

Abstract
The proteasome has emerged as the primary target for the treatment of multiple myeloma. Unfortunately, nearly all patients develop resistance to competitive-type proteasome inhibitors, such as bortezomib. Herein, we describe the optimization of non-competitive proteasome inhibitors to yield derivatives that exhibit nanomolar potency (compound 46, IC50 130 nM) towards proteasome inhibition and overcome bortezomib resistance. These studies illustrate the feasibility of the development of non-competitive proteasome inhibitors as additives and/or alternatives to competitive proteasome inhibitors.
Keywords: Imidazolines, proteasome, non-competitive inhibition, multiple myeloma, chemotherapy
INTRODUCTION
Over the past decade, the proteasome has emerged as a clinical target for the chemotherapeutic treatment of certain cancers, and the first-of-its class proteasome inhibitor bortezomib is currently approved for the treatment of multiple myeloma (MM) and relapsing mantle cell lymphoma (MCL).1 However, all clinically relevant proteasome inhibitors, including bortezomib and all second generation proteasome inhibitors under clinical evaluation, elicit their activity via the same mechanism, which is the formation of a covalent bond to the N-terminal threonine in the catalytic sites of the enzyme.2 In these cases the electrophilic head group of the inhibitor interacts with the N-terminal threonine of the β1, β2 and β5 catalytic domains of the enzyme.3 This type of binding blocks total proteasome-dependent proteolysis, which subsequently results in cell death and translates into potent anti-cancer efficacy.2a However, it has also been well documented that, due to this particular mechanism of binding, these agents exhibit permanent abrogation of global protein degradation, lack of specificity, low systemic tissue distribution, resistance and severe off-target effects.3–4 The use of these inhibitors in the clinic is therefore limited to a few types of blood related cancers, and most of those patients (>97%) become resistant or intolerant to the treatment within a few years, after which survival is typically less than one year.5
Non-competitive modulation of enzymatic activity is characterized by the interaction of a ligand with the enzyme at a site distinct from the catalytic site, effectively modulating the binding of some substrates to the enzyme’s catalytic site(s).6 Aside from overcoming acquired resistance to active site inhibitors, this type of modulation of enzyme activity may limit off-target effects, thus limiting toxicity.6b, 6e, 7 Examples of small molecules that act as non-competitive proteasome inhibitors are very scarce and typically exhibit activity at high concentrations and/or non-physiologically relevant concentrations.8,9
We previously reported the activity of the non-competitive proteasome inhibitor, TCH-013, in purified protein systems, cell culture, and in vivo models of multiple myeloma10 and arthritis.11 Although these non-competitive inhibitors exhibit good in vivo efficacy, the discovery of agents with increased potency would be a significant step toward the potential clinical development of this new type of proteasome modulation. Herein, we report the optimization of the imidazoline scaffold as non-competitive proteasome inhibitors with IC50 values in the nanomolar range.
RESULTS AND DISCUSSION
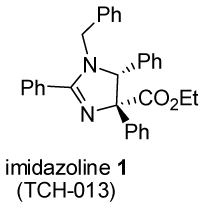
The imidazoline scaffold 1 (TCH-013, Table 1) was first reported to inhibit NF-κB- mediated gene transcription,12 and more recent studies identified the human proteasome as its molecular target.10 The mechanism of proteasome inhibition by these imidazolines proceeded via non-competitive kinetics, which makes this class of compounds unique amongst proteasome inhibitors.3, 10 Our goal in this study was to modify the imidazoline scaffold to increase its efficacy for proteasome inhibition. As the proteasome is required for the activation of NF-κB mediated gene transcription, we first compared the previously reported cellular EC50 values of NF-κB inhibition13 with the IC50 values of inhibition of the purified 20S proteasome to validate their direct correlation.
Table 1.
