–1.24 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 173.7, 136.2, 128.3, 128.2, 128.0, 108.3, 65.8, 61.9, 60.4, 47.9, 32.8, 30.5, 28.7, 25.7, 23.7, 20.9; IR (thin film, cm-1) 2956, 2865, 1733; Anal. Calcd for C18H24O4: C, 71.03; H, 7.95. Found: C, 71.12; H, 7.94; HRMS (ES) m/z: [M + Na]+ Calcd for C18H24O4Na 327.1572; Found: 327.1564.
3-(6,10-Dioxaspiro[4.5]dec-1-yl)-N-[(1R)-1-phenylbut-3-en-1-yl]propionamide (S8)
Following general procedure D, S8 (1.66 g, 4.85 mmol, 97%) was obtained as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.30–7.22 (m, 5H), 6.41 (app br d, J = 20.8 Hz, 1H), 5.71–5.63 (m, 1H), 5.08–5.05 (m, 3H), 3.87–3.80 (m, 4H), 2.53–2.51 (m, 2H), 2.48–2.41 (m, 1H), 2.29–2.22 (m, 1H), 2.18–2.10 (m, 1H), 2.02–1.74 (m, 5H), 1.69–1.49 (m, 3H), 1.41–1.36 (m, 1H), 1.31–1.19 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 173.19, 173.16, 142.3, 142.2, 134.5, 134.4, 128.74, 128.69, 128.6, 127.4, 127.33, 127.27, 126.8, 126.7, 126.6, 118.2, 118.03, 118.00, 109.0, 108.9, 62.39, 62.36, 62.1, 60.5, 52.6, 52.4, 47.5, 47.4, 40.9, 35.3, 34.3, 31.0, 30.9, 29.6, 29.5, 26.2, 25.0, 21.34, 21.25; IR (thin film, cm-1) 3288, 2956, 1638, 1542, 1248; HRMS (ESI) m/z: [M + Na]+ Calcd for C21H29NO3Na 366.2045; Found: 366.2037.
N-[3-(6,10-Dioxaspiro[4.5]dec-1-yl)-propyl]-(1R)-1-phenylbut-3-en-1-amine (16)
Following general procedure E, 16 (1.27 g, 3.84 mmol, 88%) was obtained as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.30–7.27 (m, 4H), 7.21–7.18 (m, 1H), 5.72–5.64 (m, 1H), 5.06–4.99 (m, 2H), 3.87–3.79 (m, 4H), 3.62 (t, J = 6.9 Hz, 1H), 2.43–2.36 (m, 4H), 2.06–2.00 (m, 1H), 1.95–1.88 (m, 1H), 1.84–1.68 (m, 6H), 1.34 (dq, J = 7.9, 2.6 Hz, 1H), 1.26–1.19 (m, 1H), 1.17–1.08 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 144.31, 144.26, 135.8, 128.4, 127.39, 127.37, 127.0, 117.5, 108.9, 62.9, 62.8, 62.11, 62.10, 60.8, 48.9, 48.8, 48.4, 48.3, 43.2, 43.1, 30.71, 30.68, 29.1, 29.02, 28.99, 28.9, 26.34, 26.27, 26.1, 21.12, 21.09; IR (thin film, cm-1) 2952, 2860, 1453, 1246, 1109; HRMS (ESI) m/z: [M + H]+ Calcd for C21H32NO2 330.2433; Found: 330.2433.
Benzyl 3-(2-oxocyclohexyl)propanoate (S9)
Ketone S9 was prepared using an adaptation of the procedure of Cotarco and co-workers.35 A stirring solution of cyclohexanone (24 mL, 0.21 μmol), 4-methoxyphenol (0.17 g, 1.4 mmol), cyclohexylamine (1.7 mL, 15 mmol) and acetic acid (0.15 mL, 2.5 mmol) was heated to 80 °C for 10 min, then warmed to 130 °C over 30 min. Benzyl acrylate (16 mL, 0.11 mol) was added via syringe pump at a rate of 6.8 mL/h. Upon complete addition, the reaction was cooled to 25 °C and the residue was purified directly on silica gel by flash chromatography (1:9–1:4 EtOAc:hexanes) to provide S9 (23 g, 87 mmol, 83%) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.32–7.24 (m, 5H), 5.06 (s, 2H), 2.45–2.1 (m, 5H), 2.09–1.95 (m, 3H), 1.83–1.74 (m, 1H), 1.64–1.49 (m, 3H), 1.31 (dq, J = 8.9, 3.7 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 211.0, 173.0, 135.8, 128.2, 128.0, 127.9, 65.8, 49.3, 41.8, 33.8, 31.5, 27.7, 24.8, 24.5; IR (thin film, cm−1) 2935, 2861, 1733, 1708; Anal. Calcd for C16H20O3: C, 73.82; H, 7.72. Found: C, 73.88; H, 7.72; HRMS (ES) m/z: [M + Na]+ Calcd for C16H20O3Na 283.1310; Found: 283.1297.
Benzyl 3-(1,5-dioxaspiro[5.5]undecan-7-yl)propanoate (S10)
Following general procedure C, S10 (4.1 g, 13 mmol, 78%) was obtained as a colorless oil; changes from the standard procedure include the exposure of this substrate to reaction conditions at 25 °C for 3 h. 1H NMR (500 MHz, CDCl3) δ 7.36–7.27 (m, 5H), 5.08 (s, 2H), 3.98 (app td, J = 11.5, 2.7 Hz, 1H), 3.85 (app td, J = 11.3, 2.7 Hz, 1H), 3.77–3.72 (m, 2H), 2.49–2.40 (m, 1H), 2.39–2.30 (m, 1H), 2.22–2.13 (m, 1H), 1.93–1.83 (m, 1H), 1.62–1.46 (m, 6H), 1.41–1.26 (m, 3H), 1.25–1.12 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 174.0, 136.2, 128.4, 128.1, 128.0, 98.9, 65.9, 58.8, 58.7, 44.0, 32.9, 28.0, 27.3, 25.5, 24.2, 23.4, 22.2; IR (thin film, cm-1) 2935, 2861, 1735; Anal. Calcd for C19H26O4: C, 71.67; H, 8.23. Found: C, 71.62; H, 8.39.
3-(1,5-Dioxaspiro[5.5]undec-7-yl)-N-[(1R)-1-phenylbut-3-en-1-yl]propionamide (S11)
Following general procedure D, S11 (1.45 g, 4.03 mmol, 99%) was obtained as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.34–7.24 (m, 5H), 6.51 (app t, J = 9.4 Hz, 1H), 5.76–5.67 (m, 1H), 5.17–5.08 (m, 3H), 4.07 (tdd, J = 11.9, 5.9, 3.0 Hz, 1H), 3.92 (td, J = 11.7, 2.8 Hz, 1H), 3.82–3.73 (m, 2H), 2.62–2.59 (m, 1H), 2.57–2.52 (m, 2H), 2.44–2.38 (m, 1H), 2.25–2.14 (m, 2H), 2.02–1.90 (m, 1H), 1.66–1.50 (m, 4H), 1.44–1.11 (m, 7H); 13C NMR (125 MHz, CDCl3) δ172.8, 172.7, 142.4, 142.3, 134.7, 134.5, 128.7, 128.6, 127.4, 127.3, 126.8, 126.7, 118.0, 99.53, 99.52, 59.19, 59.17, 59.1, 52.51, 52.45, 41.0, 40.8, 35.3, 35.2, 28.4, 28.1, 28.0, 25.9, 24.3, 24.2, 22.44, 22.4; IR (thin film, cm-1) 3305, 2935, 1640, 1541, 1448; HRMS (ESI) m/z: [M + H]+ Calcd for C22H32NO3 358.2382; Found: 358.2390.
N-[3-(1,5-Dioxaspiro[5.5]undec-7-yl)-propyl]-(1R)-1-phenylbut-3-en-1-amine (17)
Following general procedure E, 17 (1.17 g, 3.41 mmol, 89%) was obtained as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.34–7.31 (m, 4H), 7.25–7.22 (m, 1H), 5.76–5.67 (m, 1H), 5.10–5.03 (m, 2H), 4.01 (td, J = 11.4, 3.1 Hz, 1H), 3.88 (td, J = 10.5, 2.7 Hz, 1H), 3.79–3.75 (m, 2H), 3.65 (t, J = 6.8 Hz, 1H), 2.49–2.41 (m, 5H), 1.91–1.83 (m, 1H), 1.78–1.70 (m, 1H), 1.66–1.62 (m, 1H), 1.57–1.46 (m, 4H), 1.44–1.30 (m, 3H), 1.27–1.20 (m, 3H), 1.14–1.06 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 144.2, 135.8, 128.5, 127.43, 127.40, 127.1, 117.6, 99.3, 62.8, 62.7, 59.10, 59.09, 59.0, 48.3, 48.1, 43.1, 28.7, 28.4, 28.34, 28.29, 27.33, 27.25, 25.8, 25.2, 25.1, 24.3, 22.5; IR (thin film, cm-1) 2933, 2860, 1453, 1244, 1107; HRMS (ESI) m/z: [M + H]+ Calcd for C22H34NO2 344.2590; Found: 344.2581.
General Procedure F for 2-Aza-Cope Rearrangement and Cbz Protection to Yield 1-Azabicyclic Products ent-8 and 18–20
Preparation of (3aR, 6aS)-benzyl 6a-allylhexahydrocyclopenta[b]pyrrole-1(2H)-carboxylate (18)
A stirring mixture of 15 (228 mg, 0.722 mmol), TFA (56.0 μL, 0.722 mmol) and morpholine (6.30 μL, 0.0722 mmol) was heated to 120 °C. Dimedone (253 mg, 1.81 mmol) was added and the solution was maintained at 120 °C for 2 h. After cooling to room temperature, the reaction mixture was dissolved in CHCl3 (5 mL). A saturated aqueous solution of Na2CO3 (2.4 mL), water (2.4 mL) and benzyl chloroformate (310 μL, 2.17 mmol) was added and the biphasic solution was stirred at room temperature for 18 h. The aqueous layer was extracted with CH2Cl2 (3 × 10 mL) and the combined organic layer was dried (Na2SO4), filtered, and concentrated in vacuo. The crude residue was purified by flash chromatography (1:20 EtOAc:hexanes) to afford 18 as a colorless oil (196 mg, 95%). HPLC analysis indicated an enantiomeric excess of 92% [CHIRALCEL OJ column; flow: 1.0 mL/min; 96% n-hexane/4% i-PrOH; λ = 220 nm; major enantiomer tR = 6.7 min; minor enantiomer tR = 8.5 min]. . 1H NMR (500 MHz, DMSO-d6, 388 K) δ 7.36 (app d, J = 4.5 Hz, 4H), 7.32–7.28 (m, 1H), 5.79–5.71 (m, 1H), 5.13–5.00 (m, 2H), 3.53–3.42 (m, 2H), 2.81–2.77 (m, 2H), 2.50–2.44 (m, 1H), 2.37 (dd, J = 18.8, 7.5 Hz, 1H), 2.16 (dt, J = 12.3, 6.3 Hz, 1H), 1.93 (dq, J = 12.6, 8.1 Hz, 1H), 1.87–1.80 (m, 1H), 1.72–1.65 (m, 1H), 1.64–1.51 (m, 3H), 1.45–1.38 (m, 1H); 13C NMR (125 MHz, DMSO-d6, 388 K) δ 152.6, 136.7, 134.2, 127.4, 126.7, 126.6, 116.4, 73.1, 64.9, 47.3, 47.0, 40.5, 40.0, 39.8, 39.7, 39.5, 39.3, 39.2, 39.0, 37.3, 31.1, 27.8, 24.0; IR (thin film, cm-1) 2946, 1696, 1405, 1355; HRMS (ESI) m/z: [M + Na]+ Calcd for C18H23NO2Na 308.1626; Found: 308.1622.
(3aR, 7aS)-Benzyl 7a-allyloctahydro-1H-indole-carboxylate (Cbz-ent-8)
Following general procedure F, (164 mg, 0.549 mmol, 85%) was obtained as a colorless oil; changes from the standard procedure include heating 9a at 120 °C for 5 h and purification by flash chromatography on silica gel (20:1 hexanes:Et2O). HPLC analysis indicated an enantiomeric excess of 89% [CHIRALCEL OJ column; flow: 1.0 mL/min; 96% n-hexane/4% i-PrOH; λ = 220 nm; major enantiomer tR = 7.8 min; minor enantiomer tR = 11.6 min]. . 1H NMR (500 MHz, DMSO-d6, 398 K) δ 7.36–7.28 (m, 5H), 5.78–5.70 (m, 1H), 5.12–5.00 (m, 4H), 3.62–3.57 (m, 1H), 3.34–3.29 (m, 1H), 2.72 (dd, J = 15.0, 7.1 Hz, 1H), 2.50–2.44 (m, 1H), 2.14–2.09 (m, 1H), 1.94–1.60 (m, 5H), 1.51–1.28 (m, 5H); 13C NMR (125 MHz, DMSO-d6, 398 K) δ 152.9, 136.8, 133.8, 127.4, 126.6, 126.5, 116.3, 64.7, 63.1, 45.4, 39.0, 38.8, 31.3, 25.4, 24.8, 20.7, 20.4; IR (thin film, cm-1) 2927, 1696, 1401, 1337; HRMS (ESI) m/z: [M + Na]+ Calcd for C19H25NO2Na 322.1783; Found: 322.1777.
