

Abstract
G protein coupled receptors (GPCRs) are a large superfamily of integral cell surface plasma membrane proteins that play key roles in transducing extracellular signals, including sensory stimuli, hormones, neurotransmitters, or paracrine factors into the intracellular environment through the activation of one or more heterotrimeric G proteins. Structural alterations provoked by mutations or variations in the genes coding for GPCRs may lead to misfolding, altered plasma membrane expression of the receptor protein and frequently to disease. A number of GPCRs regulate reproductive function at different levels; these receptors include the gonadotropin-releasing hormone receptor (GnRHR) and the gonadotropin receptors (follicle-stimulating hormone receptor and luteinizing hormone receptor), which regulate the function of the pituitary-gonadal axis. Loss-of-function mutations in these receptors may lead to hypogonadotropic or hypergonadotropic hypogonadism, which encompass a broad spectrum of clinical phenotypes. In this review we describe mutations that provoke misfolding and failure of these receptors to traffick from the endoplasmic reticulum to the plasma membrane. We also discuss some aspects related to the therapeutic potential of some target-specific drugs that selectively bind to and rescue function of misfolded mutant GnRHR and gonadotropin receptors, and that represent potentially valuable strategies to treat diseases caused by inactivating mutations of these receptors.
Keywords: G protein-coupled receptors, intracellular trafficking, gonadotropin-releasing hormone receptor, luteinizing hormone receptor, follicle-stimulating hormone receptor, pharmacological chaperon, hypogonadism, protein misfolding
1. Introduction
G-protein coupled receptors (GPCRs) constitute a large superfamily of integral membrane proteins that mediate cellular responses from structurally diverse extracellular signals, including hormones, sensory stimuli, neurotransmitters, proteinases, chemokines, and drugs, by coupling to a repertoire of heterotrimeric G-proteins (Fredriksson et al., 2003; Lagerstrom and Schioth, 2008; Ulloa-Aguirre and Conn, 1998). In turn, activated G proteins regulate specific downstream effectors, such as phospholipases, adenylyl cyclase, protein kinases, and ion channels. Although G protein-coupled receptors vary considerably in molecular size, all share a common molecular topology that consists of a single polypeptide chain which traverses the lipid bilayer seven times, forming characteristic transmembrane (TM) α-helices connected by alternating extracellular and intracellular loops (eL and iL, respectively), with extracellular NH2-terminus and intracellular COOH-terminal (Ctail) sequences of variable length. This particular type of receptors plays essential roles on a wide range of biological functions (Ulloa-Aguirre and Conn, 2009), and because of their relevance in function and as a cause of disease, they are important therapeutic targets for an array of diseases, as reflected by the fact that ~60–70% of all approved drugs are proposed to selectively target these particular receptors (Lagerstrom and Schioth, 2008; Overington et al., 2006; Schlyer and Horuk, 2006).
The life cycle of GPCRs begins at the endoplasmic reticulum (ER), were synthesis, folding and assembly of proteins occur. Properly folded receptors are then targeted to the ER-Golgi intermediate complex and thereafter to the Golgi apparatus and trans-Golgi network where processing of the protein is completed before delivery to the cell surface plasma membrane (PM); here they become accessible to their cognate ligands (Broadley and Hartl, 2009) (Fig. 1). Interaction between GPCRs and agonists at the PM, provokes endocytosis of the receptor-agonist complex, which is then followed by either recycling of the receptor back to the PM or targeting to the lysosomes and/or proteasomes for degradation. Thus, the balance between trafficking from the ER to the PM and the endocytosis-recycling pathway determines the net amount of receptor protein available to interact with agonist and elicit a measurable biological response. As with any newly synthesized protein, GPCRs are subjected to a strict quality control system (QCS) that monitors and is sensitive to the correctness of nascent protein folding into a three-dimensional structure (Ulloa-Aguirre and Conn, 2009). By monitoring the structural and conformational features of newly synthesized proteins, the QCS determines which proteins must be retained at the ER and eventually degraded or routed to the Golgi apparatus and thereafter to the secretory pathway or appropriate compartment within the cell. Thus, the QCS prevents accumulation of misfolded proteins that may aggregate and interfere with cell function. G protein-coupled receptor export from the ER to the Golgi is modulated by interaction of the proteins with specialized folding factors, escort proteins, retention factors, enzymes, and members of the molecular chaperone families, which belong to the ER QCS and the so-called proteostasis network (Hartl et al., 2011; Hutt et al., 2009; Ron and Walter, 2007). In particular, molecular chaperones are essential components of the ER QCS that evaluate native receptor conformation and promote delivery from the ER to the Golgi where the protein molecule is processed before final delivery of the mature form to the PM (Brooks, 1999; Hartl and Hayer-Hartl, 2002; Morello et al., 2000a). Molecular chaperones are an important quality control mechanism that not only recognizes, but also retains and targets misfolded, non-native protein conformers for their eventual degradation via the polyubiquitination/proteasome pathway (Chevet et al., 2001; Klausner and Sitia, 1990; Schubert et al., 2000; Werner et al., 1996). Some general structural features such as exposure of hydrophobic shapes, unpaired cysteines, immature glycans, and distinct sequence motifs embedded within the receptor sequence are factors important for determining chaperone-protein association (Angelotti et al., 2010; Broadley and Hartl, 2009; Hartl and Hayer-Hartl, 2009; Schubert et al., 2000; Werner et al., 1996). Some of the effects of molecular chaperones on protein folding may be mimicked by the so-called chemical chaperones (Arakawa et al., 2006; Loo and Clarke, 2007; Robben et al., 2006; Sampedro and Uribe, 2004) and pharmacological chaperones (or pharmacoperones) (Bernier et al., 2004b), which may rescue misfolded receptors in vitro and in vivo, and thus represent therapeutic options for an array of diseases caused by protein misfolding (Bernier et al., 2006; Conn and Ulloa-Aguirre, 2010; Conn et al., 2007; Ulloa-Aguirre, 2011). Despite their potential application for treating misfolded diseases, there are some drawbacks for the use of chemical chaperones in vivo. Effective folding of mutant proteins requires high concentrations of chemical chaperones and although chemical chaperones can rescue some misfolded proteins, they are nonspecific and might in parallel increase secretion or intracellular retention of many different proteins, potentially leading to inappropriate changes in the levels and/or bioactivity, thereby compromising cell function and/or local homeostasis (Bernier et al., 2004b; Castro-Fernandez et al., 2005). In contrast to chemical chaperones, pharmacoperones are target-specific and have the advantage of selective association with the misfolded protein, without interfering with the fate of other proteins. Pharmacoperones are small, often hydrophobic, compounds that enter cells and serve as specific molecular templates, promoting correct folding and allowing the target protein to pass the ER QCS and be expressed at the cellular loci where they may function (Arakawa et al., 2006; Bernier et al., 2004b; Conn and Janovick, 2011; Conn and Ulloa-Aguirre, 2011; Loo and Clarke, 2007). Pharmacoperones bind the target protein with high affinity and have been used experimentally in a number of experimental disease models provoked by misfolding and/or abnormal aggregation of particular proteins (Estrada and Soto, 2007, 2006; Hammarstrom et al., 2003; Ishii et al., 2007; Sigurdsson et al., 2000; Soto, 2001), including GPCRs (Bernier et al., 2004a; Bernier et al., 2004b; Bernier et al., 2006; Conn and Ulloa-Aguirre, 2010; Conn et al., 2007).
Figure 1.
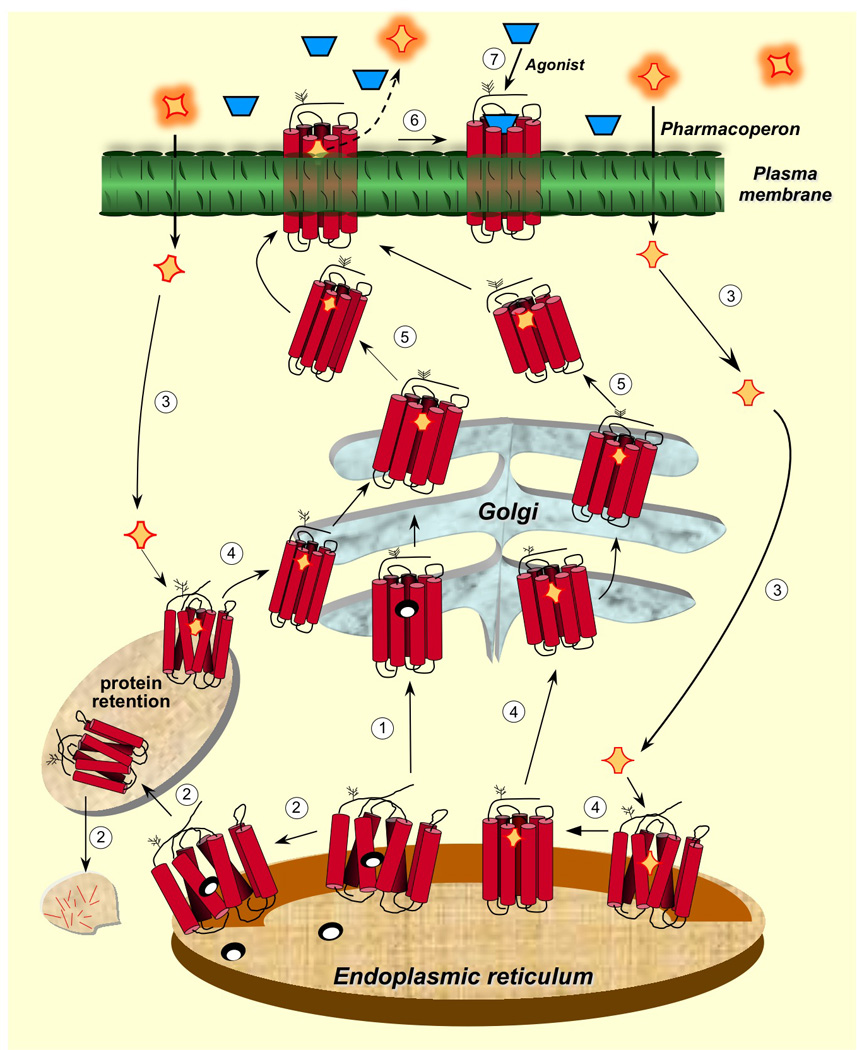
Intracellular trafficking of GPCRs from the ER to the plasma membrane and relation to the cellular quality control system. Newly synthesized proteins are translocated to the lumen of the ER where folding is both facilitated and/or corrected by molecular chaperones (oval structures). The correctly folded protein is thereafter exported to the Golgi complex for further processing (eg. glycosylation) (step 1 in clear circles) and routing to the plasma membrane. When folding fails, misfolded proteins are retained in the ER and eventually targeted for degradation through the polyubiquitination/proteasome pathway (step 2). Pharmacoperones (orange rhomboid structures) diffuse into the cell (step 3) and selectively bind the misfolded protein to correct misfolding promoting its export to the Golgi complex (step 4). Previously synthesized misfolded proteins, retained by the QCS, may still be rescued by pharmacoperones (steps 3 and 4 at the left). Mature processed proteins are then delivered to the cell surface PM (step 5), where the pharmacoperone can dissociate from the target protein (step 6) allowing the receptor to interact with agonist (step 7). Development of pharmacoperones that dissociate functions or rescue from agonism is currently needed as many pharmacoperone drugs are also receptor agonists/antagonists. Modified from Conn and Ulloa-Aguirre (2011), with permission of the publisher.
Defective trafficking of the GPCR from the ER to the PM due to misfolding is now widely recognized as a cause of a large number of loss-of-function diseases, including retinitis pigmentosa, congenital hypothyroidism and obesity, X-linked nephrogenic diabetes insipidus, and reproductive disorders, among others, in which misfolding leads to retention, increased intracellular degradation, and hence, deficient upward trafficking of the misfolded mutant to the PM (Castro-Fernandez et al., 2005; Ulloa-Aguirre and Conn, 2009). Although loss-of-function mutations in GPCRs may modify domains involved in key functions of the receptor (binding to agonist, receptor activation, and interaction with cognate G proteins and/or accessory/scaffold proteins), they also may affect sequences essential for proper folding and traffic of the protein to the PM. Thus, misfolding may give rise to proteins that retain intrinsic function but that are misrouted and thus cannot reach the PM, leading to disease. The fact that some misfolded proteins may retain function, offers the therapeutic opportunity to directly correct misrouting and rescue, partially or completely, function of the mutant receptors, potentially curing disease (Bernier et al., 2006; Conn and Janovick, 2009; Janovick et al., 2002; Loo and Clarke, 2007; Nakamura et al., 2010; Ulloa-Aguirre et al., 2003; Ulloa-Aguirre et al., 2006).
This review focuses on mutations that lead to misfolding and defective trafficking of GPCRs involved in reproductive function in humans. A number of GPCRs regulate reproductive function at different levels; these receptors include the prokineticin-2 receptor (PROKR2), involved in the migration of GnRH-producing neurons from the olfactory bulb to the hypothalamus during embryogenesis; the neurokinin-B receptor and GPR54, which play a pivotal role in the function of the hypothalamic gonadotropin- releasing hormone (GnRH) pulse generator; and the GnRH, follicle-stimulating hormone (FSH) and luteinizing hormone (LH) receptors (GnRHR, FSHR and LHR, respectively), which are directly involved in the regulation of gonadal function. Although mutations in all these GPCRs may result in misfolding and defective intracellular trafficking of the altered receptor (Francou et al., 2011; Huhtaniemi and Alevizaki, 2007; Martens et al., 2002; Monnier et al., 2009; Nimri et al., 2011; Rannikko et al., 2002; Ulloa-Aguirre et al., 2004), this review focuses on the three latter receptors (the GnRHR, FSHR and LHR), which represent valuable models for the development of pharmacoperone drugs potentially applicable to treat diseases caused by GPCR misrouting.
