

Abstract
Late Onset Alzheimer Disease (LOAD) constitutes the majority of AD cases (~90%). Amyloidosis and tau pathology, which are present in AD brains, appear to be sporadic in nature. We have previously shown that infantile lead (Pb) exposure is associated with a change in the expression and regulation of the amyloid precursor protein (APP) and its beta amyloid (Aβ) products in old age. Here we report that infantile Pb exposure elevated the mRNA and protein levels of tau as well as its transcriptional regulators namely specificity protein 1 and 3 (Sp1 and Sp3) in aged primates. These changes were also accompanied by an enhancement in site-specific tau phosphorylation as well as an increase in the mRNA and protein levels of cyclin dependent kinase 5 (cdk5). There was also a change in the protein ratio of p35/p25 with more Serine/Threonine phosphatase activity present in aged primates exposed to Pb as infants. These molecular alterations favored abundant tau phosphorylation and immunoreactivity in the frontal cortex of aged primates with prior Pb exposure. These findings provide more evidence that neurodegenerative diseases may be products of environmental influences that occur during the development.
Keywords: aging, Alzheimer’s disease, cdk5, hyperphosphorylation, lead, tau protein
1. Introduction
Alzheimer’s disease (AD) is the most common form of dementia that affects aging individuals. This age-related disease is characterized by changes in brain anatomy, biology, and function (Kitazawa et al., 2009). Mutations in three genes; the amyloid precursor protein (APP), presenilin 1 (PSEN 1), presenilin 2 (PSEN2) have been implicated in the familial early onset (<65years) autosomal dominant form of AD (EOAD) (Liddell et al., 2001). On the other hand, Late onset AD (LOAD) represents over 90% of the cases (>65 years) is sporadic in nature and of unknown cause. The unexplained etiology of LOAD maybe linked to environmental factors acting during the formation of the brain to predetermine disease outcomes later in life (Migliore and Coppede, 2009).
Epidemiological observations have provided evidence showing that in addition to genetic and life-style factors, environmental exposures during the perinatal and infant phases of life influence the etiology of some adult chronic non-communicable diseases (Gluckman and Hanson, 2004). This is postulated by the “Developmental Origins of Adult Disease” (DOAD) doctrine, which states that reprogramming of genes at the developmental stage, plays a pivotal role in the latent expression of specific genes thus contributing to adult pathogenesis (Barker, 1999, Barker et al., 1989).
While no conclusive epidemiological evidence has been published to confirm the connection between early life Pb exposure and AD, evidence for the association between exposure to Pb and cognitive decline in humans has been established in several longitudinal and cross-sectional studies in the elderly (Nordberg et al., 2000, Weisskopf et al., 2007). Mild cognitive impairment likely occurs at an intermediate stage on the way to AD. Furthermore, recent studies have demonstrated that rodents that were exposed to Pb Postnatally performed poorly on cognitive tests as aged adults (Bihaqi et al., 2013).
In addition to cognitive decline in animals with prior Pb exposure, rodents and primates exhibit latent overexpression of genes and proteins associated with the amyloid pathway, and enhancement of AD pathology is seen in primates (Basha et al., 2005a, Wu et al., 2008). Furthermore, genome-wide mRNA expression profiling and methylomic analysis revealed that a set of genes are reprogrammed by Pb exposure during development thus altering their expression in the aging brain via epigenetic mechanisms (Alashwal et al., 2012, Dosunmu et al., 2012).
Neurofibrillary degeneration and β-amyloidosis are both essential features of AD; however, each one of these lesions can exist without the other (Iqbal et al., 2010). Neurofibrillary degeneration in the absence of β-amyloid is also seen in several tauopathies such as Guam parkinsonism dementia complex, dementia pugilistica, corticobasal degeneration, Pick’s disease and FTDP-17 tau (frontotemporal dementia with parkinsonism linked with chromosome 17 and tau mutations) and progressive supranuclear palsy (Iqbal et al., 2010). Furthermore, neurofibrillary pathology not β-amyloidosis correlates best with the presence of dementia in humans (Burns et al., 1997).
While earlier studies from our lab focused on APP and its amyloidogenic Aβ product, the effect of environmental exposure on tau pathology, was unclear. Non human primates express amyloid plaques and tau tangles in a manner homologous to humans (Price and Sisodia, 1994, Voytko, 1998). In order to study the effect of developmental Pb exposure on the expression of tau and its related proteins later in life, we examined the cerebral cortex of aged Cynomolgus monkeys who were exposed to Pb as infants (Wu et al., 2008).
Materials and Methods
2.1. Animal exposure
In 1980, a cohort of female monkeys (Macaca fascicularis) were randomly assigned at birth to one of two exposure groups: one received 1.5 mg/kg/day of Pb-acetate (Sigma-aldrich, MO) from birth until 400 days of age via infant formula and vehicle after weaning (n=5), whereas the other group served as a control group and received formula or vehicle only (n=4). No overt signs of toxicity or health-related problems were observed in the animals as a result of Pb exposure (Rice, 1990, 1992). The primates were then transferred to the National Institutes of Health (NIH) facility until termination in 2003 at ~23 years of age. Animals were sacrificed following CO2 exposure and multiple organ tissues, including the brain, were collected and stored at −80°C until further use. Previous studies reported the blood Pb levels of these animals at 400 days of age to average 19–26 μg/dl in Pb-exposed primates compared with 3–6 μg/dl in the controls (Rice, 1990, 1992). Levels of Pb in the brain tissue at age 23 years were measured by us using Inductively Coupled Plasma Mass Spectrometry (ICP-MS) and found to be the same (background level) in both control and developmentally-exposed animals (Wu et al., 2008). All animal procedures were conducted under the supervision of a licensed veterinarian according to a National Institute on Environmental Health Sciences–NIH-Approved animal protocol.