(4aR, 7aS)-Benzyl 7a-allyloctahydro-1 H-cyclopenta[b]pyridine-1-carboxylate (19)
Following general procedure F, 19 (155 mg, 0.517 mmol, 80%) was obtained as a colorless oil; changes from the standard procedure include heating at 120 °C for 5 h and purification of the product by silica gel column chromatography (10:1 hexanes:Et2O). HPLC analysis indicated an enantiomeric excess of 91% [CHIRALCEL OJ column; flow: 1.0 mL/min; 96% n-hexane/4% i-PrOH; λ = 220 nm; major enantiomer tR = 7.3 min]. . . 1H NMR (500 MHz, DMSO-d6, 298 K) δ 7.38–7.29 (m, 5H), 5.80 (ddt, J = 16.7, 10.4, 7.5 Hz, 1H), 5.08–4.96 (m, 4H), 3.81 (ddd, J = 13.4, 6.4, 3.3 Hz, 1H), 2.96 (ddd, J = 13.4, 11.2, 5.0 Hz, 1H), 2.61 (dd, J = 13.8, 7.4 Hz, 1H), 2.26 (dd, J = 13.8, 7.6 Hz, 1H), 2.20–2.14 (m, 1H), 2.04–1.97 (m, 2H), 1.78–1.71 (m, 1H), 1.67–1.34 (m, 7H); 13C NMR (125 MHz, DMSO-d6, 298 K) δ 155.3, 137.1, 134.7, 128.3, 127.6, 127.5, 117.5, 66.6, 65.7, 41.7, 40.9, 40.1, 37.5, 28.7, 24.0, 21.1, 20.7; IR (thin film, cm-1) 2948, 1702, 1410, 1218; HRMS (ESI) m/z: [M + Na]+ Calcd for C19H25NO2Na 322.1783; Found: 322.1778.
(4aR, 8aS)-Benzyl 8a-allyloctahydroquinoline-1(2H)-carboxylate (cis-20) and (4aR, 8aR)-benzyl 8a-allyloctahydroquinoline-1(2H)-carboxylate (trans-20)
Octahydroquinoline cis-20 was isolated as a colorless oil (80 mg, 0.25 mmol, 41%) and trans-20 was isolated as a colorless oil (66 mg, 0.21 mmol, 34%). Changes from the standard procedure include heating at 120 °C for 5 h and separation of the products by flash chromatography on silica gel (20:1 hexanes:Et2O). cis-20: HPLC analysis indicated an enantiomeric excess of 93% [CHIRALCEL OJ column; flow: 1.0 mL/min; 96% n-hexane/4% i-PrOH; λ = 220 nm; major enantiomer tR = 6.2 min; minor enantiomer tR = 7.9 min]. . 1H NMR (500 MHz, DMSO-d6, 348 K) δ 7.39-7.28 (m, 5H), 5.76 (ddt, J = 17.5, 10.0, 7.3 Hz, 1H), 5.07–5.00 (m, 4H), 3.79 (dt, J = 13.2, 4.7 Hz, 1H), 3.26–3.20 (m, 1H), 2.68 (dt, J = 7.3, 1.2 Hz, 1H), 2.33 (ddd, J = 12.7, 8.3, 3.8 Hz, 1H), 1.83–1.64 (m, 4H), 1.60–1.48 (m, 5H), 1.46–1.27 (m, 3H); 13C NMR (125 MHz, DMSO-d6, 348 K) δ 154.9, 136.9, 133.8, 127.8, 127.1, 127.0, 116.9, 65.3, 60.1, 40.8, 38.3, 35.7, 32.2, 27.3, 23.1, 21.9, 21.7, 20.6; IR (thin film, cm-1) 2934, 1697, 1398, 1261; HRMS (ESI) m/z: [M + Na]+ Calcd for C20H27NO2Na 336.1939; Found: 336.1938. trans-20: HPLC analysis indicated an enantiomeric excess of 93% [CHIRALCEL OJ column; flow: 1.0 mL/min; 96% n-hexane/4% i-PrOH; λ = 220 nm; major enantiomer tR = 6.5 min; minor enantiomer tR = 8.0 min]. . 1H NMR (500 MHz, DMSO-d6) δ 7.37–7.28 (m, 5H), 5.69 (ddt, J = 17.2, 10.1, 7.2 Hz, 1H), 5.10 (d, J = 17.1 Hz, 1H), 5.00–4.93 (m, 3H), 3.97 (dt, J = 14.0, 4.4 Hz, 1H), 3.08 (ddd, J = 14.3, 11.4, 3.0 Hz, 1H), 2.88 (br d, J = 13.1 Hz, 1H), 2.64 (dd, J = 15.0, 7.6 Hz, 1H), 2.40 (dd, J = 14.8, 6.9 Hz, 1H), 1.64–1.52 (m, 5H), 1.51–1.30 (m, 5H), 1.29–1.18 (m, 2H); 13C NMR (125 MHz, DMSO-d6, 298 K) δ 155.1, 137.3, 134.0, 128.3, 127.6, 127.4, 117.2, 65.5, 62.5, 45.2, 41.2, 35.0, 30.7, 29.6, 26.6, 25.3, 24.8, 22.7; IR (thin film, cm-1) 2930, 1702, 1391, 1160; HRMS (ESI) m/z: [M + Na]+ Calcd for C20H27NO2Na 336.1939; Found: 336.1947.
Preparation and Characterization of Aminoketals Employed in the Transformations Reported in Table 3
The direct precursors of 1-azabicyclic products 31a and 34a have been reported previously.9
N-((S)-2-Methylbut-3-en-1-yl)-2-(6,10-dioxaspiro[4.5]decan-1-yl)acetamide) (S12)
Following general procedure D, S12 (63 mg, 0.24 mmol, 85%) was obtained as a colorless solid; changes to the general procedure involve adding Et3N (42 μL) to the amide-coupling step. 1H NMR (500 MHz, CDCl3) δ 6.35 (broad s, 1H), 5.72–5.69 (m, 1H), 5.08–5.03 (m, 2H), 3.94–3.87 (m, 4H), 3.32–3.29 (m, 1H), 3.09–3.08 (m, 1H), 2.62 (dd, J = 15.6, 7.0 Hz, 1H), 2.38–2.31 (m, 1H), 2.28–2.21 (m, 1H), 2.14–2.07 (m, 2H), 2.02–1.85 (m, 3H), 1.70–1.65 (m, 2H), 1.42–1.25 (m, 2H), 1.03 (d, J = 6.8 Hz, 3H); 13C NMR (125 MHz) δ 173.3, 141.83, 141.78, 115.1, 115.0, 108.3, 62.4, 60.8, 60.8, 46.19, 46.15, 44.67, 44.65, 38.2, 38.1, 36.2, 30.6, 29.85, 28.82, 26.06, 26.05, 21.3, 17.79, 17.75; IR (thin film, cm-1) 3304, 3079, 2962, 1735, 1103; HRMS (ESI) m/z: [M + H]+ Calcd for C15H26NO3 268.1913; Found: 268.1915.
(2S)-N-(2-(6,10-dioxaspiro[4.5]decan-1-yl)ethyl)-2-methylbut-3-en-1-amine (S13)
Following general procedure E, S13 (28 mg, 0.11 mmol, 46%) was obtained as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 5.70–5.63 (m, 1H), 5.07–4.99 (m, 2H), 3.93–3.84 (m, 4H), 2.72–2.61 (m, 2H), 2.56–2.46 (m, 2H), 2.39–2.33 (m, 1H), 2.11 (dddd, J = 7.7, 7.7, 7.7, 7.7 Hz, 1H), 2.02–1.97 (m, 1H), 1.87–1.79 (m, 3H), 1.68-1.57 (m, 3H), 1.43–1.25 (m, 4H), 1.00 (d, J = 6.7 Hz, 3H); 13C NMR (125 MHz) δ 143.0, 114.6, 114.5, 108.9, 62.2, 60.79, 55.77, 55.76, 49.1, 48.9, 47.1, 47.0, 38.5, 38.4, 30.70, 30.66, 29.4, 29.3, 29.2, 26.2, 21.3, 18.50, 18.46; IR (thin film, cm-1) 3315, 3075, 2955, 1111; HRMS (ESI) m/z: [M + H]+ Calcd for C15H28NO2 254.2120; Found: 254.2127.
N-((R)-2-phenylbut-3-enyl)-2-(1,5-dioxaspiro[5.5]undecan-7-yl)acetamide (S14)
Following general procedure D, S14 (307 mg, 0.893 mmol, 55%) was obtained as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.32 (t, J = 7.5 Hz, 2H), 7.25–7.21 (m, 3H), 6.00–5.93 (m, 2H), 5.15–5.09 (m, 2H), 3.98 (t, J = 11.7 Hz, 1H), 3.86 (t, J = 11.7 Hz, 1H), 3.74–3.58 (m, 3H), 3.55–3.44 (m, 2H), 2.72 (d, J = 14.6 Hz, 1H), 2.59 (d, J = 11.7 Hz, 1H), 1.95 (broad s, 1H), 1.89–1.75 (m, 2H), 1.57 (app d, J = 9.9 Hz, 3H), 1.39–1.22 (m, 4H), 1.12 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 173.9, 173.8, 141.54, 141.46, 139.3, 128.9, 128.03, 127.99, 127.0, 116.4, 116.3, 98.8, 98.7, 59.13, 59.10, 49.7, 49.6, 43.7, 43.6, 36.9, 36.8, 29.1, 28.9, 28.1, 25.8, 25.0, 22.4; IR (thin film, cm-1) 3301, 2933, 1644, 1108; HRMS (ESI) m/z: [M + Na]+ Calcd for C21H29NO3Na 366.2045; Found: 366.2051.
(2R)-N-(2-(1,5-dioxaspiro[5.5]undecan-7-yl)ethyl)-2-phenylbut-3-en-1-amine (S15)
Following general procedure E, S15 (228 mg, 0.692 mmol, 80%) was obtained as a light yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.32 (t, J = 7.9 Hz, 2H), 7.24–7.21 (m, 3H), 6.01–5.94 (m, 1H), 5.15–5.10 (m, 2H), 4.01 (tt, J = 11.4, 3.2 Hz, 1H), 3.88 (t, J = 10.1 Hz, 1H), 3.78–3.76 (m, 2H), 3.55 (q, J = 7.6 Hz, 1H), 2.95–2.87 (m, 2H), 2.72–2.65 (m, 1H), 2.62–2.55 (m, 1H), 2.46 (app broad s, 1H), 1.99–1.86 (m, 2H), 1.59–1.52 (m, 3H), 1.45–1.22 (m, 8H); 13C NMR (125 MHz, CDCl3) δ 142.7, 140.40, 140.37, 128.9, 128.8, 127.94, 127.92, 126.77, 126.75, 116.0, 115.9, 99.2, 59.14, 59.08, 54.7, 54.6, 50.1, 48.92, 48.86, 28.6, 28.3, 28.1, 25.8, 24.5, 22.5; IR (thin film, cm-1) 3319, 3080, 2933, 1108; HRMS (ESI) m/z: [M + H]+ Calcd for C21H32NO2 330.2433; Found: 330.2437.
N-((S)-2-Methylbut-3-en-1-yl)-2-(1,5-dioxaspiro[5.5]undecan-7-yl)acetamide (S16)
Following general procedure D, S16 (83 mg, 0.29 mmol, 80%) was obtained as a colorless solid; changes to the general procedure involve the addition of Et3N (56.0 μL) to the amide coupling step. 1H NMR (500 MHz, CDCl3) δ 5.92 (broad s, 1H), 5.71–5.64 (m, 1H), 5.07–5.05 (m, 2H), 4.05 (app dt, J = 11.8, 2.8 Hz, 1H), 3.93 (app dt, J = 11.8, 2.8 Hz, 1H), 3.80–3.78 (m, 2H), 3.35–3.27 (m, 1H), 3.07–2.98 (m, 1H), 2.76 (dd, J = 14.3, 4.6 Hz, 1H), 2.61 (broad d, J = 12.9 Hz, 1H), 2.35–2.29 (m, 1H), 2.04–1.95 (m, 1H), 1.94–1.87 (m, 2H), 1.65–1.58 (m, 3H), 1.41–1.18 (m, 5H), 1.01 (d, J = 6.9 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 173.80, 173.79, 141.84, 141.82, 115.14, 115.09, 98.8, 59.20, 59.15, 44.51, 44.48, 38.34, 38.26, 36.9, 36.8, 29.03, 28.98, 28.2, 25.8, 25.0, 22.4, 17.8, 17.7; IR (thin film, cm-1) 3079, 2933, 1643; HRMS (ESI) m/z: [M + Na]+ Calcd for C16H27NO3Na 304.1889; Found: 304.1881.
2S)-N-(2-(1,5-Dioxaspiro[5.5]undecan-7-yl)ethyl)-2-methylbut-3-en-1-amine (S17)
Following the general procedure, S17 (24 mg, 0.095 mmol, 74%) was obtained as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 5.65–5.61 (m, 1H), 5.06–4.98 (m, 2H), 4.01 (app dt, J = 11.5, 2.8 Hz, 1H), 3.88 (app dt, J = 11.2, 2.1 Hz, 1H), 3.78–3.76 (m, 2H), 2.66–2.29 (m, 1H), 2.57–2.43 (m, 4H), 2.36–2.33 (m, 1H), 1.96–1.88 (m, 2H), 1.60–1.55 (m, 4H), 1.42–1.24 (m, 7H), 0.99 (d, J = 6.7 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 143.0, 114.6, 114.5, 99.2, 59.2, 59.1, 55.83, 55.76, 49.2, 49.0, 38.5, 38.4, 28.7, 28.6, 28.4, 28.3, 28.1, 25.8, 24.4, 22.5, 18.53, 18.48; IR (thin film, cm-1) 2933, 2862, 1446; HRMS (ESI) m/z: [M + H]+ Calcd for C16H30NO2 268.2277; Found: 268.2271.