2. GnRHR trafficking-defective mutants leading to HH
The GnRHR is a GPCR that belongs to family A (rhodopsin/β-adrenergic-like family) of GPCRs (Millar et al., 2004; Ulloa-Aguirre and Conn, 1998). This receptor binds GnRH, which is a decapeptide produced by the hypothalamus and is released to the anterior pituitary where it acts to engender gonadotropin synthesis and secretion (Conn and Crowley, 1994; Ulloa-Aguirre and Timossi, 2000). GnRH signals mainly through activation of the G11/q protein. Besides several unique structural features that distinguish the mammalian type I GnRHR from other GPCRs (Millar et al., 2004), two particular determinants dramatically influence trafficking of the GnRHR: i. the lack of a Ctail peptide which projects into the cytosol (Millar et al., 2004) and ii. the presence of a lysine residue at position 191 (K191) in the eL2. Other species, such as fish, reptiles, birds, and the primate type II GnRHR contain a Ctail peptide, which plays an important role in determining differential physiological receptor regulation (Blomenrohr et al., 1999; Heding et al., 1998; Lin et al., 1998; McArdle et al., 1999; Millar, 2003). Insertion of the piscine Ctail sequence to the wild-type (Wt) human (h) GnRHR, dramatically increases its PM expression (Janovick et al., 2003b; Maya-Nunez et al., 2011), an effect that has allowed the study of mechanisms that differentially regulate trafficking of this particular receptor (Janovick et al., 2003b; Maya-Nunez et al., 2011; Maya-Nunez et al., 2002). On the other hand, at position K191 is frequently found glutamic acid or glycine (ocassionally) in non-primate mammals instead of lysine (Janovick et al., 2006; Ulloa-Aguirre et al., 2006); in fact, in rodents (rat and mice) GnRHRs the orthologous amino acid is absent, thereby yielding a structure that is one residue smaller (327 amino acid residues). The absence of lysine in this position confers the rodent GnRHRs with an increased PM expression (Arora et al., 1999), whereas its presence in the hGnRHR (328 amino acids) limits the number of GnRHR molecules that reach the PM after synthesis. The mechanism subserving the effect of K191 on PM expression of the primate receptor, includes interfering with formation of the C14-C200 disulfide bridge, which is an essential structural requirement to stabilize the hGnRHR in a conformation compatible with ER export (Arora et al., 1999; Janovick et al., 2006; Jardon-Valadez et al., 2009; Maya-Nunez et al., 2002; Ulloa-Aguirre et al., 2006). Several motifs in multiple domains of the hGnRHR control the destabilizing influence of K191 on this bridge and limit its PM expression (Janovick et al., 2006; Ulloa-Aguirre et al., 2006); identification of these motifs in the hGnRHR sequence explains why insertion of K191 to the rat sequence does not perturb PM expression of the receptor. In fact, in the rat GnRHR the C14-C199 bridge is not essential for receptor expression as replacement of either of the cysteine residues of the bridge does not affect PM expression or GnRH-stimulated cellular signaling (Janovick et al., 2006). These data unveiled a novel and underappreciated mechanism for posttranslational control of a GPCR by altering its interaction with the ER QCS and provided a biochemical explanation for the pathogenesis of some disease-causing mutations of this receptor (Conn et al., 2006).
Thirty one inactivating mutations (including deletions of two large sequences) in the hGnRHR have been described as a cause of hypogonadotropic hypogonadism (HH) (Beranova et al., 2001; Conn and Ulloa-Aguirre, 2010; Gianetti et al., 2012; Ulloa-Aguirre et al., 2004), a disease characterized by decreased or apulsatile gonadotropin release and reproductive failure (Gianetti et al., 2012; Ulloa-Aguirre et al., 2004). Eight homozygous and ~18 heterozygous combinations of hGnRHR mutants have been detected to be expressed by individuals exhibiting either partial or complete forms of HH (Conn and Ulloa-Aguirre, 2010; Gianetti et al., 2012). Patients harboring some of these hGnRHR mutations, also exhibit alterations in other HH-causing genes, such as the PROKR2 or the fibroblast growth factor receptor 1 gene (Gianetti et al., 2012). Expression of these hGnRHR mutants in in vitro heterologous systems resulted in cells that minimally bind GnRH agonists and thus that exhibit no, or marginal response to agonist stimulation by effector activation. Initially, these observations suggested that such mutations provoked structural alterations in motifs involved in agonist binding, receptor activation or interaction with coupled effectors. Nevertheless, the majority (~90%) of the hGnRHR mutants whose function has been examined to date have showed to be related to defects in intracellular trafficking, as revealed by mutational studies and/or response to pharmacologic manipulations (Conn and Ulloa-Aguirre, 2011; Leanos-Miranda et al., 2002; Maya-Nunez et al., 2011). The clinical phenotypes expressed by homozygous or compound heterozygous patients bearing these particular mutations have been summarized previously (Beranova et al., 2001; Gianetti et al., 2012; Karges et al., 2003; Leanos-Miranda et al., 2005; Ulloa-Aguirre et al., 2004).Because reproductive failure is not life-threatening, it is likely that many partial forms of HH go undiagnosed and individual mutants, if severe in phenotype, are not inherited by progeny (Beranova et al., 2001; Gianetti et al., 2012). Such misfolded mutants frequently exhibit gain (eg. the Y108C mutant) or loss (eg. the C200Y mutant) of either cysteine residues (necessary to form bridges associated with the tertiary structure of proteins), a change in residue charge (eg. the E90K hGnRHR mutant, see below), or a proline (an amino acid residue frequently associated with an inflexible turn in the protein backbone fold, eg. the P320L mutant). These structural characteristics, as well as motifs involved in the control of the destabilizing influence of K191 on the C14-C200 bridge formation (see above), explain some of the pathogenic mechanisms whereby mutations in the hGnRHR lead to defective intracellular traffic. For example, in the misfolded E90K mutant (located at the TM2), the dramatic change in amino acid charge blocks the formation of an E90-K121 salt bridge required to stabilize the interaction between the TMs 2 and 3, which apparently is a requisite to pass the ER QCS (Janovick et al., 2009b). Furthermore, the native E90K hGnRHR conformation results in a constitutively active receptor as unveiled by pharmacoperone and genetic (eg. deletion of K191) interventions, which allowed for plasma membrane localization (Janovick and Conn, 2010). This indicated that the intact TM2-TM3 salt bridge required for correct trafficking is an evolutionary sentinel employed by the ER QCS to protect the cell against PM expression of a constitutively active receptor.
As mentioned above, the amino acid residue K191 is important from the evolutionary point of view since the absence of K191 confers the rodent GnRHRs with an increased efficiency of PM expression, whereas its presence in the human GnRHR limits the number of GnRHR molecules that reach the PM after synthesis (Arora et al., 1999). Consequently, and in contrast to humans, mice homozygous for the E90K mutation, exhibit a much milder phenotype due to partial PM expression of the mutant GnRHR. Whereas male patients harboring this mutation in both alleles exhibit a complete form of hypogonadism, extremely low serum gonadotropin levels, and unresponsiveness to exogenous GnRH stimulation (Soderlund et al., 2001), in male mice harboring the Gnrhr E90K mutation both gonadotropins are only modestly reduced in serum and increase after GnRH agonist administration (Stewart et al., 2012). Further, these mice are fertile and only exhibit reduced (albeit histologically normal) testis size. In contrast to males, females homozygous for the mutation are infertile; although they exhibit folliculogenesis, they fail to ovulate and to form corpora lutea, most probably due to reduced sensitivity to endogenous GnRH and inability to decode GnRH pulse frequency and amplitude to produce the gonadotropin response necessary to trigger ovulation (Stewart et al., 2012).
In the case of the C200Y mutant, the substitution prevents formation of the C14-C200 bridge required for the hGnRHR to pass the ER QCS (Janovick et al., 2006; Jardon-Valadez et al., 2009; Ulloa-Aguirre et al., 2006). Interestingly, the markedly reduced PM expression of several misfolded mutants (C200Y, C14A or C14S mutants) may be completely restored by pharmacoperones and/or by deleting K191 (Janovick et al., 2006; Jardon-Valadez et al., 2009). Further, molecular dynamics simulations have shown that when K191 is removed from the hGnRHR, the distance between the sulfur or oxygen-sulfur groups of cysteine 14 and 200 is shorter and more stable, and the conformation of both the eL2 and the NH2-terminal domain become less fluctuating than in the receptor bearing K191 (Jardon-Valadez et al., 2009). Contrary to this mutant, the Y108C mutation provokes formation of an aberrant C108 (at the eL1)- C200 (at the eL2) disulfide bridge, leading to a gross distortion in the conformation of the receptor, preventing its traffic to the PM (Maya-Nunez et al., 2011). It is possible to partially rescue function of this mutant by deletion of K191 or pharmacoperone treatment, although complete rescue may be only achieved when pharmacoperone action and deletion of K191 are combined (Maya-Nunez et al., 2011). Finally, mutations at positions 168 (S168K) and 217 (S217K) in the TMs 4 and 5, respectively, are thermodynamically unfavored substitutions that provoke twisting of the corresponding a-helices, moving eL2 away from the NH2-terminal domain, and preventing formation of the C14-C200 bridge. Therefore, these mutant receptors never pass the ER QCS and are completely resistant to genetic or pharmacologic rescue approaches (Janovick et al., 2003a; Janovick et al., 2006; Leanos-Miranda et al., 2002; Ulloa-Aguirre et al., 2006).
It is widely acepted that most GPCRs self associate to form dimers. This proclivity can have detrimental effects in some conditions, such as in autosomal dominant inherited diseases. In such a case, defective PM expression of completely or partially functional receptors might be attributed or amplified by the dominant-negative effect of oligomerization of the defective, misfolded receptor with its Wt counterpart (Conn et al., 2007; Le Gouill et al., 1999; Zhu and Wess, 1998). In the case of the hGnRHR, K191 plays an important role in mediating the inhibitory effects of misfolded hGnRHR mutants on Wt receptor PM expression, an effect that is mediated by intracellular retention and aggregation of the mutant-Wt complexes (Brothers et al., 2004). This dominant negative effect might potentially worsen the disease in simple or compound heterozygous patients showing complete or partial forms of HH (Leanos-Miranda et al., 2005; Leanos-Miranda et al., 2003). In this regard, folding of hGnRHR mutants and Wt receptor PM expression may be promoted by pharmacoperones, preventing the dominant negative effect of the former and rescuing Wt receptor function (Leanos-Miranda et al., 2005).
In order to understand the mechanism of action of pharmacoperones on misfolded hGnRHRs, we have recently employed a combined strategy that includes mutagenesis, confocal microscopy, and computer modeling and ligand docking. We have found that a number of different chemical classes of compounds [indoles and quinolones, and the distinctly different erythromycin macrolide, A177775, and TAK-013 (Janovick et al., 2003a; Ulloa-Aguirre, 2011)] stabilize the E90K hGnRHR mutant by promoting formation of a salt bridge associating D98 (at the extracellular face of TM1) and K121 (at the TM3) (Conn and Janovick, 2009; Janovick et al., 2009b; Jardon-Valadez et al., 2008). This pharmacoperone-mediated bridge functions as a surrogate bond for the highly conserved E90-K121 salt bridge (Jardon-Valadez et al., 2008), allowing stabilization of the TM2-TM3 conformation, which is a potential structural requirement for passing the ER QCS and for rescuing function of the hGnRHR to the PM. Since D98 and K121 are contact points for the natural ligand of the GnRHR (Millar et al., 2004), it is possible that different competitors of GnRH may interact at or near the ligand binding site, which is located in the lateral plane of the PM, a region rich in hydrophobic residues (Jardon-Valadez et al., 2008). The finding that all pharmacoperones tested rescue a number of hGnRHR mutants to different extents (Janovick et al., 2003a; Leanos-Miranda et al., 2002; Maya-Nunez et al., 2011), despite their wide distribution along the receptor, indicates that in addition to the C14-C200 disulfide bridge, the E90-K121 salt bridge may be also recognized by the ER QCS. It should be emphasized that the efficiency of pharmacoperones to rescue function of misfolded receptors depends on the structure of the pharmacoperone (that determines selectivity for a given protein), the complexity of misfolding, and the potential alterations in receptor function provoked by the mutation (Morello et al., 2000a; Morello et al., 2000b; Ulloa-Aguirre et al., 2004, 2003). For this reason, functional rescue of misfolded receptors cannot be expected if the mutation affects key domains involved in receptor function.
3. Trafficking-defective mutants of the gonadotropin receptors leading to hypergonadotropic hypogonadism
The FSHR and LHR are the target receptors for the pituitary gonadotropins, which play a key role in reproductive function. These glycoprotein hormone receptors, together with the thyroid-stimulating hormone receptor (TSHR), belong to family A of the GPCR superfamily. FSHR and LHR are expressed by specific cells in the gonads (Vassart et al., 2004), whereas the TSHR is expressed in the thyroid follicular cell. The FSHR is mainly expressed in ovarian granulosa cells where its action is essential for FSH induced maturation of ovarian follicles and granulosa cell progesterone and estrogen production; in the testis, the FSHR is expressed by the Sertoli cells in the seminiferous tubules, where it supports Sertoli cell growth and metabolism, initiating spermatogenesis. In males, the LHR is expressed mainly in the testicular Leydig cells nests between the seminiferous tubules, where LH induces testosterone production. In females, LHR is expressed in the ovarian theca cells which line the developing follicle and where LH induces androgen production that serves as a substrate for FSH induced P450 aromatase, the enzyme that converts androgen to estrogen. LH also is responsible for the follicular breakage and the subsequent release of the oocyte (ovulation).
In contrast to the GnRHR, glycoprotein hormone receptors exhibit a large NH2-terminal extracellular domain (ECD) that bind their cognate ligands (Ascoli et al., 2002; Dias et al., 2002; Simoni et al., 1997; Ulloa-Aguirre and Conn, 1998). Amino acid sequences of the extracelllar domain of the gonadotropin receptors are 46% identical; they exhibit a NH2-terminal cysteine-rich region, a central region bearing a structural motif of nine imperfect leucine-rich repeats (LRRs) that is shared with a number of other membrane receptors involved in ligand selectivity and specific protein-protein interactions (Bogerd, 2007), and a carboxyl-terminal cysteine-rich sequence, the so-called hinge region, which structurally links the leucine-rich ECD with the serpentine transmembrane domain of glycoprotein hormone receptors (Fan and Hendrickson, 2005; Jiang et al., 2012). Depending on the glycoprotein hormone receptor, this latter region may be involved in high-affinity hormone binding, receptor activation, intramolecular signal transduction, and/or silencing the basal activity of the receptor in the absence of ligand (Mueller et al.). The mature human hFSHR protein consists of 678 amino acid residues long, whereas the processed LHR is 675 amino acids long (Dias et al., 2002).
