

Abstract
A highly conserved threonine in the I-helix of cytochrome P450s has been suggested to play an important role in dioxygen activation, a critical step for catalytic turnover. However, subsequent studies with some P450s in which this highly conserved threonine was replaced by another residue such as alanine showed that significant catalytic activities were still retained when the variants were compared with the wild type enzymes. These results make the role of this residue unclear. We provide data here that suggest a novel role for this highly conserved threonine (Thr303) in the function of P450 2E1. We found that the P450 2E1 T303A mutant undergoes rapid auto-inactivation in the reconstituted system during catalytic turnover when the electrons are provided by NADPH. This inactivation was much faster than that of the wild type P450 2E1 and was prevented by catalase. Both the P450 2E1 wild type and T303A mutants produce hydrogen peroxide during the incubations. The inactivation was accompanied by heme destruction with part of the heme becoming covalently attached to protein. The heme destruction was prevented by catalase or by the presence of substrate. Interestingly, this inactivation occurred much more rapidly in the presence of both an electron transfer system and hydrogen peroxide externally added to enzyme. This accelerated inactivation during catalytic turnover was also found with a 2B4 T302A mutant, which corresponds to 2E1 T303A. Our results suggest that the conserved threonine in these P450s prevents rapid auto-inactivation during the catalytic cycle, and that this residue may be highly conserved in P450s since it allows them to remain catalytically active for longer periods of time.
The cytochrome P450 enzymes belong to a superfamily of heme-containing mono-oxygenases that play an important role in steroid biosynthesis, drug metabolism, and detoxification of xenobiotics (1, 2). Cytochrome P450s exhibit a large diversity in terms of substrate specificity (3), but the overall tertiary structure between bacterial and mammalian P450s is consistent (4). In general, structures closest to the heme or active site are more highly conserved. This is especially true for the I and L helices, which are in direct contact with the heme (4). Some residues located in the active sites of P450s are highly conserved and are considered to be catalytically important (5). A threonine residue in the I-helix located on the distal side of the heme is one of the most highly conserved residues in the P450s (6). Since the initial solution of the crystal structure of bacterial P450cam (6), this highly conserved threonine in the I-helix (Thr252), has been thought to play an important role in dioxygen activation during catalytic turnover (7, 8). This residue has also been implicated as playing a role in the proton transfer network that promotes oxygen-oxygen bond cleavage to produce the oxenoid-iron that is thought to be a catalytically active species (7). Schlichting et al. (8) suggested, from observations of the crystal structure of P450cam, that Thr252 provides a hydrogen bond to the dioxygen complex via catalytic water molecules. Because dioxygen activation is a critical step in the catalytic cycle of P450s, this highly conserved threonine residue corresponding to Thr252 in P450cam has been a target of mutation studies in bacterial (P450cam, P450BM3) and mammalian P450s. The results of the studies using these mutants suggested that this conserved threonine residue may be associated with dioxygen stabilization (9, 10), proton delivery (11–13), and substrate recognition (14, 15).
This above-mentioned experimental evidence provided strong arguments for the importance of the conserved threonine in P450 function; thus, it has been surprising that some mutants where the conserved threonine residue was replaced with an aliphatic residue exhibit significant NADPH consumption rates and/or catalytic activities for substrate oxidation when compared to the wild type enzymes. Kimata et al. reported that a mutant of P450cam where Thr252 was replaced with methoxy threonine, an unnatural amino acid that masks the hydroxyl group of threonine, exhibited unaltered catalytic activity, indicating that the OH group of Thr252 is not important in the catalytic turnover (16). In addition, although substrate specificity and regio-selectivity were altered by mutations of mammalian cytochrome P450s where the conserved threonine was replaced with other residues, significant catalytic activities have still been observed in these mutants (13–15, 17–21). Recently, Nagano and Poulos reported that, as with the wild type enzyme, the two catalytic water molecules involved in the hydrogen bonding network with the iron-dioxygen complex were retained in the active site of the crystal structure of the P450cam T252A mutant (22). This result indicated that Thr252 is not important for the retention of the catalytic water molecules involved in hydrogen bonding. These authors suggested that Thr252 may not serve as a hydrogen bond or proton donor in the activation of dioxygen, as suggested before, although it remains a possibility that Thr252 can accept a hydrogen bond from the hydroperoxo-iron intermediate. Thus, these recent data examining the catalytic activities and crystal structures of mutants where the conserved threonine has been replaced with other residues that cannot form hydrogen bonds or donate protons call into question why it is so highly conserved. In general, residues that are highly conserved across all species have been found to play an important role in the function of P450s. In addition, the close proximity of the conserved threonine to the heme suggests a crucial role for this residue; however, its precise function is still unclear.
Cytochrome P450 2E1 T303A, in which this highly conserved threonine residue was mutated to alanine, has previously been used to investigate the role of the conserved threonine in catalysis (13, 19–21, 23). This mutant showed higher catalytic activities for several substrates in comparison to the wild type enzyme (19–21). Coon and coworkers proposed that this mutation may lead to an increase in the steady-state levels of the hydroperoxo-iron intermediate due to the failure of proton delivery for O-O bond cleavage, and that this hydroperoxo-iron intermediate can act as an active oxygen species (19). We have previously investigated the role that T303 plays in the inactivation of P450 2E1 by mechanism-based inactivators, such as tert-butyl acetylene (13, 21). These studies revealed that a mutation of T303 resulted in either protein- or heme-modification by these inactivators. Studies using tert-butyl acetylene suggested that the T303 residue might be involved in the proton delivery network to the active site (13, 21).
Further studies in our laboratory on the inactivation of wild type P450 2E1 and the T303A mutant by mechanism-based inactivators have shown that an accelerated auto-inactivation of 2E1 T303A mutant occurs during incubation with reductase, even in the absence of inactivators. As presented here, this inactivator-independent auto-inactivation of the mutant was much faster than that seen with the wild-type enzyme and could be prevented by co-incubation with catalase. The auto-inactivation was accompanied by heme destruction and part of the heme became covalently attached to the apoprotein. Interestingly, this accelerated inactivation during catalytic turnover was also found with the 2B4 T302A mutant, which corresponds to 2E1 T303A. Our present results suggest that the conserved threonine might stabilize the P450 or decrease production of a reactive heme-oxygen intermediate to prevent the accelerated auto-inactivation which can occur in the presence of hydrogen peroxide during electron transfer from the reductase under aerobic conditions. We propose here a novel concept for the role of this highly conserved threonine and an explanation of why P450s may require this highly conserved threonine residue in the active site.
Experimental Procedures
Materials
L-α-Dilauroyl-phosphatidylcholine (DLPC), NADP+, NADPH, catalase, superoxide dismutase (SOD), reduced glutathione (GSH), N-acetylcysteine, desferroxamine, 4-methylpyrazole (4MP), and bovine serum albumin (BSA) were purchased from Sigma Chemical Co. (St. Louis, MO). Mannitol and thiourea were purchased from Fisher Scientific Co. (Fair Lawn, NJ). 7-Ethoxy-4-(trifluoromethyl)-coumarin (7-EFC) was obtained from Molecular Probes, Inc. (Eugene, OR). HPLC-grade acetonitrile was from Fisher (Pittsburgh, PA), and trifluoroacetic acid (TFA) was from Pierce (Rockford, IL).
Enzymes
The cDNAs for rabbit cytochromes P450 2E1 and 2E1 T303A that encodes a protein lacking amino acids 3–29 and rabbit cytochromes P450 2B4 and 2B4 T302A were cloned into pCW vector (provided by M. J. Coon, The University of Michigan) were expressed in Escherichia coli MV1304 cells. Expression and purification of the proteins was carried out according to published methods (24) with some modifications (25). NADPH-P450 reductase, cloned into pIN-III-ompA3 vector, was purified after expression in E. coli Topp3 cells as previously described (26).
Enzyme Activity Assays for P450 2E1 and 2E1 T303A
Purified P450 2E1 and 2E1 T303A were reconstituted with reductase and lipid for 45 min at 4 °C as previously described (13, 20). Each P450 enzyme mixture consisting of 1 μM P450, 1 μM reductase, and 166 μg/mL DLPC in 50 mM potassium phosphate buffer (pH 7.4) was divided into equal samples (120 μL) that contained either no NADPH, 1 mM NADPH or 1 mM NADPH together with 2000 units/mL catalase. The samples (primary incubation mixture) were incubated at 30°C. At the indicated time points, a 10 μL aliquot of the P450 primary incubation mixture was transferred into 990 μL of a secondary reaction mixture containing 100 μM 7-EFC, 0.2 mM NADPH, 400 units/mL catalase and 40 μg/mL BSA in 50 mM potassium phosphate buffer (pH 7.4). The secondary reaction mixtures were incubated for 10 min at 30 °C in a shaking water bath and the enzyme activity was terminated by the addition of 350 μL of acetonitrile. Enzymatic activity was assessed spectrofluorometrically by measuring the extent of O-deethylation of 7-EFC to 7-HFC on a Shimadzu model RF-5301PC spectrofluorometer (Shimadzu Scientific Instruments, Columbia, MD) with excitation at 410 nm and emission at 510 nm (27). For longer time incubations of the P450 2E1 wild-type enzyme up to 90 min, an NADPH regenerating system consisting of 0.1 mM of NADP+, 10 mM MgCl2, 10 mM glucose-6-phosphate, and 1 unit/mL glucose-6-phosphate dehydrogenese was used to maintain the NADPH levels. Where indicated, the following reagents were added to the primary incubation mixture: hydrogen peroxide at a final concentration of 0.1 or 1 mM, 200 μM 4MP, 500 units/mL of SOD, 100 mM mannitol, 1 mM thiourea, 2 mM GSH, 2 mM N-acetylcysteine and 100 μM desferroxamine. The appropriate volume of water was added to the control samples instead of the reagents indicated.