Inhibition of the chymotryptic-like activity of purified human 20S proteasome by compounds 1–5.
| compound IC50(μM) |
|
|
|
|
|
|---|---|---|---|---|---|
| proteasome | 2.5 | 1.4 | >10 | >10 | >10 |
| NF-κB-luc | 2.5 | 4.6 | >10 | >10 | >10 |
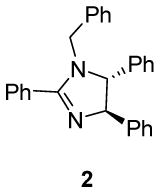
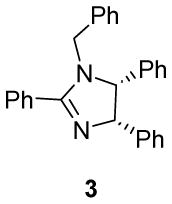
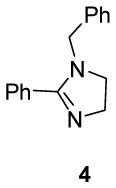
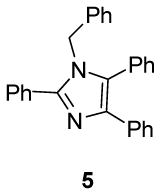
We first investigated the requirements of the trans-aryl moieties and their relative stereochemistry (Table 1) for inhibition of both the proteasome and NF-κB activity. Imidazoline scaffolds 1–5 were prepared as previously described13a and their ability to inhibit the 20S proteasome was determined in vitro using purified human 20S proteasome and the fluorogenic peptide Suc-LLVY-AMC as substrate for chymotryptic-like activity.14 The rates of hydrolysis were monitored by fluorescence increase at 37° C over 30 minutes and the linear portion of the curves was used to calculate the IC50 values. The results from Table 1 clearly indicate the requirement of the trans-aryl stereochemistry seen in compounds 1 and 2, as the cis (compound 3) and dearylated scaffold, (compound 4) as well as the planar imidazole 5 were all depleted of activity. This data corresponded well with our previously reported NF-κB inhibition of these compounds in cell culture as the inactive proteasome inhibitors were inactive as NF-κB inhibitors.13 Interestingly, upon standing, imidazolines 2 and 3, slowly isomerised and dehydrogenated to the inactive aromatic imidazole 5, which compromised the further development of the di-substituted trans scaffold 2.13b This cis-trans isomerization has been described previously in the literature and is conveniently blocked by the ester in compound 1, which was subsequently chosen as our lead compound for optimization.13b, 15
The synthesis of the imidazolines is accomplished via a dipolar cycloaddition reaction of an oxazol-5(4H)-one and a subsequent esterification of the carboxylic acid (Scheme 1). Acylation of phenylglycine with the appropriate acid chloride followed by cyclic dehydration using trifluoroacetic anhydride provided the oxazolone (I). The oxazolone can undergo a münchnone-type cycloaddition reaction in the presence of Lewis acids, such as TMSCl, with imines to render the functionalized imidazoline scaffold (II) as a single diastereomer.16 The cycloaddition reaction places a carboxylic acid in the C-4 position of the imidazoline scaffold (II). The free carboxylic acid is metabolically unstable,13b thus efforts were directed towards the functionalization of this group. Various compounds including esters 7, 8 and 10 and the amide 9 were prepared and tested for their ability to inhibit the proteasome in vitro (Table 2). The formation of esters (III) was accomplished using the carbodiimide EDCI and dimethylaminopyridine (DMAP) to yield compounds 6–8, and prevented the decarboxylation and aromatization. The imidazolines were able to tolerate multiple different ester groups (6–8) without affecting activity. As shown in Table 2, the activity of the compounds 1,6–10 against the purified proteasome corresponded very well with the reported cellular inhibition of NF-κB transcription. However, incorporation of an amide rendered a decrease of activity. Therefore, subsequent optimization studies were focused around the ethyl ester (III) as the leading scaffold. After having established the strong correlation between the inhibition of cellular NF-κB activity and the inhibition of proteasome activity in the purified protein assays, we evaluated modification of the respective R1-R3 domains (Scheme 1) in order to optimize the potency of the imidazoline scaffold towards proteasome inhibition.
Scheme 1.
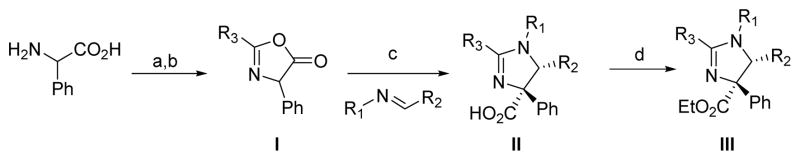
Synthesis of imidazolines via a dipolar cycloaddition reaction. a) Na2CO3, H2O/1,4-dioxane, R3COCl, rt; b) TFAA, DCM, rt; c) imine, TMSCl, DCM, reflux; d) EDCI.HCl, DMAP, EtOH, DCM, rt.
Table 2.