N-((R)-2-Phenylbut-3-enyl)-3-(6,10-dioxaspiro[4.5]decan-1-yl)propanamide (S18)
Following general procedure D, S18 (408 mg, 1.19 mmol, 73%) was obtained as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.32 (t, J = 7.5 Hz, 2H), 7.24–7.21 (m, 3H), 5.99–5.92 (m, 2H), 5.14–5.09 (m, 2H), 3.88 (tt, J = 11.7, 2.4 Hz, 1H), 3.83–3.78 (m, 2H), 3.77–3.72 (m, 1H), 3.70–3.61 (m, 1H), 3.54–3.46 (m, 2H), 2.41–2.35 (m, 1H), 2.24–2.17 (m, 1H), 2.14–2.08 (m, 1H), 1.98–1.87 (m, 1H), 1.85–1.66 (m, 4H), 1.64–1.52 (m, 3H), 1.34 (dt, J = 13.2, 2.4 Hz, 1H), 1.29–1.19 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 173.98, 173.97, 141.5, 139.34, 139.28, 128.84, 128.82, 128.02, 127.98, 127.0, 116.33, 116.31, 108.8, 62.31, 62.28, 60.5, 49.62, 49.57, 47.5, 43.5, 35.2, 30.81, 30.77, 29.55, 29.54, 26.1, 25.1, 21.29, 21.27; IR (thin film, cm-1) 3299, 3083, 2958, 1646; HRMS (ESI) m/z: [M + Na]+ Calcd for C21H29NO3Na 366.2045; Found: 366.2044.
(2R)-N-(3-(6,10-Dioxaspiro[4.5]decan-1-yl)propyl)-2-phenylbut-3-en-1-amine (S19)
Following general procedure E, S19 (330 mg, 1.00 mmol, 88% yield) was obtained as light yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.32 (t, J = 7.6 Hz, 2H), 7.24–7.21 (m, 3H), 6.01–5.94 (m, 1H), 5.15–5.11 (m, 2H), 3.93–3.82 (m, 4H), 3.55 (q, J = 7.4 Hz, 1H), 2.90 (d, J = 7.4 Hz, 2H), 2.68–2.59 (m, 2H), 2.11–2.05 (m, 1H), 2.03–1.93 (m, 1H), 1.89–1.37 (m, 9H), 1.30–1.12 (m, 3H); 13C NMR (125 MHz, CDCl3) δ 142.6, 140.3, 128.9, 127.9, 126.8, 116.1, 109.0, 62.2, 60.8, 54.5, 50.3, 50.1, 48.9, 30.7, 29.0, 28.9, 26.3, 26.2, 21.2; IR (thin film, cm-1) 3332, 3081, 2863, 1148, 1109; HRMS (ESI) m/z: [M + H]+ Calcd for C21H32O2N 330.2433; Found: 330.2429.
N-((S)-2-Methylbut-3-en-1-yl)-3-(6,10-dioxaspiro[4.5]decan-1-yl)propanamide (S20)
Following general procedure D, S20 (59 mg, 0.21 mmol, 72%) was obtained as a colorless solid; changes in the procedure include the addition of Et3N (45 μL) to the amide-coupling step. 1H NMR (500 MHz, CDCl3) δ 5.95 (broad s, 1H), 5.73–5.66 (m, 1H), 5.08–5.03 (m, 2H), 3.95–3.87 (m, 4H), 3.32 (ddd, J = 12.7, 6.0, 6.0 Hz, 1H), 3.08–3.03 (m, 1H), 2.41–2.39 (m, 1H), 2.31–2.29 (m, 1H), 2.26–2.22 (m, 1H), 2.18–2.12 (m, 1H), 2.02–2.00 (m, 1H), 1.90–1.81 (m, 3H), 1.78–1.76 (m, 1H), 1.66–1.60 (m, 3H), 1.39 (d, J = 13.3 Hz, 1H), 1.31–1.27 (m, 1H), 1.02 (d, J = 6.8 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 173.9, 141.8, 141.7, 115.1, 115.0, 108.9, 62.3, 60.5, 47.72, 47.71, 44.5, 44.4, 38.2, 38.1, 35.4, 30.9, 30.8, 29.50, 29.46, 26.1, 25.1, 25.0, 21.29, 21.27, 17.72, 17.68; IR (thin film, cm-1) 3298, 3080, 2960, 1644; HRMS (ESI) m/z: [M + Na]+ Calcd for C16H27NO3Na 304.1889; Found: 304.1882.
(2S)-N-(3-(6,10-Dioxaspiro[4.5]decan-1-yl)propyl)-2-methylbut-3-en-1-amine (S21)
Following general procedure E, S21 (26 mg, 0.097 mmol, 46%) was obtained as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 5.69–5.62 (m, 1H), 5.11–5.00 (m, 2H), 3.93–3.85 (m, 4H), 2.62–2.52 (m, 3H), 2.49–2.45 (m, 1H), 2.38–2.35 (m, 1H), 2.11–2.05 (m, 1H), 1.98–1.96 (m, 1H), 1.89–1.78 (m, 3H), 1.65–1.51 (m, 5H), 1.39 (d, J = 14.7 Hz, 1H), 1.32–1.12 (m, 2H), 1.00 (d, J = 6.7 Hz, 3H), 0.89–0.88 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 142.8, 114.69, 114.68, 108.9, 62.1, 60.6, 55.50, 55.49, 60.4, 48.87, 48.85, 38.3, 36.8, 30.6, 30.4, 29.8, 28.9, 28.8, 28.6, 26.2, 26.1, 24.9, 23.7, 21.1, 18.5, 18.4, 17.8; IR (thin film, cm-1) 3076, 2955, 2863; HRMS (ESI) m/z: [M + H]+ Calcd for C16H30NO2 268.2277; Found: 268.2271.
2-(11-Methyl-1,5-dioxaspiro[5.5]undecan-7-yl)-N-((S)-2-methylbut-3-en-1-yl)acetamide (S22)
Following general procedure D, S22 (31 mg, 0.11 mmol, 70%) was obtained as a colorless solid; changes to the general procedure include addition Et3N (23 μL) to the amide coupling step. 1H NMR (500 MHz, CDCl3) δ 5.85 (broad d, J = 26.6 Hz, 1H), 5.69–5.66 (m, 1H), 5.07–5.04 (m, 2H), 3.95–3.80 (m, 4H), 3.34–3.29 (m, 1H), 3.04–3.01 (m, 1H), 2.78–2.58 (m, 1H), 2.50–2.44 (m, 1H), 2.35–2.29 (m, 1H), 2.19–2.10 (m, 1H), 2.03 (dd, J = 14.3, 4.6 Hz, 1H), 1.98–1.90 (m, 1H), 1.85–1.81 (m, 1H), 1.67–1.52 (m, 2H), 1.52–1.42 (m, 1H), 1.39–1.32 (m, 3H), 1.02 (d, J = 6.6 Hz, 3H), 0.96 (d, J = 7.0 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 172.0, 141.7, 141.6, 115.24, 115.16, 58.7, 58.6, 48.13, 48.11, 45.6, 44.50, 44.46, 44.4, 38.31, 38.30, 38.24, 38.19, 37.4, 37.12, 37.11, 36.8, 35.58, 35.56, 25.51, 25.48, 19.7, 17.69, 17.65, 14.6, 13.7; IR (thin film, cm-1) 3296, 3078, 2963, 1640; HRMS (ESI) m/z: [M + Na]+ Calcd for C17H29NO3Na 318.2045;, Found: 318.2047.
(2S)-2-Methyl-N-(2-(11-methyl-1,5-dioxaspiro[5.5]undecan-7-yl)ethyl)but-3-en-1-amine (S23)
Following general procedure E, S23 (42 mg, 0.15 mmol, 48%) was obtained as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 5.71–5.63 (m, 1H), 5.08–5.00 (m, 2H), 3.84 (broad s, 4H), 2.71–2.63 (m, 1H), 2.60–2.45 (m, 3H), 2.39–2.34 (m, 1H), 1.90–1.51 (m, 7H), 1.44–1.36 (m, 5H), 1.01 (d, J = 6.6 Hz, 3H), 0.93 (d, J = 6.8 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 143.0, 114.61, 114.56, 100.7, 58.7, 58.5, 55.74, 55.67, 49.1, 48.9, 38.5, 38.4, 25.6, 19.7, 18.5, 18.4, 13.9; IR (thin film, cm-1) 3076, 2931, 2861; HRMS (ESI) m/z: [M + H]+ Calcd for C17H32NO2 282.2433; Found: 282.2428.
Preparation and Characterization Data of 1-Azabicyclic Products Reported in Table 3
Characterization data for azabicyclic compounds 31a, 32a, 33a, and 34a have been described.9
General Procedure G for the 2-Aza-Cope Rearrangement and Subsequent Cbz Protection to Provide Products Reported in Tables 3 and 4
(3aS,6aR)-Benzyl 6a-((E)-but-2-en-1-yl)hexahydrocyclopenta[b]pyrrole-1(2H)-carboxylate (31b)
A mixture of aminoketal S13 (28 mg, 0.12 mmol), morpholine (1 μL, 0.011 mmol), TFA (13 μL, 0.11 mmol), and dimedone (29 mg, 0.28 mmol) was stirred at 120 °C in a sealed 1-dram vial using an aluminum heating block. After 30 min, the reaction vessel was cooled in an ice bath for approximately 1 min. The reaction mixture was allowed to warm to room temperature, dissolved in CHCl3 (1 mL) and transferred to a larger reaction vessel. To this flask was added saturated aqueous Na2CO3 (0.5 mL), H2O (0.5 mL), and benzyl chloroformate (57 μL, 0.34 mmol). The reaction was vigorously stirred at room temperature for 24 h, then extracted with CH2Cl2 (3 × 5 mL). The combined organic extracts were dried (MgSO4), filtered, and concentrated in vacuo. The crude residue was purified on silica gel (1:19 EtOAc:hexanes) to provide 24 mg (0.081 mmol, 73%) of 31b as a colorless oil. HPLC analysis indicated an enantiomeric excess of 95% [CHIRALCEL AD column; flow: 1 mL/min; 98% n-hexane/2% i-PrOH; λ = 210 nm; major enantiomer tR = 11.25 min; minor enantiomer tR = 13.89 min]. . 1H NMR (500 MHz, DMSO-d6, 393K) δ 7.36–7.28 (m, 5H), 5.46–5.32 (m, 2H), 5.11 (d, J = 12.7 Hz, 1H), 5.06 (d, J = 12.7 Hz, 1H), 3.52–3.46 (m, 1H), 3.43–3.39 (m, 1H), 2.69 (dd, J = 12.1, 6.3 Hz, 1H), 2.43 (dddd, J = 8.6, 8.6, 5.3, 5.3 Hz, 1H), 2.27 (dd, J = 15.8, 8.4 Hz, 1H), 2.15–2.11 (m, 1H), 1.93–1.86 (m, 1H), 1.81 (dddd, J = 15.4, 7.3, 7.3, 7.3 Hz, 1H), 1.72–1.45 (m, 4H), 1.62 (d, J = 5.8 Hz, 3H), 1.43–1.37 (m, 1H); 13C NMR (125 MHz, DMSO-d6, 393K) δ 152.8, 136.9, 127.5, 126.8, 126.70, 126.67, 125.8, 125.0, 73.4, 64.9, 47.3, 47.1, 37.3, 31.2, 27.8, 24.1, 16.7; IR (thin film, cm-1) 2948, 1699; HRMS (ESI) m/z: [M + H]+ Calcd for C19H26NO2 300.1964; Found: 300.1960.
(3aS,7aR)-Benzyl 7a-((E)-but-2-en-1-yl)octahydro-1H-indole-1-carboxylate) (32b)
Following general procedure G, 32b (25 mg, 0.078 mmol, 75%) was obtained as a colorless oil. HPLC analysis indicated an enantiomeric excess of 95% [CHIRALCEL AD column; flow: 1.0 mL/min; 98% n-hexane/2% i-PrOH; λ = 210 nm; major enantiomer tR = 13.47 min; minor enantiomer tR = 17.22 min]. . 1H NMR (500 MHz, DMSO-d6, 393K) δ 7.38–7.27 (m, 5H), 5.41–5.37 (m, 1H), 5.30–5.29 (m, 1H), 5.07 (d, J = 12.7 Hz, 1H), 5.01 (d, J = 12.2 Hz, 1H), 3.56–3.52 (m, 1H), 3.24 (app dd, J = 16.3, 7.9 Hz, 1H), 2.43 (dd, J = 16.2, 6.0 Hz, 1H), 2.33 (dd, J = 13.6, 7.8 Hz, 1H), 2.06–2.02 (m, 1H), 1.89–1.64 (m, 4H), 1.58 (d, J = 5.6 Hz, 3H), 1.46–1.26 (m, 6H); 13C NMR (125 MHz, DMSO-d6, 393K) δ 152.9, 137.0, 127.6, 126.8, 126.7, 126.3, 124.9, 64.8, 63.3, 45.5, 40.1, 37.4, 31.5, 25.4, 24.8, 21.0, 20.6, 16.8; IR (thin film, cm-1) 3029, 2928, 2857, 1701; HRMS (ESI) m/z: [M + Na]+ Calcd for C20H27NO2Na 336.1939; Found: 336.1934.