2.2. Western blot analysis
Brain protein content was measured using the BCA kit (Pierce Biotechnology, IL). Total protein (40 μg) was separated on 4–15% Mini-Protean TGX™ precast gels (BIO-RAD, MA) and then transferred to polyvinylidiene diflouride (PVDF) membranes. Immunoblotting for total tau, to the following antibodies diluted at 1:1000: mouse anti-Tau 46, anti-phospho-Tau [rabbit anti pThr-212 (Sigma Aldrich, MO), rabbit anti p-Thr-181, mouse anti pSer-396 (Cell Signaling technology, MA), rabbit anti p-Ser 235 (Abcam, MA)], rabbit anti-cdk5, and rabbit anti p35/25 (C64B10) (Cell Signaling technology, MA). On the following day, membranes were washed and exposed for 1 h to goat anti mouse/goat anti rabbit IRDye® 680LT infrared dye (LI-COR Biotechnology, NE), diluted at 1:10,000. The images were developed using Odyssey infrared imaging system (Model- 9120, LI-COR Biotechnology, NE). As a control for equal protein loading, membranes were stripped and reprobed for 2 h with mouse GAPDH antibody diluted at 1:5000 (Sigma-Aldrich, MO) at room temperature followed by washing and re-exposure to goat anti mouse IRDye® 680LT infrared dye. After transferring to a PVDF membrane, the gel was stained with Bio-Safe Coomassie blue stain (Bio-Rad, CA) to assess the equal loading of the samples.
2.3. Total RNA isolation, synthesis of cDNA, and real-time PCR
RNA from various control and exposed monkey frontal cortical tissues was isolated according to the TRIzol method (Invitrogen, CA). First-strand cDNA was synthesized from 1.5 μg of total RNA using the iScript cDNA synthesis kit (Bio-Rad, CA). cDNA was then amplified using real-time PCR. The SYBR Green qRT-PCR assay was performed in 25 μl reactions in triplicates using 1.5 μl of cDNA template, 1X SYBR Green master mix, 0.4 μM forward and reverses primers, and deionized H2O. The following primers were used for tau, cdk5, Sp1, Sp3 and GAPDH: tau, sense- 5′-GGT GGT CCG TAC TCC ACC TA-3′, antisense- 5′-TTT GAG CCA CAC TTG GAC TG -3′; cdk5, sense- 5′-TCA TAG CCG CAA TGT GCT AC -3′, antisense- 5′-TCG ATG GAC GTG GAG TAC AG -3′; Sp1, sense- 5′-CAA GCC CAA ACA ATC ACC TT-3′, antisense- 5′-CAA TGG GTG TGA GAG TGG TG-3′; Sp3, sense-5′-CCA GGA TGT GGT AAA GTC TA-3′, antisense-5′-CTC CAT TGT CTC ATT TCC AG-3′; GAPDH, sense- 5′-TGA AGC AGG CGT CGG AGG G -3′, antisense- 5′-CGA AGG TGG AAG AGT GGG TG-3′. Each sample was done in triplicate. Amplification was carried out for all above genes with respective primer pairs in a 7500 Real-Time PCR System (Applied Biosystems, CA) following standard protocol. The initial step was 50°C for 2 min followed by 95°C for 10 min, then 42 cycles of 95°C for 15 sec and 60°C for 1 min. Results were analyzed with system software Sequence Detection Software (SDS) version 1.3, and expression was reported relative to GAPDH mRNA with 2−ΔΔCt method.
2.4. Ser/Thr phosphatase assay
Evaluation of serine/threonine phosphatase (Ser/Thr) activity in aged primate cortex brain samples was examined using the Ser/Thr phosphatase assay kit 1 (Millipore, CA). Total protein (10 μg) and 200 mM peptide (KRpTIRR) substrate were used per well to determine enzyme activity after a 15 min reaction. The enzyme reaction was terminated with 100 μl Malachite Green solution and a subsequent 15 min was allowed for color development. Absorbance was measured at 650 nm with a plate reader (SpectraMax M5; Molecular Devices, CA). Enzyme activity was calculated from the amount of released phosphate in pmol phosphate/min/mg based on a phosphate standard curve.
2.5. Tissue preparation and Immunohistochemistry
The primate brains were dissected out, cut along the mid-sagittal plane, and each hemisphere was cut into 10 μm sections and rapidly immersion fixed in 10% neutral buffered formalin. Prior to the embedding in the paraffin the brain sections containing the frontal association cortex were rinsed and taken through varying ethanol concentrations (Subaiea et al., 2013).
In order to identify neurofibrillary degeneration, immunohistochemistry was undertaken by us. Endogenous peroxidase activity was removed by briefly washing the paraffin embedded section in buffered saline and 3% Hydrogen peroxide (H2O2) for a duration of 30 min. After rinsing the sections were incubated in PBS with 2% Bovine Serum Albumin (BSA) and 1% Triton X-100 blocking solution for 30 min, followed by rinsing with PBS. The section were incubated overnight at 4°C with 1:200 AT8 antibody (Innogenetics, GA), which recognizes phosphorylated tau at Ser 198 and Ser 209 regions. On the following, day the section were washed with PBS and incubated with biotinylated secondary antibody, goat Anti-Mouse IgG Antibody (Vector Labs, Burlingame, CA) of 1:500 dilution for 30 min and then incubated with horseradish peroxidase HRP-conjugated streptavidin (Vector Labs, Burlingame, CA) for 30 min. After washing the sections with PBS the immunoreactivity was detected and visualized using the substrate 3-3, diaminobenzidine (DAB) (Vector Labs, Burlingame, CA). Cover slips on the sections were mounted with permanent mounting medium (Vector Labs, Burlingame, CA).
2.6. Statistical analysis
Western blot bands were quantified by using the LI-COR/Odyssey infrared image system (LI-COR Biosciences, NE). All the measurements were made in triplicates and all values were represented as mean ± S.E.M. The significance of difference between means of experimental groups was determined via one-way ANOVA, Tukey-Kramer multiple comparison post hoc test and student Newman-Keuls comparison post hoc test, using Graph pad Prism 3.0 computer software (company/state). The level of significance was set at p< 0.05.
