

Abstract
A 3-pyridyl ether scaffold bearing a cyclopropane-containing side chain was recently identified in our efforts to create novel antidepressants that act as partial agonists at α4β2-nicotinic acetylcholine receptors. In this study, a systematic structure-activity relationship investigation was carried out on both the azetidine moiety present in compound 3 and its right-hand side chain, thereby discovering a variety of novel nicotinic ligands that retain bioactivity and feature improved chemical stability. The most promising compounds 24, 26, and 30 demonstrated comparable or enhanced pharmacological profiles compared to the parent compound 4, and the N-methylpyrrolidine analogue 26 also exhibited robust antidepressant-like efficacy in the mouse forced swim test. The favorable ADMET profile and chemical stability of 26 further indicate this compound to be a promising lead as a drug candidate warranting further advancement down the drug discovery pipeline.
Introduction
Neuronal nicotinic acetylcholine receptors (nAChRs) belong to the ligand-gated ion channel super-family of neurotransmitter receptors, which are widely distributed in the central and peripheral nervous system.1 They are thought to be vital players modulating major brain functions and participating in pathophysiological processes, and the dysfunction of this family of receptors has been implicated in a variety of nervous system disorders such as Alzheimer’s disease, Parkinson’s disease, schizophrenia, nicotine addiction, and depression.1 Brain nAChRs are homomeric or heteromeric pentamers assembled from diverse combinations of subunits (α2–α10 and β2–β4), and different subtype stoichiometries allow the assembly of varied pentamers with a wide range of physiological and pharmacological profiles.2 Increasing evidence has implied that the α4β2*-nAChRs (the asterisk denotes the possible integration of other subunits aside from those specified into the pentamer), the most abundant and widespread nAChRs in the brain, may play a role in mood and reward. Numerous preclinical studies in murine models have illustrated that administration of α4β2-nAChR partial agonists or antagonists potentiates antidepressant-like effects of conventional antidepressants, suggesting possible human use as an adjunctive therapy, but also indicate that nicotinic ligands acting alone can elicit antidepressant-like effects.3 The antidepressant effects of mecamylamine, and even those of the tricyclic antidepressant amitriptyline, are abolished in nAChR β2 subunit knockout mice that lack α4β2*-nAChRs.4, 5 Moreover, results of clinical trials in depressed patients also demonstrate that the marketed drug varenicline, an α4β2-nAChR partial agonist for smoking cessation pharmacotherapy, has positive effects on relieving the symptoms of depression.6 Given that more than one-third of depressed individuals are resistant to currently available drug treatments,7 selective α4β2-nAChR ligands could offer hope as mechanistically novel antidepressant medications. In addition, considerable progress has recently been made in understanding nAChRs containing the α6 subunit regarding their anatomical distribution, subunit composition, and physiological function. Although their expression in the brain is relatively restricted, these receptors are prevalent in midbrain dopaminergic regions and are thought to influence physiological mechanisms associated with mood and drug dependence.8, 9
Azetidine is a four-membered nitrogen-containing saturated heterocycle possessing reasonable chemical stability. With the increasing number of methods that have been developed for the synthesis of four-membered ring systems,10 azetidine-containing building blocks have received considerable interest by the medicinal chemistry community as components in various drugs or bioactive compounds. For instance, azetidine-2-one (β-lactam ring) is the key structural element of β-lactam antibiotics that were discovered more than 80 years ago and are still widely used clinically, as exemplified by penicillins, cephalosporins, and pemems.11 Azelnidipine, a long-acting calcium channel blocker containing a azetidinyl ether unit, has been approved in Japan as an antihypertensive drug.12 Additionally, the azetidine moiety has also been introduced by Abbott laboratories for the development of neuronal nAChR ligands for treating specific CNS disorders with reduced side effect liabilities.13 Among them, tebanicline (1) is a highly potent α4β2-nAChR agonist with improved selectivity over other subtypes in comparison with the parent compound epibatidine (2). A chiral azetidinylmethoxy group was used to replace the 7-azabicyclo[2.2.1]heptane structure in compound 2 providing a protonated nitrogen atom as an essential pharmacophoric element of nicotinic ligands (Figure 1). Although compound 1 was abandoned after completing Phase II clinical trials for the treatment of neuropathic pain due to an unacceptably narrow therapeutic index,14 the 3-pyridyl ether scaffold offers new perspectives on the design of α4β2-nAChR ligands and holds promise for α4β2-nAChR modulators to be advanced to the clinic as novel therapeutic agents.
Figure 1.
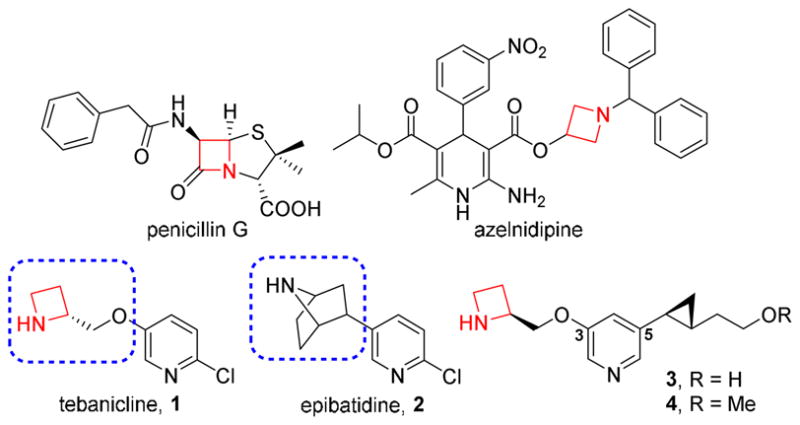
Selected azetidine-containing compounds and nicotinic ligands.
Previously, in our efforts to design nAChR ligands as potential antidepressants, a series of selective α4β2-nAChR partial agonists were identified based on the 3-pyridyl ether scaffold.15 The chiral cyclopropane-containing side chain appended to the 5-position of the pyridine ring confers substantial nAChR subtype selectivity to these ligands compared to the 5-unsubstituted compound A-85380, particularly over ganglionic α3β4*-nAChRs that are thought to be associated with unwanted side effects in vivo,16 while not affecting their high affinity for α4β2-nAChRs. Some of the most promising compounds (3 and 4) demonstrate favorable antidepressant-like effects in the mouse forced swim test as well as acceptable ADMET profiles, and may thus serve as suitable lead compounds for the development of novel antidepressant agents. However, it is noteworthy that the hydrochloride salt of compound 3 was hygroscopic and partially decomposed after the absorption of water to give considerable quantities of a major byproduct. On the basis of analysis of LC-MS results and NMR spectra, the structure of the decomposition product was identified as a dimer of the parent compound, which resulted from attack of one azetidine nitrogen atom on the less-substituted α-methylene group of another azetidine ring. Similar intermolecular dimerization was also observed by Abbott chemists during the process of salt screening and selection for compound 1.17 Although this potential stability issue can be prevented by changing the salt form of these azetidine-containing ligands,17 replacement of the azetidine unit by other ring systems having improved chemical stability is a logical first choice. Thus, in continuation of our efforts to develop α4β2-nAChR ligands for treating depression, a novel series of nAChR ligands with different ring systems replacing the azetidine ring found in the lead compound 4 were designed and synthesized for pharmacological evaluation. Selected compounds were further assessed in behavioral tests used to gauge antidepressant drug action and preliminary ADMET studies were carried out. Moreover, we detailed additional structural modifications of the side chain attached to the 5-position of the pyridine ring.
CHEMISTRY
In our previous study of the isoxazolylpyridine ether analogues, we found that the length of the side chain was crucial to their biological activity.18 We thus chose to synthesize cyclopropane ligands bearing a shorter or longer side chain in comparison with compound 3 to explore the optimal length of the side chain for the cyclopropane series. In addition, in order to better understand structure-activity relationship (SAR) of the right-hand side chain, analogues featuring a terminal carboxylic acid, trifluoromethoxy group, carbamate function, or six-membered ring were designed. The syntheses of the cyclopropane analogues 7, 10, 11, 14, 17a-c, 19a-b, and 21 are outlined in Scheme 1. The optically pure alcohol 5 was acylated with isobutyric anhydride, and the benzyl group was removed by hydrogenolysis, followed by installation of the azetidine moiety by a modified Mitsunobu reaction to obtain the intermediate 6. Successive removal of the isobutyryl group and the Boc group from 6 gave the desired product 7. For compound 10 with a longer side chain, alcohol 5 was subjected to standard Swern oxidation and Wittig reaction to furnish the α,β-unsaturated ester 8. After removal of the benzyl group and saturation of the double bond by catalytic hydrogenation in the same step, the azetidine moiety was installed to give the ester 9. Reduction of the ester group in the intermediate 9 and subsequent removal of the Boc group gave compound 10. Hydrolysis of the ester 9 and then removal of the Boc group furnished the carboxylic acid compound 11. For the terminal trifluoromethoxy compound 14, the starting alcohol 12 was converted to dithiocarbonate, followed by reaction with 70% HF/pyridine to give hydroxypyridine 13 after removal of the benzyl group.19 Installation of the azetidine moiety and subsequent removal of Boc group gave the desired product 14. The carbamate analogues 19a and 19b were prepared from the previously reported alcohol 15,15 which was transformed to the corresponding iodide, followed by nucleophilic substitution with sodium azide to give the intermediate 18. Reduction of the azide group in compound 18 and subsequent reaction with various chloroformates afforded the desired products after removal of the Boc protecting group. Similarly, the iodide 16 was substituted by morpholine, piperidine, or piperazine and the Boc group then removed to give 17a, 17b, and 17c, respectively. Compound 21, the diastereoisomer of 17b, was synthesized from alcohol 20 by the same sequence of steps. All target compounds were isolated as their trifluoroacetate salts.
Scheme 1a.