(4aS,7aR)-Benzyl 7a-((E)-but-2-en-1-yl)octahydro-1H-cyclopenta[b]pyridine-1-carboxylate (33b)
Following general procedure G, 33b (42 mg, 0.13 mmol, 71%) was obtained as a colorless oil. HPLC analysis indicated an enantiomeric excess of 95% [CHIRALCEL OJ column; flow: 0.5 mL/min; 99.9% n-hexane/0.1% i-PrOH; λ = 210 nm; major enantiomer tR = 44.40 min; minor enantiomer tR = 5.46 min (for the opposite enantiomer as shown above)]. . 1H NMR (500 MHz, CDCl3) δ 7.35–7.31 (m, 5H), 5.46–5.38 (m, 2H), 5.15 (d, J = 12.4 Hz, 1H), 3.96 (d, J = 12.6 Hz, 1H), 3.96 (app dd, J = 13.4, 5.7 Hz, 1H), 2.97 (app dt, J = 13.2, 4.8 Hz, 1H), 2.62 (dd, J = 12.7, 4.4 Hz, 1H), 2.19–2.12 (m, 3H), 2.10–2.08 (m, 1H), 1.77–1.67 (m, 3H), 1.64 (d, J = 4.9 Hz, 3H), 1.57–1.42 (m, 5H); 13C NMR (125 MHz, CDCl3) δ 156.6, 137.3, 128.53, 128.52, 128.49, 128.2, 128.0, 127.9, 127.1, 67.6, 66.7, 42.3, 41.4, 39.5, 38.3, 29.5, 24.7, 21.8, 21.4, 18.2; IR (thin film, cm-1) 3031, 2922, 2851, 1745; HRMS (ESI) m/z: [M + Na]+ Calcd for C20H27NO2Na 336.1939; Found: 336.1931.
(3aS,7R,7aS)-Benzyl 7a-((E)-but-2-en-1-yl)-7-methyloctahydro-1H-indole-1-carboxylate (34b)
Following general procedure G, 34b (12 mg, 0.035 mmol, 70%) was obtained as a colorless oil. HPLC analysis indicated an enantiomeric excess of 91% [CHIRALCEL AD column; flow: 1.0 mL/min; 98% n-hexane/2% i-PrOH; λ = 210 nm; major enantiomer tR = 9.02 min; minor enantiomer tR = 20.11 min (for the opposite enantiomer as shown above)]. ; data is for the opposite enantiomer as shown above). 1H NMR (500 MHz, DMSO-d6, 393K) δ 7.42–7.34 (m, 5H), 5.49–5.47 (m, 1H), 5.23–5.19 (m, 1H), 5.13 (d, J = 12.1 Hz, 1H), 5.07 (d, J = 12.6 Hz, 1H), 3.54–3.51 (m, 1H), 3.40–3.33 (m, 1H), 3.26–3.22 (m, 1H), 2.21–2.14 (m, 2H), 1.98–1.90 (m, 2H), 1.71–1.42 (m, 7H), 1.61 (d, J = 23.9 Hz, 3H), 1.00 (broad s, 3H); 13C NMR (125 MHz, DMSO-d6, 393K) δ 154.4, 128.7, 128.5, 128.2, 128.1, 127.8, 127.64, 127.55, 125.7, 67.4, 65.7, 46.4, 37.1, 31.8, 29.9, 24.9, 24.7, 24.4, 20.2, 17.8, 17.1; IR (thin film, cm-1) 3028, 2826, 2885, 1704; HRMS (ESI) m/z: [M + Na]+ Calcd for C21H29NO2Na 350.2096; Found: 350.2098.
Synthesis and Characterization Data of 1-Azabicyclics Reported in Table 4 and Their Precursors
N-((R)-2-phenylbut-3-enyl)-3-(1,5-dioxaspiro[5.5]undecan-7-yl)propanamide (S24)
Following general procedure D, S24 (496 mg, 1.39 mmol, 86%) was obtained as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.32 (t, J = 7.5 Hz, 2H), 7.27–7.21 (m, 3H), 6.00–5.93 (m, 2H), 5.15–5.10 (m, 2H), 4.03–3.97 (m, 1H), 3.88 (tt, J = 11.7, 3.1 Hz, 1H), 3.77–3.60 (m, 3H), 3.53–3.46 (m, 2H), 2.54 (app d, J = 12.7 Hz, 1H), 2.35–2.28 (m, 1H), 2.15–2.02 (m, 2H), 1.96–1.83 (m, 1H), 1.59–1.53 (m, 3H), 1.39–1.07 (m, 7H); 13C NMR (125 MHz, CDCl3) δ 173.65, 173.63, 141.54, 141.52, 139.29, 139.25, 128.9, 128.8, 128.01, 127.97, 126.99, 126.98, 116.41, 116.37, 99.3, 59.15, 59.13, 59.0, 49.6, 49.5, 44.7, 43.7, 35.4, 35.3, 28.2, 28.0, 25.8, 24.8, 24.43, 24.41, 22.4; IR (thin film, cm-1) 3299, 3083, 2861, 1644; HRMS (ESI) m/z: (M + Na]+ Calcd for C22H31NO3Na 380.2202; Found: 380.2200.
(2R)-N-(3-(1,5-dioxaspiro[5.5]undecan-7-yl)propyl)-2-phenylbut-3-en-1-amine (S25)
Following general procedure E, S25 (229 mg, 0.667 mmol, 49%) was obtained as a light yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.32 (t, J = 7.6 Hz, 2H), 7.23–7.22 (m, 3H), 5.99–5.93 (m, 1H), 5.14–5.11 (m, 2H), 4.02 (t, J = 11.2 Hz, 1H), 3.89 (dt, J = 11.3, 2.3 Hz, 1H), 3.78 (app broad s, 2H), 3.54 (q, J = 7.5 Hz, 1H), 2.90 (d, J = 7.4 Hz, 2H), 2.64–2.60 (m, 2H), 2.45 (broad d, J = 8.8 Hz, 1H), 1.90–1.10 (m, 15H); 13C NMR (125 MHz, CDCl3) δ 142.6, 140.3, 128.8, 128.0, 127.9, 126.8, 116.0, 99.3, 59.1, 59.0, 54.5, 50.3, 50.1, 28.5, 28.3, 27.3, 25.8, 25.2, 24.4, 22.5; IR (thin film, cm-1) 3315, 3081, 2933; HRMS (ESI) m/z: [M + H]+ Calcd for C22H34NO2 344.2589; Found: 344.2583. Characterization data for azabicyclic compound 35 has been described.9
Benzyl 2-(2-oxocyclododecyl)acetate (S26)
Following general procedure B, S26 (825 mg, 2.50 mmol, 64%) was obtained as a colorless solid. mp 64–65 °C; 1H NMR (500 MHz, CDCl3) δ 7.38–7.31 (m, 5H), 5.13–5.06 (m, 2H), 3.18–3.13 (m, 1H), 2.96–2.87 (m, 2H), 2.32 (dd, J = 16.7, 5.2 Hz, 1H), 2.25 (ddd, J = 17.7, 5.8, 3.6 Hz, 1H), 2.04–1.98 (m, 1H), 1.89–1.83 (m, 1H), 1.60–1.53 (m, 1H), 1.42–1.17 (m, 14H), 1.08–1.06 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 212.4, 172.7, 136.1, 128.8, 128.43, 128.40, 66.6, 47.3, 37.6, 34.3, 28.6, 26.3, 24.1, 23.3, 22.7, 22.4, 21.8, 21.2; IR (thin film, cm-1) 3033, 2931, 2864, 1735, 1709, 1164; HRMS (ES) m/z: [M + Na]+ Calcd for C21H30O3Na 353.2093; Found: 353.2084.
(1,5-Dioxa-spiro[5.11]heptadec-7-yl)-acetic acid benzyl ester (S27)
Following general procedure C, ketal S27 was obtained as a colorless oil (430 mg, 1.10 mmol, 91%). This product was purified by flash chromatography on silica gel (100:0 to 98:2 to 95:5 hexanes:EtOAc). 1H NMR (500 MHz, CDCl3) δ 7.40–7.29 (m, 5H), 5.15–5.08 (m, 2H), 4.00 (t, J = 11.5 Hz, 1H), 3.78–3.73 (m, 2H), 3.53 (dd, J = 11.0, 4.1 Hz, 1H), 2.82 (dd, J = 15.3, 8.7 Hz, 1H), 2.43 (t, J = 8.7 Hz, 1H), 2.30–2.23 (m, 1H), 2.10 (dd, J = 15.3, 2.4 Hz, 1H), 1.98–1.90 (m, 1H), 1.61–1.22 (m, 19H), 1.11–1.06 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 174.7, 136.9, 128.5, 128.3, 128.0, 102.4, 65.8, 59.6, 59.5, 39.1, 34.4, 26.43, 26.37, 25.8, 25.4, 24.6, 23.6, 22.9, 22.7, 22.4, 22.2, 19.3; IR (thin film, cm-1) 3063, 2931, 2862, 1736, 1248, 1142; HRMS (ES) m/z: [M + Na]+ Calcd for C24H36O4Na 411.2511; Found: 411.2515.
N-((R)-2-Phenylbut-3-enyl)-2-(1,5-dioxaspiro[5.11]heptadecan-7-yl)acetamide (S28)
Following general procedure D, S28 (171 mg, 0.400 mmol, 71%) was obtained as an amorphous colorless solid. 1H NMR (500 MHz, CDCl3) δ 7.32 (t, J = 7.9 Hz, 2H), 7.23–7.21 (m, 3H), 6.31 (d, J = 18.8 Hz, 1H), 5.96 (dddd, J = 17.2, 9.9, 6.9, 2.5 Hz, 1H), 5.14–5.07 (m, 2H), 3.99 (t, J = 12.1 Hz, 1H), 3.80–3.69 (m, 3H), 3.64–3.42 (m, 4H), 2.78 (dt, J = 15.1, 5.8 Hz, 1H), 2.25 (ddd, J = 14.8, 11.7, 7.5 Hz, 1H), 2.06 (t, J = 7.7 Hz, 1H), 1.93 (ddd, J = 15.3, 7.6, 2.6 Hz, 1H), 1.88–1.77 (m, 2H), 1.56–1.20 (m, 17H), 1.09–1.04 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 174.9, 174.8, 141.64, 141.58, 139.6, 139.5, 128.87, 128.85, 128.03, 127.99, 126.9, 116.2, 116.1, 102.9, 59.64, 59.58, 59.3, 49.6, 49.4, 43.5, 43.4, 39.5, 39.4, 37.5, 37.4, 27.2, 27.1, 26.4, 26.3, 25.5, 24.5, 23.6, 22.74, 22.65, 22.47, 22.45, 22.1, 19.3; IR (thin film, cm-1) 3323, 2931, 2863, 1647, 1140, 1117, 1089; HRMS (ESI) m/z: [M + Na]+ Calcd for C27H41NO3Na 450.2984; Found: 450.2972.
(2R)-N-(2-(1,5-Dioxaspiro[5.11]heptadecan-7-yl)ethyl)-2-phenylbut-3-en-1-amine (S29)
Following general procedure E, S29 (81 mg, 0.20 mmol, 58%) was obtained as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.32 (t, J = 7.5 Hz, 2H), 7.25–7.20 (m, 3H), 6.01–5.95 (m, 1H), 5.14–5.10 (m, 2H), 4.02 (t, J = 11.9 Hz, 1H), 3.84 (q, J = 11.4 Hz, 1H), 3.77–3.69 (m, 2H), 3.57 (q, J = 7.3 Hz, 1H), 2.92 (d, J = 7.3 Hz, 2H), 2.72–2.69 (m, 2H), 2.32–2.26 (m, 1H), 2.07–1.93 (m, 2H), 1.56–1.23 (m, 22H), 1.12–1.08 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 142.77, 142.75, 140.52, 140.48, 128.8, 127.94, 127.90, 126.7, 115.9, 115.8, 103.31, 103.27, 59.5, 59.4, 54.6, 50.3, 50.2, 50.1, 39.9, 39.8, 29.93, 29.91, 27.1, 27.0, 26.6, 26.4, 25.6, 25.11, 25.09, 24.0, 23.14, 23.10, 23.0, 22.6, 22.3, 19.5; IR (thin film, cm-1) 3327, 3026, 2929, 2861, 1467, 1245, 1112; HRMS (ESI) m/z: [M + H]+ Calcd for C27H44O2N 414.3372; Found: 414.3362.
