

Amphiphilic di-block or tri-block copolymers that are capable of self-assembling into thermodynamically stable nano-structures with a hydrophobic core and hydrophilic corona, referred to as micelles, have become increasingly important in pharmaceutical applications.[1– 5] The hydrophobic core can house hydrophobic drugs [6] and the hydrophilic corona functions as a steric barrier to prevent micelle aggregation ensuring micelle solubility in an aqueous environment.[7] To date, multiple synthetic amphiphilic micelle systems (Genexol-PM, [8, 9] NK012, [10] SP1049C, [11, 12] NC-6004, [13] and NK105 [14, 15]) are under clinical evaluation for delivering hydrophobic anti-cancer drugs. The size of these micelles typically range from 10 to 100 nm in diameter, and are composed of a hydrophilic corona, which prevents opsonization resulting in relatively long plasma retention times and reduced uptake by the reticuloendothelial system (RES).[16] The small size of these micelles allows them to accumulate in tumors through an enhanced permeability and retention (EPR) effect via the leaky microvasculature of tumors, which are characterized by fenestration cutoffs or large gaps ranging from 380 nm up to 1.2 μm.
In recent years, fluorescent micelles have gained significant attention at the forefront of theranostic (therapeutic + diagnostic) nanomedicine.[4, 17, 18] For example, fluorescent micelles have been used as in vivo tracers for cancer detection[19, 20] and as probes for investigating the cellular internalization and intracellular trafficking of particles, drugs, and genes.[21] The existing strategies to confer fluorescent properties to micelles are centered on conjugating or encapsulating fluorescent organic dyes (rhodamine, Cyanines, and fluorescein),[22–25] quantum dots (QDs),[26] or gold particles[27, 28] on or within the micelles. However, conjugation or encapsulation of these materials is often associated with low dye-to-micelle ratios, increased particle sizes, inferior photobleaching-resistance (organic dyes), and significant cytotoxicity (QDs and other metallic particles), which are inevitable concerns for these fluorescent micelle systems.[29] Furthermore, premature leakage of the dye molecules into surrounding tissues may interfere with the detection of samples of interest.[30]
Recently, our group has made progress in the development of biodegradable photoluminescent polymers (BPLPs), which present intriguing fluorescent properties without the conjugation of any organic dyes or QDs.[31] BPLPs display superior biocompatibility both in vitro and in vivo, high quantum yields (up to 79%), photobleaching resistance, and tunable emission up to near infrared wavelengths. The monomers used in the synthesis of BPLPs, citric acid (a metabolite in the Krebs cycle), polyethylene glycol (PEG), aliphatic diols, and all 20 α-amino acids, are all compounds used in many Food and Drug Administration (FDA)-regulated devices.[31, 32] The use of different α-amino acids results in photostable BPLPs with tunable fluorescence properties in terms of their fluorescent intensity, excitation, emission, quantum yield, etc. BPLPs are referred to as BPLP-Cys and BPLP-Ser when L-cysteine and L-Serine are used in the syntheses, respectively. One noteworthy advantage of BPLPs over the traditional organic dyes or QDs is that BPLPs are implantable polymers, and can be fabricated into medical devices, such as nanoparticles and cellular scaffolds, while also functioning as imaging probes. In contrast, organic dyes and QDs are solely imaging probes, which have to be ancillary to other materials and devices.
Given the growing needs for theranostic micelle systems, we describe the synthesis and characterization of photostable fluorescent amphiphilic copolymers based on BPLPs, referred to as amphiphilic BPLPs (ABPLPs) (Figure 1A). Supporting information, Scheme S1 shows the synthesis schematic of amphiphilic biodegradable photoluminescent polymers (ABPLPs). BPLPs were used as the hydrophobic block and methoxy poly(ethylene glycol) (MPEG) (MW 750, 2000, and 5000) was used as the hydrophilic block due to its broad acceptance in many micelle systems.[33–35] In step 1, MPEG was first converted to MPEG-COOH by reacting with succinic anhydride. Characteristic bond vibrations illustrated in Fourier transform infrared (FTIR) spectra (Supporting Information, Figure S1) and chemical shifts observed in 1H-NMR spectrum (Figures S2A and S2B) confirmed the successful modification of MPEG to MPEG-COOH.
