

Abstract
In recent years the role of acetylation has gained ground as an essential modulator of intermediary metabolism in skeletal muscle. Imbalance in energy homeostasis or chronic cellular stress, due to diet, aging or disease, translate into alterations in the acetylation levels of key proteins which governs bioenergetics, cellular substrate use and/or changes in mitochondrial content and function. For example, cellular stress induced by exercise or caloric restriction can alter the coordinated activity of acetyltransferases and deacetylases to increase mitochondrial biogenesis and function in order to adapt to low energetic levels. The natural duality of these enzymes, as metabolic sensors and effector proteins, have helped biologists understand how the body can integrate seemingly distinct signaling pathways to control mitochondrial biogenesis, insulin sensitivity, glucose transport, reactive oxygen species handling, angiogenesis and muscle satellite cell proliferation/differentiation. Our review will summarize the recent developments related to acetylation dependent responses following metabolic stress in skeletal muscle.
Keywords: Acetylation, Sirtuins, Muscle adaptation, Energy stress, Oxidative Metabolisms, glucose homeostasis
Skeletal muscle is an essential organ for the maintenance of mobility and whole body metabolic health. The skeletal muscle demonstrates plasticity in mitochondrial content and function, vascular remodeling, glucose mobilization, and myogenesis/atrophy following metabolic stressors such as diet or exercise. As such, metabolic stress can have both acute and chronic effects on skeletal muscle homeostasis. Metabolic sensing is therefore a crucial component of skeletal muscle adaptation to various stressors and is imperative for whole body health.
This review will examine the essential metabolic sensing pathways that are regulated by posttranslational acetylation or deacetylation modifications that define the activity level and/or subcellular translocation of key proteins in muscle. Lysineacetyltransferases (KATs; previously known as HATs) and -deacetylases (KDACs; also known as HDACs) regulate many proteins involved in intermediate metabolism (Guan & Xiong 2011; Patel et al. 2011), often by “sensing” the redox state and/or the energetic state of the cell. These two states, although seemingly similar, describe different processes within the cell. The redox state often implies the NAD+/NADH or NADP+/NADPH ratio in a cell, which is correlated to the type of substrates being used to generate energy for the cell (Graham & Saltin 1989). Muscle cells that rely on glycolysis would typically be defined as having a lower NAD+/NADH ratio, while myocytes that depend on fatty acid oxidation for energy production have a higher NAD+/NADH quotient (Guarente 2012). Not so divergent from this description, but an important distinction, the energetic state of a muscle conveys the state of available energy to perform work, in the form of ATP, and yet is also inherently integrated with the redox state of a cell. Given that the muscle is a large organ, ultimately, whole body energy homeostasis relies to a large extent on muscle mitochondrial function for aerobic ATP production and maintenance of physiological redox conditions. This reliance on muscle, in turn, can prevent insulin resistance which can predispose to obesity, diabetes and cardiovascular disease, all of which contribute to accelerated aging (Patti et al. 2003; Joseph et al. 2012).
The known and potential roles of sirtuin proteins in muscle energy homeostasis
The sirtuin proteins (SIRT1 to SIRT7) are a major subfamily of the KDACs. Sirtuins have become increasingly regarded as regulators of metabolism [reviewed in (Guarente 2012; Houtkooper et al. 2012)]. In particular, the SIRT1 homolog has been identified as a metabolically regulated protein capable of sensing cellular redox and energy status that can then illicit mitochondrial and redox defense adaptations (Cantó et al. 2012). SIRT2, a predominantly cytosolic protein, has not been studied in muscle but has been shown to regulate autophagy in mammalian cells (Zhao et al. 2010) and differentiation in adipocytes (Jing et al. 2007). Recent evidence has also linked SIRT2 with a potential role in cardiac protection from ischemic injury (Narayan et al. 2012). The mitochondrially located sirtuins, SIRT3, SIRT4 and SIRT5, are known to modulate the acetylation status of mitochondrial proteins and may therefore directly influence muscle metabolism and stress responses [reviewed in (Pirinen et al. 2012)]. SIRT6 and SIRT7 are preferentially nuclear proteins. Of these nuclear proteins, SIRT6 expression is elevated with exercise and plays a role in glycolytic regulation that will be discussed further in this review (Koltai et al. 2010; Xiao et al. 2010). In contrast, SIRT7 has a low expression level in skeletal muscle, but it may play a role in cardiac muscle function and plasticity (Ford 2006). Although not discussed in this review, there is also recent evidence that non-sirtuin deacetylases can act as metabolic sensors and may regulate metabolism and other processes in a metabolite sensitive manner that is dependent on changes in conenzyme A (CoA) derivatives as well as NADPH (Vogelauer et al. 2012).
Despite the recent surge in sirtuin research, there are still many questions to be answered concerning the influence of these energy-sensing proteins on muscle metabolism and homeostasis. Elucidation of sirtuin pathways will certainly lead to a better understanding of the metabolic response of muscle to exercise or degenerative processes brought about by physiological stressors such as disease, aging or unhealthy diets.
The SIRT1 and AMPK nexus regulates metabolic-dependent acetylation
An understanding of the redox state and energetic state of muscle is important during any physiological condition, since the balance of these states determines the coordinated response of various energy sensors/effectors to regain muscle homeostasis. For instance, during both aerobic exercise training and caloric restriction (CR), skeletal muscle bioenergetics is coordinated by metabolic sensing proteins, which independently detect changes in both the ratio of redox and energetic metabolites/substrates, more specifically the NAD+/NADH and AMP/ATP ratios. These energy sensing proteins include NAD+-dependent deacetylase sirtuin proteins (Imai et al. 2000), with our focus here being SIRT1, SIRT3 and SIRT6 (Houtkooper et al. 2010), and the AMP-activated protein kinase (AMPK) (Hardie et al. 2012). These effector proteins are activated upon changes in the abundance of the corresponding metabolites NAD+ and AMP. The level of these metabolites depend on a complex series of energetic (Xiao et al. 2007; Hardie et al. 2012) and redox pathways (Cantó & Auwerx 2012; Mouchiroud et al. 2013), along with a reliance on the quantity, type and location of available energy substrate stores.
During exercise, muscle requires an abundant supply of ATP to maintain the rapid cycling of crossbridges between actin and myosin and the oscillatory changes in intracellular Ca2+ between the extracellular space and the sarcoplasmic reticulum. Additionally, during aerobic metabolism NADH is extensively used to transfer electron loads from intermediary substrates to the electron transport chain to synthesize ATP via oxidative phosphorylation (OXPHOS). It is therefore evident that both AMPK and SIRT1, as AMP- and NAD+-sensors, are central for detecting energy stress situations during muscle metabolism.
It is through an interaction of SIRT1 and AMPK signaling pathways that muscle metabolism is tuned to adapt to the demands of energetic stress. Initial evidence demonstrating the similarity of these two effector proteins was revealed in longevity studies. In particular, early Sir2p (the yeast SIRT1 homolog) studies suggested that this protein increased longevity in a NAD+-dependent manner (Tissenbaum & Guarente 2001; Anderson et al. 2003; Lin et al. 2004). Lifespan extension also occurred in C. elegans overexpressing the AMPKα subunit (AAK-2) of the AMPK hetero-trimeric complex (Apfeld et al. 2004). During periods of energy deprivation, AMPK is responsible for the phosphorylation of many proteins, including metabolic enzymes, such as acetyl-CoA carboxylase (ACC) for fatty acid oxidation, or the peroxisome proliferator-activated receptor-gamma coactivator 1α (PGC-1α), a coactivator for the transcription of nuclear encoded mitochondrial proteins (Winder et al. 2000; Nemoto 2005; Jager et al. 2007; Rodgers et al. 2008). During energy deprivation there is also a rise in the NAD+/NADH ratio, which enables SIRT1 to deacetylate up to 13 potential acetyl-acceptor lysine sites on PGC-1α, resulting in the activation of PGC-1α (Nemoto 2005; Rodgers et al. 2005; Lerin et al. 2006).
The integration of AMPK and SIRT1 signaling, and thus the connection between AMPK activity and regulation of protein acetylation, is evident in pharmacological experiments where AICAR, an activator of AMPK, was shown to induce SIRT1 deacetylase activity (Cantó et al. 2009; Gurd et al. 2011). Although not yet shown in the muscle, experiments performed in vivo and in vitro demonstrated that AMPK activation can lead to the dissociation of the inhibitor deleted in breast cancer 1 (DBC1) from SIRT1, resulting in its activation (Nin et al. 2012). Furthermore, CR of mice and glucose restriction of primary myoblasts both culminate in the AMPK-mediated induction of nicotinamide phosphoribosyltransferase (NAMPT) transcription (Fulco et al. 2008; Cantó et al. 2009), the rate-limiting enzyme that converts NAM to NAD+ (Revollo et al. 2004; Houtkooper et al. 2010). Elevations in NAMPT expression increases the NAD+/NADH ratio (Preiss & Handler 1957; Kosaka et al. 1971; Bieganowski & Brenner 2004), thereby activating SIRT1 (Revollo et al. 2004; Hayashida et al. 2010), and thus providing a clear mechanistic link between the activation of AMPK and an increase in SIRT1 activity. Conversely, AMPKγ3-KO animals have lower levels of NAD+, NAMPT and deacetylated PGC-1α following exercise (Cantó et al. 2010). The evidence is therefore convincing that AMPK controls the activity of SIRT1. In a reciprocal manner, some evidence has demonstrated that SIRT1 can also control the activity of AMPK. Through the use of an inducible SIRT1-KO model, AMPK-directed stimulation of muscle mitochondrial biogenesis by resveratrol was shown to be dependent on SIRT1 (Price et al. 2012). In addition, human cultured myotubes that carry a point mutation in SIRT1 exhibit reduced AMPK activation (Biason-Lauber et al. 2013). In addition, glucose induced changes in AMPK activity appear to depend on SIRT1 abundance or activity in liver cells (Suchankova et al. 2009). Therefore, it appears that these two pathways can be independently activated but may posses overlapping and potentiating properties.