(3aS,13aR)-Benzyl 13a-((Z)-3-phenylallyl)tetradecahydro-1H-cyclododeca[b]pyrrole-1-carboxylate (36)
Following general procedure E, 36 (41 mg, 89 μmol, 74%) was obtained as a colorless amorphous solid (9:1 mixture of diastereomers by 1H NMR). The major diastereomer was partially purified by preparatory TLC (9:1 hexanes/EtOAc) for characterization. HPLC analysis indicated an en-antiomeric excess of 99% [CHIRALCEL OD column; flow: 1.0 mL/min; 99.5% n-hexane/0.5% i-PrOH; λ = 254 nm; major enantiomer tR = 19.16 min; minor enantiomer tR = 27.21 min]. 1H NMR (500 MHz, DMSO-d6, 393 K) δ 7.32–7.25 (m, 9H), 7.19–7.18 (m, 1H), 6.28 (d, J = 15.4 Hz, 1H), 6.15–6.11 (m, 1H), 5.12 (d, J = 12.6 Hz, 1H), 5.03 (d, J = 12.6 Hz, 1H), 3.51 (t, J = 9.8 Hz, 1H), 3.30–3.24 (m, 1H), 2.82–2.68 (m, 2H), 2.32 (dd, J = 14.0, 7.4 Hz, 1H), 2.26–2.22 (m, 1H), 2.09–2.07 (m, 1H), 1.90 (td, J = 12.8, 7.1 Hz, 1H), 1.69 (t, J = 11.8 Hz, 1H), 1.51–1.27 (m, 18H); 13C NMR (125 MHz, DMSO, 393 K) δ 137.2, 131.1, 127.9, 127.7, 127.1, 126.3, 125.2, 67.2, 65.0, 46.7, 26.3, 25.9, 25.1, 23.5, 21.59, 21.56, 21.4, 21.1, 17.9; IR (thin film, cm-1) 3027, 2925, 2852, 1702, 1459, 1402; HRMS (ESI) m/z: [M + Na]+ Calcd for C31H41NO2Na 482.3035; Found: 482.3034. The ring fusion geometry was assigned by 1H NOE analysis as depicted in the drawing below.

Benzyl 2-(2-oxocycloheptyl)acetate (S30)
Keto ester S30 was prepared using an adapted procedure from Cotarco and co-workers.35 A 250 mL round bottom flask with stir bar fitted with a Dean Stark trap was charged with cycloheptanone (11 mL, 89 mmol), pyrrolidine (6.4 g, 89 mmol), and benzene (60 mL) and heated to reflux. After 20 h, the reaction was concentrated in vacuo at 55 °C for 1 h. The resultant red oil was dissolved in benzene (45 mL) and 2-bromobenzylacetate (8.0 mL, 12 mmol) was added and the reaction was heated to reflux. After 24 h, the reaction was allowed to cool to 22 °C, concentrated in vacuo, taken up in MeOH (30 mL) and water (6 mL), heated to reflux for 2 h, allowed to cool to 22 °C, and partitioned between Et2O (400 mL) and 1 M HCl (200 mL). The organic layer was washed with 1 M HCl (2 × 100 mL), saturated aqueous sodium bicarbonate (1 × 50 mL) and brine (1 × 15 mL), dried (MgSO4), filtered, concentrated in vacuo and the residue was purified by flash chroma-tography on silica gel (2:25–3:25 EtOAc:hexanes) to give S30 (6.3 g, 24 mmol, 45%) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.36–7.27 (m, 5H), 5.12–5.05 (m, 2H), 3.10 (dddd, J = 10.9, 8.5, 5.7, 2.9 Hz, 1H), 2.87 (dd, J = 16.8, 8.4 Hz, 1H), 2.59 (dt, J = 16.3, 7.8 Hz, 1H), 2.50–2.38 (m, 1H), 2.33 (dd, J = 16.8, 5.7 Hz, 1H), 1.92–1.76 (m, 3H), 1.76–1.63 (m, 2H), 1.57–1.47 (m, 1H), 1.40–1.22 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 213.8, 172.2, 135.8, 128.4, 128.02, 127.98, 66.1, 47.2, 43.2, 36.5, 31.0, 29.1, 28.8, 23.3; IR (thin film, cm-1) 2925, 2854, 1733, 1702; Anal. Calcd for C16H20O3: C, 73.82; H, 7.72. Found: C, 73.49; H, 7.73.
Benzyl 2-(1,5-dioxaspiro[5.6]dodecan-7-yl)acetate (S31)
Following general procedure C, S31 (5.2 g, 16 mmol) was isolated as a colorless oil in 84% yield; changes from the general procedure include the exposure of the substrate to the reaction conditions for 2 h. 1H NMR (500 MHz, CDCl3) δ 7.37–7.25 (m, 5H), 5.15 (d, J = 12.4 Hz, 1H), 5.07 (d, J = 12.4 Hz, 1H), 3.94 (td, J = 11.9, 2.9 Hz, 1H), 3.81 (td, J = 11.7, 2.4 Hz, 1H), 3.73–3.67 (m, 2H), 2.92–2.84 (m, 1H), 2.27–2.18 (m, 2H), 2.09 (ddd, J = 14.8, 9.6, 2.2 Hz, 1H), 1.96–1.80 (m, 2H), 1.76–1.63 (m, 2H), 1.58–1.35 (m, 6H), 1.28 (dt, J = 10.9, 2.2 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 173.8, 136.2, 128.1, 127.9, 127.7, 101.2, 65.5, 59.04, 58.96, 45.8, 35.8, 29.4, 28.0, 27.8, 26.6, 25.3, 20.3; IR (thin film, cm-1) 2931, 2863, 1733; Anal. Calcd for C19H26O4: C, 71.67; H, 8.23. Found: C, 71.45; H, 8.34.
N-((R)-2-Phenylbut-3-enyl)-2-(1,5-dioxaspiro[5.6]dodecan-7-yl)acetamide (S32)
Following general procedure D, S32 (369 mg, 1.03 mmol, 66%) was obtained as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.32 (t, J = 7.4 Hz, 2H), 7.25–7.21 (m, 3H), 6.00–5.93 (m, 1H), 5.83 (app broad d, J = 5.3 Hz, 1H), 5.15–5.10 (m, 2H), 3.96 (t, J = 11.7 Hz, 1H), 3.82 (t, J = 11.6 Hz, 1H), 3.76–3.59 (m, 3H), 3.56–3.45 (m, 2H), 2.66 (dd, J = 14.6, 4.0 Hz, 1H), 2.10–2.04 (m, 2H), 2.01–1.95 (m, 2H), 1.91–1.76 (m, 2H), 1.70–1.61 (m, 2H), 1.52–1.32 (m, 6H); 13C NMR (125 MHz, CDCl3) δ 173.90, 173.87, 141.50, 141.46, 139.3, 128.9, 128.0, 127.0, 116.39, 116.36, 102.1, 59.5, 59.43, 59.41, 49.7, 49.6, 46.43, 46.38, 43.7, 43.6, 38.14, 38.09, 29.79, 29.77, 28.4, 28.1, 28.0, 27.1, 25.6, 20.6; IR (thin film, cm-1) 3296, 3080, 2928, 1643; HRMS (ESI) m/z: [M + Na]+ Calcd for C22H31NO3Na 380.2202; Found: 380.2201.
(2R)-N-(2-(1,5-dioxaspiro[5.6]dodecan-7-yl)ethyl)-2-phenylbut-3-en-1-amine (S33)
Following general procedure E, S33 (225 mg, 0.655 mmol, 70%) was obtained as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.32 (t, J = 7.5 Hz, 2H), 7.24–7.21 (m, 3H), 6.02–5.95 (m, 1H), 5.15–5.11 (m, 2H), 3.97 (tt, J = 11.3, 2.9 Hz, 1H) 3.86 (t, J = 11.0 Hz, 1H), 3.81–3.74 (m, 2H), 3.56 (q, J = 7.5 Hz, 1H), 2.97–2.88 (m, 2H), 2.71 (tt, J = 10.1, 4.8 Hz, 1H), 2.64–2.57 (m, 1H), 2.25–2.18 (m, 1H), 1.97–1.84 (m, 2H), 1.77–1.72 (m, 4H), 1.61–1.26 (m, 9H); 13C NMR (125 MHz, CDCl3) δ 142.7, 140.4, 128.84, 128.82, 127.94, 127.92, 126.8, 126.7, 116.0, 115.9, 102.6, 59.5, 59.4, 54.73, 54.71, 50.2, 49.11, 49.08, 46.23, 46.19, 30.51, 30.49, 30.3, 28.74, 28.72, 27.20, 27.17, 27.0, 26.9, 25.7, 20.9; IR (thin film, cm-) 3027, 2929, 2861, 2809, 1454, 1108; HRMS (ESI) m/z: [M + H]+ Calcd for C22H34O2N 344.2590; Found: 344.2581.
Characterization data for azabicyclic compound 37 has been described previously.9
Benzyl 5-(2-oxocyclopentyl)pentanoate (S34)
Following general procedure B, S34 (385 mg, 1.40 mmol, 55%) was obtained as a colorless oil after flash chromatography on silica gel (19:1 hex-anes:EtOAc to 9:1 hexanes:EtOAc to 4:1 hexanes:EtOAc). H NMR (500 MHz, CDCl3) δ 7.37-7.32 (m, 5H), 5.12 (s, 2H), 2.37 (t, J = 7.4 Hz, 2H), 2.29 (dd, J = 18.7, 8.4 Hz, 1H), 2.22–2.16 (m, 1H), 2.13–2.05 (m, 1H), 2.02–1.96 (m, 2H), 1.80–1.74 (m, 2H), 1.71–1.61 (m, 2H), 1.48 (qd, J = 10.8, 6.7 Hz, 1H), 1.37 (app quintet, J = 7.9 Hz, 2H), 1.26 (dq, J = 16.1, 8.4 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 221.5, 173.6, 136.3, 128.7, 128.37, 128.36, 66.3, 49.1, 38.3, 34.3, 29.7, 29.4, 27.2, 25.0, 20.9; IR (thin film, cm-1) 3033, 2940, 2860, 1735, 1158; HRMS (ES) m/z: [M + Na]+ Calcd for C17H22O3Na 297.1467; Found: 297.1467.
5-(6,10-Dioxa-spiro[4.5]dec-1-yl)-pentanoic acid benzyl ester (S35)
Following general procedure C, ketal S35 was obtained as a colorless oil (622 mg, 1.87 mmol, 64%). 1H NMR (500 MHz, CDCl3) δ 7.39–7.32 (m, 5H), 5.12 (s, 2H), 3.93–3.82 (m, 4H), 2.38 (t, J = 7.6 Hz, 2H), 2.09–2.05 (m, 1H), 2.05–1.88 (m, 1H), 1.86–1.76 (m, 3H), 1.70–1.59 (m, 5H), 1.41–1.19 (m, 5H); 13C NMR (125 MHz, CDCl3) δ 174.0, 136.4, 128.7, 128.4, 128.3, 109.0, 66.2, 62.0, 60.8, 48.9, 34.6, 30.7, 29.0, 28.2, 28.1, 26.2, 25.6, 21.2; IR (thin film, cm-1) 3066, 3034, 2950, 1737, 1151, 1106; HRMS (ES) m/z: [M + Na]+ Calcd for C20H28O4Na 355.1885; Found: 355.1884.
N-((R)-2-phenylbut-3-enyl)-5-(6,10-dioxaspiro[4.5]decan-1-yl)pentanamide (S36)
Following general procedure D, S36 (242 mg, 0.651 mmol, 72%) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.31 (t, J = 7.5 Hz, 2H), 7.23–7.18 (m, 3H), 5.98–5.91 (m, 1H), 5.54 (broad s, 1H), 5.14–5.09 (m, 2H), 3.90–3.82 (m, 4H), 3.59–3.58 (m, 1H), 3.51–3.48 (m, 2H), 2.11–2.03 (m, 3H), 1.97–1.93 (m, 1H), 1.85–1.73 (m, 3H), 1.64–1.54 (m, 5H), 1.38–1.14 (m, 5H); 13C NMR (125 MHz, CDCl3) δ 173.3, 141.3, 139.0, 128.8, 127.9, 127.0, 116.5, 108.9, 62.0, 60.7, 49.5, 48.8, 43.6, 36.9, 30.6, 28.9, 28.13, 28.09, 26.2, 26.1, 21.0; IR (thin film, cm-1) 3295, 3078, 2944, 1645; HRMS (ESI) m/z: [M + Na]+ Calcd for C23H33O3NNa 394.2358; Found: 394.2365.
N-((R)-2-phenylbut-3-enyl)-5-(6,10-dioxaspiro[4.5]decan-1-yl)pentan-1-amine (S37)
Following general procedure E, S37 (146 mg, 0.408 mmol, 66%) was obtained as a light-yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.32 (t, J = 7.4 Hz, 2H), 7.23–7.18 (m, 3H), 6.00–5.93 (m, 1H), 5.14–5.11 (m, 2H), 3.92–3.85 (m, 5H), 3.54 (q, J = 6.9 Hz, 1H), 2.89 (d, J = 7.4 Hz, 2H), 2.60 (t, J = 7.2 Hz, 2H), 2.10–0.90 (m, 17H); 13C NMR (125 MHz, CDCl3) δ 142.5, 140.3, 128.8, 127.8, 126.8, 116.0, 109.1, 62.1, 60.8, 54.6, 50.1, 49.9, 48.9, 30.7, 30.1, 28.9, 28.5, 28.4, 27.9, 26.2, 21.1; IR (thin film, cm-1) 2930, 2858, 1453, 1149, 1111; HRMS (ESI) m/z: [M + H]+ Calcd for C23H36NO2 358.2746; Found: 358.2741.
Preparation and Characterization Data for 1-Azabicyclic Reported in Scheme 8 and Their Aminoketal Precursors
Scheme 8. Enantioselective Synthesis of cis-exahydrocyclopenta[b]pyrrole Carbamates Having Chiral Allylic Side Chains.