Figure 1.

Syntheses and characterization of ABPLP particles. A) Chemical structure of ABPLP polymer chain. B) ABPLP copolymers composed of fluorescent hydrophobic blocks can self assemble into core-shell (micelle) structures encapsulating hydrophobic drugs within their core in an aqueous solution. Camera photograph of ABPLP-3 solution (in acetone) and ABPLP micelle (in water) taken under Ultraviolet lamp demonstrating their bright fluorescence. C) Representative transmission electron microscopy (TEM) image of ABPLP-3 micelles in aqueous solution. D) Sizes of ABPLP-3 micelles measured by dynamic light scattering. E) Size comparison of ABPLP micelles with BPLP nanoparticles at concentration below (2 mg/L) and above (200 mg/L) CMC. F) Summary of composition and particle properties of ABPLP (CMC: Critical Micelle Concentration).
In step 2, the hydrophobic blocks were synthesized similar to the procedures reported in our previous publication.[31] Citric acid was reacted with poly(propylene glycol) (PPG) to form a polyester backbone and further condensed with L-cysteine via the pendent carboxyl group and germinal hydroxyl group of the citrate units to create a 6-membered ring. The 6-membered planar rings pendant on the BPLP polymer backbones are composed of amide and ester bonds with different R groups from the various amino acids, which is believed to cause the polymer fluorescence through the hyperconjugation theory.[31] The R groups pendant to the α-C in the amino acids likely influence the degree of hyperconjugation, propensity for cyclization, and provide slight perturbations in the associated energy levels resulting in the different emission maxima and quantum yields observed for the BPLP-amino acids.
In step 3, the -COOH terminal hydrophilic MPEG chains were conjugated with the -OH terminal hydrophobic BPLP chains through DCC/DMAP chemistry to form ABPLPs. Various molecular weights of PPG (425, 725, and 2000 Da) and MPEG (750, 2000, and 5000) were used in the synthesis protocol to understand variation of chain lengths on the properties of ABPLP copolymer.
As amphiphilic copolymers, ABPLPs have the ability to self-assemble into nano-sized micelles (Figure 1B and 1C) in an aqueous medium. ABPLPs can be dissolved in solvents such as acetone, tetrahydrofuran (THF), dimethylformamide (DMF), methyl chloride, and dimethylsulfoxide (DMSO). Under transmission electron microscopy (TEM), ABPLP-3 micelles were spherical in shape and 60 nm in diameter, which was in an agreement with the size measurements from dynamic light scattering (an average diameter of 68 nm and a polydispersity index of 0.17) (Figure 1D). No particle aggregation was observed, and ABPLPs synthesized using low molecular weight PPG, as in the case of ABPLP-1 and ABPLP-2, demonstrated higher particle sizes of 178 and 107 nm, respectively. However, ABPLPs synthesized with high molecular weight PPG, as in the case of ABPLP-3, exhibited smaller particle sizes of 68 nm. In addition, particle size can be further reduced with an increase in the hydrophilic block molecular weight as in the case of ABPLP-4 (53 nm) and ABPLP-5 (48 nm).