Spectrophotometric Quantitation
Wild-type P450 2E1 and P450 2E1 T303A were reconstituted and incubated as described above. The primary incubation mixture of each P450 sample contained either no NADPH, 1 mM NADPH, 1 mM NADPH with 2000 units/mL catalase, or 1 mM NADPH with 200 μM 4MP and was incubated for 10 min at 30°C. Then, 200 μL aliquots of the primary incubation mixture were removed and diluted with 400 μL of ice-cold 50 mM potassium phosphate buffer (pH 7.4), containing 400 units/mL catalase to prevent further inactivation. Dithionite was added, carbon monoxide was bubbled through the samples, and the reduced CO spectra were recorded from 400 to 500 nm on a UV spectrophotometer (Shimadzu Scientific Instruments, Columbia, MD) (28). The maximal absorbance at 450 nm was used to quantify P450 heme remaining.
HPLC Analysis
Wild-type P450 2E1 and the P450 2E1 T303A mutant were reconstituted with reductase and lipid and incubated as described above. For the heme and protein analysis following incubation, the primary incubation mixtures of each P450 were injected onto a 250 mm x 4.60 mm C4 reverse phase HPLC column (Phenomenex, Torrance, CA) (solvent A, H2O and 0.1% TFA; solvent B, 100% acetonitrile and 0.1% TFA). The flow rate was 1 mL/min and a linear gradient from 35% B to 95% B over 30 min was used. The elution of proteins and heme were monitored using diode array detection. For detection of heme-protein adducts, P450s and reductase were incubated in the reconstituted system as described and then injected onto a 150 mm x 4.60 mm POROS R2/10 column (Applied Biosystems, Foster City, CA) (solvent A, H2O and 0.1% TFA; solvent B, 100% acetonitrile and 0.1% TFA). The flow rate was 1 mL/min and the components were resolved using a linear gradient from 30% B to 95% B over 30 min. The eluate was monitored using a diode array detector.
SDS–Polyacrylamide Gel Electrophoresis
Protein-bound heme adducts were detected using the procedure described by Vuletich and Osawa (29). Briefly, the P450 2E1 wild-type and the P450 2E1 T303A mutant were reconstituted with reductase and lipid and incubated as described above. After incubation of the primary mixture, sample buffer containing 5% SDS, 20% glycerol, 50 mM Tris (2-carboxyethyl)-phosphine hydrochloride (Pierce, Rockford, IL) and 0.02% bromophenol blue in 125 mM Tris–HCl, pH 6.8, was added to the primary mixture, and they were incubated for 30 min at room temperature. The samples were first subjected to electrophoresis on a 10% SDS–polyacrylamide gel and then transferred to nitrocellulose membranes (0.2 mm; Bio-Rad, Hesperia, CA) using a Mini-Trans-Blot Electrophoresis Transfer Cell (Bio-Rad, Hercules, CA). ECL reagent (Super Signal, Pierce, Rockford, IL) and Kodak BioMax MS film (Eastman Kodak, Rochester, NY) were used as described by the manufacturers to detect the peroxidase activity of the protein-bound heme adduct. Films were exposed to the nitrocellulose membranes for 30 min before developing.
Measurement of H2O2 Concentration
Wild-type P450 2E1 and the P450 2E1 T303A mutant were reconstituted with reductase and lipid, and incubated as described above. For the time course studies of hydrogen peroxide formation, each P450 enzyme was incubated with 1 mM NADPH. At indicated times, a 150 μL sample was removed and the reaction was terminated by adding 30 μL of a solution of 60% TCA followed by incubating the samples at room temperature for 10 min. Then, the H2O2 concentration was measured using the ferricyanide method according to previously described procedures (30). For the other studies, the incubation mixture of each P450 enzyme was also divided into equal samples that either contained no NADPH, 1 mM NADPH, 1 mM NADPH with 2000 units/mL catalase, or 1 mM NADPH with 200 μM 4MP. The samples were incubated for 10 min and the H2O2 concentrations were measured as described above.
Enzyme Activity Assays for P450 2B1 and 2B1 T302A
Purified P450s 2B4 and 2B4 T302A were reconstituted with reductase and lipid for 45 min at 4 °C as previously described (19). The P450 enzyme mixture consisting of 1 μM P450, 1 μM reductase, and 166 μg/mL DLPC in 50 mM potassium phosphate buffer (pH 7.4) was divided into equal samples that contained either no NADPH, 1 mM NADPH, 1 mM NADPH with 2000 units/mL catalase or 1 mM NADPH with and without 300 μM hydrogen peroxide in a final volume of 120 μl. The samples (primary incubation mixture) were incubated at 30°C, and the enzymatic activities for O-deethylation of 7-EFC were measured as described above.
Results
Inactivation of P450 2E1 and 2E1 T303A in the Presence of NADPH
The time-dependent loss in activity of P450 2E1 and P450 2E1 T303A in the presence of reductase and NADPH was studied by measuring the 7-EFC O-deethylation activity. In these experiments, neither substrate nor inactivator for P450 2E1 was added to the incubation mixture. As shown in Figure 1, the activity of P450 2E1 T303A in the reconstituted system decreased in a time-dependent manner following the addition of NADPH and the half-life for the loss of P450 2E1 T303A activity was approximately 11 min under the conditions described in Experimental Procedures. Interestingly, the activity loss of the T303A mutant in the presence of NADPH could be prevented by co-incubation with catalase, as shown in Figure 1B. In contrast to the mutant, the loss in activity of the wild-type P450 2E1 in the presence of 1 mM NADPH was very small under the same conditions used with the P450 2E1 T303A mutant. As shown in the inset to Figure 1-A, longer incubation times of the wild-type enzyme with the NADPH generating system resulted in an obvious loss in activity which also could be prevented by co-incubation with catalase. P450s are known to be auto-inactivated during catalytic turnover (31, 32). The present data indicated that the wild-type enzyme was gradually auto-inactivated when electrons were provided by the NADPH-reductase system, but that auto-inactivation was greatly accelerated when the T303 residue was mutated to alanine. The apparent inactivation rate constants (kinact) for the wild-type and T303A mutant in the presence of NADPH were 0.0102 and 0.0607 min−1, respectively, indicating an approximately 6-fold increase in the rate of inactivation of the mutant. When catalase was included in the incubation the inactivation of both the wild-type and mutant enzymes was prevented. These results suggest a role for hydrogen peroxide, which might be produced through an uncoupling reaction during catalytic turnover. Inclusion of cytochrome b5 during the reconstitution step had no effect on this loss in activity (data not shown).
Figure 1.

Time-dependent loss in activity for P450 2E1 wild-type and the T303A mutant. P450 2E1 wild-type (A) or P450 2E1 T303A (B) were incubated with reductase in the absence of NADPH (●), in the presence of 1 mM NADPH (□) or in the presence of 1 mM NADPH and 2000 units/mL catalase (▲). The inset to A shows the activity loss for wild-type P450 2E1 incubated with reductase and the NADPH generating system for longer times. The incubation conditions and measurements of 7-EFC O-deethylation activity are described under Experimental Procedures. The data represent the mean and standard deviations from three to six separate experiments.
Analysis of Heme using Carbon Monoxide Binding and HPLC
As shown in Figure 2, the reduced CO-spectra of the P450s in the reconstituted systems incubated in the presence or absence of NADPH were determined to evaluate the P450 content. The quantitative values for the spectral data are summarized in Table 2. The incubation time for determination of the reduced CO-spectra was 10 min, at which time there was approximately a 50% loss in the catalytic activity of the T303A mutant. With wild type P450 2E1, there was very little change in the spectrum after 10 min incubation with NADPH when compared to control samples incubated without NADPH. When 4MP, a substrate of 2E1 (33), and catalase were co-incubated with wild-type P450 2E1 and NADPH, no effect on the reduced CO-spectrum was observed. In contrast to the wild-type enzyme, the 2E1 T303A mutant showed a drastic decrease in the spectral peak at 450 nm (64% loss) in the presence of NADPH. The spectral loss was not accompanied by a shift in the maximum absorption of the peak, indicating that the heme of P450 2E1 T303A was destroyed during incubation with NADPH. This decrease in the amount of P450 as measured by the CO-spectrum was comparable to the loss of the catalytic activity (47%). Similar to the activity data shown in Figure 1, the decrease in the spectral peak of the reduced CO complex of the T303A mutant in the presence of NADPH could also be prevented by co-incubation with catalase, indicating that heme losses as well as activity loss were both related to hydrogen peroxide formation during the incubation. In addition, incubation with 4MP also prevented the decrease in the spectral peak of the mutant (Figure 2 and Table 2). The catalytic activity remaining for the wild-type and mutant enzymes (see Table 2) could not be evaluated in the presence of 4MP because of the potent competitive inhibition of 7-EFC O-deethylation by 4MP. However, the protection by 4MP indicated that it occupied the active sites of P450 2E1 T303A and of the 2E1 wild-type enzyme and protected against inactivation.