Upon agonist binding, the activated FSH and LH receptors stimulate a number of intracellular signaling pathways. The canonical Gαs/cAMP/PKA signaling pathway has been recognized as a key effector mechanism of LH and FSH biological action for more than 20 years (Hunzicker-Dunn and Maizels, 2006). However, gonadotropin receptors have also been reported to couple to other G protein subtypes and activate a number of distinct effector enzymes [reviewed in (Ulloa-Aguirre et al., 2011)], depending on the particular developmental stage of the host cells (Musnier et al., 2009).
Similar to other GPCRs, the gonadotropin receptors have to be correctly folded into a conformation to pass the QCS and be compatible with ER export (Ulloa-Aguirre and Conn, 2009). In fact, mutations that affect the folding process of these receptors result in intracellular retention of the folding intermediates, and eventually in disease. Several GPCR interacting proteins that support folding and trafficking to the PM have been identified to interact with the gonadotropin receptors during their residency at the ER. Coimmunoprecipitation experiments have shown that the folding process of the FSHR and LHR precursors involves interactions with the chaperones calnexin and calreticulin, which bind a broad range of glycoproteins facilitating proper folding of intermediate molecules (Mizrachi and Segaloff, 2004; Rozell et al., 1998), as well as with the protein disulfide isomerase PDI (Mizrachi and Segaloff, 2004), which is an ER-resident enzyme involved in disulfide bond formation of folding intermediates, and that probably acts as a co-chaperone with calnexin and calreticulin during their association with the glycoprotein hormone receptors. No interaction of these receptors with the chaperones BiP (a chaperone that maintains proteins in a state competent for subsequent folding and oligomerization, and that mediates retrograde translocation of misfolded conformers for proteosomal degradation), Grp94 (involved in folding and assembly of membrane proteins of the secretory pathway) or Erp57 (a thiol-disulfide oxidoreductase that catalyzes disulfide bond formation and isomerization, and which is thought to be a cochaperone that physically associates with calnexin and calreticulin in the nascent polypeptide chain) (Gething, 1999; Ulloa-Aguirre and Conn, 2009; Weekes et al., 2012), were detected in these studies. Studying the interaction of gonadotropin receptor mutants with these chaperones has been important to detect differences in folding among mutants (see below).
Properly folded and assembled secretory proteins are segregatd from ER-resident proteins into COPII-coated vesicles for exporting to the Golgi to be further processed before being sent to their final destination. Several recently identified motifs have been shown to be involved in GPCR exit from the ER and the Golgi. Among these motifs is the F(X)6LL sequence identified in the Ctail of several GPCRs, including the gonadotropin receptors (Duvernay et al., 2004). In the hFSHR, this export motif is located between amino acid residues 633 and 641, whereas in the hLHR this motif is located between residues 630 and 638 (Ascoli et al., 2002; Dias et al., 2002; Ulloa-Aguirre and Timossi, 1998). The Ctail peptide of the hFSHR contains the minimal BBXXB motif reversed in its juxtamembrane region (residues 631 to 635) (Timossi et al., 2004), which in other GPCRs is involved in G protein coupling (Okamoto and Nishimoto, 1992). The last two residues of this motif (R634 and R635) and the preceding F633 constitute the NH2-end of the highly conserved F(X)6LL motif, and thus it was not surprising that mutations in these residues impaired receptor trafficking and cell surface PM localization of the receptor (Timossi et al., 2004; Zarinan et al., 2010). Further, some of these laboratory-manufactured mutant hFSHRs (eg. R635A) exert potent dominant negative effects on Wt receptor PM expression (Zarinan et al., 2010), as it has also been observed for some naturally occurring hLHR mutants (Tao et al., 2004). The iL3 of the hFSH and hLH receptors also contains this BXXBB motif (residues 569 to 573) and either deletion or replacement of the basic residues of this motif with alanine impaired PM expression of the modified receptors (Schulz et al., 1999; Timossi et al., 2004). Human FSHRs with point mutations in this sequence (eg. hFSHR R573A) are retained intracellularly, and when co-transected with the Wt receptor also exert strong dominant negative effects, probably by forming misfolded mutant:wild-type receptor complexes (Zarinan et al., 2010) (see below). Another motif identified in all glycoprotein receptors is the AFNGT motif (amino acid residues 193 to 197 in the hLHR and 189 to 193 in the hFSHR), which bears a potential glycosylation site (N195GT and N191GT, in the human LH and FSH receptors, respectively). Although mutations in this motif might interfere with the integrity of the LRRs and thus with binding of agonists, it has been shown that mutations in this region rather influence receptor folding and trafficking to the PM (Gromoll et al., 2002; Rannikko et al., 2002)(see below). In fact, mutations in the first three amino acid residues of this motif lead to altered agonist-stimulated second messenger production without grossly affecting ligand binding affinity (Gromoll et al., 2002). Further mutation of the putative glycosylation site present in this motif (N195I in the hLHR and N191I in the hFSHR) resulted in reduced cAMP production by both receptors, but was more prominent for the hFSHR (Gromoll et al., 2002), thus emphasizing on the importance of glycosylation at this particular residue in the latter receptor.
Two posttranslational modifications are particularly important for intracellular trafficking of the gonadotropin receptors: glycosylation and palmitoylation. Glycosylation occurs during biosynthesis and facilitates folding of protein precursors by increasing their solubility and stabilizing protein conformation (Helenius and Aebi, 2004). The ECD of the hFSHR bears four potential glycosylation sites (as defined by the consensus sequence N-X-Ser/Thr, where X is any amino acid except proline) at positions 191, 199, 293, and 318 (Dias et al., 2002), whereas the hLHR exhibits six glycosylation sites (at positions 99, 174, 195, 291, 299, and 313). The only direct biochemical evidence that exists as to which sites are glycosylated in the hFSHR comes from the crystal structure of the ECD residues 25–250 (Fan et al., 2005; Jiang et al., 2012). The structures show that carbohydrate is attached at residue N191 which protrudes into solvent, whereas no carbohydrate is attached at residue N199 which protrudes from the flat beta sheet into the hormone-receptor binding interface. No structural information is available for residues 293 and 318 at this time. Studies with the rat FSHR have suggested that the FSHR is glycosylated at two of three glycosylation consensus sequences (191, 199, and 293) and that the presence of carbohydrates at either one of these residues (N191 or N293) is sufficient for receptor folding and trafficking to the PM (Davis et al., 1995). In contrast, abrogation of glycosylation of the rat LHR, did not modify agonist binding, suggesting that the deglycosylated receptor folded properly and trafficked to the PM. These studies underline the differential effects of glycans on the LH and FSH receptor with respect to folding and PM expression.
Another post-translational modification important for GPCR trafficking to the PM is palmitoylation. Cysteine residues in the COOH-terminus of several GPCR have been shown to be the target for S-acylation with palmitic acid; in some receptors this posttranslational modification is often required for efficient delivery of the protein to the cell membrane (Blanpain et al., 2001; Fukushima et al., 2001; Percherancier et al., 2001; Resh, 2006; Tanaka et al., 1998). The hFSHR exhibits in its Ctail two conserved cysteine residues (at positions 646 and 672) and one non-conserved cysteine residue at position 644. Although the hFSHR is palmitoylated at all cysteine residues, regardless of their location in the Ctail of the receptor (Uribe et al., 2008), S-acylation at C627 and C655 is not essential for efficient FSHR cell surface membrane expression, whereas at C629 it is, as replacement of this residue with glycine or alanine reduced detection of the mature form of the receptor by ~40–70%. Further, when all palmitoylation sites are removed from the FSHR, cell surface PM expression is reduced to ~10–30% of that shown by the Wt receptor (Uribe et al., 2008). The hLHR is palmitoylated at two conserved cysteine residues (643 and 644) (Kawate and Menon, 1994) but in contrast to the hFSHR, palmitoylation of this receptor is not important for trafficking to the PM, as abrogation of palmitoylation did not appear to affect PM expression and agonist binding (Kawate and Menon, 1994; Munshi et al., 2005).
As with many other GPCRs, gonadotropin receptors constitutively form dimers or oligomers during receptor biogenesis (Guan et al., 2009; Tao et al., 2004; Thomas et al., 2007), which may impact on the intracellular trafficking of the receptor to the PM as well as for signal diversification through coupling to multiple G proteins (Breitwieser, 2004; Marshall et al., 1999; Nechamen et al., 2007; Szidonya et al., 2008). Although the mechanisms and extents of the hLHR and hFSHR self-association are still incompletely known, studies on these and other GPCRs suggest that association between receptors might occur at multiple contact sites, including the ECD and the TM regions (Fan and Hendrickson, 2007, 2005; Fan et al., 2005; Guan et al., 2010; Hebert et al., 1996; Jiang et al., 2012; Lee et al., 2003; Ng et al., 1996; Zarinan et al., 2010). As discussed above for the hGnRHR, misfolded hFSH and hLH receptors also associate with their immature Wt counterparts, and prevent Wt receptor traffickng to the PM (Tao et al., 2004; Zarinan et al., 2010). In fact, cotransfection of the A593P or S616Y misfolded hLHR mutants with the Wt receptor in HEK-293 cells, resulted in a 40–50% reduction in cell surface PM expression as compared to cells transfected only with the Wt receptor (Tao et al., 2004). Likewise, cotransfection of the the Wt receptor with increasing amounts of the laboratory-manufactured R573A (at the iL3) or R635A (at the Ctail) hFSHR mutants led to a dose dependent reduction in both ligand binding and signal transduction. Interestingly, the dominant negative effect of the mutants was prevented when the transfection mix included also Wt hFSHR fragments involving transmembrane domains or the transmembrane domain 7 and/or the Ctail peptide (Fig. 2)(Zarinan et al., 2010), strongly suggesting that these regions were involved in the intracellular mutant-Wt receptor association. The fact that individuals who are heterozygous for misfolded mutations in the gonadotropin receptors do not exhibit detectable reproductive abnormalities, suggest that the decrease in PM expression of the Wt receptor that results from the dominant negative effect of the misfolded mutant is not low enough to impact on cell function, given that occupancy of only a low fraction of gonadotropin receptors per cell is sufficient to elicit normal responses (Huhtaniemi et al., 1982). In this regard, it is interesting that heterozygous subjects bearing misfolded TSHR mutants (eg. C41S, L467P, and C600R) expressed a clinical phenotype of partial TSH resistance presumably due to the dominant negative effect of the mutant receptors on Wt receptor PM expression (Calebiro et al., 2005), underlining the potential effects of misfolded receptors on the dominant transmission of partial GPCR resistance in heterozygous individuals.
Figure 2.

Dominant negative effect of the laboratory-manufactured R573A mutant hFSHR on Wt hFSHR PM expression (A and B) and function (C), and effect of co-transfecting the Wt hFSHR L526-V599 fragment (inset, black circles) cDNA on the dominant negative effects of the mutant. (A) Representative autoradiogram from an immunoblot of the hFSHR present in protein extracts from HEK-293 cells transfected with the cDNAs shown at the top of the blot. The autoradiograph was overexposed in order to show the expression levels of the mutant receptor species and the immature forms of the hFSHR. (B) HEK-293 cells were co-transfected with the cDNAs shown at the bottom of the graph and specific [125I]-FSH binding was determined in the presence or absence of the L526-V599 fragment. Note that in contrast to the L526-V599 fragment, co-transfection with the hFSHR A607-N695 cDNA fragment (encoding the TM7 and the entire Ctail of the Wt receptor), did not affect expression of the Wt receptor co-transfected with the R573A mutant. (C) Maximal FSH-stimulated total cAMP accumulation in HEK-293 cells co-transfected with the cDNAs shown at the bottom of the graph; note that the L526-V599 fragment cDNA was co-transfected in increasing concentrations. The Wt and mutant hFSHR cDNAs were hosted by the pSG5 vector whereas the hFSHR fragments were in pcDNA3.1. The results shown in B and C are the mean ± SEM from three independent experiments. *p < 0.05 vs all other conditions; ¶p < 0.01 vs all other conditions; §p < 0.05 vs Wt + L526-V599 fragment + empty vector. (m)FSHR, mature form of the hFSHR; (i)FSHR, immature form of the hFSHR. Co-transfection of the mutant and Wt hFSHRs resulted in decreased PM expression and function of the mature form of the hFSHR as well as reduced specific [125I]-FSH binding. Co-transfection of the Wt and mutant FSHRs with the L526-V599 fragment led to almost complete functional recovery and PM expression of the hFSHR. Reproduced from Zariñán et al., (2010), with permission of Elsevier Ireland LTD.
Loss-of-function mutations in the gonadotropin receptor genes may lead to disease, whenever both alleles are affected by a mutation, as it occurs in individuals who are homozygous or compound heterozygous for mutations in the hFSHR or hLHR genes. Several naturally occurring inactivating mutations (which include point mutations, amino acid insertions or deletions, or premature truncations) scattered throughout the polypeptide chain of the hLH and hFSH receptor molecules have been described. In 46, XY individuals, loss-of-function mutations in the LHR gene leads to an array of phenotypes, from severe genital ambigüity [due to unresponsiveness of the fetal Leydig cell to CG stimulation –the so-call Type 1 Leydig cell hypoplasia (LCH)-] to cryptorchidism and micropenis (Type II LCH), depending on the severity of the functional deficit provoked by the mutation (Table 1). Women with inactivating mutations in the hLHR display pubertal development, but frequently exhibit primary or secondary amenorrhea and infertility. Meanwhile, inactivating mutations of the hFSHR in men lead to impaired quality of spermatogenesis with normal androgen production, which probably contributes to fertility preservation (Tapanainen et al., 1997). In women bearing inactivating FSHR mutations, the panorama is completely different and comprises an array of phenotypes ranging from lack of pubertal development and primary amenorrhea, with arrest of follicular maturation between primordial and preantral stage and complete resistance to FSH stimulation, to secondary amenorrhea and premature ovarian failure (Aittomaki et al., 1995; Huhtaniemi and Alevizaki, 2007) (Table 2). In either case, the level of residual, functional receptors at the PM, has been shown to correlate with the severity of the clinical phenotype expressed by the patients bearing inactivating mutations in these receptors (Huhtaniemi and Alevizaki, 2007; Huhtaniemi and Themmen, 2005; Touraine et al., 1999), which is an important determinant for the response to exogenous gonadotropins (Vaskivuo et al., 2002).