The thermodynamic stability of micelles can be determined by the critical micelle concentration (CMC). A low CMC is important in maintaining micelle stability and integrity since a major difference between in vitro and in vivo conditions is the effect of dilution after intravenous administration.[36, 37] It is well known that the CMC values are mainly dictated by the relative lengths of hydrophobic and hydrophilic blocks. Typically, when the length of the hydrophilic block is held constant, an increase in the length of the hydrophobic blocks results in decreased CMC values.[38] CMC values of 0.012, 0.006 and 0.004 mg/mL for ABPLP-1, ABLP-2 and ABPLP-3, respectively, in aqueous solution were seen to decrease as the fraction of the PPG hydrophobic block in the amphiphilic copolymers increased (Supporting Information, Figure S4).[39] On the other hand, the CMC values for ABPLP-3, ABPLP-4, and ABPLP-5 were calculated as 0.004, 0.005, and 0.010 mg/mL, respectively (Figure 1F). When the length of the hydrophobic block remained constant, the CMC values increased in relation to an increasing hydrophilic block (MPEG) length due to enhanced hydrophilicity.[40] These CMC values are lower than other amphiphilic polymeric micelles reported (1.1 mg/mL for PEO-b-PHB-b-PEO,[41] 0.07 mg/mL for PDMAEMA-b-PTHF-b-PDMAEMA, and 0.03 mg/mL for PDMAEMA-b-PBD-b-PDMAEMA[42]). The lower CMC values for ABPLPs suggest that the ABPLP micelles are potentially more stable after intravenous administration. Based on the “salt out” effect,[43] an even lower CMC value may be expected for micelles in ionic blood solution. To verify the micelle formation, the sizes of ABPLP micelles were measured by DLS at concentrations above and below the CMC for various ABPLP micelles and compared to the nanospheres of BPLPs. Upon dilution below the CMC (0.002 mg/mL), all the ABPLP micelles completely disassembled and the sizes could not be detected by DLS, whereas BPLP nanospheres displayed stable solid colloidal solutions even at very low concentrations (Figure 1E). In addition, ABPLP micelles displayed a negative zeta potential in deionized water in the range of −20 to −27 mV (Figure 1F), which also contributes to the micelle stability since the strong electrostatic repulsion minimizes micelle aggregation.[44] ABPLP micelles are a unique biomaterial system due to their unique fluorescent properties inherited from the hydrophobic BPLP blocks.[31] In this study, we primarily investigated the fluorescent properties of a representative ABPLPs, ABPLP-Cys (0.2 molar ratio). As compared to BPLP-Cys, ABPLP-Cys also showed a strong fluorescence emission within the range of 390–550 nm with a peak wavelength at 446 nm (Figure 2A). Interestingly, the fluorescent intensity of ABPLP-Cys was significantly reduced in micelle form when compared to that of polymer solution in 1,4-dioxane (Figure 2B). This was further evidenced by the quantum yields measured for all ABPLP copolymers (Figure 2C, Figure 2G). For example, the quantum yields of ABPLP-1, ABPLP-2, ABPLP-3, ABPLP-4, and ABPLP-5 were 0.453, 0.443, 0.447, 0.434, and 0.424, respectively, in a 1,4-dioxane solution. In micelle form, the quantum yields significantly reduced to 0.046, 0.072, 0.158, 0.256, and 0.266, respectively (Figure 2G). This might be due to the fact that the random chains of amphiphilic block copolymers were fully solvated in the organic solvent system for polymer solutions. In contrast, the fluorescent hydrophobic blocks (fluorophore) of the polymeric chains were drawn together through hydrophobic interactions in micelles, which brings a possibility of fluorescence quenching. This potential fluorescence quenching may contribute to a significant reduction in their quantum yields. On the other hand, the molar absorption coefficients were found to be significantly higher for all ABPLPs in micelle form when compared to that in 1,4-dioxane solution. This result indicates that ABPLPs have a high capacity for light absorption even though they exhibit relative low quantum yields when in a micelle form.
Figure 2.

Photoluminescent properties of ABPLPs. A) Excitation and emission spectra of ABPLP- 3 micelles in water. B) Emission spectra of ABPLP solution in 1, 4-dioxane and ABPLP micelles in water excited at 350 nm. C) Intensity-absorbance curve of ABPLP-1, ABPLP-2, and ABPLP-3 for quantum yield measurements. D) Photostability evaluation of ABPLP-1, ABPLP-2, and ABPLP-3 and control organic dye Rhodamine, E) Emission spectra of ABPLP-Ser, and F) Fluorecent intensity of ABPLP-1, ABPLP-2, and ABPLP-3 at various time periods when incubated in Phosphate Buffer Solution (pH 7.4). G) Summary of fluorescent properties of ABPLP micelles (QY: Quantum Yield, ε: molar absorption coefficient).