Figure 2.
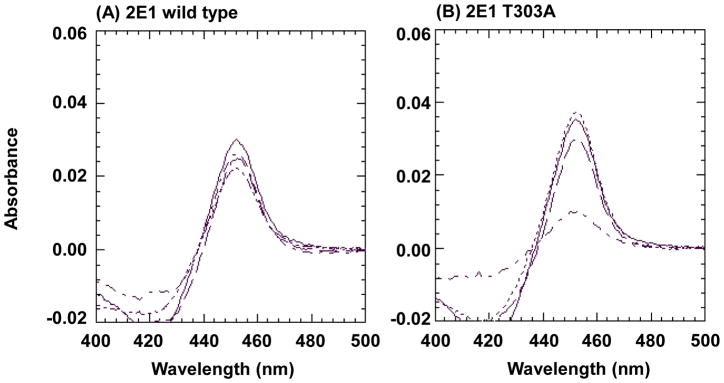
Reduced CO difference spectra of wild-type cytochrome P450s 2E1 and the 2E1 T303A mutant after incubation with reductase. Wild-type P450 2E1 (A) or P450 2E1 T303A (B) were incubated with reductase as described under Experimental Procedures. Spectra for wild-type P450 2E1 and the P450 T303A mutant are shown by ----for incubations without NADPH, -·-·- for incubations with 1 mM NADPH,– – –for incubations with NADPH and 2000 units/mL catalase, and —— for incubations with NADPH and 200 μM 4MP.
Table 2.
Remaining catalytic acitivity and heme content of cytochrome P450 2E1 and 2E1 T303A after incubation with reductase.
Wild-type P450 2E1 and T303A mutant were incubated for 10 min with reductase in the absence and presence of 1mM NADPH and in the presence of NADPH and 2000 units/ml catalase or 200 mM 4MP as described under Experimental Procedures. After incubation for 10 min, the 7-EFC O-deethylation activity was measured. Reduced CO-spectra were measured to quantify the P450 content. Heme content and P450 protein recovered were measured by HPLC monitored at 398 nm and 220 nm, respectively. The data represent the mean and standard deviations from three separate incubations.
| Primary reaction mixture | % remaining
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Activity
|
Reduced CO
|
Heme
|
P450 Protein recovery
|
|||||
| 2E1 | T303A | 2E1 | T303A | 2E1 | T303A | 2E1 | T303A | |
| −NADPH without incubation | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| −NADPH | 91 ± 3 | 88 ± 6 | 92 ± 1 | 93 ± 2 | 99 ± 1 | 100 ± 1 | 100 ± 3 | 101 ± 1 |
| +NADPH | 87 ± 5 | 53 ± 8 | 82 ± 5 | 36 ± 12 | 80 ± 1 | 47 ± 6 | 94 ± 5 | 67 ± 6 |
| +NADPH, +Catalase | 93 ± 2 | 87 ± 3 | 88 ± 1 | 77 ± 2 | 85 ± 6 | 93 ± 2 | 96 ± 4 | 98 ± 5 |
| +NADPH, +4MP | NA | NA | 101 ± 2 | 93 ± 5 | 88 ± 3 | 93 ± 3 | 96 ± 5 | 102 ± 5 |
NA: not able to measure catalytic activities due to the strong competitive inhibiton by 4MP.
In addition to determining the heme remaining by the reduced CO-spectra, we also used HPLC to measure the amount of the native heme remaining after incubation with NADPH (Table 2). The elution profile was monitored at 398 nm for intact heme and at 220 nm for protein. Under the HPLC conditions described in Experimental Procedures, heme is released from the apoprotein during the separation and elutes earlier than the apoprotein. As shown in Table 2, the heme loss assayed by HPLC monitored at 398 nm paralleled the loss of the catalytic activity as well as the CO-spectral data. Together, the data presented in Table 2 suggest that the enzymatic activity loss was consistent with the loss of heme, indicating that incubation of the mutant with NADPH caused destruction of the heme, which in turn was responsible for the loss in enzymatic activity. P450 2E1 and reductase eluted off the C4 column at 16 min and at 18 min, respectively. In the inactivated sample of the mutant, the peak area for the P450 apoprotein decreased by approximately 30%. This decrease was prevented by co-incubation with catalase and 4MP, suggesting that the mutant protein was partially denatured or aggregated by the inactivation and therefore, may be retained on the C4 column.
Formation of Heme Protein Adducts
As described above, a decrease in the intact heme peak monitored at 398 nm was observed during incubation of the T303A mutant with NADPH. In addition to the decrease in the size of the peak for the native heme, a new peak absorbing at 398 nm was observed in the inactivated sample of the mutant (Figure 3). The retention time of this new peak overlapped with that of the P450 apoprotein monitored at 220 nm (data not shown). Under the denaturing solvent conditions used for the separation of the components of the reconstituted system by reverse-phase chromatography, the heme moiety is released from the apoprotein and elutes earlier. Therefore, the peak at 398 nm that overlaps with the protein peak appears to be heme that has become covalently attached to the protein. The formation of this heme-protein cross-linked species was further analyzed using a different HPLC column (Poros R2) with a larger pore size to avoid the loss of modified proteins that may become stuck to the top of other columns due to aggregation or oligomerization (see column for protein recovery in Table 2). As shown in Figure 3, the T303A mutant, which was incubated for 10 min in the absence of NADPH under conditions that did not inactivate the enzyme, exhibited a relatively small peak at 398 nm with a retention time of 24 min. This peak eluted later than the native heme and overlapped with protein peak detected at 220 nm. This same peak was also observed at a similar level in samples that had not been incubated (data not shown), indicating that the native P450 2E1 T303A may already have a very small amount of heme covalently bound to the apoprotein. As shown in Fig. 3, the modified heme peak that overlapped with the protein peak increased approximately 7-fold when the mutant was incubated with NADPH. This increase in the protein-associated peak area could be prevented and remained at the control levels when the samples were incubated in the presence of 4MP.
Figure 3.

HPLC profiles for P450 2E1 T303A incubated with or without NADPH. P450 2E1 T303A was incubated with reductase for 10 min in the absence (A) or presence (B) of 1 mM NADPH, and in the presence of NADPH together with 200 μM 4MP (C). After incubation, samples were subjected to HPLC using a Poros R2/10 column. The incubation conditions were as described under Experimental Procedures. Intact heme eluted at 11 min and was monitored at 398 nm. P450 and reductase eluted together at 24 min and were monitored at 220 nm. The modified heme peak exhibited the same retention time as the protein peak.
In order to determine if the heme peak that overlapped with the P450 protein peak contained heme that had been cross-linked to the P450 protein, we employed SDS–polyacrylamide gel electrophoresis followed by an ECL-detection method that has been described by Vuletich and Osawa (29). This method can detect heme which is covalently bound to protein using the peroxidase-like activity of the heme. This method is specific for the identification of protein-bound heme adducts, and native heme or non-covalently bound heme is completely dissociated from the P450 during electrophoresis (29). As shown in Figure 4, heme-bound P450 protein was not detected in samples obtained from the P450 T303A mutant that had been incubated without NADPH. In contrast, an obvious band representing the heme cross-linked to the P450 was detected in the samples incubated with NADPH. A protein band with covalently bound heme could first be observed after 5 min of incubation with NADPH and the amount increased at 10 min. However, no further increase could be seen after 20 min of incubation. This band was not detected in the P450 T303A sample that had been incubated with 4MP in the presence of NADPH. The small peak of the heme-protein adduct that was detected by HPLC in samples without NADPH and samples incubated with NADPH and 4MP was probably too weak a signal to be detected in the electrophoresis method with ECL detection. Wild-type P450 2E1 incubated without and with NADPH again exhibited a very slight cross-linked band. In each case, the intensity of the heme-protein cross-linked band seen with the T303A mutant incubated with NADPH was much stronger than that of wild-type enzyme, again indicating that heme-protein adduct formation in P450 T303A occurs during inactivation.
Figure 4.