aReagents and conditions: (a) isobutyric anhydride, cat. 4-(dimethylamino)pyridine (DMAP), Et3N, CH2Cl2, rt; (b) 10% Pd/C, H2, EtOAc/MeOH, rt; (c) 1-(tert-butoxycarbonyl)-(2S)-azetidinylmethanol, azodicarbonyldipiperidide (ADDP), P(n-Bu)3, PhMe, 0 °C to rt; (d) NaOMe, MeOH, 40 °C; (e) CF3COOH, CH2Cl2, rt; (f) i. (COCl)2, DMSO, Et3N, CH2Cl2, −78 °C; ii. Ph3P=CHCO2CH3, THF, 0 °C to rt; (g) LiAlH4, THF, rt; (h) 2N NaOH, MeOH/THF (1:1), rt; (i) i. NaH, CS2, DMF, 0 °C to rt; ii. MeI, rt; (j) 70% HF/pyridine, 1,3-dibromo-5,5-dimethylhydantoin (DBH), CH2Cl2, −78 °C; (k) I2, PPh3, imidazole, 0 °C to rt; (l) morpholine, piperidine, or piperazine, CH3CN; (m) NaN3, DMF, 60 °C; (n) 10% Pd/C, H2, ethanol, rt; (o) ethyl or isopropyl chloroformate, Et3N, 0 °C to rt.
On the other hand, pyrrolidine, a higher homolog of azetidine and a common heterocyclic building block in medicinal chemistry, was chosen to replace the azetidine ring and thus furnish a variety of pyrrolidine-containing derivatives. The target molecules 23-31 were prepared through a previously reported route employing the hydroxypyridine intermediate 22 as the starting material (Scheme 2).15 Coupling of 22 with various alcohols to afford the corresponding ethers was brought about using the Mitsunobu reaction. Removal of the Boc group from the coupling intermediates, or reduction of the Boc or acetyl group to the corresponding methyl or ethyl groups respectively, gave the desired products as their trifluoroacetate salts. Compound 33, the diastereoisomer of 26, was synthesized from alcohol 32 by the same sequence of steps.
Scheme 2a.
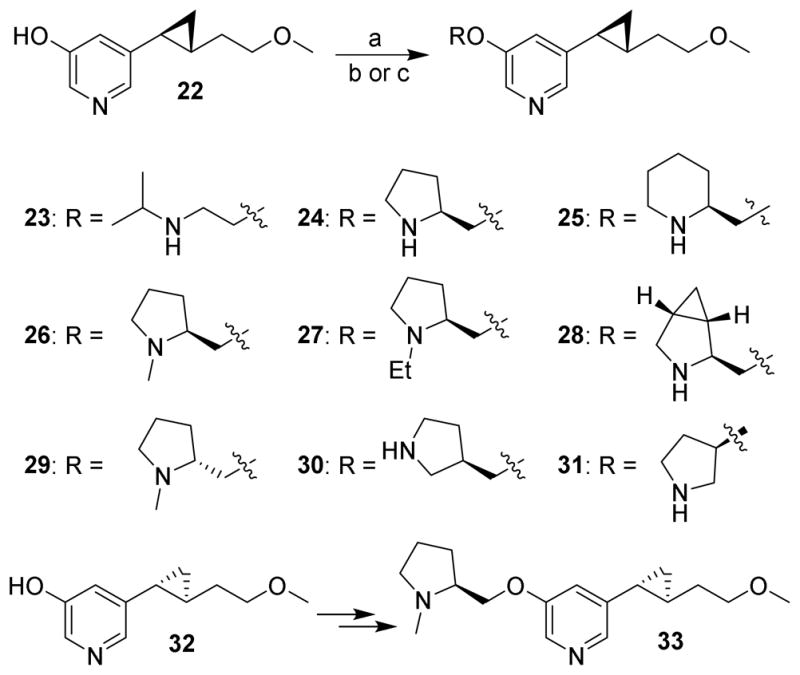
aReagents and conditions: (a) azodicarbonyldipiperidide (ADDP), P(n-Bu)3, alcohol, PhMe, 0 °C to rt; (b) CF3COOH, CH2Cl2, rt; (c) LiAlH4, THF, reflux.
RESULTS AND DISCUSSION
In Vitro Characterization: Radioligand Binding Studies
All the synthesized compounds were first assayed for [3H]epibatidine binding competition at seven, heterologously expressed, rat nAChR subtypes. As shown in Table 1, these compounds generally demonstrated favorable subtype selectivity for β2*-nAChRs (α2β2-, α3β2-, α4β2-, and α4β2*-nAChRs) over β4*-nAChRs (α2β4 -, α3β4-, and α4β4-nAChRs) compared to nicotine. Compound 7 exhibited subnanomolar binding affinity at both α4β2- and α4β2*-nAChRs, while the Ki values were slightly higher for compound 3. In contrast, the activity for α4β2- or α4β2*-nAChRs was retained when extending the side chain in compound 3 by one more carbon atom (compound 10). In view of the existing pharmacological data for the cyclopropane ligands15 combined with the knowledge obtained from the isoxazolylpyridine ether analogues,18 a two-carbon alkyl linker between the chiral cyclopropane and the terminal functional groups was optimal for biological activity. In reference to the terminal hydroxyl group, an at least 2-fold decline in binding affinities at β2*-nAChRs was observed when converting the hydroxyl group in compound 3 to a carboxylic acid (compound 11). The O-methylation of compound 3 did not alter binding affinities and selectivity for β2*-nAChRs. Moreover, this group would preclude the possible metabolic liability of the hydroxyl group, which can undergo oxidation and/or bioconjugation reactions.20 The replacement of the methoxy group in compound 4 with a more lipophilic and electron-withdrawing trifluoromethoxy moiety (compound 14) caused a 7-fold decrease in binding affinity for α4β2- or α4β2*-nAChRs and significantly decreased the subtype selectivity for β2*- over β4*-nAChRs. As previously reported, derivation of the hydroxyl group in compound 3 to a variety of carbamate groups in general maintained the high binding affinities at β2*-nAChRs. The reversed carbamate derivatives 19a and 19b also exhibited subnanomolar to low nanomolar binding affinities at β2*-nAChRs, and binding affinities decreased as the size of substituents at the carbamate oxygen increased. Introduction of a saturated six-membered heterocycle at the end of the side chain to replace the hydroxyl group (compounds 17a, 17b, and 17c) resulted in at least 2-fold less potency at β2*-nAChRs, while a more polar heterocyclic ring was preferred in this position (potency at α4β2-nAChRs: piperazine > morpholine > piperidine). In addition, compound 21, bearing a (1R,2S)-configured cyclopropane ring, was found to be about 10-fold less active than its diastereoisomer 17b at both α4β2- and α4β2*-nAChRs. Collectively, as shown in Table 1, these compounds bearing a varied cyclopropane-containing side chain generally were very potent at both α4β2- and α4β2*-nAChRs, suggesting that variations in the terminal substituents on the right-hand side chain do not significantly affect their binding affinities, which is consistent with the previous finding21 that the cyclopropane-containing side chain points outward from the binding pocket without engaging in any significant polar interactions based on the analysis of the co-crystal structure of the acetylcholine binding protein from Capitella teleta in complex with compound 3.
Table 1.
Affinities of compounds 7, 10, 11, 14, 17a-c, 19a-b, and 21 for Rat nAChR Subtypes Defined by Competition for [3H]Epibatidine Binding
| Compd. |
Ki (nM) a
|
||||||
|---|---|---|---|---|---|---|---|
| α2β2 | α2β4 | α3β2 | α3β4 | α4β2 | α4β2* b | α4β4 | |
| 7 | 0.2 | 233.4 | 2.8±0.5 | 6119 | 0.2 | 0.7±0.1 | 78.1±11.9 |
| 10 | 0.1 | 211.8 | 3.8±0.6 | 7514 | 0.1 | 0.4±0.1 | 88.1±10.4 |
| 11 | 0.5±0.1 | 318 | 10.2±1.9 | 4290 | 0.2 | 1.6±0.3 | 75.7±16 |
| 14 | 1.1±0.1 | 20.2±4.1 | 17±3.3 | 612.7 | 0.8±0.1 | 3.5±0.6 | 4.2±0.3 |
| 17a | 15.3±1.1 | NA | 224 | NA | 11.3±0.9 | 9.8±2.1 | NA |
| 17b | 14.9±3.5 | >104 | 403 | NA | 4.6±0.4 | 10.4±1.4 | 2930 |
| 17c | 65.3±14.4 | NA | 6.5±1.9 | NA | 2.1±0.3 | 1.7±0.5 | 6237 |
| 19a | 0.5±0.1 | 203 | 8.9±2.3 | 8830 | 0.4±0.1 | 1.0±0.1 | 125 |
| 19b | 0.6±0.1 | 453 | 11.1±3.7 | NA | 0.5±0.1 | 1.4±0.2 | 158 |
| 21 | 89.7±16 | 7850 | 1190 | NA | 56.1±7.8 | 91.5±14.3 | 2700 |
| 3 c | 0.1 | 249 | 3.0 | 6520 | 0.1 | 0.5 | 82.6 |
| 4 c | 0.1 | 236 | 2.4 | NA | 0.1 | 0.3 | 50.2 |
| nicotine d | 5.5 | 70 | 29 | 260 | 4.9 | 9.8 | 23 |
| saz-A e | 0.087 | 210 | 0.38 | 1900 | 0.062 | 0.17 | 52 |
See Experimental Section. SEM values are not provided for Ki values > 100 nM.
α4β2*, prepared from rat forebrain.
Ki values for compounds 3 and 4 are obtained from Reference 15.
Ki values for nicotine are taken from the PDSP Assay Protocol Book (http://pdsp.med.unc.edu/).
Ki values for sazetidine-A were obtained from the literature.22
NA: not active, defined as < 50% binding in the primary assay at 10 μM.