Table 1.
Location within the hLHR and clinical phenotype of patients bearing trafficking-defective loss-of-function mutations (in bold letter type) in the hLHR. The clinical phenotypes correspond to those described in the original reports.
| hLHR mutation | Location | Phenotype | Reference |
|---|---|---|---|
|
In-frame 33 bp insertion/ C545Stop (or W491X) (compound heterozygous) |
Exon 1, signal peptide (33 bp insertion) |
Type I LCH: female phenotype and undescended gonads. |
(Richter-Unruh et al., 2002; Wu et al., 1998) |
| V144F | Exon 5, ECD | Type I LCH: female phenotype, slight clitoral enlargement, and inguinal testes. |
(Richter-Unruh et al., 2004) |
| F194V | Exon 7, ECD | Type I LCH: female phenotype, absent virilization, and inguinal testes. |
(Gromoll et al., 2002) |
|
C343S/C543R (compound heterozygous) |
Exon 11, ECD and TM5 | Type I LCH: female phenotype, inguinal testes, abscence of secondary sex characteristics at pubertal age. |
(Martens et al., 2002) |
| A593P | Exon 11, TM6 | Type I LCH: female phenotype, absence of pubertal development. |
(Kremer et al., 1995; Martens et al., 1998) |
|
In women: primary amenorrhea with pubertal development (full maturation of secondary sexual characteristics). |
(Toledo et al., 1996) | ||
| DelL608-V609 | Exon 11, TM7 | Type I LCH: Female phenotype, pubic hair (Tanner stage 4), testis in labia majora. |
(Latronico et al., 1998) |
|
In women: Normal pubertal development, oligoamenorrhea, infertility, ovarian cysts. |
|||
| S616Y | Exon 11, TM7 | Type II LCH: male phenotype with micropenis, descended testis, and infertility. |
(Latronico et al., 1996) |
| I625K | Exon 11, TM7 | Type II LCH: male phenotype, micropenis and abscence of puberty. |
(Martens et al., 1998; Richter-Unruh et al., 2002) |
Table 2.
Location of trafficking-defective loss-of-function mutations (in bold letter type) within the human FSHR gene and protein, and clinical phenotypes of patients bearing such mutations. The clinical phenotypes correspond to those described in the original reports.
| hFSHR mutation | Location | Phenotype | Reference |
|---|---|---|---|
|
I160T/R573C (compound heterozygous) |
Exon 6, ECD | Partial phenotype of ovarian failure: Normal puberty, secondary amenorrhea, and infertility. |
(Beau et al., 1998) |
| A189V | Exon 7, ECD | Women: Complete phenotype of ovarian failure: Primary amenorrhea, delayed and variable development of secondary sex characteristics, infertility. |
(Aittomaki et al., 1996) |
|
In men: Normal virilization, low-normal to low testicular volume, abnormal semen parameters, fertility. |
(Tapanainen et al., 1997) | ||
|
N191I (simple heterozygous) |
Exon 7, ECD | Normal ovarian function and fertility. | (Gromoll et al., 1996) |
|
D224V/L601V (compound heterozygous) |
Exon 9, ECD | Partial phenotype of ovarian failure: Normal puberty, primary amenorrhea. |
(Touraine et al., 1999) |
| P519T | Exon 10, eL2 | Complete penotype of ovarian failure: Primary amenorrhea, delayed puberty, infertility |
(Meduri et al., 2003) |
Among the 24 hLHR mutants described so far, at least nine are trafficking defective receptors in which the net amount of functional receptors expressed at the PM is decreased to a variable extent. These trafficking defective/misfolded receptors bear mutations either in their ECD [V144F, F194V, C343S, and a 33-bp insertion between amino acid residues 18 and 19, immediately upstream of the signal peptide cleavage site (Gromoll et al., 2002; Martens et al., 1998; Richter-Unruh et al., 2002; Richter-Unruh et al., 2004; Wu et al., 1998)] or the TMs 5–7 [C543R, A593P, delL608-V609, S616Y, and I625K (Kremer et al., 1995; Latronico et al., 1996; Latronico et al., 1998; Laue et al., 1996; Martens et al., 2002; Martens et al., 1998; Richter-Unruh et al., 2002; Toledo et al., 1996)]. Some of these mutant hLHRs are interesting from the structural and functional points of view. The F194V misfolded mutant bears the substitution in a highly conserved motif of the gonadotropin receptors (193AFNGT197 at the LHR ECD) that contains the N195GT glycosylation motif. This mutation severely impairs trafficking of the mutant receptor to the PM without significantly altering agonist affinity (Gromoll et al., 2002), and leads to LCH. In the case of the inactivating A593P and S616Y mutant hLHRs, which provoke severe or moderate forms of LCH, respectively, both exhibited normal ligand binding affinity but the response to agonist was absent or severely impaired due to misfolding and intracellular retention of the mutant receptors (Martens et al., 2002; Mizrachi and Segaloff, 2004). Further, it was shown that these particular mutants are conformationally distinct and display different folding conformations during their maturation at the ER as suggested by their differential association with molecular chaperones in co-immunoprecipitation studies (Mizrachi and Segaloff, 2004). Whereas similar to the Wt hLHR, the A593P and S616Y mutants were found associated with calnexin, only S616Y was coimmunoprecipitated with PDI; further, although both A593P and S616Y mutants appeared to interact with BiP only the former was found to interact with Grp94 (Mizrachi and Segaloff, 2004). Some mutant hLHRs exhibiting further defects, in addition to misfolding, have also been described. This is the case of the delLeu608/Val609 misfolded mutant, which in spite of being expressed at low levels at the PM and to exhibit normal binding affinity, it is unable to activate the Gs protein upon exposure to agonist (Latronico et al., 1998). The same appears to occur with the I625K mutant, which leads to partial LCH; in vitro studies showed that in addition to low PM expression, the mutant exhibited decreased coupling efficiency. Since in addition to provoke defective folding and intracellular retention of the protein, the mutations also affect an intrinsic function of the receptor (eg. signal transduction), it might be expected that the benefit of pharmacoperone treatment to correct function will be limited (see below).
In contrast to the LHR, which seems insensitive to effects on hormone binding when trafficking is impaired by mutagenesis, the FSHR is particularly sensitive to mutations of the primary sequence. When mutations impair trafficking of this receptor from the ER to the PM (Rozell, 1995; Song et al., 2001), hormone binding activity is severely impaired (Nechamen and Dias, 2003; Rozell, 1995; Song et al., 2001), which may be due to differences in stability between these receptors. In fact, naturally occuring mutations in the ECD of this receptor (eg. I160T, A189V, N191I, D224V), drastically impair targeting of the receptor to the PM, and thereby limit hormone binding and agonist-stimulated intracellular signaling. Meanwhile, the majority of mutations at or near the transmembrane region (eg. A419T, R573C, L601V) have modest effects on FSH binding capacity but impair to variable extent signal transduction, suggesting that, in general, the location of the mutation is a strong determinant of the functional response.
Loss-of-function mutations of the hFSHR are less frequent than those in the hLHR. In fact, only 12 inactivating mutations in the hFSHR receptor have been reported to date (Achrekar et al., 2010; Aittomaki et al., 1995; Allen et al., 2003; Beau et al., 1998; Gromoll et al., 1996a; Kotlar et al., 1997; Meduri et al., 2003; Nakamura et al., 2008; Tapanainen et al., 1997; Touraine et al., 1999). As it has been observed for the inactivating hLHR mutants, there is in general a good correlation between the residual activity exhibited by the mutant hFSHRs and the severity of the clinical phenotype expressed by the patients bearing the mutation(s) (Aittomaki et al., 1996; Huhtaniemi and Alevizaki, 2007; Huhtaniemi and Themmen, 2005). The exception is the F591S mutation for which no clinical phenotype has been described; this particular mutation, which leads to severely impaired agonist binding and intracellular signaling, was found to be associated with the development of sex cord tumors (Kotlar et al., 1997). Among the 12 mutant hFSHRs reported to date, at least five are trafficking defective receptors which have been detected as intracellular retained molecules [I160T, A189V, N191I, D224V, at the ECD (Beau et al., 1998; Gromoll et al., 2002; Gromoll et al., 1996b; Touraine et al., 1999); and P519T at the eL2 (Meduri et al., 2003)] by in vitro studies. The functional defects of the V221G (at the ECD) (Nakamura et al., 2008) and A575V (at the TM6) (Achrekar et al., 2010) mutant hFSHRs have not been studied in detail, whereas in the case of the P348R hFSHR (located at the hinge region of the receptor), both ligand binding and agonist-stimulated signaling were severely impaired (Allen et al., 2003). Whether these three hFSHR mutations interfere with proper trafficking of the receptor from the ER to the PM is still unknown.
The naturally occurring mutation A189V cause a profound defect in targeting of the receptor protein to the cell membrane (Huhtaniemi and Themmen, 2005; Rannikko et al., 2002), confirming the importance of the AFNGT motif for intracellular trafficking of the receptor. As described above, A189 is the first amino acid in the perfectly conserv+ed stretch of five amino acids in the gonadotropin receptors (189AFNGT193 in the hFSHR), which is also the locus of loss-of-function mutations in the hLHR (Gromoll et al., 2002) (see above). Valine in this position as well as isoleucine in position 191 may interfere with structural integrity of the LRRs which hosts the glycosylation site, and perturbation of this structure likely impairs proper receptor LRR formation, particularly its a-helical portion. Although the putative loss of glycosylation may affect folding and trafficking of the mutant receptor to the PM, it has yet to be determined whether this form of the receptor is glycosylated or not at the N191 site. When the A189V mutant is overexpressed in vitro, only a very small proportion of the mutated receptor is present at the PM but is functional (Rannikko et al., 2002; Tranchant et al., 2011). Instead, most of the mutated receptor is sequestered inside the cell, explaining the inactivation mechanism (Rannikko et al., 2002). Interestingly, the reduced level of PM expression of the A189V hFSHR confers preferential coupling to the β-arrestin-mediated signaling pathway, similar to that observed when the Wt receptor is expressed at low PM densities, indicating that the selective signaling is due to the low level of PM expression rather than to the mutation itself (Tranchant et al., 2011). This observation might help to clarify why mutations of FSHβ are more deleterious to male fertility than the hFSHR A189V mutation (Layman et al., 2002; Lindstedt et al., 1998; Tapanainen et al., 1997), which preserves a fraction of its receptor signaling repertoire.
The case of the N191I mutation is also interesting. This mutation was originally detected in the heterozygous state in a phenotypically normal women (Gromoll et al., 1996b). Although the mutation affects the putative glycosylation site at position 191 of the receptor, curiously no studies on the effect of this mutation on the binding capacity and PM expression of the FSHR have been performed. Further, in vitro studies have been somehow contradictory regarding the impact of the N191I mutation on agonist-stimulated signaling: in one study, FSH-stimulated cAMP production was severely impaired (Gromoll et al., 1996b), whereas in a later study performed by the same group, it was found only modestly decreased (by ~40% relative to that exhibited by the Wt receptor) (Gromoll et al., 2002), suggesting that a fraction of the transfected receptor was, in fact, present at the PM. Given that glycosylation in only one position at the ECD is sufficient for folding and trafficking of the FSHR to the PM (Davis et al., 1995), these studies suggest that the limited PM expression of the mutant receptor is mainly due to the alteration in the structural integrity of the AFNGT motif, rather than to the absence of glycosylation at this particular site.
Another interesting hFSHR mutant is R573C; this mutation does not affect PM expression of the receptor but partially impairs agonist-stimulated signaling (Beau et al., 1998). Nevertheless, when R573 is replaced with alanine (Zarinan et al., 2010), the PM expression of the mutant receptor is severely impaired, thus indicating that the location of the mutation and the nature of the amino acid substitution are both strong determinants of the functional features exhibited by mutant hFSHRs. In fact, the P519T mutation in the center of the eL2, leads to complete failure to bind agonist and trigger intracellular signaling; women homozygous for this mutation express a severe phenotype including absence of puberty, primary amenorrhea and small ovaries (Meduri et al., 2003). The effects of this particular mutation contrast with those provoked by other mutations in the serpentine region of the hFSHR (A419T, R573C, and L601V), which usually result in partial receptor inactivation, with minimal effects on FSH binding (Beau et al., 1998; Doherty et al., 2002; Touraine et al., 1999). Therefore, it seems that the loss of a proline at position 519 provokes a severe conformational defect that leads to trapping of the receptor at the ER (Meduri et al., 2003). Because the peptide backbone of proline is constrained in a ring structure, occurrence of this amino acid is associated with a forced turn in the protein sequence, which is likely lost by the substitution with the more reactive threonine; it is thus possible that the abrupt turn at the middle of the eL2 is probably a requisite not only for activity (Dupakuntla et al., 2012) but also for routing.
As mentioned above, the success of pharmacological treatment of misfolded GPCRs with pharmacoperones depends on several factors, including the partial or complete integrity of domains involved in ligand binding, receptor activation and/or coupling to effectors. As demonstrated in the hGnRHR mutants, misfolded hFSH and hLH receptors may also be rescued in vitro by pharmacoperone treatment that correct folding and promote trafficking of the intracellularly trapped receptors from the ER to the cell surface PM. In the case of the misfolded A189V hFSHR mutant (which is the prototype of a misfolded hFSHR due to a point mutation), we tested the effect of Org41841, which is a thienopyr(im)idine molecule reported to bind a conserved region of the hLHR without competing for the LH binding site and the site for interaction with effector (ie. an allosteric modulator)(van Straten et al., 2002). Exposure of COS-7 cells transfected with the A189V hFSHR expression plasmid to Org41841 increased almost by two-fold PM expression and FSH-stimulated cAMP production of the A189V mutant without significantly altering mARN expression of the receptor nor its ligand binding affinity (Janovick et al., 2009a) (Fig. 3A). Shortly after this report, Newton and colleagues (Newton et al., 2011) found that another cell-permeant, allosterically binding small molecule agonist (Org 42599) rescued folding, PM expression and function of two hLHR misfolded mutants (A593P and S616Y) that lead to LCH (Fig. 3B). Major challenges include determining the molecular mechanisms by which these allosteric compounds stabilize and rescue these particular misfolded receptors, and to identify new, highly specific molecules that do not interfere with agonist binding or activation of the misfolded receptor and that may function well in vivo. Meanwhile, these studies indicate that treatment of patients bearing mutations that lead to misfolding of gonadotropin receptors may be feasible, particularly in those cases with mild clinical phenotypes.