3. Results
3.1. Total tau and phosphorylated tau levels
Our results indicate that primates with developmental exposure to Pb displayed a significant (p<0.05) increase in the protein level of total tau at 23 years of age compared to age-matched controls (Fig. 1). The site-specific phosphorylation of tau was examined using four different phosphorylation-dependent antibodies to specific residues of tau in Western blots of the cerebral cortex; these include threonine 212 (Thr-212), threonine 181(Thr-181), serine 396 (Ser-396), and serine 235 (Ser-235). Aged primates developmentally exposed to Pb, showed a significant (p<0.05, p<0.001) threefold increase in tau phosphorylation at 23 years of age in comparison to age-matched controls (Fig. 2A–D).
Fig. 1. Expression of total tau in aged primates exposed to Pb as infants.
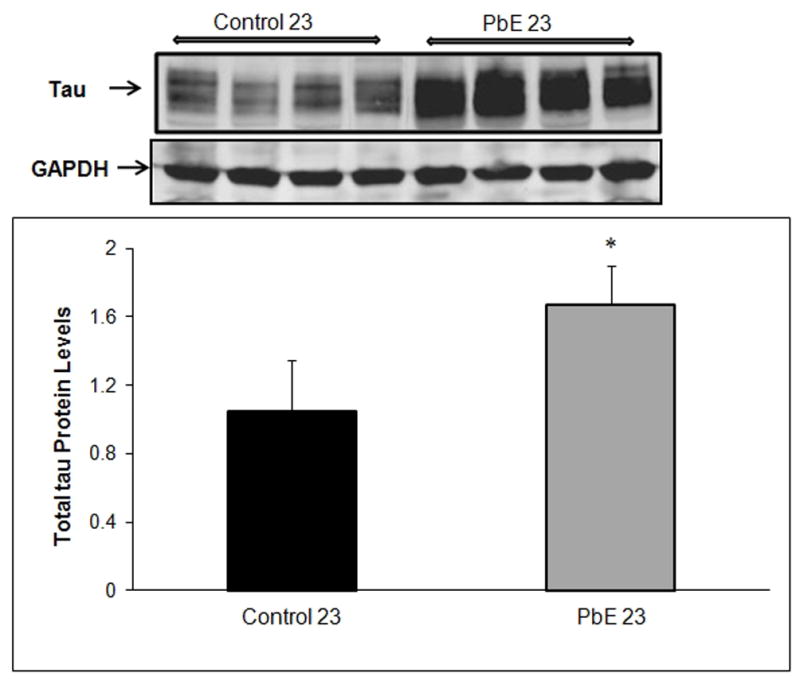
Changes in tau protein levels in the cerebral cortex of control and Pb-exposed primate brains (PbE, exposed as infants only) were monitored using Western blot. (Top) Immunoblots of total tau protein; (Bottom) mean +/− SEMs (n=4). Western blot results were normalized against GAPDH. *p<0.05 as compared to control 23-year olds.
Fig. 2. Phosphorylated tau expression in aged primate brains with developmental exposure to Pb.
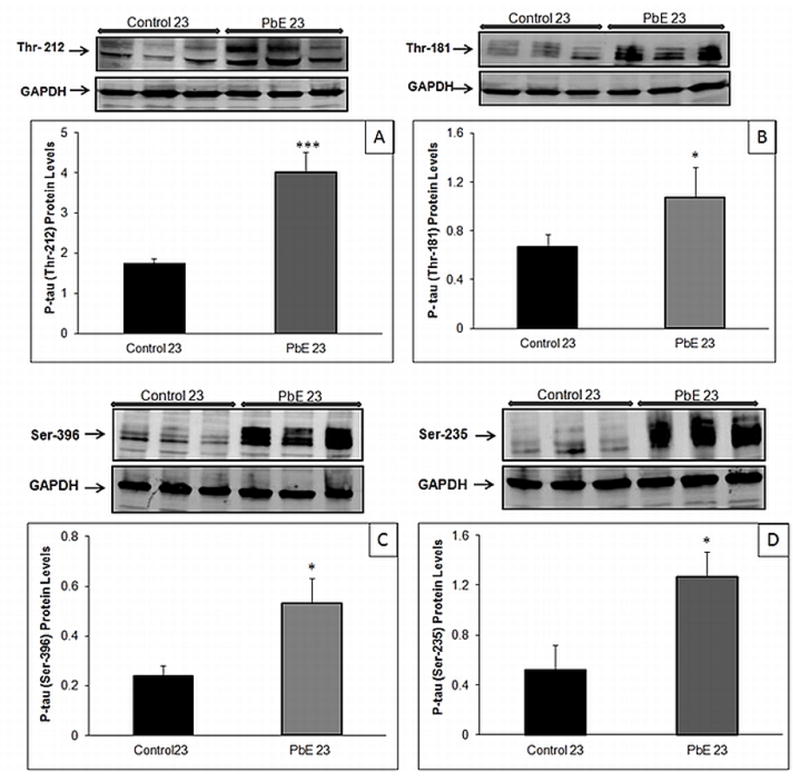
Changes in tau phosphorylation in the cerebral cortex of control and Pb-exposed primate brains (PbE, exposed as infants only) were monitored using Western blot analysis. Relative phosphorylated tau levels (A) Thr-212 (B) Thr-181 (C) Ser-396 (D) Ser-235. Diagrams represents mean +/− SEMs (n=3). Western blot results were normalized against GAPDH. *p<0.05, ***p<0.001 as compared to control 23-year olds.
3.2. Cdk5 and p35/25 protein levels
The increased phosphorylation on many residues in cortical tissue of aged primates developmentally exposed to Pb could be caused by the activation of kinases and other activators. Western blot examination revealed an increase in the protein level of cdk5 and a subsequent decrease in the levels of p35, a neuron-specific activator of cdk5 (p<0.05) in the frontal cortical region of aged primates with infantile Pb exposure as compared to aged-matched controls (Fig 3A). Furthermore, our results indicated a significant (p<0.001) increase in the levels of p25 (truncated form of p35) in primates with infantile Pb exposure as compared to aged matched control (Fig 3B).
Fig. 3. Cdk5 and p35/p25 expression in aged primates with infantile exposure to Pb.