Next, we fixed the structure of the right-hand side chain and explored a series of functional groups to replace the somewhat labile azetidine unit. As shown in Table 2, replacement of the azetidine ring as in compound 4 with an aliphatic amine (compound 23) resulted in the complete loss of activity at all of the nAChR subtypes tested with the exception of weak binding affinity at α4β2-nAChRs (2.2 μM), suggesting that the conformationally restricted azetidine ring plays an important role in maintaining high binding affinity for the nAChRs. As the smallest saturated azaheterocycle aziridine is highly reactive and thus an unsuitable replacement for the azetidine, we thus chose to examine the higher homolog of azetidine, and thus prepared the pyrrolidine analogue 24. This compound exhibited subnanomolar to low nanomolar binding affinity at the α4β2- and α4β2*-nAChRs and good selectivity for β2*-nAChRs, similar to the parent compound 4. Further ring expansion of the pyrrolidine ring to a piperidine ring (compound 25) caused more than a 150-fold drop in binding affinities at β2*-nAChRs. The presence of an N-methyl group, as in compound 26, increased binding at the α4β2- or α4β2*-nAChRs relative to compound 24, however an N-ethyl group (compound 27) remarkably reduced binding affinity for all seven nAChRs. The fusion of a cyclopropane ring to the pyrrolidine ring (compound 28) also caused a sharp decrease in binding potency toward all nAChR subtypes tested, indicating that the space in the cation-binding pocket accommodating the pyrrolidine ring is limited. With respect to stereoselectivity, compound 26 was at least 70-fold or 10-fold more potent at the α4β2- and α4β2*-nAChRs than its diastereoisomers 29 and 33, respectively, indicating that an (S)-configuration of the N-methyl-pyrrolidine and a cyclopropane ring with the (1S,2R)-configuration were preferred. Shifting the nitrogen atom in the pyrrolidine ring of compound 24 to the less-substituted α-methylene position gave compound 30, having the highest binding affinity at β2*-nAChRs in this series, while exhibiting weak activity at α3β4-nAChRs (2 μM). Modification of compound 30 by deletion of the methylene group connecting the pyrrolidine ring with the 3-pyridyl ether framework led to compound 31 which showed a significant decrease in binding potency at all nAChR subtypes tested. In spite of the fact that some of the compounds listed in Table 2 had only modest activity at α4β2-nAChRs, most of them generally presented good subtype-selectivity for β2*-nAChRs over β4*-nAChRs compared to nicotine. On the basis of previous findings that α4β2-nAChR ligands featuring distinct chemical scaffolds can engage in similar receptor interactions,21 we anticipate that the cyclopropane-containing side chain could be appended to other α4β2-nAChR ligands with diverse scaffolds to improve their subtype-selectivity.
Table 2.
Affinities of compounds 23-31 and 33 for Rat nAChR Subtypes Defined by Competition for [3H]Epibatidine Binding
| Compd. |
Ki (nM) a
|
||||||
|---|---|---|---|---|---|---|---|
| α2β2 | α2β4 | α3β2 | α3β4 | α4β2 | α4β2* b | α4β4 | |
| 23 | NA | NA | NA | NA | 2176 | NA | NA |
| 24 | 0.5±0.1 | 612 | 17.3±5.3 | NA | 0.4±0.1 | 1.1±0.2 | 161 |
| 25 | 18.1±2.3 | >104 | 2534 | NA | 38.8±7 | 221 | >104 |
| 26 | 0.2 | 129.5 | 21±5.7 | NA | 0.3 | 0.5±0.1 | 584.8 |
| 27 | 167.9 | NA | NA | NA | 49.5±8.9 | 173.7 | NA |
| 28 | 43.8±10.7 | NA | 2094 | NA | 28.5±5.6 | 193.8 | NA |
| 29 | 12.9±1.7 | 9959 | 472.5 | NA | 21.7±6.6 | 86.6±15.3 | 9475 |
| 30 | 0.04 | 50.8±10.5 | 0.8±0.2 | 2074 | 0.05 | 0.2 | 30±4.3 |
| 31 | 6.1±0.6 | NA | 191.5 | NA | 90.9±27 | 40.7±5.3 | NA |
| 33 | 3.5±0.5 | 546.6 | 137.3 | NA | 3.3±1 | 15.7±2.8 | 234.1 |
| 4 c | 0.1 | 236 | 2.4 | NA | 0.1 | 0.3 | 50.2 |
| nicotine d | 5.5 | 70 | 29 | 260 | 4.9 | 9.8 | 23 |
| saz-A e | 0.087 | 210 | 0.38 | 1900 | 0.062 | 0.17 | 52 |
See Experimental Section. SEM values are not provided for Ki values > 100 nM.
α4β2*, prepared from rat forebrain.
Ki values for compound 4 are obtained from Reference 15.
Ki values for nicotine are taken from the PDSP Assay Protocol Book (http://pdsp.med.unc.edu/).
Ki values for sazetidine-A were obtained from the literature.22
NA: not active, defined as < 50% binding in the primary assay at 10 μM.
In Vitro Functional Characterization
In functional studies, the most promising compounds 24, 26, and 30 were tested using 86Rb+ ion flux assays in SH-EP1-hα4β2 (expressing human α4β2-nAChRs), SH-EP1-h(α6/3)β2β3 (expressing human (α6/3)β2β3-nAChRs; referred to as α6*-nAChRs), SH-SY5Y (α3β4*-nAChRs), or TE671/RD (α1β1γδ-nAChRs) cells. Abilities of ligands to stimulate ion flux were characterized as EC50 values with efficacies normalized to that of carbamylcholine. Abilities of chronically administered ligands to inhibit ion flux were characterized as inactivation IC50 values following the 10 min preincubation used to characterize receptor stimulation.
These pyrrolidine analogues were found to be highly potent, partial agonists at the mixture of high sensitivity (HS) and low sensitivity (LS) α4β2-nAChRs and to have weak if any agonist activity at α3β4*- or α1β1γδ-nAChRs. Their EC50 values were comparable to that found for parent compound 4, with the exception of compound 30 possessing an EC50 value in the single-digit nanomolar range, and the trend was consistent with results observed in the binding studies (Table 3 and Figure S2). Response magnitude (efficacy) was 29 to 92% relative to a maximally efficacious concentration of the full agonist, carbamylcholine, for action at HS α4β2-nAChRs. At LS α4β2-nAChRs, these ligands had no measurable efficacy as agonists. Their inactivation of nAChR function was most potent for α4β2-nAChRs and weak or absent for actions at α3β4*- or α1β1γδ-nAChRs. IC50 values for compounds 26 and 30 were 4-fold higher or similar to that of compound 4 respectively, while compound 24 was 20-fold less potent. The absence or weakness of agonist or antagonist activity at ganglionic α3β4*- or muscle-type α1β1γδ-nAChRs indicates that the occurrence of peripheral side effects is unlikely. All of the pyrrolidine analogues had agonist activity at α6*-nAChRs with EC50 values in the range of 9.0 to 51.5 nM. Their agonism efficacies were relatively low, ranging from 4.9 to 12% of the maximum response to carbamylcholine. This low-efficacy agonism correlates with inactivation efficacies that are much higher than that seen for nicotine (Table 3). Inactivation IC50 values of these ligands were lower than 24 nM, with the exception of compound 24, having an IC50 value greater than 220 nM.
Table 3.
Potencies and Efficacies of Ligand Agonism and Inactivation of Human α4β2- and α6*-nAChRsa
| Compd. | Agonism | Inactivation | ||||||
|---|---|---|---|---|---|---|---|---|
| α4β2 | α6* | α4β2 | α6* | |||||
| EC50 (nM) | Efficacy at HS (%)b,c | EC50 (nM) | Efficacy (%) | IC50 (nM) | Efficacy (%) b | IC50 (nM) | Efficacy (%) | |
| 24 | 23 | 29 ± 2.5 | 51.5 | 4.9 ± 0.6 | 108 | 84 ± 4.6 | 223 | 79 ± 5.6 |
| 26 | 14.4 | 49 ± 5.2 | 9.0 | 6.5 ± 0.4 | 20 | 88 ± 2.7 | 23.9 | 87 ± 1.5 |
| 30 | 6.2 | 92 ± 1.5 | 23.5 | 12 ± 0.6 | 5.5 | 70 ± 3.3 | 8.3 | 76 ± 0.7 |
| 4 | 17.5 | 60 ± 5.9 | 7.4 | 6.2 ± 0.3 | 5.6 | 71 ± 5.5 | 9.7 | 80 ± 3.5 |
| nicotine | 290 | 125 ± 10 | 127 | 56 ± 0.8 | 430 | 92 ± 2.1 | 84.8 | 26 ± 1.0 |
See Experimental Section.
Efficacies were measured in a mixture of HS and LS α4β2-nAChRs.
Efficacy values for actions at HS α4β2-nAChR were extrapolated from results obtained using sazetidine-A as a full agonist at HS α4β2-nAChR (see Supporting Information for details).
In Vivo Behavioral Pharmacology
To determine whether the excellent potency of the pyrrolidine analogs at α4β2-nAChRs would translate into antidepressant-like efficacy in a behavioral model, compounds 24, 26, and 30 were investigated in the mouse forced swim test,23 an assay in which mice are placed into a beaker of water and the time the mouse spends passively floating in the water (immobility) is recorded. Most traditional antidepressants decrease the amount of time mice spent immobile. Mice were administered nicotinic ligands or the selective serotonin reuptake inhibitor, sertraline, as a positive control (20 mg/kg). Compound 24 showed no significant effects in the forced swim test when administered intraperitoneally at a dose of 1, 3, or 10 mg/kg (Figure S3), likely due to its lower inhibitory potency at both α4β2- and α6*-nAChRs relative to the parent compound 4. On the other hand, the N-methylpyrrolidine analogue 26 demonstrated an antidepressant-like effect when administered intraperitoneally or orally, with a significant reduction in immobility at the minimal dose of 1 mg/kg. It appears that the N-methyl group in compound 26 is crucial to maintaining in vivo activity. Intraperitoneal administration of the most potent compound 30 in vitro, however, produced abnormal behavioral effects, such as sedation and tremor at 5 mg/kg, and resulted in death of the mice during the pretreatment or trial at 10 mg/kg.