(R,E)-tert-Butyl 2-phenylpent-3-enylcarbamate (S38)
Diphenylphosphoryl azide (0.33 mL, 1.5 mmol) was added dropwise via syringe to a solution of (R,E)-3-phenylhex-4-enoic acid36 (0.29 g, 1.5 mmol) and Et3N (0.21 mL, 1.5 mmol) in t-BuOH (2.2 mL) at room temperature. The resulting solution was heated at 85 °C for 48 h, cooled to room temperature, diluted with EtOAc (50 mL), and washed sequentially with H2O (10 mL), saturated aqueous NaHCO3 (10 mL), and brine (10 mL). The organic layer was dried (MgSO4), filtered, and concentrated in vacuo. Purification of the crude residue by flash chromatography on silica gel (19:1 hexanes: EtOAc to 9:1 hexanes:EtOAc) gave S38 (0.31 g, 1.2 mmol, 77%) as a colorless solid (mp 47–48 °C). HPLC analysis indicated an enantiomeric excess of 84%. The product was recrystallized with n-hexane/i-PrOH to an enantiomeric excess of 87% [CHIRALCEL OD-H column; flow: 1.0 mL/min; 99.7% n-hexane/0.3% i-PrOH; λ = 220 nm; major enantiomer tR = 44.75 min; minor enantiomer tR = 55.41 min]. ; 1H NMR (500 MHz, CDCl3) δ 7.33 (t, J = 7.4 Hz, 2H), 7.25–7.20 (m, 3H), 5.58–5.55 (m, 2H), 4.50 (broad s, 1H), 3.42–3.36 (m, 3H), 1.70 (d, J = 4.5 Hz, 3H), 1.43 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 156.0, 142.3, 131.9, 128.9, 127.9, 127.4, 126.9, 79.4, 49.2, 45.5, 28.6, 18.3; IR (thin film, cm-1) 3357, 2975, 2931, 1702, 1506, 1171; HRMS (ESI) m/z: [M + Na]+ Calcd for C16H23NO2Na 284.1626; Found: 284.1628.
(R,E)-2-Phenylpent-3-en-1-amine (S39)
To a solution of N-Boc amine S38 (180 mg, 0.690 mmol) in CH2Cl2 (3.4 mL) was added TFA (0.530 mL, 6.90 mmol) dropwise via syringe at 0 °C. The resulting solution was stirred at room temperature for 1.5 h and quenched with 10% aqueous NaOH at 0 °C until a pH of 14 was reached. The mixture was allowed to warm to room temperature and extracted with Et2O (3 × 15 mL). The organic layers were combined, dried (MgSO4), filtered and concentrated in vacuo to provide S39 (140 mg) as a yellow oil that was used in subsequent reactions without further purification.
N-((R,E)-2-Phenylpent-3-enyl)-2-(6,10-dioxaspiro[4.5]decan-1-yl)acetamide (S40)
Following general procedure D, S40 (35 mg, 0.10 mmol, 16%) was obtained as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.32 (t, J = 7.9 Hz, 2H), 7.24–7.21 (m, 3H), 6.26 (broad s, 1H), 5.57–5.54 (m, 2H), 3.87–3.75 (m, 4H), 3.63–3.44 (m, 3H), 2.56 (dd, J = 14.9, 6.6 Hz, 1H), 2.20–2.08 (m, 1H), 2.08–2.04 (m, 2H), 1.87–1.80 (m, 3H), 1.69 (d, J = 5.2 Hz, 3H), 1.65–1.62 (m, 1H), 1.33–1.26 (m, 3H); 13C NMR (125 MHz, CDCl3) δ 173.25, 173.23, 142.5, 142.4, 132.19, 132.16, 131.9, 128.9, 128.8, 127.93, 127.88, 127.2, 127.1, 127.0, 126.8, 108.3, 108.2, 62.30, 62.28, 60.74, 60.71, 48.8, 48.7, 46.03, 45.98, 44.23, 44.17, 36.05, 36.04, 30.47, 30.46, 29.6, 29.5, 26.0, 25.99, 21.2, 18.3; IR (thin film, cm-1) 3299, 2961, 2932, 2867, 1642, 1544; HRMS (ESI) m/z: [M + Na]+ Calcd for C21H29NO3Na 366.2045; Found: 366.2044.
(2R,E)-N-(2-(6,10-Dioxaspiro[4.5]decan-1-yl)ethyl)-2-phenylpent-3-en-1-amine (39a)
Following general procedure E, 39a (33 mg, 0.10 mmol, 98%) was obtained as a light-brown oil. 1H NMR (500 MHz, CDCl3) δ 7.31 (t, J = 7.7 Hz, 2H), 7.23–7.19 (m, 3H), 5.58–5.55 (m, 2H), 3.92–3.82 (m, 4H), 3.49 (q, J = 7.2 Hz, 1H), 2.88–2.85 (m, 2H), 2.69–2.64 (m, 2H), 2.10–2.06 (m, 1H), 2.00–1.91 (m, 1H), 1.84–1.74 (m, 5H), 1.69–1.62 (m, 8H), 1.40–1.36 (m, 2H), 1.31–1.26 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 143.59, 143.55, 133.2, 133.1, 128.77, 128.75, 127.81, 127.79, 126.8, 126.7, 126.56, 126.55, 108.9, 62.2, 60.8, 55.2, 49.3, 48.9, 47.1, 30.6, 30.5, 29.9, 29.3, 29.1, 26.1, 21.3, 18.3; IR (thin film, cm-1) 3305, 3206, 2954, 2861, 1451, 1149, 1110; HRMS (ESI) m/z: [M + H]+ Calcd for C21H32NO2 330.2433; Found: 330.2423.
(3aS,6aS)-Benzyl 6a-((R,E)-4-phenylbut-3-en-2-yl)hexahydrocyclopenta[b]pyrrole-1(2H)-carboxylate (40)
Following general procedure G, 40 (28 mg, 0.075 mmol, 75%) was obtained as a colorless oil. HPLC analysis indicated an enantiomeric excess of 87% [CHIRALCEL OD-H column; flow: 1.0 mL/min; 99.8% n-hexane/0.2% i-PrOH; λ = 254 nm; major enantiomer tR = 43.90 min; minor enantiomer tR = 40.20 min]. 1H NMR (500 MHz, CDCl3) δ 7.38–7.29 (m, 9H), 7.20 (t, J = 7.3 Hz, 1H), 6.43 (d, J = 15.8 Hz, 1H), 6.17 (dd, J = 15.8, 8.5 Hz, 1H), 5.14–5.09 (m, 2H), 3.60–3.47 (m, 2H), 3.34 (m, 1H), 2.67–2.50 (m, 1H), 2.14–2.10 (m, 1H), 2.00–1.92 (m, 2H), 1.76–1.72 (m, 1H), 1.60–1.55 (m, 2H), 1.50–1.38 (m, 2H), 0.88 (d, J = 7.3 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 152.7, 137.0, 136.8, 132.4, 129.8, 127.7, 127.6, 126.9, 126.6, 126.2, 125.3, 76.6, 65.0, 47.3, 44.1, 41.4, 36.7, 32.3, 28.6, 24.3, 15.2; IR (thin film, cm-1) 3027, 2955, 2868, 1698, 1402; HRMS (ESI) m/z: [M + Na]+ Calcd for C25H29NO2Na 398.2096; Found: 398.2097.
(R,E)-tert-Butyl (2,4-diphenylbut-3-en-1yl)carbamate (S41)
To a solution of the catalyst HG-II37 (13 mg, 0.02 mmol) in CH2Cl2 (1.3 mL) was added homoallylamine 25 (0.10 g, 0.40 mmol) and styrene (0.16 mL, 1.2 mmol). The green solution was heated to reflux for 12 h. After 12 h, the solution was cooled to room temperature and concentrated under reduced pressure to a brown solid. The crude material was purified by silica gel chromatography (100% hexanes to 4:1 hexanes:Et2O) to yield S41 as a colorless solid (92 mg, 0.28 mmol, 70%). mp 80–81 °C; ; 1H NMR (500 MHz, CDCl3) δ 7.34–7.21 (m, 10H), 6.49 (d, J = 15.9 Hz, 1H), 6.34 (dd, J = 15.9, 7.8 Hz, 1H), 4.57 (broad s, 1H), 3.69–3.68 (m, 1H), 3.57–3.56 (m, 1H), 3.52–3.48 (m, 1H), 1.43 (s, 9H); 13C NMR (125 MHz) δ 156.1, 141.7, 137.2, 131.7, 130.8, 129.0, 128.7, 128.0, 127.6, 127.1, 126.5, 79.5, 49.5, 45.5, 28.6; IR (thin film, cm-1) 3430, 3358, 3060, 2979, 1710; HRMS (ESI) m/z: [M + Na]+ Calcd for C21H25O2NNa 346.1783; Found: 346.1793.
(R,E)-2,4-Diphenylbut-3-en-1-amine (S42)
To a solution of Boc-amide S41 (89 mg, 0.28 mmol) in CH2Cl2 (1.3 mL) at 0 °C was added TFA (0.21 mL, 2.8 mmol) dropwise over 3 min. The resulting clear and colorless solution was warmed to room temperature and maintained for 1.5 h. The reaction was quenched by the addition of 10% aqueous solution of NaOH until the solution is basic (pH >10 analyzed by pH paper) at 0 °C. The solution was allowed to warm to room temperature and extracted with Et2O (3 × 50 mL). The organic layers were combined, dried with MgSO4, filtered and concentrated to provide the amine S42 as a slightly yellow oil (61 mg, 0.28 mmol). This amine was used without further purification. ; 1H NMR (500 MHz, CDCl3) δ 7.39–7.20 (m, 10H), 6.51 (d, J = 15.7 Hz, 1H), 6.25 (dd, J = 15.7, 8.1 Hz, 1H), 3.59 (broad d, J = 7.2 Hz, 1H), 3.47 (broad s, 2H), 3.10 (broad s, 2H); 13C NMR (125 MHz) δ 141.1 136.9, 132.3, 130.1, 129.2, 128.8, 127.9, 127.8, 127.4, 126.5, 50.9, 46.0; IR (thin film, cm-1) 3357, 3290, 2929; HRMS (ESI) m/z: [M + H]+ Calcd for C16H18N 224.1439; Found: 224.1445.
N-((R,E)-2,4-Diphenylbut-3-en-1-yl)-2-(6,10-dioxaspiro[4.5]decan-1yl)acetamide (S43)
Following general procedure D, S43 (45 mg, 0.11 mmol, 65%) was obtained as a colorless solid. 1H NMR (500 MHz, CDCl3) δ 7.39–7.20 (m, 10H), 6.47 (dd, J = 12.7, 4.5 Hz, 1H), 6.36–6.31 (m, 1H), 3.85–3.54 (m, 7H), 2.58 (dd, J = 14.4, 6.8 Hz, 1H), 2.21–2.13 (m, 1H), 2.11–2.00 (m, 2H), 1.91–1.71 (m, 3H), 1.65–1.54 (m, 3H), 1.29–1.22 (m, 2H); 13C NMR (125 MHz) δ 173.38, 173.36, 141.9, 141.7, 137.3, 137.2, 131.4, 131.0, 129.0, 128.72, 128.71, 128.1, 128.0, 127.63, 127.60, 127.1, 126.4, 108.2, 62.3, 60.7, 49.04, 49.01, 46.09, 46.05, 44.2, 44.1, 36.08, 36.05, 30.52, 30.45, 30.4, 29.9, 29.7, 29.6, 26.0, 21.2, 21.2; IR (thin film, cm-1) 3330, 3059, 2960, 2866, 1721; HRMS (ESI) m/z: [M + Na]+ Calcd for C26H31NO3Na 428.2202; Found: 428.2203.
(2R,E)-N-(2-(6,10-Dioxaspiro[4.5]decan-1yl)ethyl)-2,4-diphenylbut-3-en-1-amine (39b)
Following general procedure E, 39b (204 mg, 0.520 mmol, 81% yield) was obtained as a light-yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.46—7.26 (m, 10H), 6.55 (dd, J = 15.9, 4.0 Hz, 1H), 6.42 (dd, J = 15.9, 8.0 Hz, 1H), 3.97–3.76 (m, 5H), 3.08 (d, J = 7.4 Hz, 2H), 2.78–2.68 (m, 2H), 2.18–2.12 (m, 1H), 2.02–2.00 (m, 1H), 1.94–1.83 (m, 3H), 1.7–1.65 (m, 3H), 1.50–1.22 (m, 4H); C NMR (125 MHz) δ 142.71, 142.67, 137.42, 137.39, 132.1, 132.0, 131.14, 131.07, 128.89, 128.88, 128.7, 128.65, 128.59, 128.4, 128.3, 127.96, 127.94, 127.7, 127.44, 127.43, 126.8, 126.84, 126.82, 126.4, 126.3, 108.8, 62.7, 62.2, 62.1, 60.8, 54.9, 49.41, 49.39, 48.84, 48.82, 46.97, 46.96, 30.53, 30.50, 29.23, 29.21, 29.0, 26.0, 21.2; IR (thin film, cm-1) 3654, 3025, 2951, 1147; HRMS (ESI) m/z: [M + H]+ Calcd for C26H34NO2 392.2590; Found: 392.2585.