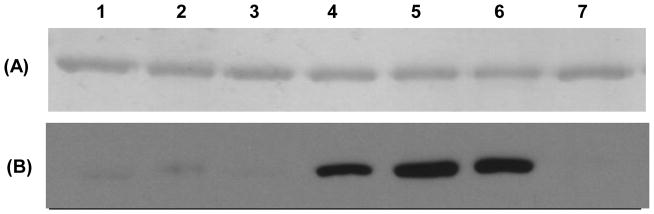
Detection of P450-heme cross-linked proteins. SDS-polyacrylamide electrophoresis was performed as described under Experimental Procedures. P450 protein was observed using Coomassie staining (A) and the heme-protein adducts were detected using ECL staining (B). Lanes 1 and 2, wild-type P450 2E1 incubated for 10 min without and with NADPH; Lane 3, P450 2E1 T303A incubated for 10 min without NADPH; Lanes 4, 5 and 6, P450 2E1 T303A incubated for 5, 10 and 20 min with NADPH; Lane 7, P450 2E1 T303A incubated for 10 min with NADPH and 4MP.
It was difficult to accurately estimate the amount of the heme-protein adduct formed by the T303A mutant because a standard for heme-protein adducts was not available. However, HPLC analysis suggests that the peak for the heme-protein adduct formed in the presence of NADPH corresponds to only about 6% of the total heme, which is much less than the overall loss in activity under these conditions. Thus, these results indicate that during the inactivation of the T303A mutant by incubation with NADPH some of the heme becomes covalently attached to the P450 protein, but that this portion only accounts for some of the heme destruction and that other pathways leading to heme destruction may be operative.
Hydrogen Peroxide Production
As shown previously, the protection by catalase provides strong evidence for the involvement of hydrogen peroxide in the inactivation of the T303A mutant during the transfer of electrons from NADPH. From these data two hypotheses were considered: 1) the mutant produces higher levels of hydrogen peroxide than the wild-type enzyme, or 2) the mutant is more sensitive to hydrogen peroxide. In order to examine the former hypothesis, the amounts of hydrogen peroxide formed during the incubations were measured. Figure 5-A shows the time-courses for hydrogen peroxide formation by the wild-type and mutant enzymes. A steady-state concentration of hydrogen peroxide was reached at approximately the same level (around 100 μM) within 10 min after adding NADPH to either of the two enzymes. P450 2E1 is well known to be highly uncoupled and to produce a large amount of hydrogen peroxide (34). These data show that analogous to the wild-type enzyme, the T303A mutant also produces significant amounts of hydrogen peroxide. These data further indicate that the rapid auto-inactivation of P450 2E1 T303A is not due to higher levels of production of hydrogen peroxide by the mutant compared to the wild-type enzyme. Incubations with catalase demonstrated that the hydrogen peroxide concentrations at 10 min for the wild-type and mutant P450s incubated with NADPH were essentially zero under the conditions used (Figure 5-B). In addition, the presence of 4MP, a compound which binds tightly to P450 2E1, resulted in a decrease in hydrogen peroxide levels by around 50% for both the wild-type and mutant P450s, which might be explained by a decrease in uncoupling during the catalytic turnover. However, significant levels of hydrogen peroxide were still produced in the presence of 4MP, and this observation suggested that the preventative effect of 4MP on heme-destruction is in all probability not due solely to the decrease in hydrogen peroxide production.
Figure 5.
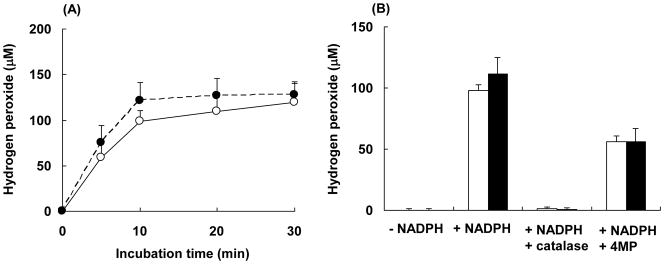
Hydrogen peroxide formation during the incubation of cytochrome P450s 2E1 or 2E1 T303A with reductase in the presence of NADPH. A, H2O2 formation by wild-type 2E1 (○) and by the T303A mutant (●)in the presence of 1 mM NADPH. B, The effect of catalase and 4MP on H2O2 formation in the presence of 1 mM NADPH. Open and filled bars represent the wild-type P450 2E1 and the P450 T303A mutant, respectively. The incubation conditions were as described under Experimental Procedures. The limit of detection for hydrogen peroxide is 2.5 μM. The data represent the mean and standard deviations from three separate experiments.
Sensitivity of P450s 2E1 and 2E1 T303A to Hydrogen Peroxide
The inactivation of wild-type P450s 2E1 and T303A by hydrogen peroxide added to the incubation mixture was determined by measuring the loss of 7-EFC O-deethylation activity with time (Figure 6). In this experiment, hydrogen peroxide was added to the reconstituted system at concentrations of 100 μM and 1 mM, consistent with and exceeding by a factor of 10 the levels formed (~100 μM) by the wild-type and mutant enzymes in the presence of NADPH, respectively (see Figure 5). The effect of added hydrogen peroxide on the catalytic activity was evaluated in the absence and presence of NADPH. With wild-type P450 2E1, even high concentrations of hydrogen peroxide (1 mM) in the absence of NADPH had little effect on the catalytic activity, and the combination of 1 mM hydrogen peroxide and NADPH only slightly inactivated the enzyme. Similarly, only a small loss in activity of P450 2E1 T303A was observed when external hydrogen peroxide was added in the absence of NADPH. This minimal loss in activity was also much less than that observed in samples of the T303A mutant incubated in the presence of NADPH but without added hydrogen peroxide. These observations indicate that hydrogen peroxide alone has a limited effect on P450 2E1 T303A and do not explain the accelerated auto-inactivation of the T303A mutant observed in the presence of NADPH. Interestingly, the combination of NADPH together with 1 mM hydrogen peroxide resulted in a substantial auto-inactivation of the mutant enzyme at a rate much faster than that seen in the presence of NADPH alone. These results demonstrate that the combination of NADPH and hydrogen peroxide cause a marked increase in the loss in activity of both enzymes over that seen with either alone. However, once again, this inactivation occurred at a much faster rate in the T303A mutant compared to the wild-type enzyme. These results demonstrate that the T303A mutant is more sensitive to hydrogen peroxide than the wild-type enzyme in the presence of NADPH. In addition, they suggest that hydrogen peroxide and NADPH cooperatively inactivate these enzymes by some unknown mechanism. The apparent rate constants (kinact) for the inactivation of P450 2E1 T303A calculated from the initial slopes of the lines in Figure 6 were 0.0218 min−1 with 1 mM hydrogen peroxide added, and 0.0672 min−1 and 0.214 min−1 in the presence of NADPH either without or with 1 mM hydrogen peroxide added, respectively. The kinact of P450 2E1 T303A in the presence of NADPH with 1 mM hydrogen peroxide added (0.214 min−1) was larger than the sum of the kinact values (0.0890 min−1) for incubations with NADPH alone (0.0772 min−1) and with added 1 mM hydrogen peroxide alone (0.0218 min−1). This clearly indicates the synergistic effect of NADPH and hydrogen peroxide for the auto-inactivation of the T303A mutant.
Figure 6.

Effect of hydrogen peroxide on the NADPH-dependent inactivation of P450s 2E1 and 2E1 T303A. Wild-type P450 2E1 (A) and the P450 T303A mutant (B) were incubated with reductase in the absence (●) or presence (□) of 1 mM NADPH. Hydrogen peroxide was added to the reaction mixture at a concentration of 100 μM (△) in the absence of NADPH and at a concentration of 1 mM in the absence (▲) or presence (○) of 1 mM NADPH. The incubation conditions and measurement of 7-EFC O-deethylation activity are described under Experimental Procedures. The data represent the mean and standard deviations from three separate experiments.
Effects of Antioxidants on the NADPH-Supported Auto-Inactivation of P4350 2E1 T303A
In order to study the involvement of reactive oxygen species in the NADPH-supported inactivation of the P450 2E1 T303A mutant, various antioxidants were incubated with the primary reaction mixture in the presence of NADPH for 20 min. As shown in Table 3, catalase almost completely abolished the NADPH-dependent inactivation suggesting the involvement of hydrogen peroxide. SOD only slightly slowed the rate of inactivation, indicating that superoxide (O2−) was unlikely to be significantly involved in the NADPH-induced inactivation. Mannitol (35) and thiourea (36), which act as hydroxyl radical scavengers, also did not prevent the auto-inactivation. Therefore, hydroxyl radicals formed through a Fenton-like reaction may not be involved in the inactivation. However, we cannot exclude the possibility that hydroxyl radicals produced in the active site of the P450 enzyme may be involved and would not be trapped by these reagents. GSH (37) and N-acetylcysteine (38) are general antioxidants which can react with reactive oxygen species (ROS) and other free radicals. As shown in Table 3, GSH but not N-acetylcysteine exhibited some protection against inactivation. One possibility is that GSH is somewhat protective due to its ability to scavenge hydrogen peroxide non-enzymatically (39). The potent iron chelator desferroxamine (40) did not prevent the inactivation. This is indicative that free iron released from the P450 heme was not involved in the inactivation. In contrast to the chemical scavengers that were studied, catalase showed the most potent protective effect against auto-inactivation. These results strongly suggest that hydrogen peroxide plays a key role in the NADPH-dependent inactivation of the 2E1 T303A mutant.