Positive regulation of acetyltransferases antagonizes the role of AMPK and SIRT1
Structural and functional studies have shown that many KATs exist as multisubunit enzymatic complexes that contain acetyl-CoA-binding domains and bromodomains. Notably, bromodomains are the only known protein modules that are able to recognize the specific lysine residues that are acetylated (Mujtaba et al. 2007). General control non-repressed protein 5 (GCN5) and P300/CBP-associated factor (PCAF) are members of the GCN5-related N-acetyltransferases (GNAT) family (Roth et al. 2001). Other important KAT families include P300/CBP (protein of 300 kDa and CREB-binding protein) (Wang et al. 2008), MYST, named after its family members (MOZ, YBF2/SAS3, SAS2, TIP60), and the steroid receptor coactivator (SRC) family that includes SRC-1 (or NCOA1), SRC-2 (TIF2, GRIP1, or NCOA2) and SRC-3 (ACTR, RAC3, AIB1, P/CIP or TRAM) (Chen et al. 1997; Onate et al. 1998). Of these KATs, GCN5, PCAF, P300/CBP, TIP60, SRC-1 and SRC-3 have been shown to immunoprecipitate with PGC-1α (Puigserver et al. 1999; Lerin et al. 2006; Coste et al. 2008), yet the inhibition PGC-1α through acetylation has only been shown for GCN5 and its close homolog PCAF (Lerin et al. 2006). GCN5 also acetylates and inactivates PGC-1β, another family member of the PGC-1 transcriptional co-activators (Kelly et al. 2009).
Various regulatory nodes have been examined that help describe the activation or deactivation of the inhibitory GCN5/PGC-1α interaction. One potential node includes the subcellular translocation of GCN5 to the cytosol to attenuate its interaction with nuclear PGC-1α, as has been shown to occur with an acute bout of exercise (Philp et al. 2011). Second, the deacetylase SIRT6 unexpectedly induces PGC-1α acetylation through the deacetylation and activation of GCN5 (Dominy et al. 2012). This effect is attenuated with overexpression of a deacetylase-inactive mutant form of SIRT6. However, this finding appears to be in theoretical contention to the observed increase of SIRT6 protein in muscle with exercise (Koltai et al. 2010; Xiao et al. 2010) and the ability of SIRT6 to inhibit HIF-1α leading to decreased glycolysis and activation of oxidative phosphorylation (Zhong et al. 2010). Furthermore, muscle cells from SIRT6-KO mice exhibit increased GLUT4 membrane translocation along with increased whole cell expression of GLUT1 (Xiao et al. 2010), thus supporting the role of SIRT6 in glycolysis. Third, GCN5 expression in skeletal muscle is increased by the transcriptional coactivator SRC-3 (Coste et al. 2008). In line with this, germline SRC-3 KO mice display increased mitochondrial function, improved glucose homeostasis and are protected against obesity. Furthermore, the expression of SRC-3, like GCN5, is sensitive to fasting and high fat diet (Coste et al. 2008). Fourth, CITED2 (CBP- and P300-interacting transactivator with glutamic acid– and aspartic acid–rich COOH-terminal domain 2) has been shown to disrupt the acetylation and inhibition of PGC-1α by GCN5 in hepatocytes (Sakai et al. 2012). Upon insulin activation of PI3K/AKT signaling the interaction between CITED2 and GCN5 is attenuated, allowing for PGC-1α acetylation, while glucagon promotes CITED2 protein expression, releasing PGC-1α from the negative control of GCN-5. Although the influence of CITED2 on GCN5/PGC-1α interaction in muscle has not been examined, CITED2 mRNA has been shown to decrease in muscle with insulin stimulation (Coletta et al. 2008). In addition, studies in mouse muscle myotubes demonstrated that CITED2 could block the effects of glucocorticoids on muscle cell atrophy. Lastly, since acetyltransferases require acetyl-CoA as a substrate for lysine acetylation, nuclear or cytosolic supplies of acetyl-CoA may regulate the activity of GCN5 or PCAF. During times of energy surplus tricarboxylic acid cyclederived citrate can be converted into acetyl-CoA by ATP citrate lyase, resulting in elevated nuclear and cytosolic acetyl-CoA, which have been shown to affect histone acetylation (Wellen et al. 2009). In analogy, increased levels of acetyl-CoA are postulated to also potentiate PGC-1α acetylation through GCN5 or PCAF, although experimental evidence for this last hypothesis is lacking at present [reviewed in (Jeninga et al. 2010)].
Defining the exact signaling network that governs the activity of GCN5/PCAF in muscle function will require extensive future work, which should include the characterization of muscle-specific GCN5/PCAF single or double KO animals and potentially studies in model organisms, such as worms, which contain a single GCN5/PCAF homolog, pcaf-1.
The commanding role of the NAD+/NADH ratio in energy homeostasis
As a metabolic cofactor and a redox determinant, NAD+ is also required as an obligatory cosubstrate for sirtuin enzymes. The balance of the oxidized form (NAD+) versus the reduced form (NADH) dictates many of the functions of NAD+-dependant enzymes. In particular, the NAD+/NADH ratio influences the activity of redox reactions during the catabolism of lipids, proteins, carbohydrates, and alcohols. In addition, NAD+ has been shown to impact on glycolysis and oxidative phosphorylation and various fundamental regulatory functions, such as transcription, DNA repair, G-protein coupled signaling and intracellular calcium signaling (Bogan & Brenner 2008; Garten et al. 2009). With all of these essential biological functions relying on NAD+, the availability of NAD+ for sirtuin activity is limited (Rodgers et al. 2005; Sun et al. 2007; Bogan & Brenner 2008; Cantó et al. 2010; Hayashida et al. 2010; Koltai et al. 2010). NAD+ is derived from the amino acid tryptophan or from nicotinic acid or nicotinamide (NAM), which are biosynthesized de novo from vitamin B3 (Magni et al. 2004; Houtkooper et al. 2010; Schenk et al. 2011; Fernandez-Marcos et al. 2012). Interestingly, the NAD+ intermediate, nicotinamide mononucleotide (NMN), can restore NAD+ levels and improve glucose tolerance in HFD-induced diabetic treated intraperitoneally with NMN (Yoshino et al. 2011). Another NAD+ precursor is NAM riboside (NR) (Rowen & Kornberg 1951), which like NAM, can increase intracellular NAD+ levels via the NAD+ salvage pathway (Bieganowski & Brenner 2004). NR-supplemented diets has been shown to increase NAD+ levels and the activity of SIRT1 and SIRT3 in mice, culminating in enhanced skeletal muscle mitochondrial function (Cantó et al. 2012). Delivery of NR into mammalian cells occurs via nucleoside transporters, such as the equilibrative nucleoside transporters (ENT) (Nikiforov et al. 2011). This was demonstrated through the use of the ENT inhibitors dipyridamole or NBTI. It is therefore predicted that NR is delivered into muscle via ENT1, ENT2 and ENT3, all of which were shown to be expressed in skeletal muscle (Lu et al. 2004).
Since NAM can inhibit the different sirtuins (Bitterman 2002), the activity of the NAD+ salvage pathway becomes very important. The rate-limiting enzyme in the salvage pathway is the enzyme NAMPT. In fact, PNC1, the yeast ortholog of mammalian NAMPT, is essential for Sir2p-dependent lifespan extension during CR (Anderson et al. 2003). NAMPT most likely plays a key role in metabolism and potentially healthspan in mammals. Evidence for the importance of NAMPT has been shown in cells deprived of glucose or serum (Yang et al. 2007) and in skeletal muscles of fasted mice (Fulco et al. 2008; Cantó et al. 2009). In addition, NAMPT expression is greatest in highly oxidative muscles and increases in expression with aerobic exercise training in both rats and humans (Costford et al. 2010; Koltai et al. 2010). These observations provide further evidence for the potential use of nutra- or pharmaceuticals to activate sirtuin-mediated deacetylation by enhancing the NAD+ salvage pathway or by introducing substrates for de novo biosynthesis of NAD+.
Protein acetylation status in muscle affects substrate metabolism
Skeletal muscle is highly reliant on oxidative phosphorylation. Lipids, which can be oxidized by mitochondria, are provided by lipolysis in adipose tissue and of intramuscular triacylglycerols. Carbohydrates, in the form of glucose, can be sequestered from the circulation in the post-prandial phase or following hepatic gluconeogenesis or glycogenolysis. Alternatively, glucose can also be generated by intramuscular glycogenolysis. Depending on exercise activity, muscle utilizes various energy stores ranging from glucose and glycogen to fats to synthesize ATP. As exercise intensity increases, there is a decrease in the proportion of energy derived from lipid oxidation and an increase in carbohydrate oxidation [reviewed in (Graham & Saltin 1989; Holloszy et al. 1998; Sahlin & Harris 2008). Conversely, the proportion of energy derived from lipid oxidation rises significantly during prolonged exercise at low and moderate intensities.