(3aS, 6aS)-Benzyl 6a-((1S, E)-1,3-diphenylallyl)hexahydrocyclopenta[b]pyrrole-1(2H)-carboxylate (41)
Following general procedure G, 41 (6 mg, 14 μmol, 65% yield) was obtained as a light colorless oil. HPLC analysis indicated an enantiomeric excess of 99% [CHIRALCEL AD column; flow: 1.0 mL/min; 98% n-hexane/2% i-PrOH; λ = 254 nm; major enantiomer tR = 29.97 min; minor en-antiomer tR = 23.72 min]. . 1H NMR (500 MHz, DMSO-d6, 393K) δ 7.50–7.40 (m, 6H), 7.35–7.30 (m, 3H), 7.24–7.15 (m, 6H), 6.73 (dd, J = 15.6, 9.9 Hz, 1H), 6.48 (d, J = 15.5 Hz, 1H), 5.22 (d, J = 12.6, 1H), 5.12 (broad d, J = 13.4 Hz, 1H), 4.50 (broad d, J = 12.8 Hz, 1H), 3.30 (ddd, J = 10.5, 8.0, 8.0 Hz, 1H), 3.02-2.97 (m, 1H), 2.69 (ddd, J = 10.7, 8.8, 4.7 Hz, 1H), 2.24 (broad s, 1H), 2.04 (ddd, J = 14.3, 7.3, 7.3 Hz, 1H), 1.90–1.84 (m, 1H), 1.60–1.46 (m, 2H), 1.42–1.37 (m, 1H), 1.31–1.28 (m, 1H), 1.18–1.12 (m, 1H); 13C NMR (125 MHz, DMSO-d6, 393K) δ 152.7, 141.4, 136.82, 136.78, 131.9, 129.5, 127.9, 127.8, 127.6, 127.1, 127.0, 126.9, 126.5, 125.6, 125.5, 77.5, 65.1, 51.6, 46.6, 43.9, 37.1, 32.6, 27.6, 24.3; IR (thin film, cm-1) 3028, 2951, 1699, 1132; HRMS (ESI) m/z: [M + Na]+ Calcd for C30H31NO2Na 460.2252; Found: 460.2243.
(R,E)-tert-Butyl (4-cyclohexyl-2-phenylbut-3-en-1-yl)carbamate (S44)
To a stirring solution of HG-II37 (23 mg, 0.036 mmol) in CH2Cl2 (1.2 mL) was added homoallylic carbamate 25 (0.090 g, 0.36 mmol) and vinylcyclohexane (0.25 mL, 1.8 mmol). The green solution was heated to reflux for 12 h. After 22 h, the solution was cooled to room temperature and concentrated under reduced pressure to a brown solid. The crude material was purified by flash chromatography on silica gel (100% hexanes to 9:1 hexanes:Et2O) to yield S44 as a colorless solid (49 mg, 0.15 mmol, 41%) that was a 9:1 mixture of E:Z double-bond isomers. mp 83–84 °C.1H NMR (600 MHz, CDCl3) δ 7.33–7.31 (m, 2H), 7.24–7.20 (m, 3H), 5.54–5.48 (m, 2H), 5.44–5.39 (m, 0.18 H), 3.41 (broad s, 1H), 3.37–3.32 (m, 2H), 3.22–3.18 (m, 0.11H), 1.97-1.93 (m, 1H), 1.74–1.69 (m, 5H), 1.65-1.63 (1.3H), 1.43 (s, 10.5 H), 1.29–1.22 (m, 2.4 H), 1.18–1.10 (m, 3.3 H); 13C NMR (125 MHz, CDCl3) δ 156.0, 142.5, 138.8, 128.8, 128.0, 127.90, 127.88, 127.8, 127.6, 126.8, 79.3, 49.0, 45.7, 40.9, 33.2, 28.6, 26.2; IR (thin film, cm-1) 3438, 3363, 2975, 1704, 1601, 1171; HRMS (ESI) m/z: [M + Na]+ Calcd for C21H31NO2Na 352.2252; Found: 352.2254.
(R,E)-4-Cyclohexyl-2-phenylbut-3-en-1-amine (S45)
To a solution of Boc-amide S44 (49 mg, 0.15 mmol) in CH2Cl2 (0.68 mL) at 0 °C was added TFA (0.12 mL, 1.5 mmol) dropwise over 3 minutes. The resulting clear and colorless solution was warmed to room temperature and maintained for 1.5 h. The reaction was quenched by the addition of 10% aqueous solution of NaOH until the solution is basic (pH >10 analyzed by pH paper) at 0 °C. The solution was warmed to room temperature and ex-tracted with Et2O (3 × 50 mL). The organic layers were combined, dried with MgSO4, filtered and con-centrated to provide the amine as a colorless oil. The crude material could be used without further puri-fication or purified to obtain an analytically pure sample (>98% E olefin isomer) by flash chromatog- raphy on silica gel (99:1 Et2O:Et3N) to yield S45 as a colorless oil (25 mg, 0.11 mmol. 73%). ; 1H NMR (500 MHz, CDCl3) δ 7.34–23 23 23 7.16 (m, 5H), 5.52–5.51 (m, 2H), 3.23–3.19 (m, 1H), 2.95–2.87 (m, 2H), 2.00–1.92 (m, 1H), 1.73–1.63 (m, 5H), 1.47 (broad s, 1H), 1.28–1.07 (m, 6H); 13C NMR (125 MHz) δ 143.3, 138.7, 128.7, 128.6, 127.9, 126.5, 53.0, 47.7, 40.9, 33.4, 33.3, 26.3, 26.2, 25.1; IR (thin film, cm-) 3366, 3284, 3026, 2920, 2850, 1074, 1029; HRMS (ESI) m/z: [M + H]+ Calcd for C16H24N 230.1909; Found: 230.1909.
N-((R,E)-4-cyclohexyl-2-phenylbut-3-en-1-yl)-2-(6,10-dioxaspiro[4.5]decan-1-yl)acetamide (S46)
Following general procedure D, S46 (27 mg, 0.065 mmol, 64% yield) was obtained as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.38–7.35 (m, 2H), 7.28–7.25 (m, 3H), 6.23 (broad d, J = 17.1 Hz, 1H), 5.55 (s, 1H), 5.54 (dd, J = 3.0, 1.2 Hz, 1H), 3.93–3.76 (m, 4H), 3.71–3.60 (m, 1H), 3.56–3.41 (m, 2H), 2.60 (dd, J = 15.1, 6.7 Hz, 1H), 2.67–2.20 (m, 1H), 2.13–2.07 (m, 2H), 2.10–1.90 (m, 1H), 1.91–1.58 (m, 12H), 1.41–1.05 (m, 5H); 13C NMR (125 MHz) δ 173.24, 173.21, 142.6, 142.5, 138.6, 138.5, 128.8, 128.3, 128.0, 127.9, 126.8, 108.3, 108.2, 62.30, 62.27, 60.73, 60.70, 48.7, 48.6, 46.1, 45.9, 44.33, 44.26, 40.9, 36.1, 33.21, 33.17, 30.51, 30.47, 29.9, 29.6, 29.5, 26.3, 26.2, 26.01, 25.99, 21.2; IR (thin film, cm-1) 3296, 3061, 2920, 1644; HRMS(ESI) m/z: [M + Na]+ Calcd for C26H37NO3Na 434.2671; Found: 434.2680.
(2R,E)-N-(2-(6,10-Dioxaspiro[4.5]decan-1-yl)ethyl)-4-cyclohexyl-2-phenylbut-3-en-1-amine (39c)
Following general procedure E, 39c (18 mg, 0.045 mmol, 67% yield) was obtained as a color-less oil. 1H NMR (500 MHz, CDCl3) δ 7.31–7.29 (m, 2H), 7.23–7.20 (m, 3H), 5.51–5.50 (m, 2H), 3.89–3.84 (m, 4H), 3.51–3.45 (m, 1H), 2.89–2.83 (m, 2H), 2.69–2.67 (m, 2H), 2.11–2.06 (m, 1H), 2.00–1.90 (m, 2H), 1.82–1.55 (m, 11H), 1.44–1.04 (m, 9H); 13C NMR (125 MHz) δ 138.4, 129.1, 128.7, 128.7, 127.8, 126.51, 126.49, 108.90, 108.88, 62.23, 62.22, 60.79, 60.77, 55.3, 55.2, 49.0, 48.82, 48.76, 47.0, 46.9, 41.0, 40.9, 33.33, 33.30, 33.24, 33.23, 30.7, 30.6, 29.31, 29.25, 29.0, 26.4, 26.27, 26.26, 26.15, 21.24, 21.23; IR (thin film, cm-1) 3430, 3313, 3052, 2923, 1264; HRMS (ESI) m/z: [M + H]+ Calcd for C26H40NO2 398.3059; Found: 398.3053.
(3aS,6aS)-Benzyl 6a-((R,E)-1-cyclohexyl-3-phenylallyl)hexahydrocyclopenta[b]pyrrole-1(2H)-carboxylate (42)
Following general procedure G, 42 (31 mg, 0.071 mmol, 49%) was obtained as a colorless oil. HPLC analysis indicated an enantiomeric excess of 99% [CHIRALCEL AD column; flow: 0.5 mL/min; 98% n-hexane/2% i-PrOH; λ = 254 nm; major enantiomer tR = 28.77 min; minor en-antiomer tR = 32.53 min]. ; 1H NMR (500 MHz, DMSO-d6, 393K) δ 7.42–7.35 (m, 6H), 7.32–7.29 (m, 3H), 7.22–7.19 (m, 1H), 6.34 (d, J = 15.8 Hz, 1H), 6.14 (dd, J = 15.7, 10.6, 1H), 5.15 (d, J = 12.7 Hz, 1H), 5.06 (d, J = 12.6 Hz, 1H), 3.62–3.51 (m, 1H), 2.97 (d, J = 9.5 Hz, 1H), 2.09 (broad s, 1H), 2.00 (dddd, J = 12.9, 8.5, 8.5, 8.5 Hz, 1H), 1.85 (dddd, J = 7.7, 7.7, 7.7, 7.7 Hz, 1H), 1.771.73 (m, 2H), 1.69–1.66 (m, 1H), 1.63–1.39 (m, 8H), 1.31–1.29 (m, 1H), 1.23–1.14 (m, 3H), 1.11–1.01 (m, 3H); C NMR (125 MHz, DMSO-d6, 393K) δ 152.7, 137.0, 136.9, 132.0, 129.4, 127.8, 127.6, 126.9, 126.8, 126.2, 125.4, 76.7, 65.0, 53.3, 47.3, 44.6, 37.9, 33.03, 32.99, 29.4, 28.4, 25.9, 25.7, 25.3, 23.8; IR (thin film, cm-1) 3028, 2925, 2851, 1699; HRMS (ESI) m/z: [M + Na]+ Calcd for C30H37NO2Na 466.2722; Found: 466.2714.
The synthesis and characterization of amino acids 43–45 have been reported previously.9
(S)-2-((3aS,6aS)-1-((Benzyloxy)carbonyl)octahydrocyclopenta[b]pyrrol-6a-yl)-2-cyclohexylacetic acid (46)
Ozone was bubbled through a solution of 42 (12 mg, 25 μmol) in MeOH (1.7 mL) at −78 °C for approximately 30 seconds until a persistent blue color was observed. The solu-tion was purged with O2 until colorless, quenched with dimethyl sulfide (81 μL, 1.1 mmol), and warmed to room temperature. After 24 h, the reaction mixture was concentrated in vacuo, dissolved in MeCN (1.8 mL) and H2O (0.17 mL), and cooled to 0 °C. Sodium chlorite (23 mg, 0.25 mmol) was added to the solution and the reaction mixture was warmed to room temperature. After stirring for 24 h, the reaction mixture was concentrated in vacuo, diluted with brine (5 mL), and extracted with EtOAc (3 × 5 mL). The combined organic extracts were dried (MgSO4), filtered, and concentrated. Purification of the crude residue by flash chromatography on silica gel (100:100:1 hexanes:Et2O:AcOH) provided 46 (10 mg, 25 μmol, 99% yield) as a colorless oil. ; 1H NMR (500 MHz, DMSO-d6, 393K) δ 11.56 (broad s, 1H), 7.36 (broad s, 4H), 7.30 (broad s, 1H), 5.16 (d, J = 1.8 Hz, 1H), 5.06 (broad m, 1H), 3.53–3.41 (broad m, 2H), 3.27 (broad s, 1H), 2.09–2.03 (broad m, 1H), 1.92–1.84 (broad m, 2H), 1.73–1.38 (broad m, 11H), 1.32–0.86 (broad m, 6H); 13C NMR (125 MHz, DMSO-d6, 393K) δ 174.6, 152.6, 137.5, 128.4, 128.3, 128.0, 127.6, 127.1, 75.7, 65.2, 53.0, 47.0, 43.4, 38.5, 36.6, 33.5, 32.7, 30.9, 29.4, 26.3, 26.1, 25.8, 25.0; IR (thin film, cm-1) 2921, 2851, 1214; HRMS (ESI) m/z: [M + Na]+ Calcd for C23H31NO4Na 408.2151; Found: 408.2153.
2,2,2-trifluoro-1l3-ethan-1-one, 6a-(3-hydroxypropoxy)-1-((S)-2-methylbut-3-en-1- yl)octahydrocyclopenta[b]pyrrol-1-ium salt (diastereomers 49 and 50)
1H NMR (500 MHz, DMSO-d6) δ 8.38–8.29 (br d, 2H), 5.73–5.66 (m, 1H), 5.13–5.05 (m, 2H), 3.85–3.74 (m, 4H), 2.98–2.84 (m, 1H), 2.84–2.83 (m, 2H), 2.52–2.48 (m, 1H), 2.11–2.06 (m, 2H), 1.81–1.69 (m, 5H), 1.55–1.50 (m, 3H), 1.34 (d, J = 2.4 Hz, 1H), 1.18–1.16 (m, 1H), 0.98 (d, J = 7.0 Hz, 3H); 13C NMR (125 MHz, DMSO-d6, contains a mixture of diastereomers, signals observed are reported): δ 258.4, 158.2, 139.8, 139.8, 139.8, 116.2, 116.2, 107.7, 107.6, 61.5, 59.9, 51.5, 46.58, 46.55, 45.97, 45.96, 34.8, 29.9, 28.4, 25.5, 24.7, 24.7, 24.6, 20.72, 20.71, 14.5.