Preliminary ADMET Study
Encouraged by the favorable biological data, we submitted compound 26 for preliminary ADMET tests.24 When incubated with human or mouse (CD-1) liver microsomes, at least 90% of compound 26 remained unchanged after 1h incubation at 0.1 μM. Incubation with human hepatocytes resulted in more than 85% of compound 26 remaining unchanged after 2h incubation at 1 μM. In the presence of compound 26 at a concentration of 10 μM, none of the CYP isoforms tested (CYP1A, CYP2C9, CYP2C19, and CYP3A) showed more than 30% inhibition, suggesting minimal adverse drug-drug interactions, with the exception of 44% inhibition of the CYP2D6 isoform. Plasma protein binding (PPB) assays were conducted with both human and mouse (CD-1) plasma at a concentration of 10 μM of compound 26. In human plasma, 31% binding was observed, while in mouse plasma the bound fraction was 11%. In the bidirectional Caco-2 cell permeability assay, compound 26 was found to exhibit high permeability, and had an acceptable efflux ratio of 0.2. In addition, the efflux ratio increased to 0.7 when compound 26 was incubated in the present of verapamil, a P-glycoprotein (P-gp) inhibitor, indicating that the test compound is not a P-gp substrate. The bacteria reversion mutation assay (Ames Test) was used to evaluate the mutagenic potential of compound 26. At all concentrations tested (5, 10, 50, and 100 μM), no positive significance was observed for compound 26 in all tested strains (TA98, TA100, TA1535, and TA1537) in the presence and absence of a liver metabolic activation system (S9 fraction), with the exception of higher concentrations (50 and 100 μM), at which compound 26 exhibited positive significance when tested using strain TA1537 in the presence of the S9 fraction. However, when the Ames genotoxicity testing was repeated using the tartrate salt of compound 26 and the TA1537 tester strain with or without metabolic activation, the compound was found not to be mutagenic at all concentrations tested (up to 3 × 105 μM). Cardiotoxicity associated with the inhibition of human hERG was also evaluated at three test concentrations (0.1, 1, and 10 μM). Compound 26 led to 4%, 7%, and 23% inhibition of tail current, respectively. Moreover, compound 26 was found to be quite chemically stable, as it could be stored at room temperature for several months showing little or no sign of decomposition.
CONCLUSION
In this report, we disclose the synthesis, pharmacological characterization, and structure-activity relationships of a series of cyclopropane-containing nAChR ligands. Modifications to both the azetidine ring present in the lead compound 3 and its right-hand side chain have been explored to improve compound druggability. We successfully identified potent and highly selective α4β2-nAChR partial agonists 24, 26, and 30 that exhibited comparable or improved pharmacological parameters in comparison to compound 3 in [3H]epibatidine binding studies as well as functional assays based on 86Rb+ ion flux measurements. In addition, minor changes such as incorporation of a methyl group (24 vs 26) or alteration the position of the nitrogen atom as in the pyrrolidines 24 vs 30 may significantly affect the in vivo activity of these nicotinic ligands. Compound 26 demonstrated favorable antidepressant-like effects in the mouse forced swim test, while possessing a favorable ADMET profile and chemical stability. Together, these findings support compound 26 as a promising lead candidate that deserves further evaluation in the design of antidepressants possessing a unique mechanism of action.
EXPERIMENTAL SECTION
General
All chemicals were purchased from Sigma-Aldrich or Chem-Impex, and solvents were used as obtained from Fisher Scientific or Sigma-Aldrich without further purification. Anhydrous THF and CH2Cl2 were obtained by distillation over sodium wire or CaH2, respectively. All non-aqueous reactions were run under an argon atmosphere with exclusion of moisture from reagents, and all reaction vessels were oven-dried. The progress of the reactions was monitored by TLC on SiO2. Spots were visualized by their quenching of the fluorescence of an indicator admixed to the SiO2 layer, or by dipping into I2/SiO2 mixture. Products were purified by column chromatography on 230–400 mesh SiO2. Proton and carbon NMR spectra were recorded at spectrometer frequencies of 400 MHz and 100 MHz, respectively. NMR chemical shifts were reported in δ (ppm) using the δ 7.26 signal of CHCl3 (1H NMR), the δ 4.80 signal of HDO (1H NMR), and the δ 77.23 signal of CDCl3 (13C NMR) as internal standards. 13C NMR spectra in D2O were not adjusted. Optical rotation was detected on an Autopol IV automatic polarimeter. Mass spectra were measured in the ESI mode at an ionization potential of 70 eV with an LC-MS MSD (Hewlett Packard). The final compounds were purified by preparative HPLC, which was carried out on an ACE 5 AQ column (150 × 20 mm), with detection at 254 and 280 nm on a Shimadzu SPD-10A VP detector; flow rate = 17.0 mL/min; gradient of 0 to 50% methanol in water (both containing 0.05 vol% of CF3COOH) in 30 min. Purities of final compounds (> 98%) were established by both elemental analysis and by analytical HPLC, which was carried out on an Agilent 1100 HPLC system with a Synergi 4 μm Hydro-RP 80A column, with detection at 254 or 280 nm on a variable wavelength detector G1314A; flow rate = 1.4 mL/min; gradient of 0 to 100% methanol in water (both containing 0.05 vol% of CF3COOH) in 18 min. See Supporting Information for detailed experimental procedures and NMR spectral data (1H and 13C) of all intermediates.
3-[(2(S)-Azetidinyl)methoxy]-5-[(1S,2S)-2-(hydroxymethyl)cyclopropyl]pyridine Trifluoroacetate (7)
1H NMR (D2O): δ 8.36 (s, 1H), 8.28 (s, 1H), 7.91 (s, 1H), 4.98 (m, 1H), 4.53 (d, J = 3.6 Hz, 2H), 4.12 (m, 2H), 3.69 (m, 1H), 3.54 (m, 1H), 2.70 (q, J = 8.4 Hz, 2H), 2.12 (m, 1H), 1.64 (m, 1H), 1.22 (t, J = 7.2 Hz, 2H); 13C NMR (D2O): δ 162.3 (TFA), 155.9, 145.2, 132.2, 128.3, 125.7, 115.8 (TFA), 67.2, 64.1, 58.2, 43.3, 25.4, 19.8, 18.1, 14.1. [α]D 20 = +27.7 (c 0.48, MeOH). Anal. Calcd for C13H18N2O2•2.15CF3COOH•0.85H2O: C, 42.0; H, 4.45; F, 24.77; N, 5.66. Found: C, 41.89; H, 4.18; F, 24.51; N, 5.55. HPLC purity = 99.1%; tR = 6.1 min.
3-[(2(S)-Azetidinyl)methoxy]-5-[(1S,2S)-2-(3-hydroxypropyl)cyclopropyl]pyridine Trifluoroacetate (10)
1H NMR (D2O): δ 8.32 (s, 1H), 8.22 (s, 1H), 7.84 (s, 1H), 4.98 (m, 1H), 4.53 (d, J = 4.0 Hz, 2H), 4.11 (m, 2H), 3.63 (t, J = 6.6 Hz, 2H), 2.70 (q, J = 8.4 Hz, 2H), 1.93 (m, 1H), 1.69 (m, 2H), 1.56-1.44 (m, 2H), 1.31 (m, 1H), 1.14 (m, 2H); 13C NMR (D2O): δ 162.4 (TFA), 155.8, 146.6, 131.9, 127.8, 125.2, 115.8 (TFA), 67.1, 60.9, 58.2, 43.3, 30.4, 29.0, 25.0, 19.8, 19.7, 16.9. [α]D 20 = +10.0 (c 1.1, MeOH). Anal. Calcd for C15H22N2O2•2.55CF3COOH•0.75H2O: C, 42.61; H, 4.63; F, 25.65, N, 4.94. Found: C, 42.70; H, 4.51; F, 25.55; N, 4.97. HPLC purity = 99.2%; tR = 6.1 min.
3-[(1S,2S)-2-[5-[((S)-Azetidin-2-yl)methoxy]pyridin-3-yl]cyclopropyl]propanoic Acid Trifluoroacetate (11)
1H NMR (D2O): δ 8.34 (s, 1H), 8.22 (s, 1H), 7.85 (s, 1H), 4.98 (m, 1H), 4.52 (d, J = 4.0 Hz, 2H), 4.12 (m, 2H), 2.70 (q, J = 8.4 Hz, 2H), 2.53 (m, 2H), 1.99 (m, 1H), 1.84 (m, 1H), 1.68 (1H), 1.33 (m, 1H), 1.17 (m, 2H); 13C NMR (D2O): δ 178.1, 162.4 (TFA), 155.8, 146.1, 132.0, 127.9, 125.4, 115.9 (TFA), 67.1, 58.2, 43.3, 33.1, 28.2, 24.2, 19.8, 19.7, 16.3. [α]D 20 = +32.5 (c, 0.36 MeOH). Anal. Calcd for C15H20N2O3•2.15CF3COOH•0.9H2O: C, 43.11; H, 4.49; F, 22.79, N, 5.21. Found: C, 42.96; H, 4.24; F, 22.53; N, 5.16. HPLC purity = 99.8%; tR = 7.0 min.
3-[(2(S)-Azetidinyl)methoxy]-5-[(1S,2R)-2-(2-trifluoromethoxyethyl)cyclopropyl]pyridine Trifluoroacetate (14)
1H NMR (D2O): δ 8.33 (s, 1H), 8.22 (s, 1H), 7.85 (s, 1H), 4.96 (m, 1H), 4.51 (d, J = 3.6 Hz, 2H), 4.18-4.03 (m, 4H), 2.68 (q, J = 8.4 Hz, 2H), 2.02 (m, 1H), 1.92 (m, 1H), 1.73 (m, 1H), 1.37 (m, 1H), 1.19 (m, 2H); 13C NMR (D2O): δ 162.5 (TFA), 156.2, 146.3, 132.4, 128.4, 125.9, 121.5 (q, JC-F = 251.1 Hz), 116.2 (TFA), 67.7, 67.6, 58.6, 43.6, 32.1, 21.8, 20.2, 19.7, 16.1; 19F NMR (D2O): δ −60.2, −75.6. [α]D 20 = +28.6 (c 0.14, MeOH); Anal. Calcd for C15H19F3N2O2•2.4CF3COOH•0.5H2O: C, 39.7; H, 3.77; F, 32.35; N, 4.68. Found: C, 39.47; H, 3.52; F, 32.14; N, 4.49. HPLC purity = 99.8%; tR = 9.7 min.