The photostability of ABPLP micelles was also investigated and compared to the widely used organic fluorescent dye, rhodamine-B. The fluorescent intensity of the ABPLP micelle solution reduced by only 1% of the initial intensity after continuous UV excitation for 3 h, which indicates excellent photostability (Figure 2D) where as rhodamine-B lost almost 10% of initial fluorescent intensity within the same experimental period. Furthermore, a family of ABPLP polymers was synthesized using different alpha-amino acids. For example, ABPLP-Ser, when synthesized with L-serine, exhibits different fluorescent colors ranging from blue to red by varying the excitation wavelength (Figure 2E) similar to BPLP-Ser. The excitation-dependent emission of ABPLP-Ser was ascribed to a well-known Red-Edge Effect (REE) where polar and rotatable fluorophores embedded in rigid and highly viscous medium can generate variable fluorescence emission.[45, 46] In our BPLP or ABPLP structure, the polymer backbones can be treated as a viscous medium for the pendent 6-membered ring fluorophores. The R group (-CH2OH) on the 6-membered ring of ABPLP-Ser is highly rotatable, which allows ABPLP-Ser to display excitation dependence. However, we have previously demonstrated that an easy H2S elimination occurs during the BPLP-Cys or ABPLP-Cys formation, which results in a double bond formation to extend the conjugation system of the 6-membered ring (Supporting Information, Figure S5). The double bond may restrict the rotation of the fluorophore. Therefore, ABPLP-Cys does not show excitation dependence for its emission.
Finally, the long-term fluorescent stability of ABPLP micelles in an aqueous system was monitored for a 6-week period (Figure 2F). In the first week, the fluorescent intensity of the micelles was nearly constant for all micelle systems, followed by an increase in the fluorescent intensity at 2 weeks, and then a continuous decrease thereafter. This result strongly suggests that ABPLP micelles maintained their thermodynamic stability in the first week and then displayed a higher fluorescent intensity due to dissociation of the polymeric chains after week 2. The continuous decrease in fluorescent intensity afterwards was due to the degradation of the BPLP chains of ABPLPs, which is also in agreement with the degradation profile of fluorescent blocks of the previously published BPLPs.[31] To demonstrate the potential of ABPLP micelles for imaging applications, the cellular uptake and fluorescent imaging of the micelles both in vitro and in vivo were conducted. ABPLP micelles were used to label NIH 3T3 fibroblasts with fluorescence colors after 3 hrs of incubation (Figure 3A and 3B). The micelles may have only accumulated on the cell surfaces at 3 hr thus the entire cell bodies seem fluorescent. When cells were imaged at 9 hr, cell nuclei were obvious and the cytoplasm was labeled with a blue color suggesting the micelles were uptaken by the cells. When incubated with NIH 3T3 fibroblasts for 24 h, ABPLP micelles did not show significant cytotoxicity to the cells at concentrations ranging from 1 to 1000 μg/mL. It was also noted that ABPLPs with smaller micelle size (as in the case of ABPLP-3, ABPLP-4, and ABPLP-5) were non-cytotoxic even at high concentrations (>500 μg/mL) (Figure 3C). To verify the potential for in vivo imaging, ABPLP micelle solutions of various concentrations were subcutaneously injected in a mouse through a 27-gauge needle. ABPLP micelles were detected using a non-invasive in vivo imaging system. As illustrated in Figure 3D, the signal intensity doubled when the concentration was increased from 0.0625 mg/mL to 1 mg/mL. Upon closer topical observation of the surrounding tissue at the injection sites, the ABPLP micelles did not induce redness or obvious irritation. These in vitro and in vivo studies suggest that ABPLP micelle systems possess great potential for in vitro cell labeling and in vivo tissue imaging.
Figure 3.