Supplementary Material
Acknowledgments
Financial support was provided by the National Institute on Neurological Disorders and Stroke (R01NS12389), National Institute of General Medical Sciences (R01GM30859), Takeda Pharmaceutical Co., and a NIH postdoctoral fellowship for T.L.M. (F32GM096690). NMR, mass spectra, and X-ray analyses were obtained at UC Irvine using instrumentation acquired with the assistance of NSF and NIH Shared Instrumentation programs. We thank Dr. Joseph Ziller and Dr. John Greaves, Department of Chemistry, UC Irvine, for their assistance with X-ray and mass spectrometric analyses.
Footnotes
Supporting Information: Copies of 1H and 13C NMR spectra for new compounds, HPLC traces used to determine enantiomeric purity, CIF files for compounds ent-8 and 18 and thermal-ellipsoid plots, and general experimental details. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Morita H, Arisaka M, Yoshida N, Kobayashi J. Tetrahedron. 2000;56:2929–2934.. Other examples of alkaloid natural products containing these structural frameworks include: huperzine B Liu JS, Whu YL, Yu CM, Zhou YZ, Han YY, Wu FW, Qi NF. Can J Chem. 1986;64:837–839.; malycorin C Hirasawa Y, Tanaka T, Kobayashi J, Kawahara N, Goda Y, Morita H. Chem Pharm Bull. 2008;56:1473–1476. doi: 10.1248/cpb.56.1473.; serratine Inubushi Y, Tsuda Y, Ishii H, Sano T, Ho-sokawa M, Harayama T. Yakugaski Zasshi. 1964;84:1108–1113. doi: 10.1248/yakushi1947.84.11_1108.
- 2.Chen C, Kozikowski AP, Wood PL, Reynolds IJ, Ball RG, Pang YP. J Med Chem. 1992;35:1634–1638. doi: 10.1021/jm00087a020. [DOI] [PubMed] [Google Scholar]
- 3.Substructure searches for 1 (n = 0–2 and m = 0–2) in SciFinder finds only 23 compounds for which an analysis criteria of biological study or use is recorded; 19 of these are 8a-substituted decahy-droquinolines.
- 4.Examples include moxifloxacin, which contains an octahydropyrrolopyridine fragment, and rami-pril, which contains a hexahydropenta[b]pyrrole unit.
- 5.For reports of the direct enantioselective construction of specific angularly substituted 1-azacyclic molecules, see: Trauner D, Schwarz JB, Danishefsky SJ. Angew Chem Int Ed. 1999;38:3542–3545. doi: 10.1002/(sici)1521-3773(19991203)38:23<3542::aid-anie3542>3.0.co;2-i.. Carson MW, Kim G, Hentemann MF, Trauner D, Danishefsky SJ. Angew Chem Int Ed. 1999;38:4450–4452. doi: 10.1002/1521-3773(20011203)40:23<4450::aid-anie4450>3.0.co;2-m.. Aldous DJ, Drew MGB, Draffin WN, Hamelin EMN, Harwood LM, Thurairatnam S. Synthesis. 2005:3271–3278.. Reggelin M, Junker B, Heinrich T, Slavik S, Bühle P. J Am Chem Soc. 2006;128:4023–4034. doi: 10.1021/ja057012a.. Xu S, Arimoto H, Uemura D. Angew Chem Int Ed. 2007;46:5746–5749. doi: 10.1002/anie.200701581.. Keck GE, Heumann SA. Org Lett. 2008;10:4783–4786. doi: 10.1021/ol801849u.. Ranatunga S, Del Valle JR. Tetrahedron Lett. 2009;50:2464–2466. doi: 10.1016/j.tetlet.2009.03.092.. Sethofer SG, Mayer T, Toste FD. J Am Chem Soc. 2010;132:8276–8277. doi: 10.1021/ja103544p.. Nama K, Inai M, Sundermeier U, Greshock TJ, Williams RM. Tetrahedron Lett. 2010;51:6557–6559. doi: 10.1016/j.tetlet.2010.10.037.. Fustero S, Mateu N, Simón-Fuentes A, Luis Aceña J Org Lett. 2010;12:3014–3017. doi: 10.1021/ol1010246.. Yuan C, Chang CT, Axelrod A, Siegal D. J Am Chem Soc. 2010;132:5924–5925. doi: 10.1021/ja101956x.. Enantiomerically enriched an-gularly substituted 1-azabicyclic molecules have been accessed also by separation of a 1:1 mixtures of diastereomers or enantiomers, see: Kozikowski AP, Chen C. Tetrahedron. 1990;31:5869–5872.. Chen C, Kozikowski AP, Wood PL, Reynolds IJ, Ball RG, Pang YP. J Med Chem. 1992;35:1634–1638. doi: 10.1021/jm00087a020.. Verbist BMP, De Borggraeve WM, Toppet S, Compernolle F, Hoornaert GJ. Eur J Org Chem. 2005:2941–2950.. Meyer U, Bisel P, Weckert E, Frahm AW. Chirality. 2006;18:383–394. doi: 10.1002/chir.20260.. Kopylova NA, Grygorenko OO, Komarov IV, Groth U. Tetrahedron: Asymmetry. 2010;21:2868–2871.
- 6.Aron ZD, Overman LE. Org Lett. 2005;7:913–916. doi: 10.1021/ol047330z. [DOI] [PubMed] [Google Scholar]
- 7.For representative examples of selective bimolecular trapping of one iminum ion isomer of a 2-aza-Cope rearrangement, see: Overman LE, Yokomatsu T. J Org Chem. 1980;45:5229–5230.. Castelhano AL, Krantz A. J Am Chem Soc. 1984;106:1877–1879.. Bennett DJ, Hamilton NM. Tetrahedron Lett. 2000;41:7961–7964.
- 8.For a recent review of dynamic kinetic resolution, see: Pellissier H. Tetrahedron. 2011;67:3769–3802.
- 9.Some of these studies were reported in a preliminary communication, see: Ito T, Overman LE, Wang J. J Am Chem Soc. 2010;132:3272–3273. doi: 10.1021/ja100607z.
- 10.Previously prepared by alkylation of a gallium enolate of cyclohexanone: Usugi S, Yori-mitsu H, Shinokubo H, Oshima K. Bull Chem Soc Jpn. 2002;75:2049–2052.; or by transesterifica-tion of commercially available methyl 2-oxocyclohexaneacetate: Meidong L, Gu Q, He Y, Huang W. Faming Zhuanli Zhuanli Shenqing. 2005 CN 150049 A 20050105.
- 11.The route utilized to prepare 11 is an optimization of previous disclosures.6,9 TASF(Me) was found to promote the alkylation to afford ketoester 11 in the highest efficiency. The use of CsF, tetrabu-tylammonium diflurotriphenylstannate, tetrabutylammonium difluorotriphenylsilicate, or benzyltrime-thylammonium fluoride as promoters afforded the product in lower yields (30%, 32%, 58%, and 60%, respectively).
- 12.Vilaivan T, Winotapan C, Banphavichit V, Shinada T, Ohfune Y. J Org Chem. 2005;70:3464–3471. doi: 10.1021/jo0477244. [DOI] [PubMed] [Google Scholar]
- 13.We found it most convenient to determine the enantiomeric purity of these primary amines by en-antioselective chromatography of the corresponding benzamides (see the Supporting Information).
- 14.Enantioselectivities were determined by enantioselective HPLC after benzolylation of ent-8; de-tails are provided in the Experimental Section and Supporting Information.
- 15.Details are provided in the Experimental Section.
- 16.Kaiser NKK, Bremberg U, Larhed M, Moberg C, Hallberg A. Angew Chem Int Ed. 2000;39:3596–3598. doi: 10.1002/1521-3773(20001016)39:20<3595::aid-anie3595>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 17.Trost BM, Hachiya I. J Am Chem Soc. 1998;120:1104–1105.. Palucki M, Um JM, Conlon DA, Yasuda N, Hughes DL, Mao B, Wang J, Reider PJ. Adv Synth Catal. 2001;343:46–50.
- 18.Krapcho AP, Weimaster JF, Eldridge JM, Jahngen EGE, Jr, Lovey AJ, Stephens WP. J Org Chem. 1978;43:138–147. [Google Scholar]
- 19.Brown E, Deroye C, Touet J. Tetrahedron: Asymmetry. 1998;9:1605–1614.. Mitsui K, Sato T, Urabe H, Sato F. Angew Chem Int Ed. 2004;43:490–492. doi: 10.1002/anie.200352769.. Gao M, Wang DX, Zheng QY, Wang MX. J Org Chem. 2006;71:9532–9535. doi: 10.1021/jo061664f.
- 20.Blanco B, Christensen J, Maurel I, Pleixats R, Serra A, Pla-Quintana A, Roglans A, Benet-Buchholz J. Synthesis. 2005:374–380. [Google Scholar]
- 21.van Zijl AW, López F, Minnaard AJ, Feringa BL. J Org Chem. 2007;72:2558–2563. doi: 10.1021/jo0625655..; (b) The enantiomeric purity of 28 was determined by enantioselective HPLC analysis of the corresponding primary alcohol obtained by an ozonolysis/reduction sequence.
- 22.This reaction could be carried out also neat; however because the reaction is heterogeneous at this lower temperature, inconsistent reaction times were observed.
-
23.The configuration assigned to the side chain stereocenter of 40 is consistent with the expectation that it would be established in the aza-Cope rearrangement step. This assignment is further supported by a strong 1H NOE enhancement observed between C3a-H and the methyl substituent of the side chain; The relative configuration of 40 was assigned by comparing the NOESY data to the lowest energy con-formation calculated using ab initio studies with DFT/B3LYP/6-31G* as implemented in Spartan 2008.
 The relative configuration of products 41 and 42 were assigned by analogy.
The relative configuration of products 41 and 42 were assigned by analogy.
- 24.For recent representative applications, see: Hanessian S, Auzzas L. Acc Chem Res. 2008;41:1241–1251. doi: 10.1021/ar8000052.. Seebach D, Gardiner J. Acc Chem Res. 2008;41:1366–1375. doi: 10.1021/ar700263g.. Newman DJ, Cragg GM. J Nat Prod. 2012;75:311–335. doi: 10.1021/np200906s.
- 25.Sayago FJ, Laborda P, Calaza MI, Jiménez AI, Cativiela C. Eur J Org Chem. 2011:2011–2028. [Google Scholar]
- 26.For a review, see: Mukherjee S, Yang JW, Hoffmann S, List B. Chem Rev. 2007;107:5471–5569. doi: 10.1021/cr0684016.
- 27.For example, Tanaka and Barbas have found that (R)-3-pyrrolidinecarboxylic acid can efficiently catalyze a highly anti-selective Mannich reactions, see: Zhang H, Mitsumori S, Utsumi N, Imai M, Garcia-Delgado N, Mifsud M, Albertshofer K, Cheong PHY, Houk KN, Tanaka F, Bar-bas CF., III J Am Chem Soc. 2008;130:875–886. doi: 10.1021/ja074907+.. See also, Armstrong A, Bhonoah Y, Colorless AJP. J Org Chem. 2009;74:5041–5048. doi: 10.1021/jo900840v.
- 28.Isotopic analysis was performed using MassLynx mass spectrometry software.
- 29.In our initial report of these transformations in the racemic series, we suggested on the basis of an experiment, which in hindsight was potentially flawed, that the allylic iminium ion sigmatropic isomers studied at that time were in equilibrium.6 As already noted, the high chirality transfer observed in the substituted allyl series that is the subject of this report rigorously establishes that the sigmatropic isomers are not in equilibrium.
- 30.See Supporting Information of reference 9 for a Scheme detailing all possible chair and boat transition structures for the 2-aza-Cope rearrangement and the corresponding products produced.
- 31.Enantiomers 47 and ent-47 exhibited baseline resolution in the enantioselective HPLC separation used in this study, allowing us to confidently say that less than 2 % of ent-47 (more likely < 1%) was formed. See the Supporting Information provided for reference 9.
- 32.Wang J. PhD Dissertation. UC Irvine: 2011. [Google Scholar]
- 33.van der Sluis M, Dalmolen J, de Lange N, Kaptein B, Kellogg RM, Broxterman QB. Org Lett. 2001;3:3943–3946. doi: 10.1021/ol016840f. [DOI] [PubMed] [Google Scholar]
- 34.Kohno K, Narasaka K. Bull Chem Soc Jpn. 1995;68:322–329. [Google Scholar]
- 35.Cotarco L, Delogu P, Maggioni P, Nardelli A, Bianchini R, Sguassero S. Synthesis. 1997:328–332. [Google Scholar]
- 36.Ocejo M, Carrillo L, Badía D. J Org Chem. 2009;74:4404–4407. doi: 10.1021/jo900397w. [DOI] [PubMed] [Google Scholar]
- 37.Garber SB, Kingsbury JS, Gray BL, Hoveyda AH. J Am Chem Soc. 2000;122:8168–8179. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.