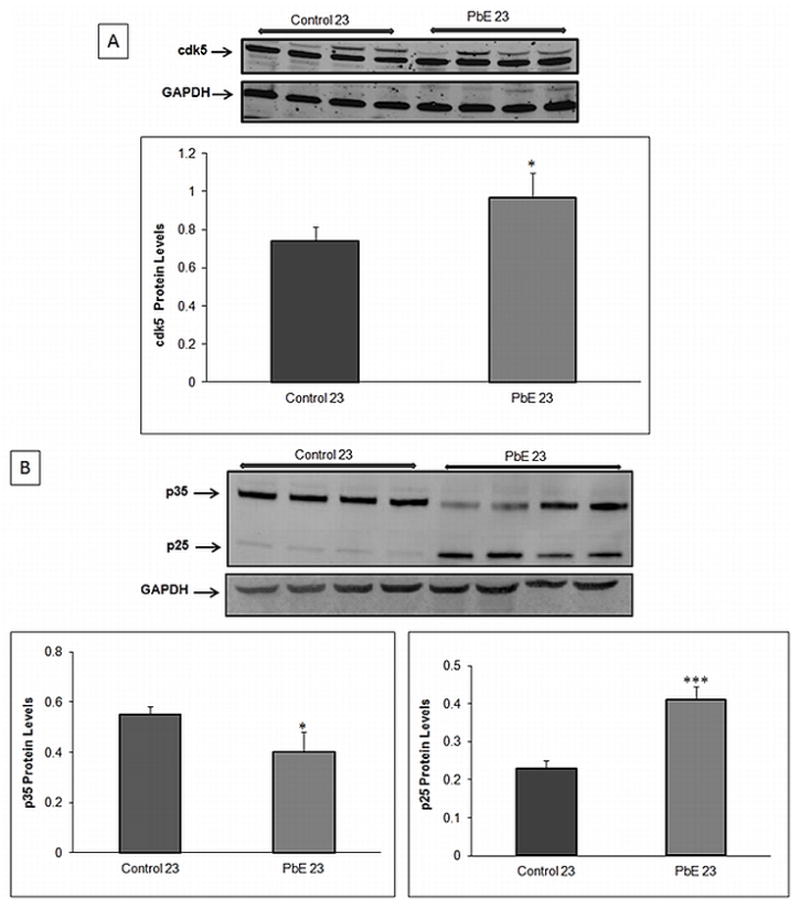
Protein expression of cdk5 and p35/p25 in the cerebral cortex of control and Pb-exposed primate brains (PbE, exposed as infants only) was monitored using Western blot. Relative protein signal (A) cdk5 (B) p35/p25. The bar diagram represents mean +/− SEMs (n=3). Western blot results were normalized against GAPDH. *p<0.05, ***p<0.001 as compared to control 23year old.
3.3. mRNA levels of tau, cdk5, Sp1 and Sp3
The latent effect of developmental exposure of Pb on the mRNA expression of tau, cdk5, Sp1, and Sp3 relative to GAPDH was examined using real-time PCR. Our results indicate a significant (p<0.001) increase in the mRNA levels of tau, cdk5, Sp1 and Sp3 in aged primates with developmental Pb exposure in comparison to control groups (Fig. 4).
Fig. 4. Changes in mRNA levels of aged primates with developmental Pb exposure.
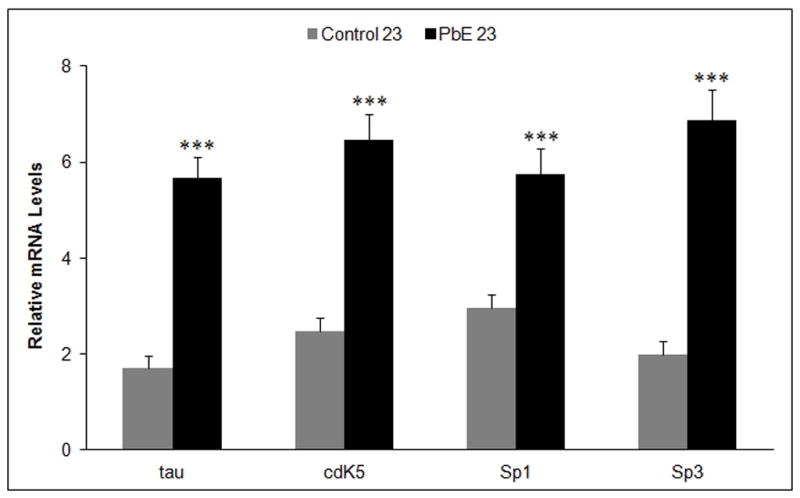
Tau, cdk5, Sp1 and Sp3 mRNA levels in the cerebral cortex of control and Pb-exposed primate brains (PbE, exposed as infants only) were analyzed using RT-PCR. The bar diagram represents mean +/− SEMs (n=3). mRNA levels were relative to GAPDH. ***p<0.001 as compared to control 23-year old.
3.4. Activity of serine/threonine phosphatases
We investigated the activity of total Ser/Thr protein phosphatases in the frontal cortex of aged control primates as well as aged primates with infantile Pb exposure. Our results show a significant (p<0.05) increase in the activity of total Ser/Thr protein phosphatases in 23-year-old primates with developmental Pb exposure as compared to age-matched controls (Fig. 5).
Fig. 5. Dysregulation of Serine/Threonine phosphatase activity in aged primates with infantile Pb exposure.
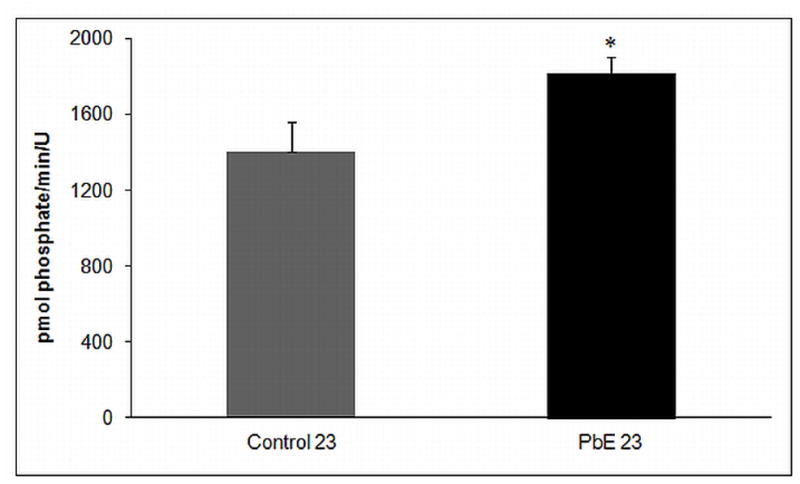
Ser/Thr Phosphatase activity in the cerebral cortex of control and Pb-exposed primate brains (PbE, exposed as infants only) were measured by using Ser/Thr phosphatase assay kit. The bar diagram represents mean +/− SEMs (n=3). *p<0.05 as compared to control 23-year old.