3-[(2(S)-Azetidinyl)methoxy]-5-[(1S,2R)-2-(2-(piperidin-1-yl)ethyl)cyclopropyl]pyridine Trifluoroacetate (17a)
1H NMR (D2O): δ 8.37 (s, 1H), 8.25 (s, 1H), 7.87 (s, 1H), 4.99 (m, 1H), 4.54 (s, 2H), 4.18-4.08 (m, 2H), 3.52 (d, J = 6.0 Hz, 2H), 3.23 (t, J = 8.0 Hz, 2H), 2.93 (t, J = 8.0 Hz, 2H), 2.67 (dd, J = 12.0, 4.0 Hz, 2H), 2.01 (m, 1H), 1.97–1.70 (m, 7H), 1.49-1.46 (m, 1H), 1.35-1.33 (m, 1H), 1.24-1.19 (m, 2H); 13C NMR (D2O): δ 162.7 (TFA), 156.2, 145.6, 132.4, 128.5, 126.0, 116.3 (TFA), 67.5, 58.6, 53.1, 27.5, 22.7, 21.5, 21.0, 20.2, 19.8, 16.3. [α]D 20 = +43.6 (c = 3.4, MeOH). Anal Calcd for C19H29N3O•3.1CF3COOH•0.45H2O: C, 44.71; H, 4.91; F, 26.1; N, 6.21. Found: C, 44.83; H, 4.77; F, 25.97; N, 6.14. HPLC purity = 99.8%; tR = 4.1 min.
3-[(2(S)-Azetidinyl)methoxy]-5-[(1S,2R)-2-(2-(morpholin-4-yl)ethyl)cyclopropyl]pyridine Trifluoroacetate (17b)
1H NMR (D2O) δ 8.33 (s, 1H), 8.21 (s, 1H), 7.82 (s, 1H), 4.96–4.94 (m, 1H), 4.50 (d, J = 4.0 Hz, 2H), 4.14–4.05 (m, 4H), 3.78 (t, J = 12.4 Hz, 2H), 3.50 (d, J = 12.8 Hz, 2H), 3.29 (t, J = 8.0 Hz, 2H), 3.16 (dt, J = 12.4, 3.2 Hz, 2H), 2.67 (q, J = 8.8 Hz, 2H), 2.05–2.03 (m, 1H), 1.91–1.85 (m, 2H), 1.30 (m, 1H), 1.21–1.15 (m, 2H); 13C NMR (D2O): δ 162.2 (TFA), 155.8, 145.1, 132.0, 128.1, 125.7, 115.9 (TFA), 67.1, 63.3, 58.2, 55.9, 51.2, 43.2, 26.7, 20.8, 19.8, 19.4, 15.9. [α]D 20 = +43.5 (c = 0.8, MeOH). Anal Calcd for C18H27N3O2•3.4CF3COOH•0.4H2O: C, 41.82; H, 4.41; F, 27.2; N, 5.9. Found: C, 41.91; H, 4.31; F, 27.09; N, 5.96. HPLC purity = 99.6%; tR = 3.9 min.
3-[(2(S)-Azetidinyl)methoxy]-5-[(1S,2R)-2-(2-(piperazin-1-yl)ethyl)cyclopropyl]pyridine Trifluoroacetate (17c)
1H NMR (D2O): δ 8.37 (s, 1H), 8.25 (s, 1H), 7.85 (s, 1H), 4.98 (m, 1H), 4.53 (d, J = 4.0 Hz, 2H), 4.17-4.05 (m, 2H), 3.65 (br s, 8H), 3.44 (t, J = 8.4 Hz, 2H), 2.71 (q, J = 8.4 Hz, 2H), 2.09 (m, 1H), 2.02-1.87 (m, 2H), 1.36 (m, 1H), 1.26-1.18 (m, 2H); 13C NMR (D2O): 162.3 (TFA), 155.9, 145.0, 132.1, 128.2, 125.7, 115.9 (TFA), 67.2, 58.2, 55.9, 48.0, 43.3, 40.2, 26.9, 20.6, 19.8, 19.4, 15.9. [α]D 20 = +36.4 (c 1.52, MeOH). Anal Calcd for C18H28N4O•4.05CF3COOH•0.45H2O: C, 39.86; H, 4.22; F, 29.36; N, 7.12. Found: C, 39.97; H, 4.23; F, 29.26; N, 7.03. HPLC purity = 99.7%; tR = 4.8 min.
Ethyl [2-[(1R,2S)-2-[5-[((S)-Azetidin-2-yl)methoxy]pyridin-3-yl]cyclopropyl]ethyl]carbamate Trifluoroacetate (19a)
1H NMR (D2O): δ 8.32 (s, 1H), 8.21 (s, 1H), 7.82 (s, 1H), 4.97 (m, 1H), 4.51 (d, J = 4.4 Hz, 2H), 4.12 (m, 2H), 3.94 (m, 2H), 3.23 (m, 2H), 2.68 (q, J = 8.4 Hz, 2H), 1.94 (m, 1H), 1.70 (m, 1H), 1.50 (m, 1H), 1.27 (m, 1H), 1.15-1.10 (m, 5H); 13C NMR (D2O): δ 162.6 (TFA), 158.7, 156.2, 146.7, 132.2, 128.2, 125.6, 116.3 (TFA), 67.5, 61.5, 58.6, 43.6, 40.0, 33.1, 23.2, 20.2, 20.0, 16.5, 13.7. [α]D 20 = +35.0 (c 0.20, MeOH). Anal. Calcd for C17H25N3O3•2.6CF3COOH•0.55H2O: C, 42.61; H, 4.62; F, 23.68, N, 6.71; Found: C, 42.75; H, 4.58; F, 23.55; N, 6.63. HPLC purity = 99.8%; tR = 6.8 min.
Isopropyl [2-[(1R,2S)-2-[5-[((S)-Azetidin-2-yl)methoxy]pyridin-3-yl]cyclopropyl]ethyl]carbamate Trifluoroacetate (19b)
1H NMR (D2O): δ 8.32 (s, 1H), 8.20 (s, 1H), 7.81 (s, 1H), 4.96 (m, 1H), 4.65 (m, 1H), 4.50 (d, J = 3.2 Hz, 2H), 4.10 (m, 2H), 3.22 (m, 2H), 2.67 (q, J = 8.4 Hz, 2H), 1.93 (m, 1H), 1.69 (m, 1H), 1.50 (m, 1H), 1.26 (m, 1H), 1.12 (m, 8H); 13C NMR (D2O): δ 162.5 (TFA), 158.3, 156.2, 146.7, 132.2, 128.2, 125.6, 116.2 (TFA), 69.3, 67.5, 58.6, 43.6, 39.9, 33.1, 23.1, 21.1, 20.2, 20.0, 16.6. [α]D 20 = +40.0 (c 0.24, MeOH). Anal. Calcd for C18H27N3O3•2.35CF3COOH•0.2H2O: C, 45.07; H, 4.96; F, 22.14, N, 6.95; Found: C, 44.99; H, 4.87; F, 22.07; N, 6.95. HPLC purity = 99.7%; tR = 7.3 min.
3-[(2(S)-Azetidinyl)methoxy]-5-[(1R,2S)-2-(2-(morpholin-4-yl)ethyl)cyclopropyl]pyridine Trifluoroacetate (21)
1H NMR (D2O) δ 8.37 (s, 1H), 8.26 (s, 1H), 7.87 (s, 1H), 5.00–4.98 (m, 1H), 4.54 (d, 2H, J = 4.0 Hz), 4.17–4.08 (m, 4H), 3.82 (t, 2H, J = 12.4 Hz), 3.54 (d, 2H, J = 12.8 Hz), 3.34 (t, 2H, J = 8.0 Hz), 3.20 (dt, 2H, J = 12.4, 3.2 Hz), 2.71 (q, 2H, J = 8.8 Hz), 2.10–2.08 (m, 1H), 1.97–1.88 (m, 2H), 1.35 (m, 1H), 1.25–1.98 (m, 2H). 13C NMR (D2O): δ 162.2 (TFA), 155.9, 145.1, 132.1, 128.1, 125.7, 115.9 (TFA), 67.1, 63.3, 58.2, 55.9, 51.2, 43.2, 26.7, 20.8, 19.8, 19.4, 15.9. [α]D 20 = −46.4 (c 0.78, MeOH). Anal. Calcd for C18H27N3O2•3.4CF3COOH•0.05H2O: C, 42.19; H, 4.35; F, 27.45; N, 5.95. Found: C, 42.22; H, 4.38; F, 27.38; N, 5.97. HPLC purity = 99.9%; tR = 3.9 min.
3-[2-(Isopropylamino)ethoxy]-5-[(1S,2R)-2-(2-methoxyethyl)cyclopropyl]pyridine Trifluoroacetate (23)
1H NMR (D2O): δ 8.14 (s, 1H), 8.07 (s, 1H), 7.66 (s, 1H), 4.35 (t, J = 4.0 Hz, 2H), 4.46-4.36 (m, 5H), 3.20 (s, 3H), 1.83 (s, 1H), 1.57 (s, 2H), 1.23 (d, J = 6.4 Hz, 6H), 1.16 (m, 1H), 1.02 (m, 2H); 13C NMR (D2O): δ 162.4 (TFA), 156.1, 146.5, 132.2, 128.2, 125.6, 116.2 (TFA), 71.8, 64.9, 57.7, 51.1, 43.3, 32.6, 22.3, 19.8, 18.0, 16.6. [α]D 20 = +33.4 (c = 0.9, MeOH). Anal. Calcd for C16H26N2O2•2.15CF3COOH•0.8H2O: C, 45.32; H, 5.57; F, 22.78; N, 5.21. Found: C, 45.06; H, 5.24; F, 22.63; N, 5.09. HPLC purity = 99.8%; tR = 9.3 min.