Defects in human skeletal muscle mitochondrial function (Simoneau et al. 1995; Simoneau & Kelley 1997; Kelley et al. 2002; Ritov et al. 2005; Samjoo et al. 2013), reduced expression of OXPHOS genes (Mootha et al. 2003; Patti et al. 2003; Samjoo et al. 2013) and aberrant mitochondrial function and morphology (Kelley et al. 2002; Morino et al. 2005), all lead to the accumulation of intramyocellular lipids, which has been proposed as a potential mechanism leading to insulin resistance and diabetes. However, a recent study that examined the effects of endurance training in lean and obese men found no definitive improvement in whole-body insulin resistance despite reporting favorable mitochondrial adaptations and suggested that intramyocellular lipid profiles in obesity may only partially contribute to insulin resistance (Samjoo et al. 2013). Clearly, obesity-induced insulin resistance is a result of perturbations that go beyond mitochondrial biogenesis and intramyocellular lipid oxidation. Since alterations in protein acetylation have strong influences on signaling pathways related to glucose homeostasis and insulin sensitivity, this posttranslational modification may contribute to these discrepancies. For example, the acetylation substrate acetyl-CoA, the product of both glucose and fatty acid breakdown, has been shown to influence mammalian cell histone acetylation, thus highlighting the link between acetylation and metabolism (Wellen et al. 2009). Although it needs to be confirmed that acetyl-CoA levels also influence directly the acetylation of non-histone proteins, the fact that the deacetylase activity of SIRT1 improves insulin sensitivity in diet-induced obese mice, by activating PGC-1α amongst others, also suggests a link between protein acetylation levels and metabolism (Lagouge et al. 2006; Sun et al. 2007; Cantó et al. 2010). Furthermore, increased expression of SIRT1, or treatments with SIRT1 agonists, were shown to be similarly beneficial (Baur et al. 2006; Smith et al. 2009). Conforming to the involvement of SIRT1 in glucose homeostasis, cultured myotubes from patients with SIRT1-L107P mutations exhibit both reduced glucose uptake and insulin sensitivity (Biason-Lauber et al. 2013). The SIRT1-mediated effect on insulin resistance occurs, in part, through the deacetylase-dependent transcriptional repression of PTP1B at the chromatin level (Sun et al. 2007). As a key insulin receptor phosphatase, PTP1B can inhibit the insulin signaling transduction cascade. Furthermore, SIRT1 positively mediates CR-induced insulin sensitivity in muscle by deacetylating STAT-3, thereby enhancing PI3K signaling and glucose uptake (Schenk et al. 2011). Therefore SIRT1 may not only increase β-oxidation of lipids through the activation of PGC-1α, but may also increase insulin sensitivity in a PTP1Band STAT-3-dependent manner to improve substrate handling during times of energy stress. SIRT1 may therefore be a potential target for the prevention or treatment of diabetes in humans through its insulin sensitization effects.
Contrary to the effects of SIRT1 deacetylase activity in the nucleus or cytosol, changes in mitochondrial protein acetylation, brought about by either liver- or muscle-specific SIRT3-KO animals, had no effect on metabolic homeostasis (Fernandez-Marcos et al. 2012). Despite this finding, germline SIRT3 KO mice exhibited both reduced hepatic and muscle fatty acid oxidation, along with reductions in hepatic ATP production, resulting in hypoglycemia, cold intolerance and impaired insulin sensitivity (Hirschey et al. 2010; Jing et al. 2011). SIRT3 may also counterbalance mitochondrial acetylation induced by the GCN5-like 1 (GCN5L1) protein, as was shown to be the case in liver cells (Scott et al. 2012). However, given the discrepant phenotypes observed between the liver- and muscle-specific SIRT3 KO mice and the germline SIRT3 KO mice, the impact of mitochondrial protein acetylation will require further attention (Hirschey et al. 2010; Fernandez-Marcos et al. 2012). Changes in mitochondrial protein acetylation were also demonstrated in the muscle-specific KO model of the mitochondrially located carnitine acetyltransferase (CRAT) (Muoio et al. 2012). These KO mice exhibit disrupted whole body glucose homeostasis due to the altered nutrient control of pyruvate dehydrogenase activity, without displaying changes in mitochondrial function or content. Although not a global acetyltransferase, CRAT specifically converts acetyl-CoA, within the mitochondrial matrix, into a membrane permeant acetylcarnitine ester. Under nutrient excess CRAT promotes the mitochondrial efflux of excess acetyl moieties that otherwise inhibit various metabolic regulatory enzymes, such as pyruvate dehydrogenase. Ultimately, the CRAT-KO mouse results in the inhibition of pyruvate dehydrogenase, which is hypothesized to slow glycolytic flux and creates a negative feedback on glucose uptake.
Evidently, mitochondrial function in muscle is important for lipid oxidation and potentially insulin sensitivity. However, evidence from the studies presented here suggest that the development of mitochondrial dysfunction only mediates part of the insulin resistance seen in muscle, with the dysregulation of insulin signaling and glucose transport via posttranslational acetylation events also being contributing factors for the development of insulin insensitivity.
Changes in the protein acetylation status during muscle normoxia and hypoxia
Skeletal muscle is a highly metabolic tissue and is therefore highly sensitive to oxygenation. As a well-known stimulus for increasing capillarization, exercise contributes to an improved aerobic capacity that is reliant on an angiogenic response system for the induction of capillary growth. An essential mediator of this pathway, vascular endothelial growth factor (VEGF), increases in response to acute exercise (Breen et al. 1996; Amaral et al. 2001). The VEGF promoter possesses a hypoxia response element for the transcription factor hypoxia inducible factor-1α (HIF-1α). However, HIF-1α may not be the only signaling pathway to activate VEGF. Important signals for the activation of myocyte VEGF may include low partial pressure of oxygen, inflammatory cytokines, high oxidative stress, and energy balance-related AMPK activity [reviewed in (Breen et al. 2008). Nonetheless, HIF-1α is an oxygen-sensitive protein that can facilitate ATP generation in mitochondrial respiration by inducing an angiogenic response that elevates VEGF expression to match the new demand of oxygen as a substrate for oxidative respiration (O’Hagan et al. 2009). HIF-1α induction of VEGF-A expression occurs with ischemic conditions in the heart and in skeletal muscle during exercise (Gustafsson et al. 1999; Ameln et al. 2005). With 4 days of chronic muscle electrical stimulation VEGF-A reaches its highest levels, while the capillary-to-muscle fiber ratio continually increases from 7- to 28-day days of muscle stimulation (Dawson & Hudlicka 1989; Hudlicka & Price 1990; Hang et al. 1995). Interestingly, following PGC-1α overexpression in primary human skeletal muscle cells and the resulting induction of mitochondrial biogenesis, there is an increase in oxygen consumption, leading to intracellular hypoxia, and the stabilization of HIF-1α (O’Hagan et al. 2009; Geng et al. 2010). However, PGC-1α can also directly regulate VEGF and angiogenesis in a HIF-1α-independent manner (Arany et al. 2008).
A sirtuin/HIF-1α balancing act: Does SIRT1/SIRT6 mediate the metabolic switch from glycolysis to OXPHOS in muscle?
Although not examined in muscle, recent findings demonstrate that SIRT1 negatively regulates HIF-1α via deacetylation, while PCAF acetylates and activates the oxygen-sensitive protein (Lim et al. 2010). In addition, in mice and cells exposed to hypoxic conditions glycolysis is activated and the tricarboxylic acid cycle is inhibited, leading to decreases in NAD+ levels, reduced SIRT1 activity and activation of HIF-1α. During normoxic conditions, NAD+ concentrations are higher resulting in SIRT1- mediated deacetylation and inactivation of HIF-1α (Lim et al. 2010). From this we hypothesize that HIF-1α may be inactivated by SIRT1 in muscle with exercise, as exercise has been shown increase the NAD+/NADH ratio and SIRT1 activity (Graham & Saltin 1989; Cantó et al. 2009; 2010; Costford et al. 2010). As discussed in the previous section, exercise may result in a temporary hypoxic environment, much like what occurs with overexpression of PGC-1α, which would activate angiogenesis to supply the new demand for oxygen fueled by the increased mitochondrial content. Thus the induction of mitochondrial biogenesis following exercise could lead to intracellular hypoxia, leading to a reduction in OXPHOS activity, NAD+ availability, and the deactivation of SIRT1 and activation of HIF-1α by PCAF (see Figure 1).
Figure 1.
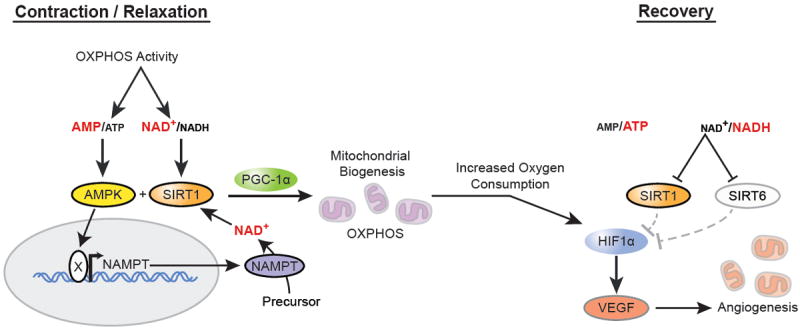
The temporally regulated mitochondrial and angiogenic adaptations following exercise: During exercise, the skeletal muscle experiences an increase in oxidative phosphorylation and an elevation in both the NAD+/NADH and AMP/ATP ratio. Consequently, SIRT1 and AMPK are activated and initiate a metabolic adaptation by increasing the activity of PGC-1α, resulting in the induction of mitochondrial biogenesis. However, these increases in mitochondria content and function have been shown to increase oxygen consumption leading to a temporary state of intracellular hypoxia and HIF-1α activation. With hypoxia comes a reduction in OXPHOS activity and NAD+ availability, which has been shown to inactivate SIRT1 and SIRT6, negative regulators of HIF-1α. Therefore, this temporal inhibition of SIRT1 and SIRT6, along with elevated HIF-1α activity, activates angiogenesis as an adaptation for the increased demand for oxygen following mitochondrial biogenesis.