3.5. Tau pathology
Immunohistochemical analysis of the frontal association cortex was undertaken to determine if the observed molecular alterations in tau levels were accompanied by qualitative changes in the pathological features of the brains of these animals. The brains of aged monkeys developmentally exposed to Pb revealed an increase in phosphorylated tau immunoreactivity and deposits compared to age-matched controls (Fig. 6).
Fig. 6. Photomicrographs showing tau pathology in the frontal association cortex of 23-year old Cynomolgus monkeys following developmental exposure to Pb.
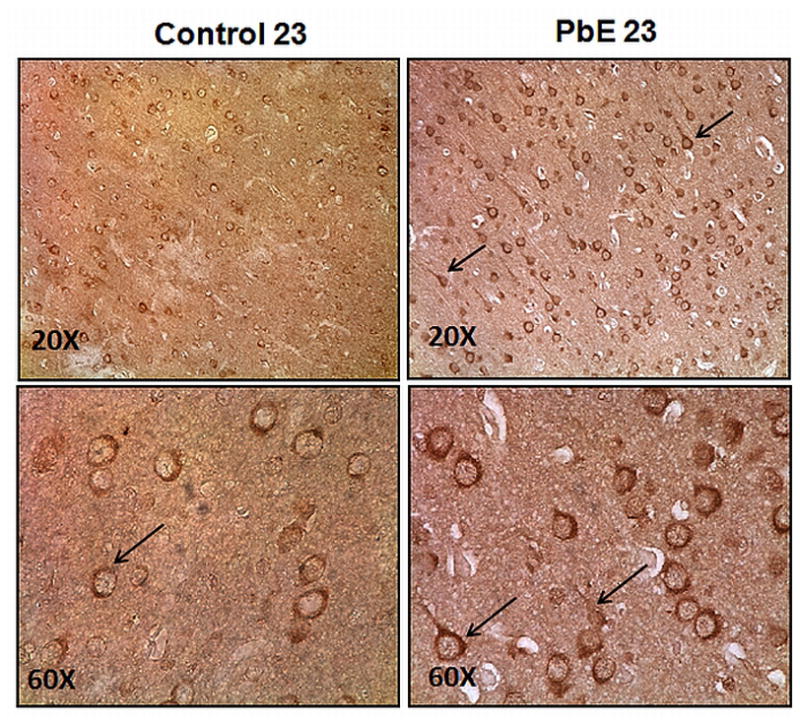
Immunohistochemical detection of enhanced phosphorylated tau reactivity by AT8 antibody in the frontal cortex of Control and Pb-exposed (PbE, exposed as infants only) 23-year old Cynomolgus monkeys. Original magnifications: 20X (top); 60X (bottom). The arrows denote phosphorylated tau immunoreactivity.
4. Discussion
The persistence of heavy metals in the environment has been of great concern and poses a a potential risk to the environment and human health (Lone et al., 2008). The heavy metal Pb is a potent neurotoxicant and children are believed to be the most vulnerable to Pb toxicity due to a causal link between low-level chronic exposure to Pb and deficiencies in intelligence quotients at blood levels below the CDC safe level of 10μg/dl (Bijoor et al., 2012, Canfield et al., 2003, Ziegler et al., 1978). While some studies have indicated that exposure to Pb in early life could have long-term effects (Needleman et al., 1990), our lab has provided convincing evidence from studies in rodents and primates indicating that indicated early life exposure has a latent impact on neurodegenerative processes (Bihaqi et al., 2011), thereby introducing the first environmental model of early life exposure that predetermines outcomes of the aging brain through epigenetic reprogramming (Bihaqi et al., 2011).
Given the important implications of these findings linking early life Pb exposure to latent increases in the expression of AD related genes; it became apparent that other AD-associated genes and proteins should also be investigated to further clarify the role of Pb in influencing the pathogenesis of AD. The protein tau is predominantly found in neuronal axons where it modulates the stability and assembly of microtubules thereby ensuring axonal growth and effective axonal transport. The loss of normal tau function can be harmful and its consequences can promote neurodegenerative disease (Gendron and Petrucelli, 2009). Our results reveal a large increase in the total tau protein and mRNA levels in the cerebral cortex of aged primates developmentally exposed to Pb.
Specificity proteins (Sp1, Sp3 and Sp4) have been associated as major transcription factor binding to GC box in neuronal cells. The promoter of the tau gene is GC (guanine-cytosine) rich and contains Sp1 binding motifs proximal to exon 1. Sp1 binding sites are thought to be involved with the control of the neuronal specific expression of tau (Citron et al., 2008, Heicklen-Klein and Ginzburg, 2000), and our lab has previously demonstrated the importance of Sp1 as mediator in Pb induced latent increases in transcriptional processes (Basha et al., 2005b). Consistent with our previous reports, primates developmentally exposed to Pb exhibited a significant increase in Sp1 and Sp3 mRNA levels, thus suggesting that both tau and its transcriptional activator may be subject to developmental epigenetic reprogramming (Mastroeni et al., 2011), which may have resulted in the up-regulation of the de novo synthesis of total tau at older ages.