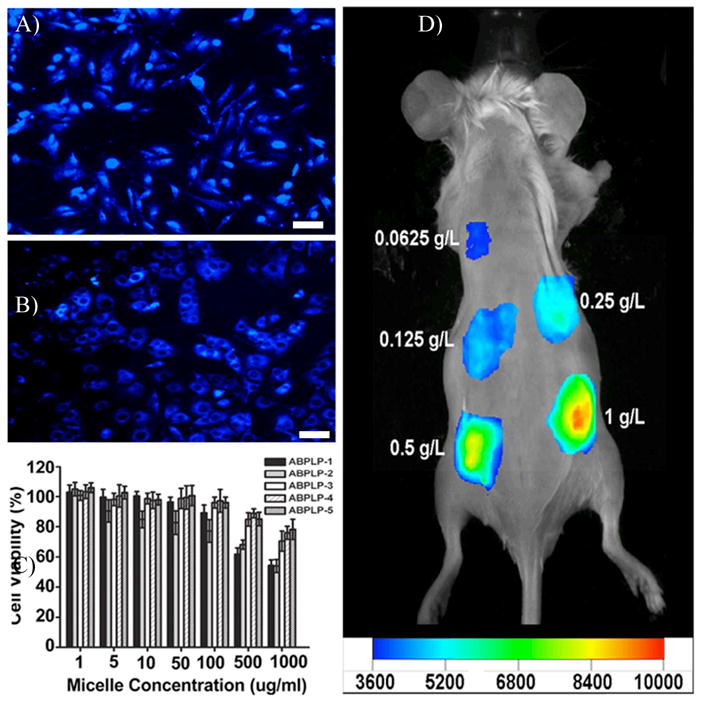
In vitro cellular uptake and in vivo fluorescence imaging of ABPLP micelles. ABPLP-3 micelle-uptaken 3T3 fibroblasts observed under fluorescent microscope with DAPI filter (4X) at A) 3 h and B) 9 hr. C) Cytotoxicity evaluation (MTT assay) of ABPLP micelles against 3T3 fibroblast at various concentrations (1–1000 μg/ml). Values were normalized to the viability of cells cultured in micelle-free medium. D) Fluorescence image of ABPLP micelles injected in a nude mouse at various concentrations (0.0625 – 1 g/L).
To demonstrate the utility of ABPLP micelles for cancer drug delivery,[47, 48] Paclitaxel (PTX) was used as a model cancer drug for in vitro drug delivery and cell culture studies.[49] It was observed that all ABPLP micelles have high drug (PTX) loading and encapsulation efficiencies of 20.6%–22.78% and 81.57%–91.12%, respectively, as shown in Figure S6 A. The PTX release profiles from various ABPLP micelles (Supporting Information, Figure S6 B) showed an initial burst release (~50% of the initial loading amount) in the first stage up to 10 h followed by a sustained release period of up to 96 h as similar to other micelle drug delivery systems.[50, 51] When incubated with a human prostrate cancer cell line (PC-3) (Supporting Information, Figure S6 C), no sign of cellular toxicity was observed for cells incubated with media containing 0.5 mg/mL of drug-free micelles. However, PTX-loaded ABPLP micelles resulted in delayed cell toxicity with the final toxicity at 24 h comparable to free PTX. These results support that ABPLP micelles may potentially serve as a viable cancer drug carrier to release a significant amount of drugs after reaching the targeting sites through the circulation in vivo.
In this work, we reported photobleaching resistant amphiphilic biodegradable photoluminescent polymers (ABPLPs) that can be self-assembled into fluorescent micelles. ABPLP micelles could label NIH 3T3 fibroblasts without inducing significant cellular toxicity. PTX-loaded ABPLP micelles could delay the release of payload to kill prostrate cancer PC3 cells. In vivo imaging studies showed that ABPLP micelles emitted strong fluorescence in vivo when injected subcutaneously. It is the first time to report biodegradable amphiphilic photoluminescent polymers without conjugating any conventional fluorescent organic dyes or quantum dots. ABPLP micelles may serve as an ideal vector in theranostic nanomedicine.
Experimental Section
The detailed experimental procedures are available in the supporting information.
Supplementary Material
Acknowledgments
This work was supported in part by a NSF CAREER award (DMR 1313553), a NIBIB R01 award (EB012575), a CPRIT High Impact/High Risk Research Award (RP110412), and a research award from National Natural Sciences Foundation of China (31228007).