Figure 3.
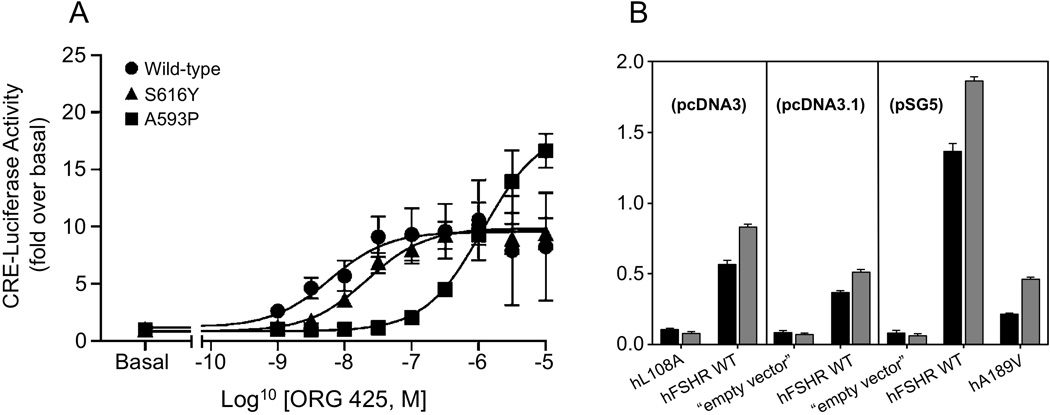
Rescue of hLHR and hFSHR misfolded mutants by pharmacoperones. (A) Measurement of cAMP accumulation by CRE-luciferase reporter gene activaton after 24 h stimulation in cells expressing Wt (●), A593P mutant (■), or S616Y (▲) mutant hLHRs was determined over a range of concentrations of Org 42599. Data were fitted by sigmoidal dose-response curves with Hill coefficients of unity and are presented as fold versus basal values for each receptor (mean ± SEM from at least three independent experiments). (B) Org41841 rescues Wt hFSHR and mutant A189V (p= 0.002), but is unable to rescue other mutants with mutation at diverse sites in the molecule (SEMs shown). After transfection and incubation in Org41841, cellular response was assessed by a challenge with 100 ng hFSH. As mutants were obtained from different laboratories and were in different expression vectors, controls are shown with the corresponding Wt hFSHR and the empty vector in each case. Figure 3A is reproduced from Newton et al. (2011), with permission of the National Academy of Sciences USA; figure 3B is reproduced from Janovick et al. (2009) with permission of Elsevier Ireland Ltd.
4. Conclusions
Intracellular retention of misfolded GPCRs is a common cause of human disease. In fact, more than 30 disorders are associated with mutations in GPCRs, which make these proteins a target for drug development (Lagerstrom and Schioth, 2008; Overington et al., 2006; Schlyer and Horuk, 2006). Although the majority of pharmacoperones identified that rescue misfolded GPCRs are mainly antagonists of the natural ligand detected from “hits” in screens designed to detect antagonists or agonists, new, fully automated high-throughput assays for identification of “pure” pharmacoperone drugs of mutant GPCRs have been recently described (Smithson et al., 2013). These approaches allow identification of structures missed in screens designed to select only agonists or antagonists, and thus offer the advantage of identifying new classes of drugs which are specific and rescue active, but do not have inherent modulatory activity (stimulatory or inhibitory). Elucidation of the crystal structure of the GPCRs discussed in this article, will undoubtedly facilitate the design of novel small molecules that may rescue misfolded receptors by specifically targeting those motifs and conformations that influence directly receptor trafficking and that may be altered by mutations in the protein molecule. Pharmacoperone compounds currently constitute the most promising therapeutic approach for treating diseases caused by misfolded GPCRs. Given that ample evidence exists to demonstrate that certain mutations found in GPCRs which bind ligands that are essential for reproductive function cause dysfunction due to aberrant trafficking of otherwise functional proteins, the mutations discussed above provide the vehicle to identify and assess such kind of drugs.
Highlights.
GPCRs are subjected to conformational scrutiny at the endoplasmic reticulum prior to processing and trafficking to the cell surface membrane.
Point mutations in genes coding the gonadotropin-releasing hormone, the luteinizing hormone and the follicle-stimulating hormone receptors, may result in misfolding and intracellular trapping of the abnormal receptor protein.
Mutations in the GnRHR and gonadotropin receptors lead to hypo- or hypergonadotropic hypogonadism in humans.
Pharmacoperones are a useful strategy to rescue function of mutant GPCRs, including the GnRHR and the gonadotropin receptors.
Acknowledgements
The authors of this article are supported by CONACyT grant 86881 (to A.U.-A) and the National Institutes of Health Grants OD012220, DK85040, OD 011092 and TW/HD-00668 (to P.M.C.), and HD18407 (to J.A.D.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Achrekar SK, Modi DN, Meherji PK, Patel ZM, Mahale SD. Follicle stimulating hormone receptor gene variants in women with primary and secondary amenorrhea. J Assist Reprod Genet. 2010;27:317–326. doi: 10.1007/s10815-010-9404-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aittomaki K, Herva R, Stenman UH, Juntunen K, Ylostalo P, Hovatta O, de la Chapelle A. Clinical features of primary ovarian failure caused by a point mutation in the follicle-stimulating hormone receptor gene. J Clin Endocrinol Metab. 1996;81:3722–3726. doi: 10.1210/jcem.81.10.8855829. [DOI] [PubMed] [Google Scholar]
- Aittomaki K, Lucena JL, Pakarinen P, Sistonen P, Tapanainen J, Gromoll J, Kaskikari R, Sankila EM, Lehvaslaiho H, Engel AR, Nieschlag E, Huhtaniemi I, de laChapelle A. Mutation in the follicle-stimulating hormone receptor gene causes hereditary hypergonadotropic ovarian failure. Cell. 1995;82:959–968. doi: 10.1016/0092-8674(95)90275-9. [DOI] [PubMed] [Google Scholar]
- Allen LA, Achermann JC, Pakarinen P, Kotlar TJ, Huhtaniemi IT, Jameson JL, Cheetham TD, Ball SG. A novel loss of function mutation in exon 10 of the FSH receptor gene causing hypergonadotrophic hypogonadism: clinical and molecular characteristics. Hum Reprod. 2003;18:251–256. doi: 10.1093/humrep/deg046. [DOI] [PubMed] [Google Scholar]
- Angelotti T, Daunt D, Shcherbakova OG, Kobilka B, Hurt CM. Regulation of G-protein coupled receptor traffic by an evolutionary conserved hydrophobic signal. Traffic. 2010;11:560–578. doi: 10.1111/j.1600-0854.2010.01033.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arakawa T, Ejima D, Kita Y, Tsumoto K. Small molecule pharmacological chaperones: From thermodynamic stabilization to pharmaceutical drugs. Biochim Biophys Acta. 2006;1764:1677–1687. doi: 10.1016/j.bbapap.2006.08.012. [DOI] [PubMed] [Google Scholar]
- Arora KK, Chung HO, Catt KJ. Influence of a species-specific extracellular amino acid on expression and function of the human gonadotropin-releasing hormone receptor. Mol Endocrinol. 1999;13:890–896. doi: 10.1210/mend.13.6.0291. [DOI] [PubMed] [Google Scholar]
- Ascoli M, Fanelli F, Segaloff DL. The lutropin/choriogonadotropin receptor, a 2002 perspective. Endocr Rev. 2002;23:141–174. doi: 10.1210/edrv.23.2.0462. [DOI] [PubMed] [Google Scholar]
- Beau I, Touraine P, Meduri G, Gougeon A, Desroches A, Matuchansky C, Milgrom E, Kuttenn F, Misrahi M. A novel phenotype related to partial loss of function mutations of the follicle stimulating hormone receptor. J Clin Invest. 1998;102:1352–1359. doi: 10.1172/JCI3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beranova M, Oliveira LM, Bedecarrats GY, Schipani E, Vallejo M, Ammini AC, Quintos JB, Hall JE, Martin KA, Hayes FJ, Pitteloud N, Kaiser UB, Crowley WF, Jr, Seminara SB. Prevalence, phenotypic spectrum, and modes of inheritance of gonadotropin-releasing hormone receptor mutations in idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2001;86:1580–1588. doi: 10.1210/jcem.86.4.7395. [DOI] [PubMed] [Google Scholar]
- Bernier V, Bichet DG, Bouvier M. Pharmacological chaperone action on G- protein-coupled receptors. Curr Opin Pharmacol. 2004a;4:528–533. doi: 10.1016/j.coph.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Bernier V, Lagace M, Bichet DG, Bouvier M. Pharmacological chaperones: potential treatment for conformational diseases. Trends Endocrinol Metab. 2004b;15:222–228. doi: 10.1016/j.tem.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Bernier V, Morello JP, Zarruk A, Debrand N, Salahpour A, Lonergan M, Arthus MF, Laperriere A, Brouard R, Bouvier M, Bichet DG. Pharmacologic chaperones as a potential treatment for X-linked nephrogenic diabetes insipidus. J Am Soc Nephrol. 2006;17:232–243. doi: 10.1681/ASN.2005080854. [DOI] [PubMed] [Google Scholar]
- Blanpain C, Wittamer V, Vanderwinden JM, Boom A, Renneboog B, Lee B, Le Poul E, El Asmar L, Govaerts C, Vassart G, Doms RW, Parmentier M. Palmitoylation of CCR5 is critical for receptor trafficking and efficient activation of intracellular signaling pathways. J Biol Chem. 2001;276:23795–23804. doi: 10.1074/jbc.M100583200. [DOI] [PubMed] [Google Scholar]
- Blomenrohr M, Heding A, Sellar R, Leurs R, Bogerd J, Eidne KA, Willars GB. Pivotal role for the cytoplasmic carboxyl-terminal tail of a nonmammalian gonadotropin-releasing hormone receptor in cell surface expression, ligand binding, and receptor phosphorylation and internalization. Mol Pharmacol. 1999;56:1229–1237. doi: 10.1124/mol.56.6.1229. [DOI] [PubMed] [Google Scholar]
- Bogerd J. Ligand-selective determinants in gonadotropin receptors. Mol Cell Endocrinol. 2007;260–262:144–152. doi: 10.1016/j.mce.2006.01.019. [DOI] [PubMed] [Google Scholar]
- Breitwieser GE. G protein-coupled receptor oligomerization: implications for G protein activation and cell signaling. Circ Res. 2004;94:17–27. doi: 10.1161/01.RES.0000110420.68526.19. [DOI] [PubMed] [Google Scholar]
- Broadley SA, Hartl FU. The role of molecular chaperones in human misfolding diseases. FEBS Lett. 2009;583:2647–2653. doi: 10.1016/j.febslet.2009.04.029. [DOI] [PubMed] [Google Scholar]
- Brooks DA. Introduction: molecular chaperones of the ER: their role in protein folding and genetic disease. Semin Cell Dev Biol. 1999;10:441–442. doi: 10.1006/scdb.1999.0314. [DOI] [PubMed] [Google Scholar]
- Brothers SP, Cornea A, Janovick JA, Conn PM. Human loss-of-function gonadotropin-releasing hormone receptor mutants retain wild-type receptors in the endoplasmic reticulum: molecular basis of the dominant-negative effect. Mol Endocrinol. 2004;18:1787–1797. doi: 10.1210/me.2004-0091. [DOI] [PubMed] [Google Scholar]
- Calebiro D, de Filippis T, Lucchi S, Covino C, Panigone S, Beck-Peccoz P, Dunlap D, Persani L. Intracellular entrapment of wild-type TSH receptor by oligomerization with mutants linked to dominant TSH resistance. Hum Mol Genet. 2005;14:2991–3002. doi: 10.1093/hmg/ddi329. [DOI] [PubMed] [Google Scholar]
- Castro-Fernandez C, Maya-Nunez G, Conn PM. Beyond the signal sequence: protein routing in health and disease. Endocr Rev. 2005;26:479–503. doi: 10.1210/er.2004-0010. [DOI] [PubMed] [Google Scholar]
- Chevet E, Cameron PH, Pelletier MF, Thomas DY, Bergeron JJ. The endoplasmic reticulum: integration of protein folding, quality control, signaling and degradation. Curr Opin Struct Biol. 2001;11:120–124. doi: 10.1016/s0959-440x(00)00168-8. [DOI] [PubMed] [Google Scholar]
- Conn PM, Crowley WF., Jr Gonadotropin-releasing hormone and its analogs. Annu Rev Med. 1994;45:391–405. doi: 10.1146/annurev.med.45.1.391. [DOI] [PubMed] [Google Scholar]
- Conn PM, Janovick JA. Pharmacoperone identification for therapeutic rescue of misfolded mutant proteins. Front Endocrinol (Lausanne) 2011;2 doi: 10.3389/fendo.2011.00006. pii: 00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn PM, Janovick JA. Drug development and the cellular quality control system. Trends Pharmacol Sci. 2009;30:228–233. doi: 10.1016/j.tips.2009.02.002. [DOI] [PubMed] [Google Scholar]
- Conn PM, Knollman PE, Brothers SP, Janovick JA. Protein folding as posttranslational regulation: evolution of a mechanism for controlled plasma membrane expression of a G protein-coupled receptor. Mol Endocrinol. 2006;20:3035–3041. doi: 10.1210/me.2006-0066. [DOI] [PubMed] [Google Scholar]
- Conn PM, Ulloa-Aguirre A. Trafficking of G-protein-coupled receptors to the plasma membrane: insights for pharmacoperone drugs. Trends Endocrinol Metab. 2010;21:190–197. doi: 10.1016/j.tem.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn PM, Ulloa-Aguirre A. Pharmacological Chaperones for MisfoldedGonadotropin-Releasing Hormone Receptors. Adv Pharmacol. 2011;62C:109–141. doi: 10.1016/B978-0-12-385952-5.00008-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn PM, Ulloa-Aguirre A, Ito J, Janovick JA. G protein-coupled receptor trafficking in health and disease: lessons learned to prepare for therapeutic mutant rescue in vivo. Pharmacol Rev. 2007;59:225–250. doi: 10.1124/pr.59.3.2. [DOI] [PubMed] [Google Scholar]
- Davis D, Liu X, Segaloff DL. Identification of the sites of N-linked glycosylation on the follicle-stimulating hormone (FSH) receptor and assessment of their role in FSH receptor function. Mol Endocrinol. 1995;9:159–170. doi: 10.1210/mend.9.2.7776966. [DOI] [PubMed] [Google Scholar]