3-[(2(S)-Pyrrolidinyl)methoxy]-5-[(1S,2R)-2-(2-methoxyethyl)cyclopropyl]pyridine Trifluoroacetate (24)
1H NMR (D2O): δ 8.26 (s, 1H), 8.19 (s, 1H), 7.77 (s, 1H), 4.53 (dd, J = 10.4, 2.4 Hz, 1H), 4.33 (m, 1H), 4.11 (m, 1H), 3.56 (t, J = 6.4 Hz, 2H), 3.39 (t, J = 7.2 Hz, 2H), 3.32 (s, 3H), 2.27 (m, 1H), 2.12-2.06 (m, 2H), 1.94 (m, 2H), 1.68 (m, 2H), 1.28 (m, 1H), 1.12 (m, 2H); 13C NMR (D2O): δ 162.6 (TFA), 156.1, 146.5, 132.3, 128.2, 125.6, 116.2 (TFA), 71.8, 67.6, 58.3, 57.7, 45.9, 32.6, 25.7, 23.4, 22.3, 19.8, 16.6. [α]D 20 = +47.3 (c 0.28, MeOH). Anal. Calcd for C16H24N2O2•2.05CF3COOH•0.05H2O: C, 47.24; H, 5.16; F, 22.86; N, 5.48. Found: C, 47.30; H, 5.24; F, 22.75, N, 5.46. HPLC purity = 99.5%; tR = 7.6 min.
3-[(2(S)-Piperidinyl)methoxy]-5-[(1S,2R)-2-(2-methoxyethyl)cyclopropyl]pyridine Trifluoroacetate (25)
1H NMR (D2O): δ 8.22 (s, 1H), 8.16 (s, 1H), 7.74 (s, 1H), 4.39 (m, 1H), 4.24 (m, 1H), 3.61 (m, 1H), 3.54 (t, J = 6.4 Hz, 2H), 3.43 (m, 1H), 3.30 (s, 3H), 3.03 (m, 1H), 1.97-1.88 (m, 4H), 1.68-1.58 (m, 5H), 1.25 (m, 1H), 1.10 (m, 2H); 13C NMR (D2O): δ 162.2 (TFA), 155.7, 146.1, 131.9, 127.8, 125.2, 115.7 (TFA), 71.4, 68.7, 57.3, 54.9, 44.2, 32.2, 23.9, 21.9, 21.2, 20.6, 19.4, 16.2. [α]D 20 = +49.3 (c 0.53, MeOH). Anal. Calcd for C17H26N2O2•2.6CF3COOH•1.05H2O: C, 44.02; H, 5.11; F, 24.46, N, 4.62. Found: C, 44.04; H, 4.89; F, 24.27; N, 4.78. HPLC purity = 98.7%; tR = 7.9 min.
3-[(1-Methyl-2(S)-pyrrolidinyl)methoxy]-5-[(1S,2R)-2-(2-methoxyethyl)cyclopropyl]pyridine Trifluoroacetate (26)
1H NMR (D2O): δ 8.30 (s, 1H), 8.23 (s, 1H), 7.81 (s, 1H), 4.61 (dd, J = 10.8, 2.8 Hz, 1H), 4.45 (m, 1H), 3.95 (m, 1H), 3.76 (m, 1H), 3.59 (t, J = 6.4 Hz, 2H), 3.34 (s, 3H), 3.25 (m, 1H), 3.04 (s, 3H), 2.40 (m, 1H), 2.18 (m, 1H), 2.09 (m, 2H), 1.98 (m, 1H), 1.71 (m, 2H), 1.31 (m, 1H), 1.15 (m, 2H); 13C NMR (D2O): δ 162.0 (TFA), 155.6, 146.2, 132.0, 127.8, 125.2, 115.7 (TFA), 71.4, 66.9, 66.0, 57.3, 56.8, 40.2, 32.2, 25.4, 21.9, 21.7, 19.4, 16.2. [α]D 20 = +37.0 (c 0.27, MeOH). Anal. Calcd for C17H26N2O2•2.4CF3COOH•0.4H2O: C, 45.83; H, 5.15; F, 23.94; N, 4.9. Found: C, 45.64; H, 4.91; F, 23.8, N, 4.9. HPLC purity = 98.9%; tR = 7.8 min.
3-[(1-Ethyl-2(S)-pyrrolidinyl)methoxy]-5-[(1S,2R)-2-(2-methoxyethyl)cyclopropyl]pyridine Trifluoroacetate (27)
1H NMR (D2O): δ 8.30 (s, 1H), 8.23 (s, 1H), 7.80 (s, 1H), 4.57 (m, 1H), 4.44 (m, 1H), 4.05 (m, 1H), 3.73 (m, 1H), 3.61-3.54 (m, 3H), 3.35 (s, 3H), 3.24 (2H), 2.34 (m, 1H), 2.09-1.97 (m, 4H), 1.71 (m, 2H), 1.38-1.32 (m, 4H), 1.15 (m, 2H); 13C NMR (D2O): δ 161.9 (TFA), 155.6, 146.2, 132.1, 127.8, 125.2, 115.9 (TFA), 71.5, 66.5, 65.5, 57.3, 53.8, 50.1, 32.2, 25.5, 22.0, 21.9, 19.4, 16.2, 9.6. [α]D 20 = +39.8 (c 0.61, MeOH). Anal. Calcd for C18H28N2O2•2.1CF3COOH•0.85H2O: C, 47.68; H, 5.73; F, 21.4, N, 5.01. Found: C, 47.4; H, 5.42; F, 21.32; N, 4.91. HPLC purity = 99.3%; tR = 8.0 min.
3-[((1S,2S,5R)-3-Azabicyclo[3.1.0]hexan-2-yl)methoxy]-5-[(1S,2R)-2-(2- methoxyethyl)cyclopropyl]pyridine Trifluoroacetate (28)
1H NMR (D2O): δ 8.25 (s, 1H), 8.17 (s, 1H), 7.76 (s, 1H), 4.67 (d, J = 14.0 Hz, 1H), 4.35-4.27 (m, 2H), 3.55-3.45 (m, 4H), 3.30 (s, 3H), 1.94-1.88 (m, 3H), 1.66 (m, 2H), 1.26 (m, 1H), 1.10 (m, 2H), 0.84 (m, 1H), 0.59 (m, 1H); 13C NMR (D2O): δ 162.4 (TFA), 156.2, 146.5, 132.2, 128.2, 125.6, 117.6 (TFA), 71.8, 67.7, 59.3, 57.7, 47.6, 32.6, 22.3, 19.8, 16.6, 15.9, 14.1, 3.9. [α]D 20 = +34.3 (c = 2.4, MeOH). Anal. Calcd for C17H24N2O2•2.15CF3COOH•0.85H2O: C, 46.61; H, 5.11; F, 22.33; N, 5.1. Found: C, 46.62; H, 4.92; F, 22.14; N, 5.13. HPLC purity = 99.5%; tR = 12.7 min.
3-[(1-Methyl-2(R)-pyrrolidinyl)methoxy]-5-[(1S,2R)-2-(2-methoxyethyl)cyclopropyl]pyridine Trifluoroacetate (29)
1H NMR (D2O): δ 8.29 (s, 1H), 8.21 (s, 1H), 7.79 (s, 1H), 4.60 (dd, J = 10.8, 2.8 Hz, 1H), 4.43 (m, 1H), 3.94 (m, 1H), 3.74 (m, 1H), 3.57 (t, J = 6.4 Hz, 2H), 3.32 (s, 3H), 3.24 (m, 1H), 3.02 (s, 3H), 2.39 (m, 1H), 2.19 (m, 1H), 2.07 (m, 2H), 1.96 (m, 1H), 1.69 (m, 2H), 1.29 (m, 1H), 1.13 (m, 2H); 13C NMR (D2O): δ 162.2 (TFA), 155.7, 146.2, 132.1, 127.9, 125.2, 115.8 (TFA), 71.4, 66.9, 66.0, 57.3, 56.8, 40.1, 32.2, 25.4, 22.0, 21.7, 19.4, 16.2. [α]D 20 = +39.5 (c 0.2, MeOH). Anal. Calcd for C17H26N2O2•2.1CF3COOH•0.75H2O: C, 46.86; H, 5.49; F, 22.03; N, 5.16. Found: C, 46.96; H, 5.14; F, 21.7, N, 5.12. HPLC purity = 99.2%; tR = 7.4 min.
3-[(3(S)-Pyrrolidinyl)methoxy]-5-[(1S,2R)-2-(2-methoxyethyl)cyclopropyl]pyridine Trifluoroacetate (30)
1H NMR (D2O): δ 8.23 (s, 1H), 8.16 (s, 1H), 7.75 (s, 1H), 4.28 (m, 1H), 4.20 (m, 1H), 3.58 (m, 3H), 3.46 (m, 1H), 3.38-3.23 (m, 5H), 2.96 (m, 1H), 2.30 (m, 1H), 1.96 (m, 2H), 1.69 (m, 2H), 1.27 (m, 1H), 1.13 (m, 2H); 13C NMR (D2O): δ 162.2 (TFA), 156.5, 145.9, 131.4, 127.8, 125.1, 115.8 (TFA), 71.5, 69.6, 57.3, 46.9, 45.1, 36.2, 32.2, 26.0, 21.8, 19.4, 16.1. [α]D 20 = +42.0 (c 0.70, MeOH). Anal. Calcd for C16H24N2O2•2.0CF3COOH•1.05H2O: C, 45.9; H, 5.41; F, 21.78, N, 5.35. Found: C, 45.52; H, 5.0; F, 21.5; N, 5.31. HPLC purity = 99.4%; tR = 7.3 min.
3-[(3(R)-Pyrrolidinyl)oxy]-5-[(1S,2R)-2-(2-methoxyethyl)cyclopropyl]pyridine Trifluoroacetate (31)
1H NMR (D2O): δ 8.23 (s, 1H), 8.15 (s, 1H), 7.75 (s, 1H), 5.37 (s, 1H), 3.68-3.46 (m, 6H), 3.29 (s, 3H), 2.34 (m, 2H), 1.91 (m, 1H), 1.66 (m, 2H), 1.24 (m, 1H), 1.09 (m, 2H); 13C NMR (D2O): δ 162.1 (TFA), 154.5, 146.2, 131.8, 128.7, 126.0, 115.8 (TFA), 77.2, 71.4, 57.3, 50.0, 43.4, 32.2, 29.3, 21.9, 19.4, 16.2. [α]D 20 = +34.0 (c 0.1, MeOH). Anal. Calcd for C15H22N2O2•2.15CF3COOH•0.1H2O: C, 45.52; H, 4.82; F, 24.06; N, 5.5. Found: C, 45.38; H, 4.72; F, 24.08, N, 5.69. HPLC purity = 99.8%; tR = 6.4 min.