Inhibition of the chymotryptic-like activity of purified human 20S proteasome by compounds 1, 6–10. N.D = not determined.
|
R | proteasome IC50 (μM) | NF-κB-Luc EC50 (μM) |
|---|---|---|---|
|
| |||
| 1 | CO2CH2CH3 | 2.5 | 2.5 |
| 6 | CO2H | N.D. | N.D. |
| 7 | CO2CH3 | 5.4 | 7.5 |
| 8 | CO2Bn | 2.4 | 3.5 |
| 9 | CONH2 | >10 | >20 |
| 10 | CO2CH2CH2OH | 3.2 | 10.9 |
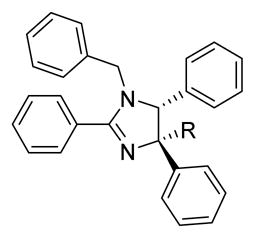
Our initial studies started by examining the requirement of the R1- moiety in the scaffold (Scheme 1). The compounds 11–25 were prepared according to the methods described previously. Hydrogenation of the R1-benzyl group of compound 1 resulted in compound 11, which was depleted of all activity. Acylation, benzylation and tosylation of 11, yielded compounds 12, 13 and 14, respectively, which were also all depleted of activity. Replacement of the benzyl group with the methylene-2-furan (15) moiety also resulted in loss of activity compared to the parent compound (1). Neither electron donating groups (16) nor withdrawing groups (17) in the para-position of the aryl group affected potency significantly. The incorporation of halogens (compounds 18–19) did increase potency following a trend of favoring lipophilicity. Unfortunately, the increase in lipophilicity came with a cost of decreased solubility as indicted for 18 (<0.5mg/mL) and 19 (<0.5 mg/mL) compared to 1 (1.91 mg/mL) in 5% dextrose in water. Thus, considering the already lipophilic nature of the imidazoline scaffolds, additional lipophilic moieties were considered unfavorable. Aryl and alkyl groups were previously found to lack cellular activity towards the inhibition of NF-κB,13a thus these were not pursued in our optimization studies.
Considering the lack of major positive changes in activity compared to the original benzyl moiety, we next focused our attention on the R2 group. Incorporation of a pyridine moiety in the R2 position rendered the imidazoline (20) inactive, whereas the more electron rich 2-furan heterocycle (21) regained its low micromolar potency. Following this trend, electron withdrawing groups (22, 24) resulted in a decrease in activity. Electron donating groups (23, 27) and lipophilic substitutions (25) retained activity but did not increase the overall potency significantly. However, benzylation of the aniline 23 yielded compound 29, which exhibited a strong increase in activity over parent compound 1 (IC50 0.5μM). This represents the first small molecule non-competitive proteasome inhibitor with IC50 values below the 1 micromolar level in purified protein assays.
Prior to following up on this new lead, we evaluated several functionalities as R3 moieties. The goal was to combine all the improved features in order to gain the highest efficacy. The R3 substituents can be incorporated by benzoylation of phenylglycine with different acyl moieties (Scheme 1). Cyclic dehydration of the benzoyl protected amino acid with trifluoroacetic anhydride provided the oxazolone I. A 1,3-dipolar cycloaddition reaction with the oxazolone and N-benzyl-1-(phenyl)methanimine was invoked using TMSCl, which generated the imidazoline II as a single diastereomer. Esterification of the carboxylic acid was accomplished using ethanol and EDCI with DMAP, to provide compound 30–33. The incorporation of an electron withdrawing group resulted in a loss of activity as indicated by compounds 32 and 33. Although the bromophenyl (30) did not significantly increased activity, the incorporation of an electron donating methoxy group resulted in an increase in activity, with the methoxyphenyl (31) being more than twice as active as the parent molecule 1.