- Dias JA, Cohen BD, Lindau-Shepard B, Nechamen CA, Peterson AJ, Schmidt A. Molecular, structural, and cellular biology of follitropin and follitropin receptor. Vitam Horm. 2002;64:249–322. doi: 10.1016/s0083-6729(02)64008-7. [DOI] [PubMed] [Google Scholar]
- Doherty E, Pakarinen P, Tiitinen A, Kiilavuori A, Huhtaniemi I, Forrest S, Aittomaki K. A Novel mutation in the FSH receptor inhibiting signal transduction and causing primary ovarian failure. J Clin Endocrinol Metab. 2002;87:1151–1155. doi: 10.1210/jcem.87.3.8319. [DOI] [PubMed] [Google Scholar]
- Dupakuntla M, Pathak B, Roy BS, Mahale SD. Extracellular loop 2 in the FSH receptor is crucial for ligand mediated receptor activation. Mol Cell Endocrinol. 2012;362:60–68. doi: 10.1016/j.mce.2012.05.008. [DOI] [PubMed] [Google Scholar]
- Duvernay MT, Zhou F, Wu G. A conserved motif for the transport of G protein-coupled receptors from the endoplasmic reticulum to the cell surface. J Biol Chem. 2004;279:30741–30750. doi: 10.1074/jbc.M313881200. [DOI] [PubMed] [Google Scholar]
- Estrada LD, Soto C. Disrupting beta-amyloid aggregation for Alzheimer disease treatment. Curr Top Med Chem. 2007;7:115–126. doi: 10.2174/156802607779318262. [DOI] [PubMed] [Google Scholar]
- Estrada LD, Soto C. Inhibition of protein misfolding and aggregation by small rationally-designed peptides. Curr Pharm Des. 2006;12:2557–2567. doi: 10.2174/138161206777698792. [DOI] [PubMed] [Google Scholar]
- Fan QR, Hendrickson WA. Assembly and structural characterization of an authentic complex between human follicle stimulating hormone and a hormone-binding ectodomain of its receptor. Mol Cell Endocrinol. 2007;260–262:73–82. doi: 10.1016/j.mce.2005.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan QR, Hendrickson WA. Structure of human follicle-stimulating hormone in complex with its receptor. Nature. 2005;433:269–277. doi: 10.1038/nature03206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan T, Varghese G, Nguyen T, Tse R, O'Dowd BF, George SR. A role for the distal carboxyl tails in generating the novel pharmacology and G protein activation profile of mu and delta opioid receptor hetero-oligomers. J Biol Chem. 2005;280:38478–38488. doi: 10.1074/jbc.M505644200. [DOI] [PubMed] [Google Scholar]
- Francou B, Bouligand J, Voican A, Amazit L, Trabado S, Fagart J, Meduri G, Brailly-Tabard S, Chanson P, Lecomte P, Guiochon-Mantel A, Young J. Normosmic congenital hypogonadotropic hypogonadism due to TAC3/TACR3 mutations: characterization of neuroendocrine phenotypes and novel mutations. PLoS One. 2011;6:e25614. doi: 10.1371/journal.pone.0025614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol. 2003;63:1256–1272. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- Fukushima Y, Saitoh T, Anai M, Ogihara T, Inukai K, Funaki M, Sakoda H, Onishi Y, Ono H, Fujishiro M, Ishikawa T, Takata K, Nagai R, Omata M, Asano T. Palmitoylation of the canine histamine H2 receptor occurs at Cys(305) and is important for cell surface targeting. Biochim Biophys Acta. 2001;1539:181–191. doi: 10.1016/s0167-4889(01)00104-5. [DOI] [PubMed] [Google Scholar]
- Gething MJ. Role and regulation of the ER chaperone BiP. Semin Cell Dev Biol. 1999;10:465–472. doi: 10.1006/scdb.1999.0318. [DOI] [PubMed] [Google Scholar]
- Gianetti E, Hall JE, Au MG, Kaiser UB, Quinton R, Stewart JA, Metzger DL, Pitteloud N, Mericq V, Merino PM, Levitsky LL, Izatt L, Lang-Muritano M, Fujimoto VY, Dluhy RG, Chase ML, Crowley WF, Jr, Plummer L, Seminara SB. When genetic load does not correlate with phenotypic spectrum: lessons from the GnRH receptor (GNRHR) J Clin Endocrinol Metab. 2012;97:E1798–E1807. doi: 10.1210/jc.2012-1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromoll J, Schulz A, Borta H, Gudermann T, Teerds KJ, Greschniok A, Nieschlag E, Seif FJ. Homozygous mutation within the conserved Ala-Phe-Asn-Glu-Thr motif of exon 7 of the LH receptor causes male pseudohermaphroditism. Eur J Endocrinol. 2002;147:597–608. doi: 10.1530/eje.0.1470597. [DOI] [PubMed] [Google Scholar]
- Gromoll J, Simoni M, Nieschlag E. An activating mutation of the follicle-stimulating hormone receptor autonomously sustains spermatogenesis in a hypophysectomized man. J Clin Endocrinol Metab. 1996a;81:1367–1370. doi: 10.1210/jcem.81.4.8636335. [DOI] [PubMed] [Google Scholar]
- Gromoll J, Simoni M, Nordhoff V, Behre HM, De Geyter C, Nieschlag E. Functional and clinical consequences of mutations in the FSH receptor. Mol Cell Endocrinol. 1996b;125:177–182. doi: 10.1016/s0303-7207(96)03949-4. [DOI] [PubMed] [Google Scholar]
- Guan R, Feng X, Wu X, Zhang M, Zhang X, Hebert TE, Segaloff DL. Bioluminescence resonance energy transfer studies reveal constitutive dimerization of the human lutropin receptor and a lack of correlation between receptor activation and the propensity for dimerization. J Biol Chem. 2009;284:7483–7494. doi: 10.1074/jbc.M809150200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan R, Wu X, Feng X, Zhang M, Hebert TE, Segaloff DL. Structural determinants underlying constitutive dimerization of unoccupied human follitropin receptors. Cell Signal. 2010;22:247–256. doi: 10.1016/j.cellsig.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarstrom P, Wiseman RL, Powers ET, Kelly JW. Prevention of transthyretin amyloid disease by changing protein misfolding energetics. Science. 2003;299:713–716. doi: 10.1126/science.1079589. [DOI] [PubMed] [Google Scholar]
- Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475:324–332. doi: 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- Hartl FU, Hayer-Hartl M. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science. 2002;295:1852–1858. doi: 10.1126/science.1068408. [DOI] [PubMed] [Google Scholar]
- Hartl FU, Hayer-Hartl M. Converging concepts of protein folding in vitro and in vivo. Nat Struct Mol Biol. 2009;16:574–581. doi: 10.1038/nsmb.1591. [DOI] [PubMed] [Google Scholar]
- Hebert TE, Moffett S, Morello JP, Loisel TP, Bichet DG, Barret C, Bouvier M. A peptide derived from a beta2-adrenergic receptor transmembrane domain inhibits both receptor dimerization and activation. J Biol Chem. 1996;271:16384–16392. doi: 10.1074/jbc.271.27.16384. [DOI] [PubMed] [Google Scholar]
- Heding A, Vrecl M, Bogerd J, McGregor A, Sellar R, Taylor PL, Eidne KA. Gonadotropin-releasing hormone receptors with intracellular carboxyl-terminal tails undergo acute desensitization of total inositol phosphate production and exhibit accelerated internalization kinetics. J Biol Chem. 1998;273:11472–11477. doi: 10.1074/jbc.273.19.11472. [DOI] [PubMed] [Google Scholar]
- Helenius A, Aebi M. Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem. 2004;73:1019–1049. doi: 10.1146/annurev.biochem.73.011303.073752. [DOI] [PubMed] [Google Scholar]
- Huhtaniemi I, Alevizaki M. Mutations along the hypothalamic-pituitary-gonadal axis affecting male reproduction. Reprod Biomed Online. 2007;15:622–632. doi: 10.1016/s1472-6483(10)60529-9. [DOI] [PubMed] [Google Scholar]
- Huhtaniemi IT, Clayton RN, Catt KJ. Gonadotropin binding and Leydig cell activation in the rat testis in vivo. Endocrinology. 1982;111:982–987. doi: 10.1210/endo-111-3-982. [DOI] [PubMed] [Google Scholar]
- Huhtaniemi IT, Themmen AP. Mutations in human gonadotropin and gonadotropin-receptor genes. Endocrine. 2005;26:207–217. doi: 10.1385/ENDO:26:3:207. [DOI] [PubMed] [Google Scholar]
- Hunzicker-Dunn M, Maizels ET. FSH signaling pathways in immature granulosa cells that regulate target gene expression: branching out from protein kinase A. Cell Signal. 2006;18:1351–1359. doi: 10.1016/j.cellsig.2006.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutt DM, Powers ET, Balch WE. The proteostasis boundary in misfolding diseases of membrane traffic. FEBS Lett. 2009;583:2639–2646. doi: 10.1016/j.febslet.2009.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii S, Chang HH, Kawasaki K, Yasuda K, Wu HL, Garman SC, Fan JQ. Mutant alpha-galactosidase A enzymes identified in Fabry disease patients with residual enzyme activity: biochemical characterization and restoration of normal intracellular processing by 1-deoxygalactonojirimycin. Biochem J. 2007;406:285–295. doi: 10.1042/BJ20070479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janovick JA, Conn PM. Salt bridge integrates GPCR activation with protein trafficking. Proc Natl Acad Sci U S A. 2010;107:4454–4458. doi: 10.1073/pnas.0914261107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janovick JA, Goulet M, Bush E, Greer J, Wettlaufer DG, Conn PM. Structure-activity relations of successful pharmacologic chaperones for rescue of naturally occurring and manufactured mutants of the gonadotropin-releasing hormone receptor. J Pharmacol Exp Ther. 2003a;305:608–614. doi: 10.1124/jpet.102.048454. [DOI] [PubMed] [Google Scholar]
- Janovick JA, Knollman PE, Brothers SP, Ayala-Yanez R, Aziz AS, Conn PM. Regulation of G protein-coupled receptor trafficking by inefficient plasma membrane expression: molecular basis of an evolved strategy. J Biol Chem. 2006;281:8417–8425. doi: 10.1074/jbc.M510601200. [DOI] [PubMed] [Google Scholar]
- Janovick JA, Maya-Nunez G, Conn PM. Rescue of hypogonadotropic hypogonadism-causing and manufactured GnRH receptor mutants by a specific protein-folding template: misrouted proteins as a novel disease etiology and therapeutic target. J Clin Endocrinol Metab. 2002;87:3255–3262. doi: 10.1210/jcem.87.7.8582. [DOI] [PubMed] [Google Scholar]
- Janovick JA, Maya-Nunez G, Ulloa-Aguirre A, Huhtaniemi IT, Dias JA, Verbost P, Conn PM. Increased plasma membrane expression of human follicle-stimulating hormone receptor by a small molecule thienopyr(im)idine. Mol Cell Endocrinol. 2009a;298:84–88. doi: 10.1016/j.mce.2008.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janovick JA, Patny A, Mosley R, Goulet MT, Altman MD, Rush TS, 3rd, Cornea A, Conn PM. Molecular mechanism of action of pharmacoperone rescue of misrouted GPCR mutants: the GnRH receptor. Mol Endocrinol. 2009b;23:157–168. doi: 10.1210/me.2008-0384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janovick JA, Ulloa-Aguirre A, Conn PM. Evolved regulation of gonadotropin-releasing hormone receptor cell surface expression. Endocrine. 2003b;22:317–327. doi: 10.1385/ENDO:22:3:317. [DOI] [PubMed] [Google Scholar]
- Jardon-Valadez E, Aguilar-Rojas A, Maya-Nunez G, Leanos-Miranda A, Pineiro A, Conn PM, Ulloa-Aguirre A. Conformational effects of Lys191 in the human GnRH receptor: mutagenesis and molecular dynamics simulations studies. J Endocrinol. 2009;201:297–307. doi: 10.1677/JOE-08-0527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jardon-Valadez E, Ulloa-Aguirre A, Pineiro A. Modeling and molecular dynamics simulation of the human gonadotropin-releasing hormone receptor in a lipid bilayer. J Phys Chem B. 2008;112:10704–10713. doi: 10.1021/jp800544x. [DOI] [PubMed] [Google Scholar]
- Jiang X, Liu H, Chen X, Chen PH, Fischer D, Sriraman V, Yu HN, Arkinstall S, He X. Structure of follicle-stimulating hormone in complex with the entire ectodomain of its receptor. Proc Natl Acad Sci U S A. 2012;109:12491–12496. doi: 10.1073/pnas.1206643109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karges B, Karges W, de Roux N. Clinical and molecular genetics of the human GnRH receptor. Hum Reprod Update. 2003;9:523–530. doi: 10.1093/humupd/dmg040. [DOI] [PubMed] [Google Scholar]
- Kawate N, Menon KM. Palmitoylation of luteinizing hormone/human choriogonadotropin receptors in transfected cells. Abolition of palmitoylation by mutation of Cys-621 and Cys-622 residues in the cytoplasmic tail increases ligand-induced internalization of the receptor. J Biol Chem. 1994;269:30651–30658. [PubMed] [Google Scholar]
- Klausner RD, Sitia R. Protein degradation in the endoplasmic reticulum. Cell. 1990;62:611–614. doi: 10.1016/0092-8674(90)90104-m. [DOI] [PubMed] [Google Scholar]
- Kotlar TJ, Young RH, Albanese C, Crowley WF, Jr, Scully RE, Jameson JL. A mutation in the follicle-stimulating hormone receptor occurs frequently in human ovarian sex cord tumors. J Clin Endocrinol Metab. 1997;82:1020–1026. doi: 10.1210/jcem.82.4.3870. [DOI] [PubMed] [Google Scholar]