Hyperphosphorylation of the cytoskeletal protein tau is responsible for the formation of neurofibrillary tangles (NFT) (Gendron and Petrucelli, 2009). Hyperphosphorylation of tau is known to contribute to impaired microtubule assembly in brain extracts from AD patients (Iqbal et al., 1986). The functional impact of tau phosphorylation depends on the specific phosphorylation sites and the extent of phosphorylation (Liu et al., 2007). In addition, hyperphosphorylated tau from AD brains has been found to segregate from normal tau, and self-assemble into tangles of paired helical/straight filaments (Alonso et al., 1994, Alonso et al., 1997). The increase in total tau content found in aged primates after infantile Pb exposure provides more substrates for phosphorylation. We show that 23 year-old primates with developmental exposure to Pb exhibit an increase in phosphorylation at the following sites; Thr 181, Thr 212, Ser 396, and Ser 235. These observations agree with studies by Hu et al., who observed increased levels of total tau and tau phosphorylated at Ser-396/Ser-404 in AD patients (Hu et al., 2002), and other studies that have found increases in phosphorylation of Thr-181 and Ser-235 in AD patients (Sengupta et al., 2006). Thr-212 is among the most readily phosphorylated sites in tau and a possible cause of tau acquiring toxic activity in AD brains (Alonso Adel et al., 2004).
The process of phosphorylation is driven by numerous kinases including the cdk5, with activators p35 and p39 directing its activity (Dhavan and Tsai, 2001). Studies have found cdk5/p35 to play a pivotal role in neuronal migration and differentiation of immature neurons as well as in neurotransmitter release (Tomizawa et al., 2002). Conversion of p35 to p25 has been implicated in the aberrant cdk5 activity advocating its role in AD. In addition, p35 is truncated to p25 in AD and the complex of cdk5 and p25 leads to mislocalization of the kinase to the soma, resulting in neuronal degeneration (Patrick et al., 1999). Recent reports have shown cytoskeletal disruptions and hyperphosphorylation of tau in transgenic mice that are similar to the AD pathology in mice expressing human p25 (Cruz et al., 2003).
Our results revealed an upregulation in the protein and mRNA levels of cdk5, a decrease in the regulatory p35 and subsequent increase in p25 (Fig. 3B). These changes were also accompanied by increases in Sp1 and Sp3 mRNA levels (Fig. 4). Furthermore, earlier reports by Valin et al., showed the importance of repeated GC box elements that bind Sp1, Sp3 and Sp4 in-vitro, to direct neuron-specific expression of the cdk5 regulator p35 (Valin et al., 2009). Thus, these posttranslational regulators of tau may also be subject to the same epigenetic reprogramming and transcriptional machinery that also impacts tau gene expression, culminating in not only an elevation of total tau but also in the enzymes involved in its phosphorylation (Bihaqi et al., 2012).
These variable changes in cdk5 accompanied by the increased phosphorylation of tau on multiple sites in primates with infantile exposure to Pb suggest that impaired activity of protein phosphatases capable of dephosphorylating many sites on tau may also contribute to tau hyperphosphorylation. Among the various Ser/Thr phosphatases present in the brain, protein phosphatase 1 (PP1), protein phosphatase 2A (PP2A) and protein phosphatase 2B (PP2B) are the major ones (Cohen, 1989, Liu et al., 2005). PP2A constitutes about 90% of total protein phosphatase activity (Gong et al., 1995). Seminal studies by Rahman et al observed that Pb exposure induces overexpression of PP1 and PP2B, and over activation of PP2A in primary human neurons. Such changes have been associated with learning and memory impairment (Rahman et al., 2011).
Consistent with earlier findings, our results revealed a significant increase in Ser/Thr phosphatase activity in primates developmentally exposed to Pb. Since tau acts as a substrate for kinases as well as phosphatases, the increased Ser/Thr phosphatase activity in aged primates exposed to Pb as infants maybe attributed to an effort to counterbalance the action of kinases. However, the overall outcome appears to favor an overabundance of phosphorylated tau as the increase in tau phosphorylation was much higher than the over activation of phosphatases.
The molecular and biochemical changes observed in 23 year old primates are also accompanied by AD like pathology in the exposed primates. Addition to changes in amyloidosis previously reported by us (Wu et al., 2008), intracellular staining revealed a qualitative enhancement in the accumulation of immunoreactivity for phosphorylated tau and tau deposits in Pb exposed primates (Fig. 6). Support for the possibility of developmental exposure to Pb resulting in the formation of tau pathology in humans was demonstrated in a patient who survived severe Pb toxicity at 2 years of age, and died of severe mental deterioration at the age of 42; his brain exhibited senile plaques and neurofibrillary tangles (NFTs) (Niklowitz and Mandybur, 1975).
Although these studies are limited by the small number of primates available for this study, the findings suggest a role for early Pb exposure in the pathogenesis of AD and other tauopathies and demonstrate the importance of the period of exposure and the developmental stage of the brain in sustaining damage that can increase susceptibility to such neurodegeneration later in life.
Highlights.
Developmental exposure to the environmental toxicant (Pb) early in life affects the expression of the tau gene and its product later in life.
An increase in the kinase activity of cdk5 that is associated with an increase in the p25 activator appears to promote an elevation in the phosphorylation of tau at specific sites which contribute to the formation of tangles.
This paper presents findings in primates that implicate an environmental agent (Pb) in the pathogenesis of AD and demonstrates that early development is an important period of vulnerability which could increase future susceptibility to neurodegeneration and AD pathology.
Acknowledgments
This research was supported by the Intramural Research Program of the National Institutes of Health (NIH), National Institute of Environmental Health Sciences (NIEHS) and by grant NIH-5RO1ES015867-03. The research core facility was funded (P20RR016457) by the National Center for Research Resources (NCRR), a component of NIH. The authors would also like to extend their thanks to Dr. Jean Harry at National Institutes of Health, Research Triangle Park, NC, USA and Deborah Rice at Maine Department of Health and Human Services, 11 State House Station, Augusta, ME, USA for their kind assistance.