Footnotes
Supporting Information is available online from Wiley InterScience or from the author)).
Contributor Information
Dipendra Gyawali, Department of Bioengineering, The University of Texas at Arlington, Arlington, TX 76010.
Shengyuan Zhou, Department of Bioengineering, The University of Texas at Arlington, Arlington, TX 76010.
Dr. Richard T. Tran, Department of Bioengineering, Materials Research Institute, Huck Institutes of The Life Sciences, The Pennsylvania State University, University Park, PA 16802
Dr. Yi Zhang, Department of Bioengineering, The University of Texas at Arlington, Arlington, TX 76010
Dr. Chao Liu, Department of Bioengineering, Materials Research Institute, Huck Institutes of The Life Sciences, The Pennsylvania State University, University Park, PA 16802
Dr. Xiaochun Bai, Academy of Orthopedics of Guangdong Province, The Third Affiliated Hospital, Southern Medical University, Guangzhou 510280, China
Prof. Jian Yang, Email: jxy30@psu.edu, Department of Bioengineering, Materials Research Institute, Huck Institutes of The Life Sciences, The Pennsylvania State University, University Park, PA 16802
References
- 1.Matsumura Y. Adv Drug Delivery Rev. 2008;60:899. doi: 10.1016/j.addr.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 2.Rosler A, Vandermeulen GWM, Klok HA. Adv Drug Delivery Rev. 2001;53:95. doi: 10.1016/s0169-409x(01)00222-8. [DOI] [PubMed] [Google Scholar]
- 3.Tyrrell ZL, Shen YQ, Radosz M. Prog Polym Sci. 2010;35:1128. [Google Scholar]
- 4.Oerlemans C, Bult W, Bos M, Storm G, Nijsen JFW, Hennink WE. Pharm Res. 2010;27:2569. doi: 10.1007/s11095-010-0233-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li XR, Yang ZL, Yang KW, Zhou YX, Chen XW, Zhang YH, Wang F, Liu Y, Ren LJ. Nanoscale Res Lett. 2009;4:1502. doi: 10.1007/s11671-009-9427-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Torchilin VP. Pharm Res. 2007;24:1. doi: 10.1007/s11095-006-9132-0. [DOI] [PubMed] [Google Scholar]
- 7.Jones MC, Leroux JC. Eur J Pharm Biopharm. 1999;48:101. doi: 10.1016/s0939-6411(99)00039-9. [DOI] [PubMed] [Google Scholar]
- 8.Kim TY, Kim DW, Chung JY, Shin SG, Kim SC, Heo DS, Kim NK, Bang YJ. Clin Cancer Res. 2004;10:3708. doi: 10.1158/1078-0432.CCR-03-0655. [DOI] [PubMed] [Google Scholar]
- 9.Lee KS, Chung HC, Im SA, Park YH, Kim CS, Kim SB, Rha SY, Lee MY, Ro J. Breast Cancer Res Treat. 2008;108:241. doi: 10.1007/s10549-007-9591-y. [DOI] [PubMed] [Google Scholar]
- 10.Matsumura Y, Kataoka K. Cancer Sci. 2009;100:572. doi: 10.1111/j.1349-7006.2009.01103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Valle JW, Armstrong A, Newman C, Alakhov V, Pietrzynski G, Brewer J, Campbell S, Corrie P, Rowinsky EK, Ranson M. Invest New Drugs. 2011;29:1029. doi: 10.1007/s10637-010-9399-1. [DOI] [PubMed] [Google Scholar]
- 12.Valle JW, Lawrance J, Brewer J, Clayton A, Corrie P, Alakhov V, Ranson M. J Clin Oncol. 2004;22:362s. [Google Scholar]