Table 3.
Effect of antioxidants on the inactivation of P450 2E1 T303A in the presence of NADPH.
P450 2E1 T303A was incubated with reductase in the presence of 1mM NADPH together with the antioxidants indicated as described under Experimetal Procedures. Samples were incubated for 20 min following the addition of NADPH and the catalytic activities were measured using the 7-EFC O-deethylation assay. The data represent the mean and standard deviations from three separate incubations.
| Primary reaction mixture | % Activity remaining |
|---|---|
| Without NADPH | 93.3 ± 4.0 |
| With NADPH | |
| + water | 26.6 ± 1.7 |
| + 2000 units/ml catalase | 93.0 ± 5.0 |
| + 500 units/ml SOD | 35.9 ± 3.9 |
| + 100 mM mannitol | 26.6 ± 1.7 |
| + 1 mM thiourea | 18.7 ± 3.0 |
| + 2 mM GSH | 43.0 ± 2.3 |
| + 2 mM N-acetyl-L-cystein | 33.7 ± 1.4 |
| + 100 μM deferroxamine | 30.4 ± 2.4 |
Inactivation of P450s 2B4 and 2B4 T302A by Hydrogen Peroxide and NADPH
Since the T303A mutant of P450 2E1 was much more susceptible to inactivation by hydrogen peroxide in the presence of NADPH than wild-type P450 2E1, similar experiments using wild-type P450 2B4 and the T302A mutant were performed to see if this mutant would also be more susceptible to inactivation (Figure 7). T302 in cytochrome P450 2B4 corresponds to the highly conserved T303 in P450 2E1 that is located in the active site (19). In this experiment, hydrogen peroxide was added to the reconstituted system at a concentration of 300 μM in the absence or presence of NADPH. The results obtained with the P450 2B4 enzymes, as shown in Figure 7, were similar to the results seen with the P450 2E1 enzymes. Activity loss of wild-type P450 2B4 in the presence of NADPH was minimal and the effect of added hydrogen peroxide was small in the absence or presence of NADPH. In contrast to the wild-type enzyme, the activity loss of the 2B4 T302A mutant was again increased when incubations were carried out in the presence of NADPH alone or when co-incubated with added hydrogen peroxide. The kinact values for the wild-type and the T303A mutant incubated with NADPH were 0.0062 and 0.0225 min−1, respectively, indicating a 3.6-fold increase in the rate of inactivation of the mutant. The kinact for P450 2B4 T302A was 0.008 min−1 in the absence of NADPH with added hydrogen peroxide, and 0.0225 and 0.0452 min−1in the presence of NADPH without and with hydrogen peroxide added, respectively. The kinact for P450 2B4 T302A in the presence of NADPH with hydrogen peroxide added (0.0452 min−1) was again larger than the sum of the kinact values (0.0305 min−1) for the NADPH system alone (0.0225 min−1) and with hydrogen peroxide added alone (0.0080 min−1). This result is similar to that we had observed with P450 2E1 T303A and again provides strong evidence for a synergistic effect of NADPH and hydrogen peroxide on the inactivation of the threonine mutants.
Figure 7.
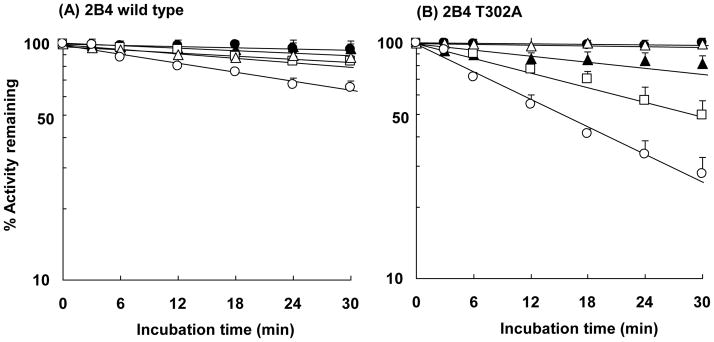
Time-dependent loss in the activity of wild-type P450 2B4 and the T302A mutant. Wild-type P450 2B4 (A) or P450 2B4 T302A (B) were incubated with reductase in the absence of NADPH (●), in the presence of 1 mM NADPH (□), and in the presence of 1 mM NADPH together with 2000 units/mL catalase (△). Hydrogen peroxide was added to the reaction mixture at a concentration of 300 μM in the absence (▲) and presence (○) of 1 mM NADPH. The incubation conditions and measurement of 7-EFC O-deethylation activity are described under Experimental Procedures. The data represent the mean and standard deviations from three separate experiments.
Discussion
Based on the results of this study we propose a novel hypothesis for the role of the highly conserved threonine in the I-helix of P450s. Our data strongly suggest that this residue functions to protect against increased auto-inactivation of the P450 enzymes during electron transfer from the reductase and subsequent oxygen activation. Based on the crystal structure of P450cam and the results of site-directed mutagenesis studies, the highly conserved threonine in the active site of P450s was suggested to be involved in dioxygen activation (6–8). Thus, in light of this hypothesis, it was surprising that some threonine mutants exhibited significant rates of NADPH consumption and catalytic activity when compared to their corresponding wild-type enzymes (16–21). These observations raised questions about the true role of this residue in the catalytic cycle. In the present study, we found that the P450 2E1 T303A mutant in which the highly conserved threonine was replaced by alanine was auto-inactivated at a significantly faster rate than the wild-type enzyme under normal conditions for catalytic turnover (Figure 1). This inactivation occurred in the presence of the electron donating system consisting of NADPH and NADPH-P450 reductase and hydrogen peroxide was formed during the incubation. The inactivation was accompanied by heme destruction and a portion of the heme became covalently attached to the P450 apoprotein (Figures 2–4 and Table 2). This heme destruction and heme-apoprotein attachment were prevented by catalase or by adding a substrate, even in the absence of catalase. Thus, the auto-inactivation of the 2E1 T303A mutant which occurs under conditions where electron transfer is occurring can be protected against by substrates and in this situation the mutant proceeds via the normal catalytic cycle either in the presence or absence of catalase. However, under normal physiological conditions, the appropriate substrates for P450s may not always be present in the proximity of the P450s and electrons may be transferred to the P450s by the reductase even in the absence of substrate. Therefore, our results suggest that P450s may require this highly conserved threonine residue in the active site in order to survive longer under cellular conditions where they may undergo reduction by NADPH and reductase in the absence of substrate.
Auto-inactivation of the wild-type mammalian liver cytochrome P450s accompanied by heme-destruction has previously been shown to occur during catalytic turnover (31, 32), and a lack of Thr303 may further accelerate this inactivation. The inactivation of P450s during catalytic turnover has been characterized using purified P450s having the conserved threonine as well as liver microsomes and the results can be summarized as follows (31, 32, 41, 42); a) the P450s are inactivated during incubation with reductase and NADPH, b) catalase prevents the inactivation, indicating an involvement of hydrogen peroxide that is formed during catalysis, c) inactivation is accompanied by heme-degradation and protein modification. d) hydrogen peroxide externally added to enzyme system had little effect on enzyme activity unless electrons were provided. In addition, modification of the heme prosthetic group of the P450s during catalytic turnover induces digestion of the P450 apoprotein by a cellular proteolysis system, and the half-lives of the apoproteins of P450 are significantly prolonged in the presence of substrates (43, 44). These previously reported results indicate that auto-inactivation during catalytic turnover generally occurs with the mammalian P450 enzymes. As shown in the inset to Figure 1, long incubation times for the wild-type P450 2E1 together with reductase in the NADPH-generating system resulted in a loss in the catalytic activity which could be prevented by co-incubation with catalase. These results are in complete support of the above concept. Interestingly, our results demonstrated that the inactivation of the P450 2E1 T303A mutant was much faster than that seen with the wild-type enzyme. The results of our studies on the inactivation of the mutant shown in Figures 1, 2 and 6 and Table 2, are consistent with features a)-d) of the inactivation of the wild-type enzyme and with the protective effect of the substrate. Therefore, the basic features involved in the inactivation of the wild-type and T303A mutant P450s appear to be similar except that the mutant may undergo much more rapid auto-inactivation.