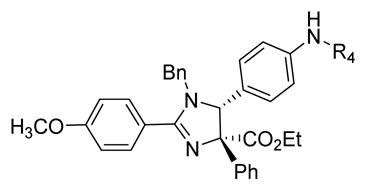
By combining the structural features with the best relative potencies, a range of derivatives were prepared that varied in the R4 position of imidazoline 37 (Table 4). Imidazoline 37 was prepared via the exposure of oxazolone 34 to TMSCl, in the presence of N-benzyl-1-(4-nitrophenyl) methanimine (Scheme 2). This generated the HCl salt of the imidazoline 35 as a single diastereomer. Esterification of the sterically hindered carboxylic acid was accomplished using ethanol and EDCI with DMAP in 86% yield. Reduction of the nitro-moiety of imidazoline 36 with zinc proceeded well to produce the aniline 37 in 93% yield. Aniline 37 was subsequently acylated with the appropriate acid chloride in the presence of pyridine to yield compounds 38 and 39. Treatment of the aniline with N,N-bis(t-butoxycarbonyl)thiourea and Hünigs base (di-isopropylethylamine, DIPEA) provided the guanidine derivative 40 after deprotection of the Boc-group with trifluoroacetic acid (TFA). Sulfonation of the aniline using the respective sulfonyl chlorides and pyridine provided analogues 41–45 in good yields. The aniline was also alkylated using reductive alkylation. Treatment of the aniline with the desired aldehyde formed the imine in situ, which was reduced with NaBH(OAc)3, to yield compounds 46–49. Yields for the formation of compounds 47 and 48 were relatively low (16% and 28%, respectively), likely due to the poor electrophilic nature of those aldehydes coupled with the poor nucleophilicity of the aniline.
Table 4.
Inhibition of the chymotryptic-like activity of purified human 20S proteasome by compounds 37–49. Data are averages of a minimum of two independent experiments, each as a technical triplicate. Individual values and standard error of the means are listed in supplemental Table S2.
|
R4 | IC50 (μM) |
|---|---|---|
|
| ||
| 37 | H | 1.97 |
| 38 | Ac | 2.17 |
| 39 | Bz | 1.44 |
| 40 |
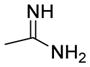
|
0.63 |
| 41 |
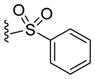
|
0.58 |
| 42 |
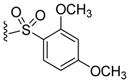
|
0.53 |
| 43 |
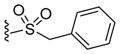
|
1.08 |
| 44 |
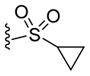
|
0.91 |
| 45 |
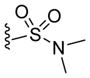
|
1.22 |
| 46 |
|
0.30 |
| 47 |
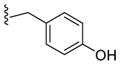
|
0.54 |
| 48 |
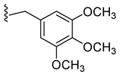
|
0.37 |
| 49 |
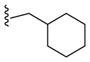
|
0.13 |
Scheme 2.

a) Na2CO3, H2O/1,4-dioxane, p-MeO-PhCO2Cl, rt, 85%; b) TFAA, DCM, rt, 88%; c) (Z)-N-benzyl-1-(4-nitrophenyl)methanimine, TMSCl, DCM, reflux, 34%; d) EDCI.HCl, DMAP, EtOH, DCM, rt, 86%; e) Zn, AcOH, rt, 93%; f) Ac2O, pyr., DCM, rt, 46%; g) BzCl, DMAP, DCE, rt, 31%; h) N,N′-bis(t-butoxycarbonyl)thiourea, DIPEA, EDCI.HCl, DCE, rt, 46%; i) 1:1 TFA:DCM, rt, 68%; j) sulfonation: RSO2Cl, pyr., DCM, 0°C at rt, 47–94%; k) reductive amination: NaBH(OAc)3, RCHO, DCE, 15–87%.
Acylation (to yield compound 38), benzoylation (to yield compound 39), sulfonation (to yield compound 41), benzylation (to yield compounds 46–48) and alkylation (to yield compound 49) of compound 37 increased the potency of the starting scaffold in all cases. Even the guanidine based-imidazoline 40 (IC50 = 0.63 μM) exhibited excellent potency. Sulfonation clearly provided several compounds with sub-micromolar activity, as indicated by imidazoline 41 (IC50 = 0.58 μM) and 42 (IC50 = 0.53 μM). However, the alkylation and benzylation (46–49) provided the first nanomolar non-competitive, non-covalent proteasome inhibitors described in the literature, with imidazoline 49 being the most potent at an IC50 of 130nM. Consistent with the biological mechanism of the parent compound 1 (TCH-013), compound 46 indicates similar kinetics for inhibition of the proteasome’s chymotryptic-like activity (supplemental information Figure S1).