- Kremer H, Kraaij R, Toledo SP, Post M, Fridman JB, Hayashida CY, van Reen M, Milgrom E, Ropers HH, Mariman E, et al. Male pseudohermaphroditism due to a homozygous missense mutation of the luteinizing hormone receptor gene. Nat Genet. 1995;9:160–164. doi: 10.1038/ng0295-160. [DOI] [PubMed] [Google Scholar]
- Lagerstrom MC, Schioth HB. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat Rev Drug Discov. 2008;7:339–357. doi: 10.1038/nrd2518. [DOI] [PubMed] [Google Scholar]
- Latronico AC, Anasti J, Arnhold IJ, Rapaport R, Mendonca BB, Bloise W, Castro M, Tsigos C, Chrousos GP. Brief report: testicular and ovarian resistance to luteinizing hormone caused by inactivating mutations of the luteinizing hormone-receptor gene. N Engl J Med. 1996;334:507–512. doi: 10.1056/NEJM199602223340805. [DOI] [PubMed] [Google Scholar]
- Latronico AC, Chai Y, Arnhold IJ, Liu X, Mendonca BB, Segaloff DL. A homozygous microdeletion in helix 7 of the luteinizing hormone receptor associated with familial testicular and ovarian resistance is due to both decreased cell surface expression and impaired effector activation by the cell surface receptor. Mol Endocrinol. 1998;12:442–450. doi: 10.1210/mend.12.3.0077. [DOI] [PubMed] [Google Scholar]
- Laue LL, Wu SM, Kudo M, Bourdony CJ, Cutler GB, Jr, Hsueh AJ, Chan WY. Compound heterozygous mutations of the luteinizing hormone receptor gene in Leydig cell hypoplasia. Mol Endocrinol. 1996;10:987–997. doi: 10.1210/mend.10.8.8843415. [DOI] [PubMed] [Google Scholar]
- Layman LC, Porto AL, Xie J, da Motta LA, da Motta LD, Weiser W, Sluss PM. FSH beta gene mutations in a female with partial breast development and a male sibling with normal puberty and azoospermia. J Clin Endocrinol Metab. 2002;87:3702–3707. doi: 10.1210/jcem.87.8.8724. [DOI] [PubMed] [Google Scholar]
- Le Gouill C, Parent JL, Caron CA, Gaudreau R, Volkov L, Rola-Pleszczynski M, Stankova J. Selective modulation of wild type receptor functions by mutants of G-protein-coupled receptors. J Biol Chem. 1999;274:12548–12554. doi: 10.1074/jbc.274.18.12548. [DOI] [PubMed] [Google Scholar]
- Leanos-Miranda A, Janovick JA, Conn PM. Receptor-misrouting: an unexpectedly prevalent and rescuable etiology in gonadotropin-releasing hormone receptor-mediated hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2002;87:4825–4828. doi: 10.1210/jc.2002-020961. [DOI] [PubMed] [Google Scholar]
- Leanos-Miranda A, Ulloa-Aguirre A, Janovick JA, Conn PM. In vitro coexpression and pharmacological rescue of mutant gonadotropin-releasing hormone receptors causing hypogonadotropic hypogonadism in humans expressing compound heterozygous alleles. J Clin Endocrinol Metab. 2005;90:3001–3008. doi: 10.1210/jc.2004-2071. [DOI] [PubMed] [Google Scholar]
- Leanos-Miranda A, Ulloa-Aguirre A, Ji TH, Janovick JA, Conn PM. Dominant-negative action of disease-causing gonadotropin-releasing hormone receptor (GnRHR) mutants: a trait that potentially coevolved with decreased plasma membrane expression of GnRHR in humans. J Clin Endocrinol Metab. 2003;88:3360–3367. doi: 10.1210/jc.2003-030084. [DOI] [PubMed] [Google Scholar]
- Lee SP, O'Dowd BF, Rajaram RD, Nguyen T, George SR. D2 dopamine receptor homodimerization is mediated by multiple sites of interaction, including an intermolecular interaction involving transmembrane domain 4. Biochemistry. 2003;42:11023–11031. doi: 10.1021/bi0345539. [DOI] [PubMed] [Google Scholar]
- Lin X, Janovick JA, Brothers S, Blomenrohr M, Bogerd J, Conn PM. Addition of catfish gonadotropin-releasing hormone (GnRH) receptor intracellular carboxyl-terminal tail to rat GnRH receptor alters receptor expression and regulation. Mol Endocrinol. 1998;12:161–171. doi: 10.1210/mend.12.2.0056. [DOI] [PubMed] [Google Scholar]
- Lindstedt G, Nystrom E, Matthews C, Ernest I, Janson PO, Chatterjee K. Follitropin (FSH) deficiency in an infertile male due to FSHbeta gene mutation. A syndrome of normal puberty and virilization but underdeveloped testicles with azoospermia, low FSH but high lutropin and normal serum testosterone concentrations. Clin Chem Lab Med. 1998;36:663–665. doi: 10.1515/CCLM.1998.118. [DOI] [PubMed] [Google Scholar]
- Loo TW, Clarke DM. Chemical and pharmacological chaperones as new therapeutic agents. Expert Rev Mol Med. 2007;9:1–18. doi: 10.1017/S1462399407000361. [DOI] [PubMed] [Google Scholar]
- Marshall FH, Jones KA, Kaupmann K, Bettler B. GABAB receptors - the first 7TM heterodimers. Trends Pharmacol Sci. 1999;20:396–399. doi: 10.1016/s0165-6147(99)01383-8. [DOI] [PubMed] [Google Scholar]
- Martens JW, Lumbroso S, Verhoef-Post M, Georget V, Richter-Unruh A, Szarras-Czapnik M, Romer TE, Brunner HG, Themmen AP, Sultan C. Mutant luteinizing hormone receptors in a compound heterozygous patient with complete Leydig cell hypoplasia: abnormal processing causes signaling deficiency. J Clin Endocrinol Metab. 2002;87:2506–2513. doi: 10.1210/jcem.87.6.8523. [DOI] [PubMed] [Google Scholar]
- Martens JW, Verhoef-Post M, Abelin N, Ezabella M, Toledo SP, Brunner HG, Themmen AP. A homozygous mutation in the luteinizing hormone receptor causes partial Leydig cell hypoplasia: correlation between receptor activity and phenotype. Mol Endocrinol. 1998;12:775–784. doi: 10.1210/mend.12.6.0124. [DOI] [PubMed] [Google Scholar]
- Maya-Nunez G, Janovick JA, Aguilar-Rojas A, Jardon-Valadez E, Leanos-Miranda A, Zarinan T, Ulloa-Aguirre A, Conn PM. Biochemical mechanism of pathogenesis of human gonadotropin-releasing hormone receptor mutants Thr104Ile and Tyr108Cys associated with familial hypogonadotropic hypogonadism. Mol Cell Endocrinol. 2011;337:16–23. doi: 10.1016/j.mce.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maya-Nunez G, Janovick JA, Ulloa-Aguirre A, Soderlund D, Conn PM, Mendez JP. Molecular basis of hypogonadotropic hypogonadism: restoration of mutant (E(90)K) GnRH receptor function by a deletion at a distant site. J Clin Endocrinol Metab. 2002;87:2144–2149. doi: 10.1210/jcem.87.5.8386. [DOI] [PubMed] [Google Scholar]
- McArdle CA, Davidson JS, Willars GB. The tail of the gonadotrophin- releasing hormone receptor: desensitization at, and distal to, G protein-coupled receptors. Mol Cell Endocrinol. 1999;151:129–136. doi: 10.1016/s0303-7207(99)00024-6. [DOI] [PubMed] [Google Scholar]
- Meduri G, Touraine P, Beau I, Lahuna O, Desroches A, Vacher-Lavenu MC, Kuttenn F, Misrahi M. Delayed puberty and primary amenorrhea associated with a novel mutation of the human follicle-stimulating hormone receptor: clinical, histological, and molecular studies. J Clin Endocrinol Metab. 2003;88:3491–3498. doi: 10.1210/jc.2003-030217. [DOI] [PubMed] [Google Scholar]
- Millar RP. GnRH II and type II GnRH receptors. Trends Endocrinol Metab. 2003;14:35–43. doi: 10.1016/s1043-2760(02)00016-4. [DOI] [PubMed] [Google Scholar]
- Millar RP, Lu ZL, Pawson AJ, Flanagan CA, Morgan K, Maudsley SR. Gonadotropin-releasing hormone receptors. Endocr Rev. 2004;25:235–275. doi: 10.1210/er.2003-0002. [DOI] [PubMed] [Google Scholar]
- Mizrachi D, Segaloff DL. Intracellularly located misfolded glycoprotein hormone receptors associate with different chaperone proteins than their cognate wild-type receptors. Mol Endocrinol. 2004;18:1768–1777. doi: 10.1210/me.2003-0406. [DOI] [PubMed] [Google Scholar]
- Monnier C, Dode C, Fabre L, Teixeira L, Labesse G, Pin JP, Hardelin JP, Rondard P. PROKR2 missense mutations associated with Kallmann syndrome impair receptor signalling activity. Hum Mol Genet. 2009;18:75–81. doi: 10.1093/hmg/ddn318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morello JP, Petaja-Repo UE, Bichet DG, Bouvier M. Pharmacological chaperones: a new twist on receptor folding. Trends Pharmacol Sci. 2000a;21:466–469. doi: 10.1016/s0165-6147(00)01575-3. [DOI] [PubMed] [Google Scholar]
- Morello JP, Salahpour A, Laperriere A, Bernier V, Arthus MF, Lonergan M, Petaja-Repo U, Angers S, Morin D, Bichet DG, Bouvier M. Pharmacological chaperones rescue cell-surface expression and function of misfolded V2 vasopressin receptor mutants. J Clin Invest. 2000b;105:887–895. doi: 10.1172/JCI8688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller S, Jaeschke H, Gunther R, Paschke R. The hinge region: an important receptor component for GPHR function. Trends Endocrinol Metab. 2009;21:111–122. doi: 10.1016/j.tem.2009.09.001. [DOI] [PubMed] [Google Scholar]
- Munshi UM, Clouser CL, Peegel H, Menon KM. Evidence that palmitoylation of carboxyl terminus cysteine residues of the human luteinizing hormone receptor regulates postendocytic processing. Mol Endocrinol. 2005;19:749–758. doi: 10.1210/me.2004-0335. [DOI] [PubMed] [Google Scholar]
- Musnier A, Heitzler D, Boulo T, Tesseraud S, Durand G, Lecureuil C, Guillou H, Poupon A, Reiter E, Crepieux P. Developmental regulation of p70 S6 kinase by a G protein-coupled receptor dynamically modelized in primary cells. Cell Mol Life Sci. 2009;66:3487–503. doi: 10.1007/s00018-009-0134-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura M, Yasuda D, Hirota N, Shimizu T. Specific ligands as pharmacological chaperones: The transport of misfolded G-protein coupled receptors to the cell surface. IUBMB Life. 2010;62:453–459. doi: 10.1002/iub.344. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Maekawa R, Yamagata Y, Tamura I, Sugino N. A novel mutation in exon8 of the follicle-stimulating hormone receptor in a woman with primary amenorrhea. Gynecol Endocrinol. 2008;24:708–712. doi: 10.1080/09513590802454927. [DOI] [PubMed] [Google Scholar]
- Nechamen CA, Dias JA. Point mutations in follitropin receptor result in ER retention. Mol Cell Endocrinol. 2003;201:123–31. doi: 10.1016/s0303-7207(02)00424-0. [DOI] [PubMed] [Google Scholar]
- Nechamen CA, Thomas RM, Dias JA. APPL1, APPL2, Akt2 and FOXO1a interact with FSHR in a potential signaling complex. Mol Cell Endocrinol. 2007;260–262:93–99. doi: 10.1016/j.mce.2006.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton CL, Whay AM, McArdle CA, Zhang M, van Koppen CJ, van de Lagemaat R, Segaloff DL, Millar RP. Rescue of expression and signaling of human luteinizing hormone G protein-coupled receptor mutants with an allosterically binding small-molecule agonist. Proc Natl Acad Sci U S A. 2011;108:7172–7176. doi: 10.1073/pnas.1015723108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng GY, O'Dowd BF, Lee SP, Chung HT, Brann MR, Seeman P, George SR. Dopamine D2 receptor dimers and receptor-blocking peptides. Biochem Biophys Res Commun. 1996;227:200–204. doi: 10.1006/bbrc.1996.1489. [DOI] [PubMed] [Google Scholar]
- Nimri R, Lebenthal Y, Lazar L, Chevrier L, Phillip M, Bar M, Hernandez-Mora E, de Roux N, Gat-Yablonski G. A novel loss-of-function mutation in GPR54/KISS1R leads to hypogonadotropic hypogonadism in a highly consanguineous family. J Clin Endocrinol Metab. 2011;96:E536–E545. doi: 10.1210/jc.2010-1676. [DOI] [PubMed] [Google Scholar]
- Okamoto T, Nishimoto I. Detection of G protein-activator regions in M4 subtype muscarinic, cholinergic, and alpha 2-adrenergic receptors based upon characteristics in primary structure. J Biol Chem. 1992;267:8342–8346. [PubMed] [Google Scholar]
- Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- Percherancier Y, Planchenault T, Valenzuela-Fernandez A, Virelizier JL, Arenzana-Seisdedos F, Bachelerie F. Palmitoylation-dependent control of degradation, life span, and membrane expression of the CCR5 receptor. J Biol Chem. 2001;276:31936–31944. doi: 10.1074/jbc.M104013200. [DOI] [PubMed] [Google Scholar]
- Rannikko A, Pakarinen P, Manna PR, Beau I, Misrahi M, Aittomaki K, Huhtaniemi I. Functional characterization of the human FSH receptor with an inactivating Ala189Val mutation. Mol Hum Reprod. 2002;8:311–317. doi: 10.1093/molehr/8.4.311. [DOI] [PubMed] [Google Scholar]
- Resh MD. Palmitoylation of ligands, receptors, and intracellular signaling molecules. Sci STKE 2006. 2006:re14. doi: 10.1126/stke.3592006re14. [DOI] [PubMed] [Google Scholar]