The inactivation of P450 2E1 T303A was accompanied by heme loss and at least two pathways for heme destruction may be involved. One pathway involves formation of a covalent heme adduct to the P450 2E1 T303A apoprotein as shown in Figures 3 and 4. The amount of this heme-protein adduct appears to be relatively low and accounts for only part of the total loss in native heme by the mutant. We think that the most likely pathway that can account for the majority of the heme loss in the mutant is heme fragmentation, which has previously been shown to be the major pathway for heme loss from P450s under conditions where electrons are provided to the system (32). This is based on our observation that the features observed in the auto-inactivation of the T303A mutant are very similar to those of P450s in general and that the absence of the T303 residue appears to primarily lead to an acceleration of this auto-inactivation. Guengerich and co-workers have previously shown that microsomal P450s are inactivated when incubated with NADPH and reductase and that the inactivation was accompanied by heme fragmentation (31, 32). The primary products of heme fragmentation were maleimides (hematinic acid and methylvinylmaleimide), and small quantities of propentdyopents (PDPs). The amount of maleimides and PDPs recovered could account only in part for the amount of the heme that was destroyed. Interestingly, in microsomes containing [14C]-labeled heme, 30–60% of the heme-derived radioactivity was covalently bound to microsomal proteins during heme destruction. It was suggested that reactive species such as methylvinylmaleimide that are formed during the heme-destruction process quickly bind to the P450 enzymes before they can dissociate away from the apoprotein. Karuzina et al. reported that P450 2B4 was auto-inactivated during incubation with reductase and NADPH and that this inactivation was accompanied by apoprotein aggregation as well as heme degradation (42). Our results indicate approximately a 30% loss in the T303A apoprotein that could be recovered by HPLC after inactivation and that loss could be prevented by co-incubation with 4MP. Our data also showed that protein modification occurs during the inactivation and that the presence of a substrate may provide a stabilizing environment in the active site which protects the heme from degradation and thereby prevents protein modification. Taken together, the denaturing of the P450 T303A apoprotein during inactivation appears to be a consequence of heme cross-linking of the modified heme to the protein and heme fragmentation.
Heme destruction may occur following the formation of one or more reactive intermediates of oxygen bound to the heme iron and the Thr303 in P450 2E1 may slow their formation or stabilize them and thereby decrease the rate of auto-inactivation. Our results also indicate a requirement for hydrogen peroxide for the inactivation of the P450 2E1 enzymes under conditions where electrons are being transferred to the P450s. However, the addition of hydrogen peroxide directly to the incubation mixture in the absence of NADPH inactivated the enzymes at a much slower rate than was seen in the presence of NADPH (Figure 6). These results indicate that hydrogen peroxide together with the transfer of electrons from NADPH through reductase co-operatively inactivated the P450s. This suggests that hydrogen peroxide may bind to the heme and form a reactive intermediate under conditions whereby electrons are transferred to the heme that ultimately leads to destabilization and modification of the heme. This mechanism may occur slowly in the wild-type P450 2E1 but appears to be markedly increased in the 2E1 T303A mutant. Scheme 1 summarizes our hypothesis based on our data on the auto-inactivation of the T303A mutant. In the presence of catalase (i.e., in the absence of hydrogen peroxide) and/or presence of substrate the upper pathway is preferred and minimal heme-destruction of P450 2E1 T303A occurs during the transfer of electrons from NADPH. Under such conditions, the intermediates of the dioxygen complex, such as the peroxo-iron (FeIII-OO-) or hydroperoxo iron (FeIII-OOH), may be formed leading to substrate oxidation. Our results also suggest that these intermediates of the dioxygen complex are unlikely to play a significant role in the auto-inactivation under these conditions. However, in the absence of both catalase (i.e., in the presence of hydrogen peroxide) and substrate, as shown by the lower pathway in Scheme 1, a reactive intermediate may be formed by the T303A mutant leading to rapid auto-inactivation accompanied by heme-destruction. Taking into consideration that auto-inactivation of the 2E1 wild-type enzyme also occurred slowly, this type of reactive intermediate might form even in the wild-type and Thr303 might be involved in stabilization of the intermediate or decreasing the rate of production of this intermediate thereby decreasing the rate of inactivation. Although the identity of the reactive heme intermediate that may be involved in the inactivation of 2E1 T303A is not clear, it is possible that this intermediate may be different from the dioxygen intermediates involved in the catalytic cycle, as mentioned previously. Guengerich and co-workers have proposed a mechanism to explain how heme-fragmentation of P450s occurs during catalytic turnover through the formation of heme intermediates in the presence of hydrogen peroxide (32). According to their mechanism, the heme iron reacts with hydrogen peroxide to form a perferryl oxygen species which can then react with the olefinic porphyrin bridge to form a glycol. Another hydrogen peroxide can then react with the heme iron leading to oxidative cleavage of the glycol to give an α-formyl pyrrole. It is possible that this mechanism would explain our results for heme degradation of the threonine mutant in the presence of NADPH and hydrogen peroxide; however, the same mechanism should be operative even when the enzyme is incubated with hydrogen peroxide without NADPH. As we have shown, heme degradation is much faster when the P450 is reduced and allowed to react with hyrogen peroxide at the same time whereas, in the absence of NADPH, heme destruction was much slower, even in the presence of high concentrations of hydrogen peroxide externally added to the reconstituted system. This suggests that the actual mechanism for fragmentation involves reaction of the external hydrogen peroxide with one or more of the CYP2E1 intermediates generated during the catalytic cycle. If so, the highly conserved threonine could serve to hinder the access of the external hydrogen peroxide to the perferryl or ferryl-oxo catalytic intermediate and its replacement with alanine would enhance access to the intermediate. Another possibility is that the threonine could stabilize the reactive intermediate produced by the reaction of hydrogen peroxide with the catalytic intermediates.
Scheme 1.
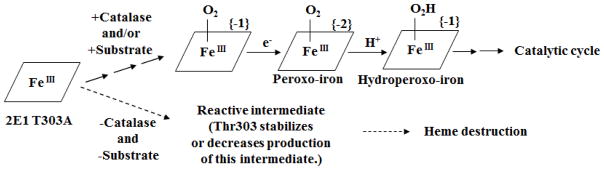
Possible pathway for the formation of the heme intermediates of P450 2E1 T303A when incubated with reductase and NADPH.
In the presence of catalase (i.e. the absence of hydrogen peroxide) and/or the presence of substrate, the top pathway is favored in which P450 2E1 T303A forms the previously described dioxygen intermediates (21, 50) leading to catalytic turnover. In this catalytic cycle there is: (i) a one-electron reduction of the P450 and binding of molecular oxygen to generate the oxyferrous P450, (ii) transfer of a second electron to form the peroxo-iron species, and (iii) addition of one proton to form the hydroperoxo-iron species which leads to further catalytic cycles. In the absence of both catalase (i.e., hydrogen peroxide is produced and remains in solution) and substrate, the bottom pathway becomes more favored and P450 2E1 T303A may form a reactive intermediate leading to heme-destruction. The reactive intermediate could be different from either the peroxo-iron or the hydroperoxo-iron species. We propose that the conserved threonine located in the active site of P450 2E1 either stabilizes this intermediate or decreases the production of the reactive heme-oxygen intermediate responsible for inactivation in order to decrease the rate of acceleration of inactivation that occurs in the presence of H2O2 under electron transfer conditions.
Covalent binding of the heme to the P450 protein as seen here with the 2E1 T303A mutant is rarely seen in other P450s except for the CYP4 family of enzymes (45–47). Ortiz de Montellano and co-workers first showed heme-protein attachment in the CYP4A enzymes. With P450 4A3, the heme was covalently bound to the protein via an ester linkage to Glu318 which is located in the I-helix (45) and which is conserved in most of CYP4 enzymes (46). This conserved glutamic acid residue in CYP4 enzymes has also been implicated to be a required residue for the covalent attachment of the heme prosthetic group (46–48). As shown in Table 1, only the CYP4 family have this required residue and this may account for the fact that only CYP4 enzymes, but no other P450s form such heme-protein adducts (45, 46). In this case, the proposed mechanism for heme-protein adduct formation requires a covalent bond between the heme 5-methyl groups and a protein carboxyl residue (46). Interestingly, during catalytic turnover of CYP4 enzymes with P450 reductase and NADPH, heme-protein attachment also increased (46, 47). This is similar to our results with the P450 2E1 T303A mutant that formed heme-protein cross-links during catalytic turnover. However, as shown in Table 1, the 2E1 T303A mutant (or wild-type 2E1) does not have the required residue that would correspond to Glu318 in CYP4A3. This suggests that the mechanism of heme-protein attachment seen with P450 2E1 T303A is quite different from that observed with the CYP4 family. Alternatively, the mechanism may be similar but another residue with a carboxylic acid side chain may be involved. In the latter case, an amino acid residue that is spatially located near T303, such as Glu302, could be the required residue because minimal covalent heme-protein attachment occurs in the 2E1 wild-type enzyme.
Table 1.
Alignment of the I-helix sequence around the highly conserved threonine in various P450s.