HIF-1α is also involved in cellular pathways related to glucose metabolism, such as glucose uptake and enzymes that regulate carbohydrate catabolism. Specifically, HIF-1α activates transcription of the glucose transporter 1, hexokinase 1 and 2, lactate dehydrogenase, pyruvate kinase and other enzymes involved in glucose metabolism [reviewed in (Semenza 2001)]. The balance between SIRT1 and HIF-1α regulation of the metabolic switch, going from oxidative to glycolytic metabolism, is corroborated by data showing that in muscle of aged rats and mice SIRT1 activity is reduced, due to low NAD+ levels, leading to an increased dependence on glycolysis (Mouchiroud et al., 2013). The reduction in SIRT1 activity releases the inhibition of HIF-1α, which supports the increasing reliance of aged muscle on glycolysis for energy production (Koltai et al. 2010). Aged animals exhibit increases in lactate and reduced glycolytic intermediates suggesting elevated anaerobic glycolysis (Houtkooper et al. 2011). However, HIF-1α action is not just controlled by SIRT1, SIRT6 also inhibits HIF-1α and glucose uptake in muscle. Deficiency of the SIRT6 protein in skeletal muscle leads to severe hypoglycemia due to heightened HIF-1α activity and glucose uptake along with reduced mitochondrial respiration (Xiao et al. 2010). Under basal conditions, SIRT6 inhibits HIF-1α leading to decreased glycolysis and activation of oxidative phosphorylation (Zhong et al. 2010). Interestingly, SIRT6 expression increases with age in unison with HIF-1α and both proteins are reduced in these animals following exercise (Koltai et al. 2010). This may indicate that SIRT6 acts as a compensation mechanism during old age to limit HIF-1α overactivity.
We propose that SIRT1 and SIRT6 coordinate their functions to regulate HIF-1α, glucose uptake and glycolysis in muscle. This concept has been demonstrated in macrophage precursors during an acute inflammatory response where the NAD+-dependent activation of SIRT1 and SIRT6 were responsible for the activation of PGC-1α and the silencing of HIF-1α transcription, respectively (Liu et al. 2012). As a result, the coordination between SIRT1 and SIRT6 induces a metabolic switch from glucose metabolism to fatty acid oxidation. Future research examining the coordination of SIRT1 and SIRT6, along with the acetyltransferases GCN5 or PCAF, will help to define the potential PGC-1α- and HIF-1α-directed metabolic switch upon changes in oxygen demands, exercise-induced mitochondrial biogenesis and other adaptations to stress in skeletal muscle.
Protein acetylation modulates muscle satellite cell fate
In contrast to what occurs within a differentiated muscle cell, SIRT1 acts as a regulator of peripheral satellite cell proliferation in hypoxic conditions (Rathbone et al. 2009). Muscle satellite cells exposed to a hypoxic environment induce a SIRT1-mediated increase in cell cycle progression through the overall repression of cyclindependent kinase inhibitors, key regulators of G1-S phase transitions. In addition increases in the activity and expression levels of the basic helix-loop-helix factor (bHLH) MYOD marks the activation of satellite cells and differentiation and is required for normal muscle regeneration (Megeney et al. 1996; Kanisicak et al. 2009). MYOD activity is controlled by multiple acetylation sites. Mice that express a mutant form of MYOD that replaces two specific lysine acetylation sites with arginine exhibit a delayed capacity for muscle regeneration in vivo (Duquet et al. 2006). In fact, the yield of myoblasts in MYOD mutant muscle was two-fold higher than that found in WT animals, yet when placed under differentiation conditions, in vitro, there was a perturbed expression of early and late differentiation markers. MYOD can be deacetylated in the cell by a SIRT1- dependent mechanism. SIRT1 associates to PCAF/GCN5 and deacetylates PCAF and MYOD to inhibit differentiation (Fulco et al. 2003). From this, we can hypothesize that in satellite cells, SIRT1 acts to protect hypoxic muscle tissue by preparing the muscle for regeneration by activating the proliferation of muscle satellite cells. Concurrently, SIRT1 inhibits the differentiation of these satellite cells until the muscle is once again in a normoxic environment. Conversely, the acetylation of MYOD and activation of differentiation occurs through CBP/300 and PCAF acetylase activity (Sartorelli et al. 1999; Polesskaya et al. 2000; Polesskaya & Harel-Bellan 2001). More recently, the ability of PCAF to activate MYOD was confirmed using a PCAF-specific inhibitor, embelin, which blocked differentiation of myoblasts into myotubes (Modak et al. 2013). Furthermore, the nuclear corepressor SMRT (silencing mediator of retinoid acid and thyroid hormone receptor), has been shown to bind and stimulate the deacetylase activity of HDAC3 towards MEF2 and PCAF causing repression of myogenesis (Grégoire et al. 2007). Likewise, the SMRT homolog NCOR1 (nuclear corepressor 1) recruits HDAC1 to the bHLH region of MYOD to repress gene transcription (Bailey et al. 1999). Importantly, muscle-specific ablation of NCOR1 in mice demonstrated enhanced exercise endurance due to an increase in muscle mass and mitochondrial biogenesis (Yamamoto et al. 2011). With the loss of NCOR1 in vivo, there was an increase in the acetylation levels and activity of MEF2, a transcription factor that potentiates the muscle inducing activity of myogenic bHLH proteins (Yamamoto et al. 2011). In opposition to SIRT1-, NCOR1- and SMRT-regulation of MYOD, the coactivator PC4 has been shown to increase the expression of MYOD, along with the ability of this protein to stimulate muscle regeneration and myoblast differentiation (Micheli et al. 2011). PC4 directs the formation of a trimolecular complex of PC4, NF-κβ and HDAC3, which deacetylates NF-κβ, an inhibitor of MYOD expression, and sequesters it to the cytosol.
These studies demonstrate that the chronological progression of deacetylase or acetyltransferase activity is important for satellite cell proliferation and differentiation. These studies also highlight the importance of the balance between these opposing enzymes when considering the development of new pharmacological approaches to activate muscle regeneration. Future studies should better define the progression and interconnections between each of the regulatory nodes of differentiation, specifically through SIRT1, PCAF/GCN5, NCOR1 and SMRT regulation of myogenic transcription factors such as MYOD and MEF2.
Reversible acetylation for the control of atrophy and autophagy
The ubiquitin-proteasome and autophagy-lysosome systems are major proteolytic pathways that contribute to muscle wasting. The ubiquitin-proteasome system may account for up to 80% of the proteolysis during muscle wasting and is activated by the muscle-specific ubiquitin ligases ATROGIN1 and MURF1. Regulation of muscle wasting through the acetylation of NF-κβ, C/EBPβ, and FOXO transcription factors have been previously reported (Gutierrez et al. 2002; Chen & Greene 2003; Perrot & Rechler 2005). The acetylation of multiple sites on NF-κβ by P300/CBP alters the capacity of NF-κβ to bind DNA, induce its transcriptional activity and interact with Iκβ inhibitors. The transcription factor C/EBPβ was shown to play a role in muscle wasting through studies on dexamethasone treatment. This synthetic glucocorticoid increases P300 binding and acetylation of C/EBPβ (Yang et al. 2005; Chamberlain et al. 2012). When active, C/EBPβ binds to a C/EBPβ-responsive cis-element in the atrogin1/MAFbx gene promoter (Zhang et al. 2011). The activation of FOXO3A in skeletal muscle causes the transcription of both the atrogin-1 and murf1 promoters, followed by the induction of the atrophy program (Sandri et al. 2004; Sandri 2010; Senf et al. 2010). SIRT1 and P300 have been shown to regulate the balance between inhibition and activation of FOXO3A via deacetylation and acetylation, respectively (Brunet et al. 2004; Motta et al. 2004; van der Horst et al. 2004). Interestingly, during glucose restriction of mouse C2C12 myotubes, FOXO3A was also shown to form a complex on mtDNA with SIRT3 and mitochondrial RNA polymerase, thus increasing mitochondrial protein expression and respiration in response to the CR-induced atrophic signal (Peserico et al. 2013).
Acetylation can also regulate the process of autophagy, as was demonstrated in the dual muscle-specific KO mouse of HDAC1 and HDAC2 (Moresi et al. 2012). These mice exhibit a progressive myopathy that is preceded by impaired autophagic flux, yet the mechanistic link still remains elusive. Furthermore, deacetylation by SIRT1 is important for activating the autophagy-related genes ATG5, ATG7 and ATG8 in an NAD+-dependent manner in mammalian cells (Lee et al. 2008). SIRT2 also appears to be involved in autophagy through the activation of FOXO1. In adipocytes, SIRT2 can bind and deacetylate FOXO1, a regulator of both muscle wasting and autophagy in mammalian cells (Jing et al. 2007; Zhao et al. 2010). During oxidative stress or serum starvation, FOXO1 is released by SIRT2, allowing for the acetylation of FOXO1 and the induction of an autophagic response (Zhao et al. 2010). Therefore, since FOXO proteins regulate key pathways during sarcopenia and muscle disuse (Sandri et al. 2004; Senf et al. 2011), SIRT2 may prove to be an interesting new avenue to manipulate autophagy and atrophy signaling in skeletal muscle. In addition, P300, which is responsible for acetylating FOXO1 in vivo and in vitro, also acts as a coactivator to potentiate FOXO1 transactivation (Perrot & Rechler 2005).