The improved analogues were screened for cytotoxicity in cell culture in human myelomonocytic THP-1 cells and a bortezomib-resistant THP-1 cell line (BTZ500).4b, 17 Considering that the mechanism of acquired resistance for bortezomib in this cell line was previously determined to be due to the overexpression of a mutated β5 catalytic subunit of the proteasome,4b, 18 compounds that inhibit the proteasome via a mechanism should be able to overcome this resistance.8, 10 The imidazolines 37, 46–49 were screened at 10 μM (Table 5) and 5 μM (supplemental Figure S2). Bortezomib was used at 0.5 μM concentration (~100X its reported IC50 of 5–8 nM for proteasome inhibition).1–3 As anticipated, the potent MM drug, bortezomib, exhibits <5% cell viability in the wild type THP-1 cell line, but is unable to induce significant cytotoxicity in the resistant cells (Table 5) at a concentration of 100X its IC50 value. Consistent with its moderate inhibition of the proteasome, analogue 37 (proteasome IC50 1.97μM) exhibits weak cytotoxic activity in both cell lines. However, the more potent proteasome inhibitors 46–49 exhibited excellent cyto-toxicity in the THP-1 wild type and mutant cell lines. Thus, similar to the parent agent TCH-013, these agents were able to overcome resistant to competitive inhibitors in the bortezomib resistant cell line BTZ500 (Table 5), further confirming their unique mechanism of proteasome inhibition.
Table 5.
Viability data of THP-1 cells in human myelomonocytic THP-1 cells and bortezomib-resistant THP-1 cell line (BTZ500) exposed to bortezomib and imidazolines for 72 hours. Viability data are means of three independent experiments. Bortezomib was used at 0.5 μM. Individual values and standard errors of the means are listed in supplemental Table S2 and Figure S2.
| Compound | Proteasome IC50 (μM) | % cell viability Wild type | (at 10μM) Resistant |
|---|---|---|---|
|
| |||
| Bortezomib (0.5 μM) | 0.01 | <5 | 68 (±10.7) |
| 37 | 1.97 | 44 (±5.4) | 98 (±1.8) |
| 46 | 0.30 | 4 (±0.4) | 19 (±2.9) |
| 47 | 0.54 | 23 (±6.8) | 36 (±8.7) |
| 48 | 0.37 | 24 (±0.6) | 34 (±17.3) |
| 49 | 0.13 | 4 (±0.4) | 5 (±2.8) |
Taken together, we have evaluated the structural features of the R1-R3 groups of the groups surrounding the imidazoline scaffold. The in vitro data obtained for inhibition of the chymotryptic activity of the 20S proteasome is consistent with the earlier findings and follows the same trend as the inhibition of NF-κB mediated gene expression in cell culture. Our new data presented herein indicates that the R4 domain is critical in achieving sub-micromolar IC50 values for proteasome inhibition. From this series, compounds 49 (IC50 = 130 nM) represents the first nanomolar, non-competitive proteasome inhibitor reported. Importantly, this data demonstrates the potential and/or viability of developing new classes of proteasome inhibitors that regulate proteasome activity via a mechanism distinct from all current leading drugs or second generation drugs under clinical evaluation.
CONCLUSION
All current therapeutics targeting the proteasome are competitive inhibitors that compete for substrate binding via the formation of a covalent and often irreversible bond with the N- terminal threonine of the proteasome’s catalytic site. Reports in the literature of small molecule, non-competitive inhibitors are scarce and are typically only effective at micromolar or non-physiologically relevant concentrations. Here we present the first report of the optimization of a non-competitive small molecule proteasome inhibitor. The imidazoline based micromolar inhibitor TCH-013 (1) was optimized for potency towards the 20S proteasome in vitro to render nanomolar non-competitive proteasome inhibitors, with imidazoline 49 being the most potent agent (IC50 = 130 nM). The R1-R3 domains surrounding the imidazoline scaffold were found to be critical for activity, yet optimization of those domains resulted in little gain in terms of overall potency. However, the R4 domain was critical in obtaining sub-micromolar activities. These studies illustrate the feasibility of the development of non-competitive proteasome inhibitors as possible additives and/or alternatives to classical competitive agents.
Supplementary Material
Table 3.