- 13.Plummer R, Wilson RH, Calvert H, Boddy AV, Griffin M, Sludden J, Tilby MJ, Eatock M, Pearson DG, Ottley CJ, Matsumura Y, Kataoka K, Nishiya T. Br J Cancer. 2011;104:593. doi: 10.1038/bjc.2011.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kato K, Hamaguchi T, Yasui H, Okusaka T, Ueno H, Ikeda M, Shirao K, Shimada Y, Nakahama H, Muro K, Matsumura Y. J Clin Oncol. 2006;24:83s. [Google Scholar]
- 15.Hamaguchi T, Kato K, Yasui H, Morizane C, Ikeda M, Ueno H, Muro K, Yamada Y, Okusaka T, Shirao K, Shimada Y, Nakahama H, Matsumura Y. J Cancer. 2007;97:170. doi: 10.1038/sj.bjc.6603855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Torchilin VP. Pharm Res. 2007;24:1. doi: 10.1007/s11095-006-9132-0. [DOI] [PubMed] [Google Scholar]
- 17.Khemtong C, Kessinger C, Gao JM. Chem Comm. 2009:3497. doi: 10.1039/b821865j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park JH, von Maltzahn G, Ong LL, Centrone A, Hatton TA, Ruoslahti E, Bhatia SN, Sailor MJ. Adv Mater. 2010;22:880. doi: 10.1002/adma.200902895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nasongkla N, Bey E, Ren JM, Ai H, Khemtong C, Guthi JS, Chin SF, Sherry AD, Boothman DA, Gao JM. Nano Lett. 2006;6:2427. doi: 10.1021/nl061412u. [DOI] [PubMed] [Google Scholar]
- 20.Sumer B, Gao JM. Nanomedicine. 2008;3:137. doi: 10.2217/17435889.3.2.137. [DOI] [PubMed] [Google Scholar]
- 21.Lu J, Owen SC, Shoichet MS. Macromolecules. 2011;44:6002. doi: 10.1021/ma200675w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luo LB, Tam J, Maysinger D, Eisenberg A. Bioconjugate Chem. 2002;13:1259. doi: 10.1021/bc025524y. [DOI] [PubMed] [Google Scholar]
- 23.Texier I, Goutayer M, Da Silva A, Guyon L, Djaker N, Josserand V, Neumann E, Bibette J, Vinet F. J Biomed Opt. 2009:14. doi: 10.1117/1.3213606. [DOI] [PubMed] [Google Scholar]
- 24.Wu DQ, Lu B, Chang C, Chen CS, Wang T, Zhang YY, Cheng SX, Jiang XJ, Zhang XZ, Zhuo RX. Biomaterials. 2009;30:1363. doi: 10.1016/j.biomaterials.2008.11.027. [DOI] [PubMed] [Google Scholar]
- 25.Lee HI, Wu W, Oh JK, Mueller L, Sherwood G, Peteanu L, Kowalewski T, Matyjaszewski K. Angew Chemie, Int Ed. 2007;46:2453. doi: 10.1002/anie.200604278. [DOI] [PubMed] [Google Scholar]
- 26.Lee J, Im JH, Huh KM, Lee Y, Shin H. J Nanosci Nanotechnol. 2010;10:487. doi: 10.1166/jnn.2010.1736. [DOI] [PubMed] [Google Scholar]
- 27.Sakai T, Alexandridis P. Langmuir. 2004;20:8426. doi: 10.1021/la049514s. [DOI] [PubMed] [Google Scholar]
- 28.Otsuka H, Akiyama Y, Nagasaki Y, Kataoka K. J Am Chem Soc. 2001;123:8226. doi: 10.1021/ja010437m. [DOI] [PubMed] [Google Scholar]
- 29.Kabanov AV, Slepnev VI, Kuznetsova LE, Batrakova EV, Alakhov VY, Meliknubarov NS, Sveshnikov PG, Kabanov VA. Biochem Int. 1992;26:1035. [PubMed] [Google Scholar]
- 30.Zhang JX, Qiu LY, Li XD, Jin Y, Zhu KJ. Small. 2007;3:2081. doi: 10.1002/smll.200700069. [DOI] [PubMed] [Google Scholar]