Here we emphasize the role of protein acetylation in atrophy and muscle wasting, along with the role of autophagy as a selective process for clearing dysfunctional proteins and organelles [reviewed in (Menzies & Hood 2012; Sandri 2013)]. The importance of examining the role of reversible acetylation in these processes is evident from a clinical standpoint and for the basic understanding of protein and organelle turnover that exists in muscle tissue.
Conclusions
In muscle, the balance between catabolism and anabolism, substrate utilization and oxygenation are tightly interwoven. From this discussion it should be apparent that acetylation tightly regulates these processes in a concerted manner. As a result the muscle cell can synchronize energy status and blood flow to changes in local muscle metabolism without compromising each of these processes or the host. With the current rise in the elderly population there is an urgent need to understand the basic mechanisms of muscle homeostasis and to delineate each of these pathways in order to rationally tackle medical problems such as age-related loss of muscle function or glucose intolerance. In addition, much like what occurs with aging, pathologies such as muscular dystrophies, amyotrophic lateral sclerosis, diabetes and cancer have also been defined by reductions in muscle mass, mitochondrial content and function, and the associated reduced capacity for adaptations to metabolic stress.
A better understanding of the involvement of reversible acetylation in mechanisms that underlie the pathogenesis of metabolic dysregulation has been the goal of many studies and is an important step for the development of new therapeutic approaches. Reversible acetylation orchestrates key regulatory mechanisms for both muscle and whole body metabolic homeostasis. It is apparent from the data presented here that in many cases acetylation or deacetylation cannot be simply categorized as activators or inhibitors of transcription, as once thought. These are complex systems that are context- and protein-specific. Therefore a better understanding of these systems may offer promising approaches for pharmacological or lifestyle interventions (i.e. exercise) to modulate the activity of this integrated system, leading to the maintenance of mitochondrial content and function, energy and oxygen supply and ultimately skeletal muscle health.
Acknowledgments
JA is the Nestlé Chair in Energy Metabolism and work in his laboratory is supported by the EPFL, the EU Ideas program (ERC-2008-ADG-231138), the NIH (1R01HL106511), the Velux Stiftung, the Swiss National Science Foundation-SNSF (31003A-140780, 310030-143748, and CRSII3-136201). KM is the recipient of a Heart and Stroke Foundation of Canada research fellowship award.
References
- Amaral SL, Papanek PE, Greene AS. Angiotensin II and VEGF are involved in angiogenesis induced by short-term exercise training. American Journal of Physiology - Heart and Circulatory Physiology. 2001;281:1163–1169. doi: 10.1152/ajpheart.2001.281.3.H1163. [DOI] [PubMed] [Google Scholar]
- Ameln H, Gustafsson T, Sundberg CJ, Okamoto K, Jansson E, Poellinger L, Makino Y. Physiological activation of hypoxia inducible factor-1 in human skeletal muscle. FASEB Journal : Official Publication of the Federation of American Societies for Experimental Biology. 2005;19:1009–1011. doi: 10.1096/fj.04-2304fje. [DOI] [PubMed] [Google Scholar]
- Anderson RM, Bitterman KJ, Wood JG, Medvedik O, Sinclair DA. Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature. 2003;423:181–185. doi: 10.1038/nature01578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apfeld J, O’Connor G, McDonagh T, DiStefano PS, Curtis R. The AMP-activated protein kinase AAK-2 links energy levels and insulin-like signals to lifespan in C. elegans. Genes & Development. 2004;18:3004–3009. doi: 10.1101/gad.1255404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arany Z, Foo S-Y, Ma Y, Ruas JL, Bommi-Reddy A, Girnun G, Cooper M, Laznik D, Chinsomboon J, Rangwala SM, et al. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature. 2008;451:1008–1012. doi: 10.1038/nature06613. [DOI] [PubMed] [Google Scholar]
- Bailey P, Downes M, Lau P, Harris J, Chen SL, Hamamori Y, Sartorelli V, Muscat GE. The nuclear receptor corepressor N-CoR regulates differentiation: N-CoR directly interacts with MyoD. Molecular Endocrinology. 1999;13:1155–1168. doi: 10.1210/mend.13.7.0305. [DOI] [PubMed] [Google Scholar]
- Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–342. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biason-Lauber A, Böni-Schnetzler M, Hubbard BP, Bouzakri K, Brunner A, Cavelti-Weder C, Keller C, Meyer-Böni M, Meier DT, Brorsson C, et al. Identification of a SIRT1 Mutation in a Family with Type 1 Diabetes. Cell Metabolism. 2013;17:448–455. doi: 10.1016/j.cmet.2013.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieganowski P, Brenner C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell. 2004;117:495–502. doi: 10.1016/s0092-8674(04)00416-7. [DOI] [PubMed] [Google Scholar]
- Bitterman KJ. Inhibition of Silencing and Accelerated Aging by Nicotinamide, a Putative Negative Regulator of Yeast Sir2 and Human SIRT1. Journal of Biological Chemistry. 2002;277:45099–45107. doi: 10.1074/jbc.M205670200. [DOI] [PubMed] [Google Scholar]
- Bogan KL, Brenner C. Nicotinic acid, nicotinamide, and nicotinamide riboside: a molecular evaluation of NAD+ precursor vitamins in human nutrition. Annual Review of Nutrition. 2008;28:115–130. doi: 10.1146/annurev.nutr.28.061807.155443. [DOI] [PubMed] [Google Scholar]
- Breen EC, Johnson EC, Wagner H, Tseng HM, Sung LA, Wagner PD. Angiogenic growth factor mRNA responses in muscle to a single bout of exercise. Journal of Applied Physiology (Bethesda, Md : 1985) 1996;81:355–361. doi: 10.1152/jappl.1996.81.1.355. [DOI] [PubMed] [Google Scholar]
- Breen E, Tang K, Olfert M, Knapp A, Wagner P. Skeletal muscle capillarity during hypoxia: VEGF and its activation. High Altitude Medicine & Biology. 2008;9:158–166. doi: 10.1089/ham.2008.1010. [DOI] [PubMed] [Google Scholar]
- Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science (New York, N Y) 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- Cantó C, Auwerx J. Targeting sirtuin 1 to improve metabolism: all you need is NAD(+)? Pharmacological Reviews. 2012;64:166–187. doi: 10.1124/pr.110.003905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantó C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantó C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y, Fernandez-Marcos PJ, Yamamoto H, Andreux PA, Cettour-Rose P, et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metabolism. 2012;15:838–847. doi: 10.1016/j.cmet.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantó C, Jiang LQ, Deshmukh AS, Mataki C, Coste A, Lagouge M, Zierath JR, Auwerx J. Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metabolism. 2010;11:213–219. doi: 10.1016/j.cmet.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain W, Gonnella P, Alamdari N, Aversa Z, Hasselgren P-O. Multiple muscle wasting-related transcription factors are acetylated in dexamethasone-treated muscle cells. Biochemistry and Cell Biology = Biochimie Et Biologie Cellulaire. 2012;90:200–208. doi: 10.1139/o11-082. [DOI] [PubMed] [Google Scholar]
- Chen H, Lin RJ, Schiltz RL, Chakravarti D, Nash A, Nagy L, Privalsky ML, Nakatani Y, Evans RM. Nuclear receptor coactivator ACTR is a novel histone acetyltransferase and forms a multimeric activation complex with P/CAF and CBP/p300. Cell. 1997;90:569–580. doi: 10.1016/s0092-8674(00)80516-4. [DOI] [PubMed] [Google Scholar]
- Chen L-F, Greene WC. Regulation of distinct biological activities of the NF-kappaB transcription factor complex by acetylation. Journal of Molecular Medicine (Berlin, Germany) 2003;81:549–557. doi: 10.1007/s00109-003-0469-0. [DOI] [PubMed] [Google Scholar]
- Coletta DK, Balas B, Chavez AO, Baig M, Abdul-Ghani M, Kashyap SR, Folli F, Tripathy D, Mandarino LJ, Cornell JE, et al. Effect of acute physiological hyperinsulinemia on gene expression in human skeletal muscle in vivo. American Journal of Physiology Endocrinology and Metabolism. 2008;294:E910–E917. doi: 10.1152/ajpendo.00607.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coste A, Louet JF, Lagouge M, Lerin C, Antal MC, Meziane H, Schoonjans K, Puigserver P, O’Malley BW, Auwerx J. The genetic ablation of SRC-3 protects against obesity and improves insulin sensitivity by reducing the acetylation of PGC-1. Proceedings of the National Academy of Sciences. 2008;105:17187–17192. doi: 10.1073/pnas.0808207105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costford SR, Bajpeyi S, Pasarica M, Albarado DC, Thomas SC, Xie H, Church TS, Jubrias SA, Conley KE, Smith SR. Skeletal muscle NAMPT is induced by exercise in humans. AJP: Endocrinology and Metabolism. 2010;298:E117–E126. doi: 10.1152/ajpendo.00318.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson JM, Hudlicka O. The effect of long-term activity on the microvasculature of rat glycolytic skeletal muscle. International Journal of Microcirculation, Clinical and Experimental / Sponsored by the European Society for Microcirculation. 1989;8:53–69. [PubMed] [Google Scholar]