These sequences show the highly conserved threonine, represented with the bold T and surrounding sequences in selected P450s from bacteria and mammals (6,15). The conserved glutamic acids in the CYP4 family, which have been demonstrated to be required for heme cross-linking to these proteins (46) are represented with the bold E.
| Protein | Partial I-helix sequence | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P450cam | L | L | L | V | G | G | L | D | T | 252 | V | V | N | F | L | S |
| P450 BM3 | T | F | L | I | A | G | H | E | T | 268 | T | S | G | L | L | S |
| P450 1A2 | D | I | F | G | A | G | F | E | T | 319 | V | T | T | A | I | F |
| P450 2B4 | S | L | F | F | A | G | T | E | T | 302 | T | S | T | T | L | R |
| P450 2E1 | D | M | F | F | A | G | T | E | T | 303 | T | S | T | T | L | R |
| P450 4A3 | T | F | M | F | E | G | H | D | T | 322 | T | A | S | G | I | S |
| P450 4F4 | T | F | M | F | E | G | H | D | T | 332 | T | A | S | G | L | S |
Cytochrome P450 2E1 (wild-type) is unique among liver microsomal P450s in terms of its high oxidase activity in the absence of substrate, leading to higher production of reactive oxygen species including hydrogen peroxide (34). This might be related to the spin state of the heme iron, where high-spin iron is observed in the absence of substrate (49). The high oxidase activity indicates a unique feature in the catalytic turnover of P450 2E1. Our results with the 2B4 T302 mutant that also exhibited accelerated auto-inactivation demonstrate that the protective role of the conserved threonine residue against auto-inactivation is not limited to P450 2E1. Here, we propose a novel hypothesis for the role of the highly conserved threonine in P450s which is to prevent increased auto-inactivation during catalytic turnover. Further studies are necessary to determine whether or not the protective effect of this highly conserved threonine residue is common among P450s and may be the reason that it is so highly conserved.
Acknowledgments
This work was supported in part by National Institutes of Health Grant CA 16954 and by Daiichi Sankyo Co., Ltd., Tokyo, Japan.
We thank Dr. Minor J. Coon for the P450 2E1, 2E1 T303A, 2B4 and 2B4 T302A plasmids. We are grateful to Chitra Sridar for her help with the purification of P450 2E1. We also thank Hsia-lien Lin for technical advice for SDS–polyacrylamide gel electrophoresis with ECL detection.
Abbreviations
- P450
cytochrome P450
- reductase
NADPH cytochrome P450 reductase
- DLPC
L-α-dilauroyl-phosphatidylcholine
- 7-EFC
7-ethoxy-4- (trifluoromethyl)coumarin
- BSA
bovine serum albumin
- TFA
trifluoroacetic acid
- HPLC
high-performance liquid chromatography
- SOD
superoxide dismutase
- GSH
reduced glutathione
- 4MP
4-methylpyrazole
- ECL
enhanced chemiluminescence
References
- 1.Gonzalez FJ. The molecular biology of cytochrome P450s. Pharmacol Rev. 1988;40:243–288. [PubMed] [Google Scholar]
- 2.Nelson DR, Koymans L, Kamataki T, Stegeman JJ, Feyereisen R, Waxman DJ, Waterman MR, Gotoh O, Coon MJ, Estabrook RW, Gunsalus IC, Nebert DW. P450 superfamily: update on new sequences, gene mapping, accession numbers and nomenclature. Pharmacogenetics. 1996;6:1–42. doi: 10.1097/00008571-199602000-00002. [DOI] [PubMed] [Google Scholar]
- 3.Danielson PB. The cytochrome P450 superfamily: biochemistry, evolution and drug metabolism in humans. Curr Drug Metab. 2002;3:561–597. doi: 10.2174/1389200023337054. [DOI] [PubMed] [Google Scholar]
- 4.Poulos TL, Johnson EF. Structures of cytochrome P450 enzymes. In: Ortiz de Montellano PR, editor. Cytochrome P450: Structure, Mechanism and Biochemistry. 3. 2005. pp. 87–114. [Google Scholar]
- 5.Munro AW, Leys DG, McLean KJ, Marshall KR, Ost TW, Daff S, Miles CS, Chapman SK, Lysek DA, Moser CC, Page CC, Dutton PL. P450 BM3: the very model of a modern flavocytochrome. Trends Biochem Sci. 2002;27:250–257. doi: 10.1016/s0968-0004(02)02086-8. [DOI] [PubMed] [Google Scholar]
- 6.Poulos TL, Finzel BC, Gunsalus IC, Wagner GC, Kraut J. The 2.6-A crystal structure of Pseudomonas putida cytochrome P-450. J Biol Chem. 1985;260:16122–16130. [PubMed] [Google Scholar]
- 7.Vidakovic M, Sligar SG, Li H, Poulos TL. Understanding the role of the essential Asp251 in cytochrome p450cam using site-directed mutagenesis, crystallography, and kinetic solvent isotope effect. Biochemistry. 1998;37:9211–9219. doi: 10.1021/bi980189f. [DOI] [PubMed] [Google Scholar]
- 8.Schlichting I, Berendzen J, Chu K, Stock AM, Maves SA, Benson DE, Sweet RM, Ringe D, Petsko GA, Sligar SG. The catalytic pathway of cytochrome p450cam at atomic resolution. Science. 2000;287:1615–1622. doi: 10.1126/science.287.5458.1615. [DOI] [PubMed] [Google Scholar]
- 9.Martinis SA, Atkins WM, Stayton PS, Sligar SG. A conserved residue of cytochrome P-450 is involved in heme-oxygen stability and activation. J Am Chem Soc. 1989;111:9252–9253. [Google Scholar]
- 10.Imai M, Shimada H, Watanabe Y, Matsushima-Hibiya Y, Makino R, Koga H, Horiuchi T, Ishimura Y. Uncoupling of the cytochrome P-450cam monooxygenase reaction by a single mutation, threonine-252 to alanine or valine: possible role of the hydroxy amino acid in oxygen activation. Proc Natl Acad Sci USA. 1989;86:7823–7827. doi: 10.1073/pnas.86.20.7823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raag R, Martinis SA, Sligar SG, Poulos TL. Crystal structure of the cytochrome P-450CAM active site mutant Thr252Ala. Biochemistry. 1991;30:11420–11429. doi: 10.1021/bi00112a008. [DOI] [PubMed] [Google Scholar]
- 12.Shimizu T, Murakami Y, Hatano M. Glu318 and Thr319 mutations of cytochrome P450 1A2 remarkably enhance homolytic O-O cleavage of alkyl hydroperoxides. An optical absorption spectral study. J Biol Chem. 1994;269:13296–13304. [PubMed] [Google Scholar]
- 13.Blobaum AL, Kent UM, Alworth WL, Hollenberg PF. Novel reversible inactivation of cytochrome P450 2E1 T303A by tert-butyl acetylene: the role of threonine 303 in proton delivery to the active site of cytochrome P450 2E1. J Pharmacol Exp Ther. 2004;310:281–290. doi: 10.1124/jpet.104.065508. [DOI] [PubMed] [Google Scholar]
- 14.Clark JP, Miles CS, Mowat CG, Walkinshaw MD, Reid GA, Daff SN, Chapman SK. The role of Thr268 and Phe393 in cytochrome P450 BM3. J Inorg Biochem. 2006;100:1075–1090. doi: 10.1016/j.jinorgbio.2005.11.020. [DOI] [PubMed] [Google Scholar]
- 15.Keizers PH, Schraven LH, de Graaf C, Hidestrand M, Ingelman-Sundberg M, van Dijk BR, Vermeulen NP, Commandeur JN. Role of the conserved threonine 309 in mechanism of oxidation by cytochrome P450 2D6. Biochem Biophys Res Commun. 2005;338:1065–1074. doi: 10.1016/j.bbrc.2005.10.066. [DOI] [PubMed] [Google Scholar]
- 16.Kimata Y, Shimada H, Hirose T, Ishimura Y. Role of Thr-252 in cytochrome P450cam: a study with unnatural amino acid mutagenesis. Biochem Biophys Res Commun. 1995;208:96–102. doi: 10.1006/bbrc.1995.1310. [DOI] [PubMed] [Google Scholar]
- 17.Imai Y, Nakamura M. The importance of threonine-301 from cytochromes P-450 (laurate (omega-1)-hydroxylase and testosterone 16 alpha-hydroxylase) in substrate binding as demonstrated by site-directed mutagenesis. FEBS Lett. 1998;234:313–315. doi: 10.1016/0014-5793(88)80106-6. [DOI] [PubMed] [Google Scholar]
- 18.Hiroya K, Murakami Y, Shimizu T, Hatano M, Ortiz de Montellano PR. Differential roles of Glu318 and Thr319 in cytochrome P450 1A2 catalysis supported by NADPH-cytochrome P450 reductase and tert-butyl hydroperoxide. Arch Biochem Biophys. 1994;310:397–401. doi: 10.1006/abbi.1994.1184. [DOI] [PubMed] [Google Scholar]
- 19.Vatsis KP, Coon MJ. Ipso-substitution by cytochrome P450 with conversion of p-hydroxybenzene derivatives to hydroquinone: evidence for hydroperoxo-iron as the active oxygen species. Arch Biochem Biophys. 2002;397:119–129. doi: 10.1006/abbi.2001.2665. [DOI] [PubMed] [Google Scholar]