- 31.Yang J, Zhang Y, Gautam S, Liu L, Dey J, Chen W, Mason RP, Serrano CA, Schug KA, Tang L. Proc Natl Acad Sci U S A. 2009;106:10086. doi: 10.1073/pnas.0900004106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wadajkar AS, Kadapure T, Zhang Y, Cui W, Nquyen KT, Samchukov M, Yang J. Adv Healthcare Mater. 2012;1:450. doi: 10.1002/adhm.201100055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gaucher G, Dufresne MH, Sant VP, Kang N, Maysinger D, Leroux JC. J Controlled Release. 2005;109:169. doi: 10.1016/j.jconrel.2005.09.034. [DOI] [PubMed] [Google Scholar]
- 34.Allemann E, Brasseur N, Benrezzak O, Rousseau J, Kudrevich SV, Boyle RW, Leroux JC, Gurny R, Vanlier JE. J Pharm Pharmacol. 1995;47:382. doi: 10.1111/j.2042-7158.1995.tb05815.x. [DOI] [PubMed] [Google Scholar]
- 35.Gao GH, Lee JW, Nguyen MK, Im GH, Yang J, Heo H, Jeon P, Park TG, Lee JH, Lee DS. J Controlled Release. 2010;123:109. doi: 10.1016/j.jconrel.2010.09.012. [DOI] [PubMed] [Google Scholar]
- 36.Fairley N, Hoang B, Allen C. Biomacromolecules. 2008;9:2283. doi: 10.1021/bm800572p. [DOI] [PubMed] [Google Scholar]
- 37.Savic R, Eisenberg A, Maysinger D. J Drug Targeting. 2006;14:343. doi: 10.1080/10611860600874538. [DOI] [PubMed] [Google Scholar]
- 38.Varshney SK, Zhong XF, Eisenberg A. Macromolecules. 1993;26:701. [Google Scholar]
- 39.Nagarajan R, Ganesh K. Macromolecules. 1989;22:4312. [Google Scholar]
- 40.Wang D, Peng ZP, Liu XX, Tong Z, Wang CYB. Ren Eur Polym J. 2007;43:2799. [Google Scholar]
- 41.Li J, Ni XP, Li X, Tan NK, Lim CT, Ramakrishna S, Leong KW. Langmuir. 2005;21:8681. doi: 10.1021/la0515266. [DOI] [PubMed] [Google Scholar]
- 42.Even M, Haddleton DM, Kukulj D. Eur Polym J. 2003;39:633. [Google Scholar]
- 43.Patist A, Kanicky JR, Shukla PK, Shah DO. J Colloid Interface Sci. 2002;245:1. doi: 10.1006/jcis.2001.7955. [DOI] [PubMed] [Google Scholar]
- 44.Kallay N, Zalac S. J Colloid Interface Sci. 2002;253:70. doi: 10.1006/jcis.2002.8476. [DOI] [PubMed] [Google Scholar]
- 45.Demchenko AP. Luminescence. 2002;17:19. doi: 10.1002/bio.671. [DOI] [PubMed] [Google Scholar]
- 46.Fletcher AN. J Phys Chem. 1968;72:2742. [Google Scholar]
- 47.Tao YH, Liu R, Chen MQ, Yang C, Liu XY. J Mater Chem. 2012;22:373. [Google Scholar]
- 48.Liang HJ, Yang QX, Deng L, Lu JZ, Chen JM. Drug Dev Ind Pharm. 2011;37:597. doi: 10.3109/03639045.2010.533276. [DOI] [PubMed] [Google Scholar]
- 49.Liu J, Lee H, Allen C. Curr Pharm Des. 2006;12:4685. doi: 10.2174/138161206779026263. [DOI] [PubMed] [Google Scholar]
- 50.Blanco E, Bey EA, Dong Y, Weinberg BD, Sutton DM, Boothman DA, Gao J. J Controlled Release. 2007;122:365. doi: 10.1016/j.jconrel.2007.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu H, Farrell S, Uhrich K. J Controlled Release. 2000;68:167. doi: 10.1016/s0168-3659(00)00247-9. [DOI] [PubMed] [Google Scholar]
- 52.Licciardi M, Cavallaro G, Di Stefano M, Pitarresi G, Fiorica C, Giammona G. Int J Pharm. 2010;396:219. doi: 10.1016/j.ijpharm.2010.06.021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.