Inhibition of the chymotryptic-like activity of purified human 20S proteasome by compounds 11–33. Data are averages of a minimum of two independent experiments, each as a technical triplicate. Individual values and standard error of the means are listed in supplemental Table S1
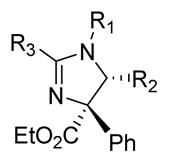
|
R1 | R2 | R3 | IC50(μM) |
|---|---|---|---|---|
|
| ||||
| 11 | H | Phenyl | Phenyl | >10 |
| 12 | Acetyl | Phenyl | Phenyl | >10 |
| 13 | Benzoyl | Phenyl | Phenyl | >10 |
| 14 | Tosyl | Phenyl | Phenyl | >10 |
| 15 | CH2-2-furan | Phenyl | Phenyl | 7.04a |
| 1 | Benzyl | Phenyl | Phenyl | 2.58 |
| 16 | 4-CH3O-benzyl | Phenyl | Phenyl | 2.73 |
| 17 | 4-F-benzyl | Phenyl | Phenyl | 3.71 |
| 18 | 4-Cl-benzyl | Phenyl | Phenyl | 2.02 |
| 19 | 4-Br-benzyl | Phenyl | Phenyl | 1.90 |
| 20 | Benzyl | 4-pyridine | Phenyl | >10 |
| 21 | Benzyl | 2-furan | Phenyl | 6.13 |
| 22 | Benzyl | 4-NO2-phenyl | Phenyl | 6.71 |
| 23 | Benzyl | 4-NH2-phenyl | Phenyl | 3.52 |
| 24 | Benzyl | 4-CF3-phenyl | Phenyl | 4.81 |
| 25 | Benzyl | 4-Cl-phenyl | Phenyl | 2.23 |
| 26 | Benzyl | 4-NH(SO2Ph)-phenyl | Phenyl | 1.08 |
| 27 | Benzyl | 4-SCH3-phenyl | Phenyl | 2.77 |
| 28 | Benzyl | 4-SO2CH3 – phenyl | Phenyl | >10 |
| 29 | Benzyl | 4-NHBenzyl-phenyl | Phenyl | 0.47 |
| 30 | Benzyl | Phenyl | 4-Br-phenyl | 3.19 |
| 31 | Benzyl | Phenyl | 4-CH3O-phenyl | 1.27 |
| 32 | Benzyl | 4-CN-phenyl | 4-CH3O-phenyl | >10 |
| 33 | Benzyl | 4-CONH2-phenyl | 4-CH3O-phenyl | >10 |
n=1
Acknowledgments
The authors gratefully acknowledge financial support in part from the Multiple Myeloma Research Foundation (MMRF) and the National Institutes of Health (CA-142644-01). In addition, LMA and JRL greatly appreciate financial support from the Michigan State University College of Osteopathic Medicine DO/PhD program and the College of Natural Science, respectively. A great deal of appreciation is given to Drs. J. Cloos, G. Jansen and J. Meerloo for the kind gift of the bortezomib resistant cell lines.
ABBREVIATIONS
- NF-κB
nuclear factor-kappaB
- AMC
aminomethylcoumarin
- Boc
tert-butoxycarbonyl
- TMSCl
trimethylsilyl chloride
- EDCI
1- ethyl-3-(3-dimethylaminopropyl)carbodiimide
Footnotes
The authors declare no competing financial interest
Supporting Information. The supporting information includes the IC50 values, kinetic analysis, cell viability values of each independent experiment and their error analysis. In addition, the supporting information describes the detailed protocols of the biological experiments, solubility evaluation and synthesis and compound characterization of all new compounds.