- Dominy JE, Lee Y, Jedrychowski MP, Chim H, Jurczak MJ, Camporez JP, Ruan H-B, Feldman J, Pierce K, Mostoslavsky R, et al. The deacetylase Sirt6 activates the acetyltransferase GCN5 and suppresses hepatic gluconeogenesis. Molecular Cell. 2012;48:900–913. doi: 10.1016/j.molcel.2012.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duquet A, Polesskaya A, Cuvellier S, Ait-Si-Ali S, Héry P, Pritchard LL, Gerard M, Harel-Bellan A. Acetylation is important for MyoD function in adult mice. EMBO Reports. 2006;7:1140–1146. doi: 10.1038/sj.embor.7400820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Marcos PJ, Jeninga EH, Cantó C, Harach T, de Boer VCJ, Andreux P, Moullan N, Pirinen E, Yamamoto H, Houten SM, et al. Muscle or liver-specific Sirt3 deficiency induces hyperacetylation of mitochondrial proteins without affecting global metabolic homeostasis. Scientific Reports. 2012;2 doi: 10.1038/srep00425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford E. Mammalian Sir2 homolog SIRT7 is an activator of RNA polymerase I transcription. Genes & Development. 2006;20:1075–1080. doi: 10.1101/gad.1399706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulco M, Cen Y, Zhao P, Hoffman EP, McBurney MW, Sauve AA, Sartorelli V. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Developmental Cell. 2008;14:661–673. doi: 10.1016/j.devcel.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulco M, Schiltz RL, Iezzi S, King MT, Zhao P, Kashiwaya Y, Hoffman E, Veech RL, Sartorelli V. Sir2 regulates skeletal muscle differentiation as a potential sensor of the redox state. Molecular Cell. 2003;12:51–62. doi: 10.1016/s1097-2765(03)00226-0. [DOI] [PubMed] [Google Scholar]
- Garten A, Petzold S, Körner A, Imai S-I, Kiess W. Nampt: linking NAD biology, metabolism and cancer. Trends in Endocrinology and Metabolism: TEM. 2009;20:130–138. doi: 10.1016/j.tem.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng T, Li P, Okutsu M, Yin X, Kwek J, Zhang M, Yan Z. PGC-1alpha plays a functional role in exercise-induced mitochondrial biogenesis and angiogenesis but not fiber-type transformation in mouse skeletal muscle. AJP: Cell Physiology. 2010;298:C572–C579. doi: 10.1152/ajpcell.00481.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham TE, Saltin B. Estimation of the mitochondrial redox state in human skeletal muscle during exercise. 1989;66:561–566. doi: 10.1152/jappl.1989.66.2.561. [DOI] [PubMed] [Google Scholar]
- Grégoire S, Xiao L, Nie J, Zhang X, Xu M, Li J, Wong J, Seto E, Yang X-J. Histone deacetylase 3 interacts with and deacetylates myocyte enhancer factor 2. Molecular and Cellular Biology. 2007;27:1280–1295. doi: 10.1128/MCB.00882-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan K-L, Xiong Y. Regulation of intermediary metabolism by protein acetylation. Trends in Biochemical Sciences. 2011;36:108–116. doi: 10.1016/j.tibs.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarente L. Sirtuins, Aging, and Metabolism. Cold Spring Harbor Symposia on Quantitative Biology. 2012;76:81–90. doi: 10.1101/sqb.2011.76.010629. [DOI] [PubMed] [Google Scholar]
- Gurd BJ, Yoshida Y, McFarlan JT, Holloway GP, Moyes CD, Heigenhauser GJF, Spriet L, Bonen A. Nuclear SIRT1 activity, but not protein content, regulates mitochondrial biogenesis in rat and human skeletal muscle. American Journal of Physiology Regulatory, Integrative and Comparative Physiology. 2011;301:R67–R75. doi: 10.1152/ajpregu.00417.2010. [DOI] [PubMed] [Google Scholar]
- Gustafsson T, Puntschart A, Kaijser L, Jansson E, Sundberg CJ. Exercise-induced expression of angiogenesis-related transcription and growth factors in human skeletal muscle. The American Journal of Physiology. 1999;276:H679–H685. doi: 10.1152/ajpheart.1999.276.2.H679. [DOI] [PubMed] [Google Scholar]
- Gutierrez S, Javed A, Tennant DK, van Rees M, Montecino M, Stein GS, Stein JL, Lian JB. CCAAT/Enhancer-binding Proteins (C/EBP) beta and delta Activate Osteocalcin Gene Transcription and Synergize with Runx2 at the C/EBP Element to Regulate Bone-specific Expression. Journal of Biological Chemistry. 2002;277:1316–1323. doi: 10.1074/jbc.M106611200. [DOI] [PubMed] [Google Scholar]
- Hang J, Kong L, Gu JW, Adair TH. VEGF gene expression is upregulated in electrically stimulated rat skeletal muscle. The American Journal of Physiology. 1995;269:H1827–H1831. doi: 10.1152/ajpheart.1995.269.5.H1827. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nature Reviews. Molecular Cell Biology. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashida S, Arimoto A, Kuramoto Y, Kozako T, Honda S-I, Shimeno H, Soeda S. Fasting promotes the expression of SIRT1, an NAD+ -dependent protein deacetylase, via activation of PPARalpha in mice. Molecular and Cellular Biochemistry. 2010;339:285–292. doi: 10.1007/s11010-010-0391-z. [DOI] [PubMed] [Google Scholar]
- Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR, et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 2010;464:121–125. doi: 10.1038/nature08778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloszy JO, Kohrt WM, Hansen PA. The regulation of carbohydrate and fat metabolism during and after exercise. Frontiers in Bioscience : a Journal and Virtual Library. 1998;3:D1011–D1027. doi: 10.2741/a342. [DOI] [PubMed] [Google Scholar]
- Houtkooper RH, Argmann C, Houten SM, Cantó C, Jeninga EH, Andreux PA, Thomas C, Doenlen R, Schoonjans K, Auwerx J. The metabolic footprint of aging in mice. Scientific Reports. 2011;1 doi: 10.1038/srep00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Cantó C, Wanders RJ, Auwerx J. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocrine Reviews. 2010;31:194–223. doi: 10.1210/er.2009-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nature Reviews. Molecular Cell Biology. 2012;13:225–238. doi: 10.1038/nrm3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudlicka O, Price S. The role of blood flow and/or muscle hypoxia in capillary growth in chronically stimulated fast muscles. Pflügers Archiv : European Journal of Physiology. 1990;417:67–72. doi: 10.1007/BF00370770. [DOI] [PubMed] [Google Scholar]
- Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- Jager S, Handschin C, St Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1. Proceedings of the National Academy of Sciences. 2007;104:12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeninga EH, Schoonjans K, Auwerx J. Reversible acetylation of PGC-1: connecting energy sensors and effectors to guarantee metabolic flexibility. Oncogene. 2010;29:4617–4624. doi: 10.1038/onc.2010.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing E, Emanuelli B, Hirschey MD, Boucher J, Lee KY, Lombard D, Verdin EM, Kahn CR. Sirtuin-3 (Sirt3) regulates skeletal muscle metabolism and insulin signaling via altered mitochondrial oxidation and reactive oxygen species production. Proceedings of the National Academy of Sciences. 2011;108:14608–14613. doi: 10.1073/pnas.1111308108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing E, Gesta S, Kahn CR. SIRT2 regulates adipocyte differentiation through FoxO1 acetylation/deacetylation. Cell Metabolism. 2007;6:105–114. doi: 10.1016/j.cmet.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph A-M, Joanisse DR, Baillot RG, Hood DA. Mitochondrial dysregulation in the pathogenesis of diabetes: potential for mitochondrial biogenesis-mediated interventions. Experimental Diabetes Research. 2012;2012:642038. doi: 10.1155/2012/642038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanisicak O, Mendez JJ, Yamamoto S, Yamamoto M, Goldhamer DJ. Progenitors of skeletal muscle satellite cells express the muscle determination gene, MyoD. Developmental Biology. 2009;332:131–141. doi: 10.1016/j.ydbio.2009.05.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes. 2002;51:2944–2950. doi: 10.2337/diabetes.51.10.2944. [DOI] [PubMed] [Google Scholar]
- Kelly TJ, Lerin C, Haas W, Gygi SP, Puigserver P. GCN5-mediated Transcriptional Control of the Metabolic Coactivator PGC-1 through Lysine Acetylation. Journal of Biological Chemistry. 2009;284:19945–19952. doi: 10.1074/jbc.M109.015164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koltai E, Szabo Z, Atalay M, Boldogh I, Naito H, Goto S, Nyakas C, Radak Z. Exercise alters SIRT1, SIRT6, NAD and NAMPT levels in skeletal muscle of aged rats. Mechanisms of Ageing and Development. 2010;131:21–28. doi: 10.1016/j.mad.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosaka A, Spivey HO, Gholson RK. Nicotinate phosphoribosyltransferase of yeast. Purification and properties. The Journal of Biological Chemistry. 1971;246:3277–3283. [PubMed] [Google Scholar]
- Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, et al. Resveratrol Improves Mitochondrial Function and Protects against Metabolic Disease by Activating SIRT1 and PGC-1α. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, Tsokos M, Alt FW, Finkel T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proceedings of the National Academy of Sciences. 2008;105:3374–3379. doi: 10.1073/pnas.0712145105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerin C, Rodgers JT, Kalume DE, Kim S-H, Pandey A, Puigserver P. GCN5 acetyltransferase complex controls glucose metabolism through transcriptional repression of PGC-1α. Cell Metabolism. 2006;3:429–438. doi: 10.1016/j.cmet.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Lim J-H, Lee Y-M, Chun Y-S, Chen J, Kim J-E, Park J-W. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Molecular Cell. 2010;38:864–878. doi: 10.1016/j.molcel.2010.05.023. [DOI] [PubMed] [Google Scholar]