3-[(1-Methyl-2(S)-pyrrolidinyl)methoxy]-5-[(1R,2S)-2-(2-methoxyethyl)cyclopropyl]pyridine Trifluoroacetate (33)
1H NMR (D2O): δ 8.30 (s, 1H), 8.22 (s, 1H), 7.80 (s, 1H), 4.60 (dd, J = 10.8, 2.8 Hz, 1H), 4.44 (m, 1H), 3.95 (m, 1H), 3.74 (m, 1H), 3.57 (t, J = 6.4 Hz, 2H), 3.33 (s, 3H), 3.25 (m, 1H), 3.03 (s, 3H), 2.40 (m, 1H), 2.20 (m, 1H), 2.08 (m, 2H), 1.97 (m, 1H), 1.70 (m, 2H), 1.30 (m, 1H), 1.14 (m, 2H); 13C NMR (D2O): δ 162.2 (TFA), 155.7, 146.2, 132.1, 127.9, 125.2, 115.9 (TFA), 71.5, 66.9, 66.0, 57.3, 56.8, 40.2, 32.2, 25.4, 22.0, 21.7, 19.4, 16.2. [α]D 20 = −33.8 (c 0.13, MeOH). Anal. Calcd for C17H26N2O2•2.05CF3COOH•0.45H2O: C, 47.61; H, 5.48; F, 21.95; N, 5.26. Found: C, 47.66; H, 5.24; F, 21.73, N, 5.19. HPLC purity = 99.8%; tR = 5.8 min.
General Procedures for Binding and Functional Studies
In vitro Binding Studies
[3H]Epibatidine competition studies and broad-range screening were carried out by the National Institute of Mental Health’s Psychoactive Drug Screening Program, Contract # HHSN-271-2008-00025-C (NIMH PDSP). For experimental details please refer to the PDSP web site http://pdsp.med.unc.edu/.
Cell Lines and Culture
Cell lines naturally or heterologously expressing specific, functional, human nAChR subtypes were used. The human clonal cell line TE671/RD naturally expresses human muscle-type α1*-nAChRs, containing α1, β1, γ, and δ subunits, with function detectable using 86Rb+ efflux assays.25 The human neuroblastoma cell line SH-SY5Y naturally expresses autonomic α3β4*-nAChRs, containing α3, β4, probably α5, and sometimes β2 subunits, and also displays function detectable using 86Rb+ efflux assays.26 SH-SY5Y cells also express homopentameric α7-nAChRs. However, their function is not detected in the 86Rb+ efflux assay under the conditions used. SH-EP1 human epithelial cells stably transfected with human α4 and β2 subunits (SH-EP1-hα4β2 cells) have been established and the human α4β2-nAChRs that they stably and heterologously express have been characterized with both ion flux and radioligand binding assays.27 Details of experimental procedures used for 86Rb+ efflux assays are as previously described.15
Also used was the recently established SH-EP1-h(α6/3)β2β3 cell line.28 The designation (α6/3) refers to a chimeric subunit that has amino acids from the extracellular, N-terminal, ligand-binding domain of the human nAChR α6 subunit fused to what otherwise is the nAChR α3 subunit. When expressed with the appropriate nAChR subunit assembly partners, in this case, human β2 and β3 subunits, functional nAChR are formed that have the pharmacological features of receptors containing wild-type α6 subunits, such as sensitivity to the α6β2β3-nAChR-selective functional antagonist, α-conotoxin MI.29, 30 However, levels of functional expression are much higher, enabling assessment using assays such as 86Rb+ efflux, probably because transmembrane and cytoplasmic domains in the nAChR α3 subunit are more permissive for assembly and/or function of receptors than the corresponding domains in nAChR α6 subunits. Briefly,28 wild-type SH-EP1 cells were transfected using a cationic polymer (Qiagen, Valencia, CA) and nAChR (α6/3), β2 and β3 subunit gene cDNAs optimized for vertebrate expression (GeneArt, Inc., Burlingame, CA) and delivered in different variants on the pcDNA3.1 vector (Invitrogen, Carlsbad, CA). Following selection of triple transfectants based on triple antibiotic resistance, clonal lines were isolated and screened for nAChR radioligand binding capacity. Promising clones were verified as consistently expressing a high level of functional (α6/3)β2β3-nAChR based on 86Rb+ assays.
TE671/RD, SH-SY5Y, and transfected SH-EP1 cell lines were maintained as low passage number (1–26 from our frozen stocks) cultures, under continuing positive selection as appropriate, to ensure stable expression of native or heterologously expressed nAChRs as previously described.25 Cells were passaged once a week by splitting just-confluent cultures 1/300 (TE671/RD), 1/10 (SH-SY5Y), or 1/20-1/40 (transfected SH-EP1) in serum-supplemented medium to maintain log-phase growth.
General Procedures for Behavioral Studies
Animals
BALB/cJ male mice (8–10 weeks old at testing) were obtained from Jackson Laboratory (Bar Harbor, ME, USA). Mice were housed four to a cage in a colony room maintained at 22 ± 2 °C on a 12 h light–dark cycle. All animal experiments were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals and the PsychoGenics Animal Care and Use Committee.
Drugs
Compounds 24, 26, and 30 were synthesized as described above, and sertraline was purchased from Toronto Research Chemicals (Ontario, Canada). All compounds were dissolved in water and administered by intraperitoneal (IP) injection or administrated orally (PO) in a volume of 10 ml/kg.
Mouse Forced Swim Test
Procedures were based on those previously described. Mice were individually placed into clear glass cylinders (15 cm tall × 10 cm wide, 1 L beakers) containing 23 ± 1 °C water 12 cm deep (approximately 800 mL). Mice were administered vehicle, the SSRI sertraline (20 mg/kg) as a positive control, or test compound 24, 26, or 30. Thirty minutes following IP or PO administration (as indicated in Figure 2), mice were placed in the water, and the time the animal spent immobile was recorded over a 6 min trial. Immobility was defined as the postural position of floating in the water.
Figure 2.
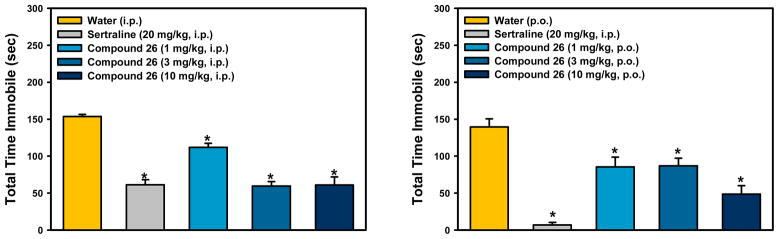
Mouse forced swim data for compound 26. The selective serotonin reuptake inhibitor, sertraline, produced the expected decrease in immobility. ANOVAs: F (4,43) = 34.20, p < 0.001 (left); F (4,44) = 20.75, p < 0.001 (right). *Fisher’s PLSD post-hoc test: ps < 0.05 vs vehicle. n = 9–10/group.
Statistical Analysis
Data were analyzed with Analysis of Variance (ANOVA) with treatment group (vehicle, sertraline, or tested compounds) as the between-group variable and total time immobile (seconds over the 6 min trial) as the dependent variable. Significant main effects were followed up with the post hoc Fisher’s LSD test.
Supplementary Material
Table 4.
ADMET Properties of Compound 26
| Assay | Compound 26 |
|---|---|
|
| |
| liver microsomes (human or mouse, 0.1 μM) | 103.3% or 90.1% |
| cryopreserved hepatocytes (human, 1 μM) | 85.7% |
|
| |
| CYP inhibition (10 μM) | |
| CYP1A | 17% |
| CYP2C9 | 30% |
| CYP2C19 | 21% |
| CYP2D6 | 44% |
| CYP3A | 10% |
|
| |
| hERG inbihition (0.1, 1, 10 μM) | 4%, 7%, 23% |
|
| |
| plasma protein binding (human or mouse, 10 μM) | 31% or 11% |
|
| |
| A-B permeability (Caco-2, pH 7.4/7.4) | 90.9 × 10−6 cm/s |
| A-B permeability (Caco-2, pH 7.4/7.4 + verapamil) | 79.2 × 10−6 cm/s |
| B-A permeability (Caco-2, pH 7.4/7.4) | 18.4 × 10−6 cm/s |
| B-A permeability (Caco-2, pH 7.4/7.4 + verapamil) | 55.2 × 10−6 cm/s |
|
| |
| Ames test (strain TA98, TA100, TA1535, TA1537, with/without S9) | Negative |
Acknowledgments
This research was supported by award No. U19MH085193 from the National Institute of Mental Health. The Phoenix research component was also supported in part by the Barrow Neurological Foundation, and work by this team was conducted in part in the Charlotte and Harold Simensky Neurochemistry of Alzheimer’s Disease Laboratory. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Mental Health or the National Institutes of Health. We thank the PDSP program for performing binding affinity assays.