References
- 1.Chen D, Frezza M, Schmitt S, Kanwar J, Dou QP. Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives. Curr Cancer Drug Targets. 2012;11:239–253. doi: 10.2174/156800911794519752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Kisselev AF, van der Linden WA, Overkleeft HS. Proteasome inhibitors: an expanding army attacking a unique target. Chem Biol. 2012;19:99–115. doi: 10.1016/j.chembiol.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kisselev AF. Joining the army of proteasome inhibitors. Chem Biol. 2008;15:419–421. doi: 10.1016/j.chembiol.2008.04.010. [DOI] [PubMed] [Google Scholar]; (c) Borissenko L, Groll M. 20S proteasome and its inhibitors: crystallographic knowledge for drug development. Chem Rev. 2007;107:687–717. doi: 10.1021/cr0502504. [DOI] [PubMed] [Google Scholar]; (d) Shah JJ, Orlowski RZ. Proteasome inhibitors in the treatment of multiple myeloma. Leukemia. 2009;23:1964–1979. doi: 10.1038/leu.2009.173. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Navon A, Ciechanover A. The 26 S proteasome: from basic mechanisms to drug targeting. J Biol Chem. 2009;284:33713–33718. doi: 10.1074/jbc.R109.018481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beck P, Dubiella C, Groll M. Covalent and non-covalent reversible proteasome inhibition. Biol Chem. 2012;393:1101–1120. doi: 10.1515/hsz-2012-0212. [DOI] [PubMed] [Google Scholar]
- 4.(a) Arastu-Kapur S, Anderl JL, Kraus M, Parlati F, Shenk KD, Lee SJ, Muchamuel T, Bennett MK, Driessen C, Ball AJ, Kirk CJ. Nonproteasomal targets of the proteasome inhibitors bortezomib and carfilzomib: a link to clinical adverse events. Clin Cancer Res. 2011;17:2734–2743. doi: 10.1158/1078-0432.CCR-10-1950. [DOI] [PubMed] [Google Scholar]; (b) Franke NE, Niewerth D, Assaraf YG, van Meerloo J, Vojtekova K, van Zantwijk CH, Zweegman S, Chan ET, Kirk CJ, Geerke DP, Schimmer AD, Kaspers GJ, Jansen G, Cloos J. Impaired bortezomib binding to mutant beta5 subunit of the proteasome is the underlying basis for bortezomib resistance in leukemia cells. Leukemia. 2012;26:757–768. doi: 10.1038/leu.2011.256. [DOI] [PubMed] [Google Scholar]; (c) Groll M, Huber R, Moroder L. The persisting challenge of selective and specific proteasome inhibition. J Pept Sci. 2009;15:58–66. doi: 10.1002/psc.1107. [DOI] [PubMed] [Google Scholar]; (d) Argyriou AA, Bruna J, Marmiroli P, Cavaletti G. Chemotherapy-induced peripheral neurotoxicity (CIPN): An update. Crit Rev Oncol Hematol. 2011;82:51–77. doi: 10.1016/j.critrevonc.2011.04.012. [DOI] [PubMed] [Google Scholar]
- 5.Alexanian R, Delasalle K, Wang M, Thomas S, Weber D. Curability of multiple myeloma. Bone Marrow Res. 2012;2012:916479. doi: 10.1155/2012/916479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Jankowska E, Gaczynska M, Osmulski P, Sikorska E, Rostankowski R, Madabhushi S, Tokmina-Lukaszewska M, Kasprzykowski F. Potential allosteric modulators of the proteasome activity. Biopolymers. 2010;93:481–495. doi: 10.1002/bip.21381. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wenner M. A new kind of drug target. Sci Am. 2009;301:70–74. 76. doi: 10.1038/scientificamerican0809-70. [DOI] [PubMed] [Google Scholar]; (c) Hauske P, Ottmann C, Meltzer M, Ehrmann M, Kaiser M. Allosteric regulation of proteases. ChemBioChem. 2008;9:2920–2928. doi: 10.1002/cbic.200800528. [DOI] [PubMed] [Google Scholar]; (d) Gaczynska M, Osmulski PA. Characterization of noncompetitive regulators of proteasome activity. Methods Enzymol. 2005;398:425–438. doi: 10.1016/S0076-6879(05)98035-X. [DOI] [PubMed] [Google Scholar]; (e) DeDecker BS. Allosteric drugs: thinking outside the active-site box. Chem Biol. 2000;7:R103–107. doi: 10.1016/s1074-5521(00)00115-0. [DOI] [PubMed] [Google Scholar]
- 7.Groebe DR. In search of negative allosteric modulators of biological targets. Drug Discov Today. 2009;14:41–49. doi: 10.1016/j.drudis.2008.09.011. [DOI] [PubMed] [Google Scholar]