- Lin S-J, Ford E, Haigis M, Liszt G, Guarente L. Calorie restriction extends yeast life span by lowering the level of NADH. Genes & Development. 2004;18:12–16. doi: 10.1101/gad.1164804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu TF, Vachharajani VT, Yoza BK, McCall CE. NAD+-dependent sirtuin 1 and 6 proteins coordinate a switch from glucose to fatty acid oxidation during the acute inflammatory response. Journal of Biological Chemistry. 2012;287:25758–25769. doi: 10.1074/jbc.M112.362343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Chen C, Klaassen C. Tissue distribution of concentrative and equilibrative nucleoside transporters in male and female rats and mice. Drug Metabolism and Disposition: the Biological Fate of Chemicals. 2004;32:1455–1461. doi: 10.1124/dmd.104.001123. [DOI] [PubMed] [Google Scholar]
- Magni G, Amici A, Emanuelli M, Orsomando G, Raffaelli N, Ruggieri S. Enzymology of NAD+ homeostasis in man. Cellular and Molecular Life Sciences. 2004;61:19–34. doi: 10.1007/s00018-003-3161-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megeney LA, Kablar B, Garrett K, Anderson JE, Rudnicki MA. MyoD is required for myogenic stem cell function in adult skeletal muscle. Genes & Development. 1996;10:1173–1183. doi: 10.1101/gad.10.10.1173. [DOI] [PubMed] [Google Scholar]
- Menzies KJ, Hood DA. The role of SirT1 in muscle mitochondrial turnover. Mitochondrion. 2012;12:5–13. doi: 10.1016/j.mito.2011.03.001. [DOI] [PubMed] [Google Scholar]
- Micheli L, Leonardi L, Conti F, Maresca G, Colazingari S, Mattei E, Lira SA, Farioli-Vecchioli S, Caruso M, Tirone F. PC4/Tis7/IFRD1 stimulates skeletal muscle regeneration and is involved in myoblast differentiation as a regulator of MyoD and NF-kappaB. Journal of Biological Chemistry. 2011;286:5691–5707. doi: 10.1074/jbc.M110.162842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modak R, Basha J, Bharathy N, Maity K, Mizar P, Bhat AV, Vasudevan M, Rao VK, Kok WK, Natesh N, et al. Probing p300/CBP Associated Factor (PCAF)-Dependent Pathways with a Small Molecule Inhibitor. ACS Chemical Biology. 2013 doi: 10.1021/cb4000597. [DOI] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson K-F, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nature Genetics. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Moresi V, Carrer M, Grueter CE, Rifki OF, Shelton JM, Richardson JA, Bassel-Duby R, Olson EN. Histone deacetylases 1 and 2 regulate autophagy flux and skeletal muscle homeostasis in mice. Proceedings of the National Academy of Sciences. 2012;109:1649–1654. doi: 10.1073/pnas.1121159109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morino K, Petersen KF, Dufour S, Befroy D, Frattini J, Shatzkes N, Neschen S, White MF, Bilz S, Sono S, et al. Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. The Journal of Clinical Investigation. 2005;115:3587–3593. doi: 10.1172/JCI25151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W, Bultsma Y, McBurney M, Guarente L. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004;116:551–563. doi: 10.1016/s0092-8674(04)00126-6. [DOI] [PubMed] [Google Scholar]
- Mouchiroud L, Houtkooper RH, Auwerx J. NAD(+) metabolism: A therapeutic target for age-related metabolic disease. Critical Reviews in Biochemistry and Molecular Biology. 2013 doi: 10.3109/10409238.2013.789479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mujtaba S, Zeng L, Zhou M-M. Structure and acetyl-lysine recognition of the bromodomain. Oncogene. 2007;26:5521–5527. doi: 10.1038/sj.onc.1210618. [DOI] [PubMed] [Google Scholar]
- Muoio DM, Noland RC, Kovalik J-P, Seiler SE, Davies MN, DeBalsi KL, Ilkayeva OR, Stevens RD, Kheterpal I, Zhang J, et al. Muscle-specific deletion of carnitine acetyltransferase compromises glucose tolerance and metabolic flexibility. Cell Metabolism. 2012;15:764–777. doi: 10.1016/j.cmet.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayan N, Lee IH, Borenstein R, Sun J, Wong R, Tong G, Fergusson MM, Liu J, Rovira II, Cheng H-L, et al. The NAD-dependent deacetylase SIRT2 is required for programmed necrosis. Nature. 2012;492:199–204. doi: 10.1038/nature11700. [DOI] [PubMed] [Google Scholar]
- Nemoto S. SIRT1 Functionally Interacts with the Metabolic Regulator and Transcriptional Coactivator PGC-1. Journal of Biological Chemistry. 2005;280:16456–16460. doi: 10.1074/jbc.M501485200. [DOI] [PubMed] [Google Scholar]
- Nikiforov A, Dölle C, Niere M, Ziegler M. Pathways and subcellular compartmentation of NAD biosynthesis in human cells: from entry of extracellular precursors to mitochondrial NAD generation. Journal of Biological Chemistry. 2011;286:21767–21778. doi: 10.1074/jbc.M110.213298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nin V, Escande C, Chini CC, Giri S, Camacho-Pereira J, Matalonga J, Lou Z, Chini EN. Role of Deleted in Breast Cancer 1 (DBC1) Protein in SIRT1 Deacetylase Activation Induced by Protein Kinase A and AMP-activated Protein Kinase. Journal of Biological Chemistry. 2012;287:23489–23501. doi: 10.1074/jbc.M112.365874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hagan KA, Cocchiglia S, Zhdanov AV, Tambuwala MM, Tambawala MM, Cummins EP, Monfared M, Agbor TA, Garvey JF, Papkovsky DB, et al. PGC-1alpha is coupled to HIF-1alpha-dependent gene expression by increasing mitochondrial oxygen consumption in skeletal muscle cells. Proceedings of the National Academy of Sciences. 2009;106:2188–2193. doi: 10.1073/pnas.0808801106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onate SA, Boonyaratanakornkit V, Spencer TE, Tsai SY, Tsai MJ, Edwards DP, O’Malley BW. The steroid receptor coactivator-1 contains multiple receptor interacting and activation domains that cooperatively enhance the activation function 1 (AF1) and AF2 domains of steroid receptors. The Journal of Biological Chemistry. 1998;273:12101–12108. doi: 10.1074/jbc.273.20.12101. [DOI] [PubMed] [Google Scholar]
- Patel J, Pathak RR, Mujtaba S. The biology of lysine acetylation integrates transcriptional programming and metabolism. Nutrition & Metabolism. 2011;8:12. doi: 10.1186/1743-7075-8-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I, Costello M, Saccone R, et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:8466–8471. doi: 10.1073/pnas.1032913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrot V, Rechler MM. The coactivator p300 directly acetylates the forkhead transcription factor Foxo1 and stimulates Foxo1-induced transcription. Molecular Endocrinology. 2005;19:2283–2298. doi: 10.1210/me.2004-0292. [DOI] [PubMed] [Google Scholar]
- Peserico A, Chiacchiera F, Grossi V, Matrone A, Latorre D, Simonatto M, Fusella A, Ryall JG, Finley LWS, Haigis MC, et al. A novel AMPK-dependent FoxO3A-SIRT3 intramitochondrial complex sensing glucose levels. Cellular and Molecular Life Sciences. 2013;70:2015–2029. doi: 10.1007/s00018-012-1244-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philp A, Chen A, Lan D, Meyer GA, Murphy AN, Knapp AE, Olfert IM, McCurdy CE, Marcotte GR, Hogan MC, et al. Sirtuin 1 (SIRT1) Deacetylase Activity Is Not Required for Mitochondrial Biogenesis or Peroxisome Proliferator-activated Receptor- Coactivator-1 (PGC-1) Deacetylation following Endurance Exercise. Journal of Biological Chemistry. 2011;286:30561–30570. doi: 10.1074/jbc.M111.261685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirinen E, Sasso Lo G, Auwerx J. Mitochondrial sirtuins and metabolic homeostasis. Best Practice & Research. Clinical Endocrinology & Metabolism. 2012;26:759–770. doi: 10.1016/j.beem.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polesskaya A, Harel-Bellan A. Acetylation of MyoD by p300 requires more than its histone acetyltransferase domain. The Journal of Biological Chemistry. 2001;276:44502–44503. doi: 10.1074/jbc.M106501200. [DOI] [PubMed] [Google Scholar]
- Polesskaya A, Duquet A, Naguibneva I, Weise C, Vervisch A, Bengal E, Hucho F, Robin P, Harel-Bellan A. CREB-binding protein/p300 activates MyoD by acetylation. The Journal of Biological Chemistry. 2000;275:34359–34364. doi: 10.1074/jbc.M003815200. [DOI] [PubMed] [Google Scholar]
- Preiss J, Handler P. Enzymatic synthesis of nicotinamide mononucleotide. The Journal of Biological Chemistry. 1957;225:759–770. [PubMed] [Google Scholar]
- Price NL, Gomes AP, Ling AJY, Duarte FV, Martin-Montalvo A, North BJ, Agarwal B, Ye L, Ramadori G, Teodoro JS, et al. SIRT1 Is Required for AMPK Activation and the Beneficial Effects of Resveratrol on Mitochondrial Function. Cell Metabolism. 2012;15:675–690. doi: 10.1016/j.cmet.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puigserver P, ADELMANT G, WU Z, Fan M, Xu J, O’Malley B, Spiegelman BM. Activation of PPARgamma coactivator-1 through transcription factor docking. Science (New York, N Y) 1999;286:1368–1371. doi: 10.1126/science.286.5443.1368. [DOI] [PubMed] [Google Scholar]