Footnotes
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alashwal H, Dosunmu R, Zawia NH. Integration of genome-wide expression and methylation data: relevance to aging and Alzheimer’s disease. Neurotoxicology. 2012;33:1450–3. doi: 10.1016/j.neuro.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso AC, Zaidi T, Grundke-Iqbal I, Iqbal K. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc Natl Acad Sci U S A. 1994;91:5562–6. doi: 10.1073/pnas.91.12.5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso AD, Grundke-Iqbal I, Barra HS, Iqbal K. Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc Natl Acad Sci U S A. 1997;94:298–303. doi: 10.1073/pnas.94.1.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso Adel C, Mederlyova A, Novak M, Grundke-Iqbal I, Iqbal K. Promotion of hyperphosphorylation by frontotemporal dementia tau mutations. J Biol Chem. 2004;279:34873–81. doi: 10.1074/jbc.M405131200. [DOI] [PubMed] [Google Scholar]
- Barker DJ. Fetal origins of cardiovascular disease. Ann Med. 1999;31 (Suppl 1):3–6. [PubMed] [Google Scholar]
- Barker DJ, Winter PD, Osmond C, Margetts B, Simmonds SJ. Weight in infancy and death from ischaemic heart disease. Lancet. 1989;2:577–80. doi: 10.1016/s0140-6736(89)90710-1. [DOI] [PubMed] [Google Scholar]
- Basha MR, Murali M, Siddiqi HK, Ghosal K, Siddiqi OK, Lashuel HA, et al. Lead (Pb) exposure and its effect on APP proteolysis and Abeta aggregation. FASEB J. 2005a;19:2083–4. doi: 10.1096/fj.05-4375fje. [DOI] [PubMed] [Google Scholar]
- Basha MR, Wei W, Bakheet SA, Benitez N, Siddiqi HK, Ge YW, et al. The fetal basis of amyloidogenesis: exposure to lead and latent overexpression of amyloid precursor protein and beta-amyloid in the aging brain. J Neurosci. 2005b;25:823–9. doi: 10.1523/JNEUROSCI.4335-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bihaqi SW, Huang H, Wu J, Zawia NH. Infant exposure to lead (Pb) and epigenetic modifications in the aging primate brain: implications for Alzheimer’s disease. J Alzheimers Dis. 2011;27:819–33. doi: 10.3233/JAD-2011-111013. [DOI] [PubMed] [Google Scholar]
- Bihaqi SW, Schumacher A, Maloney B, Lahiri DK, Zawia NH. Do epigenetic pathways initiate late onset Alzheimer disease (LOAD): towards a new paradigm. Curr Alzheimer Res. 2012;9:574–88. doi: 10.2174/156720512800617982. [DOI] [PubMed] [Google Scholar]
- Bijoor AR, Sudha S, Venkatesh T. Neurochemical and neurobehavioral effects of low lead exposure on the developing brain. Indian J Clin Biochem. 2012;27:147–51. doi: 10.1007/s12291-012-0190-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns A, Tomlinson BE, Mann DM. Observations on the brains of demented old people. B.E. Tomlinson, G. Blessed and M. Roth, Journal of the Neurological Sciences (1970) 11, 205–242 and Observations on the brains of non-demented old people. B.E. Tomlinson, G. Blessed and M. Roth, Journal of Neurological Sciences (1968) 7, 331–356. Int J Geriatr Psychiatry. 1997;12:785–90. [PubMed] [Google Scholar]
- Canfield RL, Henderson CR, Jr, Cory-Slechta DA, Cox C, Jusko TA, Lanphear BP. Intellectual impairment in children with blood lead concentrations below 10 microg per deciliter. N Engl J Med. 2003;348:1517–26. doi: 10.1056/NEJMoa022848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citron BA, Dennis JS, Zeitlin RS, Echeverria V. Transcription factor Sp1 dysregulation in Alzheimer’s disease. J Neurosci Res. 2008;86:2499–504. doi: 10.1002/jnr.21695. [DOI] [PubMed] [Google Scholar]
- Cohen P. The structure and regulation of protein phosphatases. Annu Rev Biochem. 1989;58:453–508. doi: 10.1146/annurev.bi.58.070189.002321. [DOI] [PubMed] [Google Scholar]
- Cruz JC, Tseng HC, Goldman JA, Shih H, Tsai LH. Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron. 2003;40:471–83. doi: 10.1016/s0896-6273(03)00627-5. [DOI] [PubMed] [Google Scholar]
- Dhavan R, Tsai LH. A decade of CDK5. Nat Rev Mol Cell Biol. 2001;2:749–59. doi: 10.1038/35096019. [DOI] [PubMed] [Google Scholar]
- Dosunmu R, Alashwal H, Zawia NH. Genome-wide expression and methylation profiling in the aged rodent brain due to early-life Pb exposure and its relevance to aging. Mech Ageing Dev. 2012;133:435–43. doi: 10.1016/j.mad.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendron TF, Petrucelli L. The role of tau in neurodegeneration. Mol Neurodegener. 2009;4:13. doi: 10.1186/1750-1326-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gluckman PD, Hanson MA. Living with the past: evolution, development, and patterns of disease. Science. 2004;305:1733–6. doi: 10.1126/science.1095292. [DOI] [PubMed] [Google Scholar]
- Gong CX, Shaikh S, Wang JZ, Zaidi T, Grundke-Iqbal I, Iqbal K. Phosphatase activity toward abnormally phosphorylated tau: decrease in Alzheimer disease brain. J Neurochem. 1995;65:732–8. doi: 10.1046/j.1471-4159.1995.65020732.x. [DOI] [PubMed] [Google Scholar]
- Heicklen-Klein A, Ginzburg I. Tau promoter confers neuronal specificity and binds Sp1 and AP-2. J Neurochem. 2000;75:1408–18. doi: 10.1046/j.1471-4159.2000.0751408.x. [DOI] [PubMed] [Google Scholar]
- Hu YY, He SS, Wang X, Duan QH, Grundke-Iqbal I, Iqbal K, et al. Levels of nonphosphorylated and phosphorylated tau in cerebrospinal fluid of Alzheimer’s disease patients : an ultrasensitive bienzyme-substrate-recycle enzyme-linked immunosorbent assay. Am J Pathol. 2002;160:1269–78. doi: 10.1016/S0002-9440(10)62554-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal K, Grundke-Iqbal I, Zaidi T, Merz PA, Wen GY, Shaikh SS, et al. Defective brain microtubule assembly in Alzheimer’s disease. Lancet. 1986;2:421–6. doi: 10.1016/s0140-6736(86)92134-3. [DOI] [PubMed] [Google Scholar]