- 8.Li X, Wood TE, Sprangers R, Jansen G, Franke NE, Mao X, Wang X, Zhang Y, Verbrugge SE, Adomat H, Li ZH, Trudel S, Chen C, Religa TL, Jamal N, Messner H, Cloos J, Rose DR, Navon A, Guns E, Batey RA, Kay LE, Schimmer AD. Effect of noncompetitive proteasome inhibition on bortezomib resistance. J Natl Cancer Inst. 2010;102:1069–1082. doi: 10.1093/jnci/djq198. [DOI] [PubMed] [Google Scholar]
- 9.Sprangers R, Li X, Mao X, Rubinstein JL, Schimmer AD, Kay LE. TROSY-based NMR evidence for a novel class of 20S proteasome inhibitors. Biochemistry. 2008;47:6727–6734. doi: 10.1021/bi8005913. [DOI] [PubMed] [Google Scholar]
- 10.Lansdell TA, Hurchla MA, Xiang J, Hovde S, Weilbaecher KN, Henry RW, Tepe JJ. Noncompetitive Modulation of the Proteasome by Imidazoline Scaffolds Overcomes Bortezomib Resistance and Delays MM Tumor Growth in Vivo. ACS Chem Biol. 2013;8:578–587. doi: 10.1021/cb300568r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lansdell TA, O’Reilly S, Woolliscroft T, Kahlon DK, Hovde S, McCormick JJ, Henry RW, Cornicelli JA, Tepe JJ. Attenuation of collagen-induced arthritis by orally available imidazoline-based NF-kB inhibitors. Bioorg Med Chem Lett. 2012;22:4816–4819. doi: 10.1016/j.bmcl.2012.05.056. [DOI] [PubMed] [Google Scholar]
- 12.Sharma V, Lansdell TA, Peddibhotla S, Tepe JJ. Sensitization of tumor cells towards chemotherapy: Enhancing the efficacy of camptothecin by novel imidazolines. Chem Biol. 2004;11:1689–1699. doi: 10.1016/j.chembiol.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 13.(a) Kahlon DK, Lansdell TA, Fisk JS, Tepe JJ. Structural activity relationship of functionalized trans-imidazolines as potent inhibitors of interleukin-6 production. Bioorg Med Chem. 2009;17:3093–3103. doi: 10.1016/j.bmc.2009.03.002. [DOI] [PubMed] [Google Scholar]; (b) Kahlon DK, Lansdell TA, Fisk JS, Hupp CD, Friebe TL, Hovde S, Jones AD, Dyer RD, Henry RW, Tepe JJ. Nuclear Factor-kappaB Mediated Inhibition of Cytokine Production by Imidazoline Scaffolds. J Med Chem. 2009;52:1302–1309. doi: 10.1021/jm8013162. [DOI] [PubMed] [Google Scholar]
- 14.Gaczynska M, Osmulski PA. Small-molecule inhibitors of proteasome activity. Methods Mol Biol. 2005;301:3–22. doi: 10.1385/1-59259-895-1:003. [DOI] [PubMed] [Google Scholar]
- 15.Matsuura T, Ito Y. Photoinduced reactions. LXI Photochemical cis,trans-isomerization of 2,4,5-triphenyimidazolines. Chem Lett. 1972:431–434. [Google Scholar]
- 16.(a) Sharma V, Tepe JJ. Diastereochemical diversity of imidazoline scaffolds via substrate controlled TMSCl mediated cycloaddition of azlactones. Org Lett. 2005;7:5091–5094. doi: 10.1021/ol052118w. [DOI] [PubMed] [Google Scholar]; (b) Peddibhotla S, Tepe JJ. Multicomponent synthesis of highly substituted imidazolines via a silicon mediated 1,3-dipolar cycloaddition. Synthesis. 2003;9:1433–1440. [Google Scholar]; (c) Peddibhotla S, Jayakumar S, Tepe JJ. Highly diastereoselective multicomponent synthesis of unsymmetrical imidazolines. Org Lett. 2002;4:3533–3535. doi: 10.1021/ol026703y. [DOI] [PubMed] [Google Scholar]
- 17.de Wilt LH, Jansen G, Assaraf YG, van Meerloo J, Cloos J, Schimmer AD, Chan ET, Kirk CJ, Peters GJ, Kruyt FA. Proteasome-based mechanisms of intrinsic and acquired bortezomib resistance in non-small cell lung cancer. Biochem Pharmacol. 2012;83:207–217. doi: 10.1016/j.bcp.2011.10.009. [DOI] [PubMed] [Google Scholar]
- 18.Oerlemans R, Franke NE, Assaraf YG, Cloos J, van Zantwijk I, Berkers CR, Scheffer GL, Debipersad K, Vojtekova K, Lemos C, van der Heijden JW, Ylstra B, Peters GJ, Kaspers GL, Dijkmans BA, Scheper RJ, Jansen G. Molecular basis of bortezomib resistance: proteasome subunit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood. 2008;112:2489–2499. doi: 10.1182/blood-2007-08-104950. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.