- Rathbone CR, Booth FW, Lees SJ. Sirt1 increases skeletal muscle precursor cell proliferation. European Journal of Cell Biology. 2009;88:35–44. doi: 10.1016/j.ejcb.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revollo JR, Grimm AA, Imai S-I. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. The Journal of Biological Chemistry. 2004;279:50754–50763. doi: 10.1074/jbc.M408388200. [DOI] [PubMed] [Google Scholar]
- Ritov VB, Menshikova EV, He J, Ferrell RE, Goodpaster BH, Kelley DE. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes. 2005;54:8–14. doi: 10.2337/diabetes.54.1.8. [DOI] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Gerhart-Hines Z, Puigserver P. Metabolic adaptations through the PGC-1α and SIRT1 pathways. FEBS Letters. 2008;582:46–53. doi: 10.1016/j.febslet.2007.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature Cell Biology. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Annual Review of Biochemistry. 2001;70:81–120. doi: 10.1146/annurev.biochem.70.1.81. [DOI] [PubMed] [Google Scholar]
- Rowen JW, Kornberg A. The phosphorolysis of nicotinamide riboside. The Journal of Biological Chemistry. 1951;193:497–507. [PubMed] [Google Scholar]
- Sahlin K, Harris RC. Control of lipid oxidation during exercise: role of energy state and mitochondrial factors. Acta Physiologica (Oxford, England) 2008;194:283–291. doi: 10.1111/j.1748-1716.2008.01879.x. [DOI] [PubMed] [Google Scholar]
- Sakai M, Matsumoto M, Tujimura T, Yongheng C, Noguchi T, Inagaki K, Inoue H, Hosooka T, Takazawa K, Kido Y, et al. CITED2 links hormonal signaling to PGC-1α acetylation in the regulation of gluconeogenesis. Nature Medicine. 2012;18:612–617. doi: 10.1038/nm.2691. [DOI] [PubMed] [Google Scholar]
- Samjoo IA, Safdar A, Hamadeh MJ, Glover AW, Mocellin NJ, Santana J, Little JP, Steinberg GR, Raha S, Tarnopolsky MA. Markers of skeletal muscle mitochondrial function and lipid accumulation are moderately associated with the homeostasis model assessment index of insulin resistance in obese men. PloS One. 2013;8:e66322. doi: 10.1371/journal.pone.0066322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandri M. Autophagy in skeletal muscle. FEBS Letters. 2010;584:1411–1416. doi: 10.1016/j.febslet.2010.01.056. [DOI] [PubMed] [Google Scholar]
- Sandri M. Protein breakdown in muscle wasting: Role of autophagy-lysosome and ubiquitin-proteasome. The International Journal of Biochemistry & Cell Biology. 2013 doi: 10.1016/j.biocel.2013.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117:399–412. doi: 10.1016/s0092-8674(04)00400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartorelli V, Puri PL, Hamamori Y, Ogryzko V, Chung G, Nakatani Y, Wang JY, Kedes L. Acetylation of MyoD directed by PCAF is necessary for the execution of the muscle program. Molecular Cell. 1999;4:725–734. doi: 10.1016/s1097-2765(00)80383-4. [DOI] [PubMed] [Google Scholar]
- Schenk S, McCurdy CE, Philp A, Chen MZ, Holliday MJ, Bandyopadhyay GK, Osborn O, Baar K, Olefsky JM. Sirt1 enhances skeletal muscle insulin sensitivity in mice during caloric restriction. The Journal of Clinical Investigation. 2011;121:4281–4288. doi: 10.1172/JCI58554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott I, Webster BR, Li JH, Sack MN. Identification of a molecular component of the mitochondrial acetyltransferase programme: a novel role for GCN5L1. The Biochemical Journal. 2012;443:655–661. doi: 10.1042/BJ20120118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. Hypoxia-inducible factor 1: oxygen homeostasis and disease pathophysiology. Trends in Molecular Medicine. 2001;7:345–350. doi: 10.1016/s1471-4914(01)02090-1. [DOI] [PubMed] [Google Scholar]
- Senf SM, Dodd SL, Judge AR. FOXO signaling is required for disuse muscle atrophy and is directly regulated by Hsp70. AJP: Cell Physiology. 2010;298:C38–C45. doi: 10.1152/ajpcell.00315.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senf SM, Sandesara PB, Reed SA, Judge AR. p300 Acetyltransferase activity differentially regulates the localization and activity of the FOXO homologues in skeletal muscle. AJP: Cell Physiology. 2011;300:C1490–C1501. doi: 10.1152/ajpcell.00255.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoneau JA, Kelley DE. Altered glycolytic and oxidative capacities of skeletal muscle contribute to insulin resistance in NIDDM. Journal of Applied Physiology (Bethesda, Md : 1985) 1997;83:166–171. doi: 10.1152/jappl.1997.83.1.166. [DOI] [PubMed] [Google Scholar]
- Simoneau JA, Colberg SR, Thaete FL, Kelley DE. Skeletal muscle glycolytic and oxidative enzyme capacities are determinants of insulin sensitivity and muscle composition in obese women. FASEB Journal : Official Publication of the Federation of American Societies for Experimental Biology. 1995;9:273–278. [PubMed] [Google Scholar]
- Smith JJ, Kenney R, Gagne DJ, Frushour BP, Ladd W, Galonek HL, Israelian K, Song J, Razvadauskaite G, Lynch AV, et al. Small molecule activators of SIRT1 replicate signaling pathways triggered by calorie restriction in vivo. BMC Systems Biology. 2009;3:31. doi: 10.1186/1752-0509-3-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchankova G, Nelson LE, Gerhart-Hines Z, Kelly M, Gauthier M-S, Saha AK, Ido Y, Puigserver P, Ruderman NB. Concurrent regulation of AMP-activated protein kinase and SIRT1 in mammalian cells. Biochemical and Biophysical Research Communications. 2009;378:836–841. doi: 10.1016/j.bbrc.2008.11.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Zhang F, Ge X, Yan T, Chen X, Shi X, Zhai Q. SIRT1 improves insulin sensitivity under insulin-resistant conditions by repressing PTP1B. Cell Metabolism. 2007;6:307–319. doi: 10.1016/j.cmet.2007.08.014. [DOI] [PubMed] [Google Scholar]
- Tissenbaum HA, Guarente L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature. 2001;410:227–230. doi: 10.1038/35065638. [DOI] [PubMed] [Google Scholar]
- van der Horst A, Tertoolen LGJ, de Vries-Smits LMM, Frye RA, Medema RH, Burgering BMT. FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2(SIRT1) The Journal of Biological Chemistry. 2004;279:28873–28879. doi: 10.1074/jbc.M401138200. [DOI] [PubMed] [Google Scholar]
- Vogelauer M, Krall AS, McBrian MA, Li J-Y, Kurdistani SK. Stimulation of histone deacetylase activity by metabolites of intermediary metabolism. Journal of Biological Chemistry. 2012;287:32006–32016. doi: 10.1074/jbc.M112.362467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Tang Y, Cole PA, Marmorstein R. Structure and chemistry of the p300/CBP and Rtt109 histone acetyltransferases: implications for histone acetyltransferase evolution and function. Current Opinion in Structural Biology. 2008;18:741–747. doi: 10.1016/j.sbi.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science (New York, N Y) 2009;324:1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winder WW, Holmes BF, Rubink DS, Jensen EB, Chen M, Holloszy JO. Activation of AMP-activated protein kinase increases mitochondrial enzymes in skeletal muscle. Journal of Applied Physiology (Bethesda, Md : 1985) 2000;88:2219–2226. doi: 10.1152/jappl.2000.88.6.2219. [DOI] [PubMed] [Google Scholar]
- Xiao B, Heath R, Saiu P, Leiper FC, Leone P, Jing C, Walker PA, Haire L, Eccleston JF, Davis CT, et al. Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature. 2007;449:496–500. doi: 10.1038/nature06161. [DOI] [PubMed] [Google Scholar]
- Xiao C, Kim H-S, Lahusen T, Wang R-H, Xu X, Gavrilova O, Jou W, Gius D, Deng C-X. SIRT6 deficiency results in severe hypoglycemia by enhancing both basal and insulin-stimulated glucose uptake in mice. Journal of Biological Chemistry. 2010;285:36776–36784. doi: 10.1074/jbc.M110.168039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Williams EG, Mouchiroud L, Cantó C, Fan W, Downes M, Héligon C, Barish GD, Desvergne B, Evans RM, et al. NCoR1 Is a Conserved Physiological Modulator of Muscle Mass and Oxidative Function. Cell. 2011;147:827–839. doi: 10.1016/j.cell.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Menconi MJ, Wei W, Petkova V, Hasselgren P-O. Dexamethasone upregulates the expression of the nuclear cofactor p300 and its interaction with C/EBPbeta in cultured myotubes. Journal of Cellular Biochemistry. 2005;94:1058–1067. doi: 10.1002/jcb.20371. [DOI] [PubMed] [Google Scholar]
- Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, Lamming DW, Souza-Pinto NC, Bohr VA, Rosenzweig A, et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell. 2007;130:1095–1107. doi: 10.1016/j.cell.2007.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino J, Mills KF, Yoon MJ, Imai S-I. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metabolism. 2011;14:528–536. doi: 10.1016/j.cmet.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Jin B, Li Y-P. C/EBPβ mediates tumour-induced ubiquitin ligase atrogin1/MAFbx upregulation and muscle wasting. The EMBO Journal. 2011;30:4323–4335. doi: 10.1038/emboj.2011.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Yang J, Liao W, Liu X, Zhang H, Wang S, Wang D, Feng J, Yu L, Zhu W-G. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nature Cell Biology. 2010;12:665–675. doi: 10.1038/ncb2069. [DOI] [PubMed] [Google Scholar]
- Zhong L, D’Urso A, Toiber D, Sebastian C, Henry RE, Vadysirisack DD, Guimaraes A, Marinelli B, Wikstrom JD, Nir T, et al. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell. 2010;140:280–293. doi: 10.1016/j.cell.2009.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]