- 20.Moreno RL, Goosen T, Kent UM, Chung FL, Hollenberg PF. Differential effects of naturally occurring isothiocyanates on the activities of cytochrome P450 2E1 and the mutant P450 2E1 T303A. Arch Biochem Biophys. 2001;391:99–110. doi: 10.1006/abbi.2001.2390. [DOI] [PubMed] [Google Scholar]
- 21.Blobaum AL, Lu Y, Kent UM, Wang S, Hollenberg PF. Formation of a novel reversible cytochrome P450 spectral intermediate: role of threonine 303 in P450 2E1 inactivation. Biochemistry. 2004;43:11942–11952. doi: 10.1021/bi048882s. [DOI] [PubMed] [Google Scholar]
- 22.Nagano S, Poulos TL. Crystallographic study on the dioxygen complex of wild-type and mutant cytochrome P450cam. Implications for the dioxygen activation mechanism. J Biol Chem. 2005;280:31659–31663. doi: 10.1074/jbc.M505261200. [DOI] [PubMed] [Google Scholar]
- 23.Fukuda T, Imai Y, Komori M, Nakamura M, Kusunose E, Satouchi K, Kusunose M. Replacement of Thr-303 of P450 2E1 with serine modifies the regioselectivity of its fatty acid hydroxylase activity. J Biochem (Tokyo) 1993;113:7–12. doi: 10.1093/oxfordjournals.jbchem.a124006. [DOI] [PubMed] [Google Scholar]
- 24.Larson JR, Coon MJ, Porter TD. Alcohol-inducible cytochrome P-450IIE1 lacking the hydrophobic NH2-terminal segment retains catalytic activity and is membrane-bound when expressed in Escherichia coli. J Biol Chem. 1991;266:7321–7324. [PubMed] [Google Scholar]
- 25.Hanna IH, Teiber JF, Kokones KL, Hollenberg PF. Role of the alanine at position 363 of cytochrome P450 2B2 in influencing the NADPH- and hydroperoxide-supported activities. Arch Biochem Biophys. 1998;350:324–332. doi: 10.1006/abbi.1997.0534. [DOI] [PubMed] [Google Scholar]
- 26.Shen AL, Porter TD, Wilson TE, Kasper CB. Structural analysis of the FMN binding domain of NADPH-cytochrome P-450 oxidoreductase by site-directed mutagenesis. J Biol Chem. 1989;264:7584–7589. [PubMed] [Google Scholar]
- 27.Buters JT, Schiller CD, Chou RC. A highly sensitive tool for the assay of cytochrome P450 enzyme activity in rat, dog and man. Direct fluorescence monitoring of the deethylation of 7-ethoxy-4-trifluoromethylcoumarin. Biochem Pharmacol. 1993;46:1577–1584. doi: 10.1016/0006-2952(93)90326-r. [DOI] [PubMed] [Google Scholar]
- 28.Omura T, Sato R. The carbon monoxide-binding pigment of liver microsomes. I Evidence for its hemoprotein nature. J Biol Chem. 1964;239:2370–2378. [PubMed] [Google Scholar]
- 29.Vuletich JL, Osawa Y. Chemiluminescence assay for oxidatively modified myoglobin. Anal Biochem. 1998;265:375–380. doi: 10.1006/abio.1998.2926. [DOI] [PubMed] [Google Scholar]
- 30.Thurman RG, Ley HG, Scholz R. Hepatic microsomal ethanol oxidation. Hydrogen peroxide formation and the role of catalase. Eur J Biochem. 1972;25:420–430. doi: 10.1111/j.1432-1033.1972.tb01711.x. [DOI] [PubMed] [Google Scholar]
- 31.Guengerich FP, Strickland TW. Metabolism of vinyl chloride: destruction of the heme of highly purified liver Microsomal cytochrome P-450 by a metabolite. Mol Pharmacol. 1977;13:993–1004. [PubMed] [Google Scholar]
- 32.Schaefer WH, Harris TM, Guengerich FP. Characterization of the enzymatic and nonenzymatic peroxidative degradation of iron porphyrins and cytochrome P-450 heme. Biochemistry. 1985;24:3254–3263. doi: 10.1021/bi00334a027. [DOI] [PubMed] [Google Scholar]
- 33.Koop DR. Inhibition of ethanol-inducible cytochrome P450IIE1 by 3-amino-1,2,4-triazole. Chem Res Toxicol. 1990;3:377–383. doi: 10.1021/tx00016a017. [DOI] [PubMed] [Google Scholar]
- 34.Caro AA, Cederbaum AI. Oxidative stress, toxicology, and pharmacology of CYP2E1. Annu Rev Pharmacol Toxicol. 2004;44:27–42. doi: 10.1146/annurev.pharmtox.44.101802.121704. [DOI] [PubMed] [Google Scholar]
- 35.Jennings DB, Ehrenshaft M, Pharr DM, Williamson JD. Roles for mannitol and mannitol dehydrogenase in active oxygen-mediated plant defense. Proc Natl Acad Sci USA. 1998;95:15129–15133. doi: 10.1073/pnas.95.25.15129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Whiteman M, Halliwell B. Thiourea and dimethylthiourea inhibit peroxynitrite-dependent damage: nonspecificity as hydroxyl radical scavengers. Free Radic Biol Med. 1997;22:1309–1312. doi: 10.1016/s0891-5849(96)00545-x. [DOI] [PubMed] [Google Scholar]
- 37.Hayes JD, McLellan LI. Glutathione and glutathione-dependent enzymes represent a co-ordinately regulated defence against oxidative stress. Free Radic Res. 1999;31:273–300. doi: 10.1080/10715769900300851. [DOI] [PubMed] [Google Scholar]
- 38.Aruoma OI, Halliwell B, Hoey BM, Butler J. The antioxidant action of N-acetylcysteine: its reaction with hydrogen peroxide, hydroxyl radical, superoxide, and hypochlorous acid. Free Radic Biol Med. 1989;6:593–597. doi: 10.1016/0891-5849(89)90066-x. [DOI] [PubMed] [Google Scholar]
- 39.El-Chemaly S, Salathe M, Baier S, Conner GE, Forteza R. Hydrogen peroxide-scavenging properties of normal human airway secretions. Am J Respir Crit Care Med. 2003;167:425–430. doi: 10.1164/rccm.200206-531OC. [DOI] [PubMed] [Google Scholar]
- 40.Guelman LR, Pagotto RM, Di Toro CG, Zieher LM. Desferoxamine antioxidant activity on cerebellar granule cells gamma-irradiated in vitro. Neurotoxicol Teratol. 2004;26:477–483. doi: 10.1016/j.ntt.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 41.Karuzina II, Archakov AI. Hydrogen peroxide-mediated inactivation of microsomal cytochrome P450 during monooxygenase reactions. Free Radic Biol Med. 1994;17:557–567. doi: 10.1016/0891-5849(94)90095-7. [DOI] [PubMed] [Google Scholar]
- 42.Karuzina II, Zgoda VG, Kuznetsova GP, Samenkova NF, Archakov AI. Heme and apoprotein modification of cytochrome P450 2B4 during its oxidative inactivation in monooxygenase reconstituted system. Free Radic Biol Med. 1999;26:620–632. doi: 10.1016/s0891-5849(98)00252-4. [DOI] [PubMed] [Google Scholar]
- 43.Correia MA. Hepatic cytochrome P450 degradation: mechanistic diversity of the cellular sanitation brigade. Drug Metab Rev. 2003;35:107–143. doi: 10.1081/dmr-120023683. [DOI] [PubMed] [Google Scholar]
- 44.Zhukov A, Ingelman-Sundberg M. Relationship between cytochrome P450 catalytic cycling and stability: fast degradation of ethanol-inducible cytochrome P450 2E1 (CYP2E1) in hepatoma cells is abolished by inactivation of its electron donor NADPH-cytochrome P450 reductase. Biochem J. 1999;340:453–458. [PMC free article] [PubMed] [Google Scholar]
- 45.Hoch U, Ortiz de Montellano PR. Covalently linked heme in cytochrome p 4504a fatty acid hydroxylases. J Biol Chem. 2001;276:11339–11346. doi: 10.1074/jbc.M009969200. [DOI] [PubMed] [Google Scholar]
- 46.LeBrun LA, Hoch U, Ortiz de Montellano PR. Autocatalytic mechanism and consequences of covalent heme attachment in the cytochrome P4504A family. J Biol Chem. 2002;277:12755–12761. doi: 10.1074/jbc.M112155200. [DOI] [PubMed] [Google Scholar]
- 47.LeBrun LA, Xu F, Kroetz DL, Ortiz de Montellano PR. Covalent attachment of the heme prosthetic group in the CYP4F cytochrome P450 family. Biochemistry. 2002;41:5931–5937. doi: 10.1021/bi025527y. [DOI] [PubMed] [Google Scholar]
- 48.Limburg J, LeBrun LA, Ortiz de Montellano PR. The P450cam G248E mutant covalently binds its prosthetic heme group. Biochemistry. 2005;44:4091–4099. doi: 10.1021/bi047686i. [DOI] [PubMed] [Google Scholar]
- 49.Koop DR, Morgan ET, Tarr GE, Coon MJ. Purification and characterization of a unique isozyme of cytochrome P-450 from liver microsomes of ethanol-treated rabbits. J Biol Chem. 1982;257:8472–8480. [PubMed] [Google Scholar]
- 50.Coon MJ, Vaz ADN, Mcginnity DF, Peng HM. Multiple activated oxygen species in P450 catalysis: Contributions to specificity in drug metabolism. Drug Metab Dispos. 1998;26:1190–1193. [PubMed] [Google Scholar]