- Richter-Unruh A, Martens JW, Verhoef-Post M, Wessels HT, Kors WA, Sinnecker GH, Boehmer A, Drop SL, Toledo SP, Brunner HG, Themmen AP. Leydig cell hypoplasia: cases with new mutations, new polymorphisms and cases without mutations in the luteinizing hormone receptor gene. Clin Endocrinol (Oxf) 2002;56:103–112. doi: 10.1046/j.0300-0664.2001.01437.x. [DOI] [PubMed] [Google Scholar]
- Richter-Unruh A, Verhoef-Post M, Malak S, Homoki J, Hauffa BP, Themmen AP. Leydig cell hypoplasia: absent luteinizing hormone receptor cell surface expression caused by a novel homozygous mutation in the extracellular domain. J Clin Endocrinol Metab. 2004;89:5161–5167. doi: 10.1210/jc.2004-0298. [DOI] [PubMed] [Google Scholar]
- Robben JH, Sze M, Knoers NV, Deen PM. Rescue of vasopressin V2 receptor mutants by chemical chaperones: specificity and mechanism. Mol Biol Cell. 2006;17:379–386. doi: 10.1091/mbc.E05-06-0579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Rozell TG, Davis DP, Chai Y, Segaloff DL. Association of gonadotropin receptor precursors with the protein folding chaperone calnexin. Endocrinology. 1998;139:1588–1593. doi: 10.1210/endo.139.4.5881. [DOI] [PubMed] [Google Scholar]
- Rozell TG, Wang H, Liu X, Segaloff DL. Intracellular retention of mutant gonadotropin receptors results in loss of hormone binding activity of the follitropin rreceptor but not the lutropin/choriogonadotropin receptor. Mol. Endocrinol. 1995;9:1727–1736. doi: 10.1210/mend.9.12.8614409. [DOI] [PubMed] [Google Scholar]
- Sampedro JG, Uribe S. Trehalose-enzyme interactions result in structure stabilization and activity inhibition. The role of viscosity. Mol Cell Biochem. 2004;256–257:319–327. doi: 10.1023/b:mcbi.0000009878.21929.eb. [DOI] [PubMed] [Google Scholar]
- Schlyer S, Horuk R. I want a new drug: G-protein-coupled receptors in drug development. Drug Discov Today. 2006;11:481–493. doi: 10.1016/j.drudis.2006.04.008. [DOI] [PubMed] [Google Scholar]
- Schubert U, Anton LC, Gibbs J, Norbury CC, Yewdell JW, Bennink JR. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature. 2000;404:770–774. doi: 10.1038/35008096. [DOI] [PubMed] [Google Scholar]
- Schulz A, Schoneberg T, Paschke R, Schultz G, Gudermann T. Role of the third intracellular loop for the activation of gonadotropin receptors. Mol Endocrinol. 1999;13:181–190. doi: 10.1210/mend.13.2.0233. [DOI] [PubMed] [Google Scholar]
- Sigurdsson EM, Permanne B, Soto C, Wisniewski T, Frangione B. In vivo reversal of amyloid-beta lesions in rat brain. J Neuropathol Exp Neurol. 2000;59:11–17. doi: 10.1093/jnen/59.1.11. [DOI] [PubMed] [Google Scholar]
- Simoni M, Gromoll J, Nieschlag E. The follicle-stimulating hormone receptor: biochemistry, molecular biology, physiology, and pathophysiology. Endocr Rev. 1997;18:739–773. doi: 10.1210/edrv.18.6.0320. [DOI] [PubMed] [Google Scholar]
- Smithson DC, Janovick JA, Conn PM. Therapeutic Rescue of Misfolded/Mistrafficked Mutants: Automation-Friendly High-Throughput Assays for Identification of Pharmacoperone Drugs of GPCRs. Methods Enzymol. 2013;521:3–16. doi: 10.1016/B978-0-12-391862-8.00001-6. [DOI] [PubMed] [Google Scholar]
- Soderlund D, Canto P, de la Chesnaye E, Ulloa-Aguirre A, Mendez JP. A novel homozygous mutation in the second transmembrane domain of the gonadotrophin releasing hormone receptor gene. Clin Endocrinol (Oxf) 2001;54:493–498. doi: 10.1046/j.1365-2265.2001.01211.x. [DOI] [PubMed] [Google Scholar]
- Song YS, Ji I, Beauchamp J, Isaacs NW, Ji TH. Hormone interactions to Leu-rich repeats in the gonadotropin receptors. II. Analysis of Leu-rich repeat 4 of human luteinizing hormone/chorionic gonadotropin receptor. J Biol Chem. 2001;276:3436–3442. doi: 10.1074/jbc.M003773200. [DOI] [PubMed] [Google Scholar]
- Soto C. Protein misfolding and disease; protein refolding and therapy. FEBS Lett. 2001;498:204–207. doi: 10.1016/s0014-5793(01)02486-3. [DOI] [PubMed] [Google Scholar]
- Stewart MD, Deng JM, Stewart CA, Mullen RD, Wang Y, Lopez S, Serna MK, Huang CC, Janovick JA, Pask AJ, Schwartz RJ, Conn PM, Behringer RR. Mice Harboring Gnrhr E90K, a Mutation that Causes Protein Misfolding and Hypogonadotropic Hypogonadism in Humans, Exhibit Testis Size Reduction and Ovulation Failure. Mol Endocrinol. 2012;26:1847–1856. doi: 10.1210/me.2012-1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szidonya L, Cserzo M, Hunyady L. Dimerization and oligomerization of G- protein-coupled receptors: debated structures with established and emerging functions. J Endocrinol. 2008;196:435–453. doi: 10.1677/JOE-07-0573. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Nagayama Y, Nishihara E, Namba H, Yamashita S, Niwa M. Palmitoylation of human thyrotropin receptor: slower intracellular trafficking of the palmitoylation-defective mutant. Endocrinology. 1998;139:803–806. doi: 10.1210/endo.139.2.5911. [DOI] [PubMed] [Google Scholar]
- Tao YX, Johnson NB, Segaloff DL. Constitutive and agonist-dependent self-association of the cell surface human lutropin receptor. J Biol Chem. 2004;279:5904–5914. doi: 10.1074/jbc.M311162200. [DOI] [PubMed] [Google Scholar]
- Tapanainen JS, Aittomaki K, Min J, Vaskivuo T, Huhtaniemi IT. Men homozygous for an inactivating mutation of the follicle-stimulating hormone (FSH) receptor gene present variable suppression of spermatogenesis and fertility. Nat Genet. 1997;15:205–206. doi: 10.1038/ng0297-205. [DOI] [PubMed] [Google Scholar]
- Thomas RM, Nechamen CA, Mazurkiewicz JE, Muda M, Palmer S, Dias JA. Follice-stimulating hormone receptor forms oligomers and shows evidence of carboxyl-terminal proteolytic processing. Endocrinology. 2007;148:1987–1995. doi: 10.1210/en.2006-1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timossi C, Ortiz-Elizondo C, Pineda DB, Dias JA, Conn PM, Ulloa-Aguirre A. Functional significance of the BBXXB motif reversed present in the cytoplasmic domains of the human follicle-stimulating hormone receptor. Mol Cell Endocrinol. 2004;223:17–26. doi: 10.1016/j.mce.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Toledo SP, Brunner HG, Kraaij R, Post M, Dahia PL, Hayashida CY, Kremer HTAP. An inactivating mutation of the luteinizing hormone receptor causes amenorrhea in a 46,XX female. J Clin Endocrinol Metab. 1996;81:3850–3854. doi: 10.1210/jcem.81.11.8923827. [DOI] [PubMed] [Google Scholar]
- Touraine P, Beau I, Gougeon A, Meduri G, Desroches A, Pichard C, Detoeuf M, Paniel B, Prieur M, Zorn JR, Milgrom E, Kuttenn F, Misrahi M. New natural inactivating mutations of the follicle-stimulating hormone receptor: correlations between receptor function and phenotype. Mol Endocrinol. 1999;13:1844–1854. doi: 10.1210/mend.13.11.0370. [DOI] [PubMed] [Google Scholar]
- Tranchant T, Durand G, Gauthier C, Crepieux P, Ulloa-Aguirre A, Royere D, Reiter E. Preferential beta-arrestin signalling at low receptor density revealed by functional characterization of the human FSH receptor A189 V mutation. Mol Cell Endocrinol. 2011;331:109–118. doi: 10.1016/j.mce.2010.08.016. [DOI] [PubMed] [Google Scholar]
- Ulloa-Aguirre A, Conn PM. G protein-coupled receptors and the G protein family. In: Conn PM, editor. Handbook of Physiology-Endocrinology: Section 7, Cellular Endocrinology. New York, USA: Oxford University Press; 1998. pp. 87–141. [Google Scholar]
- Ulloa-Aguirre A, Conn PM. Targeting of G protein-coupled receptors to the plasma membrane in health and disease. Front Biosci. 2009;14:973–994. doi: 10.2741/3290. [DOI] [PubMed] [Google Scholar]
- Ulloa-Aguirre A, Conn PM. Pharmacoperones: a new therapeutic approach for diseases caused by misfolded G protein-coupled receptors. Recent Pat Endocr Metab Immune Drug Discov. 2011;5:13–24. doi: 10.2174/187221411794351851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulloa-Aguirre A, Crepieux P, Poupon A, Maurel MC, Reiter E. Novel pathways in gonadotropin receptor signaling and biased agonism. Rev Endocr Metab Disord. 2011;12:259–274. doi: 10.1007/s11154-011-9176-2. [DOI] [PubMed] [Google Scholar]
- Ulloa-Aguirre A, Janovick JA, Leanos-Miranda A, Conn PM. Misrouted cell surface GnRH receptors as a disease aetiology for congenital isolated hypogonadotrophic hypogonadism. Hum Reprod Update. 2004;10:177–192. doi: 10.1093/humupd/dmh015. [DOI] [PubMed] [Google Scholar]
- Ulloa-Aguirre A, Janovick JA, Leanos-Miranda A, Conn PM. Misrouted cell surface receptors as a novel disease aetiology and potential therapeutic target: the case of hypogonadotropic hypogonadism due to gonadotropin-releasing hormone resistance. Expert Opin Ther Targets. 2003;7:175–185. doi: 10.1517/14728222.7.2.175. [DOI] [PubMed] [Google Scholar]
- Ulloa-Aguirre A, Janovick JA, Miranda AL, Conn PM. G-protein-coupled receptor trafficking: understanding the chemical basis of health and disease. ACS Chem Biol. 2006;1:631–638. doi: 10.1021/cb600360h. [DOI] [PubMed] [Google Scholar]
- Ulloa-Aguirre A, Timossi C. Biochemical and functional aspects of gonadotrophin-releasing hormone and gonadotropins. Reprod Biomed Online. 2000;1:48–62. doi: 10.1016/s1472-6483(10)61901-3. [DOI] [PubMed] [Google Scholar]
- Ulloa-Aguirre A, Timossi C. Structure-function relationship of follicle-stimulating hormone and its receptor. Hum Reprod Update. 1998;4:260–283. doi: 10.1093/humupd/4.3.260. [DOI] [PubMed] [Google Scholar]
- Uribe A, Zarinan T, Perez-Solis MA, Gutierrez-Sagal R, Jardon-Valadez E, Pineiro A, Dias JA, Ulloa-Aguirre A. Functional and Structural Roles of Conserved Cysteine Residues in the Carboxyl-Terminal Domain of the Follicle-Stimulating Hormone Receptor in Human Embryonic Kidney 293 Cells. Biol. Reprod. 2008;78:869–882. doi: 10.1095/biolreprod.107.063925. [DOI] [PubMed] [Google Scholar]
- van Straten NC, Schoonus-Gerritsma GG, van Someren RG, Draaijer J, Adang AE, Timmers CM, Hanssen RG, van Boeckel CA. The first orally active low molecular weight agonists for the LH receptor: thienopyr(im)idines with therapeutic potential for ovulation induction. Chembiochem. 2002;3:1023–1026. doi: 10.1002/1439-7633(20021004)3:10<1023::AID-CBIC1023>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Vaskivuo TE, Aittomaki K, Anttonen M, Ruokonen A, Herva R, Osawa Y, Heikinheimo M, Huhtaniemi I, Tapanainen JS. Effects of follicle-stimulating hormone (FSH) and human chorionic gonadotropin in individuals with an inactivating mutation of the FSH receptor. Fertil Steril. 2002;78:108–113. doi: 10.1016/s0015-0282(02)03148-5. [DOI] [PubMed] [Google Scholar]
- Vassart G, Pardo L, Costagliola S. A molecular dissection of the glycoprotein hormone receptors. Trends Biochem Sci. 2004;29:119–126. doi: 10.1016/j.tibs.2004.01.006. [DOI] [PubMed] [Google Scholar]
- Weekes MP, Antrobus R, Talbot S, Hor S, Simecek N, Smith DL, Bloor S, Randow F, Lehner PJ. Proteomic plasma membrane profiling reveals an essential role for gp96 in the cell surface expression of LDLR family members, including the LDL receptor and LRP6. J Proteome Res. 2012;11:1475–1484. doi: 10.1021/pr201135e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner ED, Brodsky JL, McCracken AA. Proteasome-dependent endoplasmic reticulum-associated protein degradation: an unconventional route to a familiar fate. Proc Natl Acad Sci U S A. 1996;93:13797–13801. doi: 10.1073/pnas.93.24.13797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SM, Hallermeier KM, Laue L, Brain C, Berry AC, Grant DB, Griffin JE, Wilson JD, Cutler GB, Jr, Chan WY. Inactivation of the luteinizing hormone/chorionic gonadotropin receptor by an insertional mutation in Leydig cell hypoplasia. Mol Endocrinol. 1998;12:1651–1660. doi: 10.1210/mend.12.11.0189. [DOI] [PubMed] [Google Scholar]
- Zarinan T, Perez-Solis MA, Maya-Nunez G, Casas-Gonzalez P, Conn PM, Dias JA, Ulloa-Aguirre A. Dominant negative effects of human follicle-stimulating hormone receptor expression-deficient mutants on wild-type receptor cell surface expression. Rescue of oligomerization-dependent defective receptor expression by using cognate decoys. Mol Cell Endocrinol. 2010;321:112–122. doi: 10.1016/j.mce.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Wess J. Truncated V2 vasopressin receptors as negative regulators of wild-type V2 receptor function. Biochemistry. 1998;37:15773–15784. doi: 10.1021/bi981162z. [DOI] [PubMed] [Google Scholar]