- Iqbal K, Liu F, Gong CX, Grundke-Iqbal I. Tau in Alzheimer disease and related tauopathies. Curr Alzheimer Res. 2010;7:656–64. doi: 10.2174/156720510793611592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazawa M, Cheng D, Laferla FM. Chronic copper exposure exacerbates both amyloid and tau pathology and selectively dysregulates cdk5 in a mouse model of AD. J Neurochem. 2009;108:1550–60. doi: 10.1111/j.1471-4159.2009.05901.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddell MB, Lovestone S, Owen MJ. Genetic risk of Alzheimer’s disease: advising relatives. Br J Psychiatry. 2001;178:7–11. doi: 10.1192/bjp.178.1.7. [DOI] [PubMed] [Google Scholar]
- Liu F, Grundke-Iqbal I, Iqbal K, Gong CX. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci. 2005;22:1942–50. doi: 10.1111/j.1460-9568.2005.04391.x. [DOI] [PubMed] [Google Scholar]
- Liu F, Li B, Tung EJ, Grundke-Iqbal I, Iqbal K, Gong CX. Site-specific effects of tau phosphorylation on its microtubule assembly activity and self-aggregation. Eur J Neurosci. 2007;26:3429–36. doi: 10.1111/j.1460-9568.2007.05955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lone MI, He ZL, Stoffella PJ, Yang XE. Phytoremediation of heavy metal polluted soils and water: progresses and perspectives. J Zhejiang Univ Sci B. 2008;9:210–20. doi: 10.1631/jzus.B0710633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastroeni D, Grover A, Delvaux E, Whiteside C, Coleman PD, Rogers J. Epigenetic mechanisms in Alzheimer’s disease. Neurobiol Aging. 2011;32:1161–80. doi: 10.1016/j.neurobiolaging.2010.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliore L, Coppede F. Genetics, environmental factors and the emerging role of epigenetics in neurodegenerative diseases. Mutat Res. 2009;667:82–97. doi: 10.1016/j.mrfmmm.2008.10.011. [DOI] [PubMed] [Google Scholar]
- Needleman HL, Schell A, Bellinger D, Leviton A, Allred EN. The long-term effects of exposure to low doses of lead in childhood. An 11-year follow-up report. N Engl J Med. 1990;322:83–8. doi: 10.1056/NEJM199001113220203. [DOI] [PubMed] [Google Scholar]
- Niklowitz WJ, Mandybur TI. Neurofibrillary changes following childhood lead encephalopathy. J Neuropathol Exp Neurol. 1975;34:445–55. doi: 10.1097/00005072-197509000-00006. [DOI] [PubMed] [Google Scholar]
- Nordberg M, Winblad B, Fratiglioni L, Basun H. Lead concentrations in elderly urban people related to blood pressure and mental performance: results from a population-based study. Am J Ind Med. 2000;38:290–4. doi: 10.1002/1097-0274(200009)38:3<290::aid-ajim7>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402:615–22. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- Price DL, Sisodia SS. Cellular and molecular biology of Alzheimer’s disease and animal models. Annu Rev Med. 1994;45:435–46. doi: 10.1146/annurev.med.45.1.435. [DOI] [PubMed] [Google Scholar]
- Rahman A, Brew BJ, Guillemin GJ. Lead dysregulates serine/threonine protein phosphatases in human neurons. Neurochem Res. 2011;36:195–204. doi: 10.1007/s11064-010-0300-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice DC. Lead-induced behavioral impairment on a spatial discrimination reversal task in monkeys exposed during different periods of development. Toxicol Appl Pharmacol. 1990;106:327–33. doi: 10.1016/0041-008x(90)90251-o. [DOI] [PubMed] [Google Scholar]
- Rice DC. Effect of lead during different developmental periods in the monkey on concurrent discrimination performance. Neurotoxicology. 1992;13:583–92. [PubMed] [Google Scholar]
- Sengupta A, Grundke-Iqbal I, Iqbal K. Regulation of phosphorylation of tau by protein kinases in rat brain. Neurochem Res. 2006;31:1473–80. doi: 10.1007/s11064-006-9205-9. [DOI] [PubMed] [Google Scholar]
- Subaiea GM, Adwan LI, Ahmed AH, Stevens KE, Zawia NH. Short-term treatment with tolfenamic acid improves cognitive functions in Alzheimer’s disease mice. Neurobiol Aging. 2013 doi: 10.1016/j.neurobiolaging.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomizawa K, Ohta J, Matsushita M, Moriwaki A, Li ST, Takei K, et al. Cdk5/p35 regulates neurotransmitter release through phosphorylation and downregulation of P/Q-type voltage-dependent calcium channel activity. J Neurosci. 2002;22:2590–7. doi: 10.1523/JNEUROSCI.22-07-02590.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valin A, Cook JD, Ross S, Saklad CL, Gill G. Sp1 and Sp3 regulate transcription of the cyclin-dependent kinase 5 regulatory subunit 2 (p39) promoter in neuronal cells. Biochim Biophys Acta. 2009;1789:204–11. doi: 10.1016/j.bbagrm.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voytko ML. Nonhuman primates as models for aging and Alzheimer’s disease. Lab Anim Sci. 1998;48:611–7. [PubMed] [Google Scholar]
- Weisskopf MG, Proctor SP, Wright RO, Schwartz J, Spiro A, 3rd, Sparrow D, et al. Cumulative lead exposure and cognitive performance among elderly men. Epidemiology. 2007;18:59–66. doi: 10.1097/01.ede.0000248237.35363.29. [DOI] [PubMed] [Google Scholar]
- Wu J, Basha MR, Brock B, Cox DP, Cardozo-Pelaez F, McPherson CA, et al. Alzheimer’s disease (AD)-like pathology in aged monkeys after infantile exposure to environmental metal lead (Pb): evidence for a developmental origin and environmental link for AD. J Neurosci. 2008;28:3–9. doi: 10.1523/JNEUROSCI.4405-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler EE, Edwards BB, Jensen RL, Mahaffey KR, Fomon SJ. Absorption and retention of lead by infants. Pediatr Res. 1978;12:29–34. doi: 10.1203/00006450-197801000-00008. [DOI] [PubMed] [Google Scholar]