ABBREVIATIONS USED
- nAChR(s)
nicotinic acetylcholine receptor(s)
- CNS
central nervous system
- ADMET
absorption, distribution, metabolism, excretion, and toxicity
- LC-MS
liquid chromatography–mass spectrometry
- NMR
nuclear magnetic resonance
- SAR
structure-activity relationship
- HS
high sensitivity
- LS
low sensitivity
- P-gp
P-glycoprotein
- hERG
human ether-a-go-go-related gene
- TFA
trifluoroacetic acid
Footnotes
SUPPORTING INFORMATION. Experimental details for synthesis of all compounds shown and detailed information for in vitro functional studies. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Hurst R, Rollema H, Bertrand D. Nicotinic acetylcholine receptors: From basic science to therapeutics. Pharmacol Ther. 2013;137:22–54. doi: 10.1016/j.pharmthera.2012.08.012. [DOI] [PubMed] [Google Scholar]
- 2.Gotti C, Clementi F, Fornari A, Gaimarri A, Guiducci S, Manfredi I, Moretti M, Pedrazzi P, Pucci L, Zoli M. Structural and functional diversity of native brain neuronal nicotinic receptors. Biochem Pharmacol. 2009;78:703–711. doi: 10.1016/j.bcp.2009.05.024. [DOI] [PubMed] [Google Scholar]
- 3.Philip NS, Carpenter LL, Tyrka AR, Price LH. Nicotinic acetylcholine receptors and depression: a review of the preclinical and clinical literature. Psychopharmacology (Berl) 2010;212:1–12. doi: 10.1007/s00213-010-1932-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rabenstein RL, Caldarone BJ, Picciotto MR. The nicotinic antagonist mecamylamine has antidepressant-like effects in wild-type but not beta2- or alpha7-nicotinic acetylcholine receptor subunit knockout mice. Psychopharmacology (Berl) 2006;189:395–401. doi: 10.1007/s00213-006-0568-z. [DOI] [PubMed] [Google Scholar]
- 5.Caldarone BJ, Harrist A, Cleary MA, Beech RD, King SL, Picciotto MR. High-affinity nicotinic acetylcholine receptors are required for antidepressant effects of amitriptyline on behavior and hippocampal cell proliferation. Biol Psychiatry. 2004;56:657–664. doi: 10.1016/j.biopsych.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 6.Philip NS, Carpenter LL, Tyrka AR, Whiteley LB, Price LH. Varenicline augmentation in depressed smokers: an 8-week, open-label study. J Clin Psychiatry. 2009;70:1026–1031. doi: 10.4088/jcp.08m04441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trivedi MH, Rush AJ, Wisniewski SR, Nierenberg AA, Warden D, Ritz L, Norquist G, Howland RH, Lebowitz B, McGrath PJ, Shores-Wilson K, Biggs MM, Balasubramani GK, Fava M. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry. 2006;163:28–40. doi: 10.1176/appi.ajp.163.1.28. [DOI] [PubMed] [Google Scholar]
- 8.Letchworth SR, Whiteaker P. Progress and challenges in the study of alpha6-containing nicotinic acetylcholine receptors. Biochem Pharmacol. 2011;82:862–872. doi: 10.1016/j.bcp.2011.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Quik M, Perez XA, Grady SR. Role of alpha6 nicotinic receptors in CNS dopaminergic function: relevance to addiction and neurological disorders. Biochem Pharmacol. 2011;82:873–882. doi: 10.1016/j.bcp.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brandi A, Cicchi S, Cordero FM. Novel syntheses of azetidines and azetidinones. Chem Rev. 2008;108:3988–4035. doi: 10.1021/cr800325e. [DOI] [PubMed] [Google Scholar]
- 11.von Nussbaum F, Brands M, Hinzen B, Weigand S, Habich D. Antibacterial natural products in medicinal chemistry-exodus or revival? Angew Chem Int Ed. 2006;45:5072–5129. doi: 10.1002/anie.200600350. [DOI] [PubMed] [Google Scholar]
- 12.Wellington K, Scott LJ. Azelnidipine. Drugs. 2003;63:2613–2621. doi: 10.2165/00003495-200363230-00004. [DOI] [PubMed] [Google Scholar]
- 13.Abreo MA, Lin NH, Garvey DS, Gunn DE, Hettinger AM, Wasicak JT, Pavlik PA, Martin YC, Donnelly-roberts DL, Anderson DJ, Sullivan JP, Williams M, Arneric SP, Holladay MW. Novel 3-Pyridyl ethers with subnanomolar affinity for central neuronal nicotinic acetylcholine receptors. J Med Chem. 1996;39:817–825. doi: 10.1021/jm9506884. [DOI] [PubMed] [Google Scholar]
- 14.Meyer MD. Neuronal nicotinic acetylcholine receptors as a target for the treatment of neuropathic pain. Drug Dev Res. 2006;67:355–359. [Google Scholar]
- 15.Zhang H, Tuckmantel W, Eaton JB, Yuen PW, Yu LF, Bajjuri KM, Fedolak A, Wang D, Ghavami A, Caldarone B, Paterson NE, Lowe DA, Brunner D, Lukas RJ, Kozikowski AP. Chemistry and behavioral studies identify chiral cyclopropanes as selective alpha4beta2-nicotinic acetylcholine receptor partial agonists exhibiting an antidepressant profile. J Med Chem. 2012;55:717–724. doi: 10.1021/jm201157c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sullivan JP, Bannon AW. Epibatidine: Pharmacological Properties of a Novel Nicotinic Acetylcholine Receptor Agonist and Analgesic Agent. CNS Drug Rev. 1996;2:21–39. [Google Scholar]
- 17.Lynch JK, Holladay MW, Ryther KB, Bai H, Hsiao CN, Morton HE, Dickman DA, Arnold W, King SA. Efficient asymmetric synthesis of ABT-594; a potent, orally effective analgesic. Tetrahedron: Asymmetry. 1998;9:2791–2794. [Google Scholar]
- 18.Liu J, Yu LF, Eaton JB, Caldarone B, Cavino K, Ruiz C, Terry M, Fedolak A, Wang D, Ghavami A, Lowe DA, Brunner D, Lukas RJ, Kozikowski AP. Discovery of isoxazole analogues of sazetidine-A as selective alpha4beta2-nicotinic acetylcholine receptor partial agonists for the treatment of depression. J Med Chem. 2011;54:7280–7288. doi: 10.1021/jm200855b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blazejewski JC, Anselmi E, Wakselman C. 2-trifluoromethoxyethyl triflate: a versatile trifluoromethoxyethyl carrier. J Org Chem. 2001;66:1061–1063. doi: 10.1021/jo005701t. [DOI] [PubMed] [Google Scholar]
- 20.Walsh JS, Miwa GT. Bioactivation of drugs: risk and drug design. Annu Rev Pharmacol Toxicol. 2011;51:145–167. doi: 10.1146/annurev-pharmtox-010510-100514. [DOI] [PubMed] [Google Scholar]
- 21.Zhang HK, Eaton JB, Yu LF, Nys M, Mazzolari A, van Elk R, Smit AB, Alexandrov V, Hanania T, Sabath E, Fedolak A, Brunner D, Lukas RJ, Vistoli G, Ulens C, Kozikowski AP. Insights into the structural determinants required for high-affinity binding of chiral cyclopropane-containing ligands to alpha4beta2-nicotinic acetylcholine receptors: an integrated approach to behaviorally active nicotinic ligands. J Med Chem. 2012;55:8028–8037. doi: 10.1021/jm3008739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Y, Richardson J, Tran T, Al-Muhtasib N, Xie T, Yenugonda VM, Sexton HG, Rezvani AH, Levin ED, Sahibzada N, Kellar KJ, Brown ML, Xiao Y, Paige M. Chemistry and Pharmacological Studies of 3-Alkoxy-2,5-Disubstituted-Pyridinyl Compounds as Novel Selective alpha4beta2 Nicotinic Acetylcholine Receptor Ligands That Reduce Alcohol Intake in Rats. J Med Chem. 2013;56:3000–3011. doi: 10.1021/jm4000374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Porsolt RD, Bertin A, Jalfre M. Behavioral despair in mice: a primary screening test for antidepressants. Arch Int Pharmacodyn Ther. 1977;229:327–336. [PubMed] [Google Scholar]
- 24.The preliminary ADMET studies were carried out by Cerep, Inc.
- 25.Lukas RJ, Fryer JD, Eaton JB, Gentry CL. Some methods for studies of nicotinic acetylcholine receptor pharmacology. In: Levin ED, editor. Nicotinic Receptors and the Nervous System. CRC Press; Boca Raton: 2002. pp. 3–27. [Google Scholar]
- 26.Lukas RJ, Norman SA, Lucero L. Characterization of Nicotinic Acetylcholine Receptors Expressed by Cells of the SH-SY5Y Human Neuroblastoma Clonal Line. Mol Cell Neurosci. 1993;4:1–12. doi: 10.1006/mcne.1993.1001. [DOI] [PubMed] [Google Scholar]
- 27.Eaton JB, Peng JH, Schroeder KM, George AA, Fryer JD, Krishnan C, Buhlman L, Kuo YP, Steinlein O, Lukas RJ. Characterization of human alpha4beta2-nicotinic acetylcholine receptors stably and heterologously expressed in native nicotinic receptor-null SH-EP1 human epithelial cells. Mol Pharmacol. 2003;64:1283–1294. doi: 10.1124/mol.64.6.1283. [DOI] [PubMed] [Google Scholar]
- 28.Breining SR, Melvin M, Bhatti BS, Byrd GD, Kiser MN, Hepler CD, Hooker DN, Zhang J, Reynolds LA, Benson LR, Fedorov NB, Sidach SS, Mitchener JP, Lucero LM, Lukas RJ, Whiteaker P, Yohannes D. Structure-activity studies of 7-heteroaryl-3-azabicyclo[3.3.1]non-6-enes: a novel class of highly potent nicotinic receptor ligands. J Med Chem. 2012;55:9929–9945. doi: 10.1021/jm3011299. [DOI] [PubMed] [Google Scholar]
- 29.Kuryatov A, Olale F, Cooper J, Choi C, Lindstrom J. Human alpha6 AChR subtypes: subunit composition, assembly, and pharmacological responses. Neuropharmacology. 2000;39:2570–2590. doi: 10.1016/s0028-3908(00)00144-1. [DOI] [PubMed] [Google Scholar]
- 30.McIntosh JM, Azam L, Staheli S, Dowell C, Lindstrom JM, Kuryatov A, Garrett JE, Marks MJ, Whiteaker P. Analogs of alpha-conotoxin MII are selective for alpha6-containing nicotinic acetylcholine receptors. Mol Pharmacol. 2004;65:944–952. doi: 10.1124/mol.65.4.944. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.