

Abstract
Despite recent major advances in leukemia research, the pathobiology of chronic lymphocytic leukemia (CLL) remains poorly understood. Herein we review the role chronic inflammation plays in the initiation and progression of CLL. The robust production of inflammatory cytokines and chemokines accompanied by activation of intra-cellular pro-inflammatory pathways, and the presence of somatic mutations that activate pro-inflammatory signaling pathways, suggest that chronic inflammation plays a pathophysiological role in this disease. Indeed, glucocorticoids and non-steroidal anti-inflammatory possess anti-tumor activity, and glucocorticoids have been broadly used to treat CLL and its complications. Recent clinical trials demonstrated that tyrosine kinase inhibitors, such as ibrutinib and the immune-modulatory agent lenalidomide, induced impressive clinical responses in CLL patients with relapsed or refractory disease. As those pro-inflammatory pathway inhibitors and immune modulating drugs proved to be effective in CLL, other agents with similar activities are currently investigated in clinical trials. New insights into the pathobiology of CLL and the development of novel classes of drugs will undoubtedly provide us with effective tools to treat and perhaps cure CLL.
2. Introduction
B-cell chronic lymphocytic leukemia (CLL), the most common leukemia in the Western hemisphere, is characterized by a dynamic imbalance between the proliferation and apoptosis of neoplastic B-lymphocytes co-expressing cluster of differentiation 5 (CD5) and CD19 antigens. Although approximately 20% of CLL patients are diagnosed as a result of routine blood tests [1], CLL patients may present with a wide range of symptoms typically witnessed in chronic inflammatory diseases. Fatigue, for example, might at times be so severe that it alone constitutes an indication for treatment[2], and disease progression is often associated with constitutional B symptoms such as low-grade fever, night sweats, and weight loss[2].
A paradoxical deregulation of the immune system that produces an exaggerated inflammatory response to minor insult or to self-antigens coupled with an inadequate response to infectious stimuli is typically found in patients with CLL. The breakdown of tolerance to self-antigens causes a variety of autoimmune phenomena such as autoimmune hemolytic anemia and/or thrombocytopenia (occurring in one third of CLL patients throughout the course of their disease[3]), and overt cutaneous inflammatory reactions. For example, more than 50 years ago, Weed et al.[4] described delayed hypersensitivity reactions to mosquito bites in patients with CLL and in 1998, Davis et al. described 8 patients with CLL who presented with papulovesicular lesions resembling arthropod bites whose skin biopsies showed T and B lymphocyte and prominent eosinophilic infiltrations with eosinophilic granular protein deposition[5].
Although those syndromes are caused by an amplified inflammatory reaction, the relatively high rate of infectious complications in CLL patients is the result of an inefficient immune response. Approximately 50% of patients with CLL die of infectious complications[6]. Although most serious complications are therapy related, inherent defects in mucosal, humeral, and cellular immune responses render CLL patients susceptible to infection[7]. Signs and symptoms of inflammation, detected at the onset of the disease, worsen with disease progression, as is elevated levels of C-reactive protein levels and erythrocyte sedimentation rate [8]. High beta-2 microglobulin (β2M) levels, usually detected in a broad spectrum of chronic inflammatory diseases, correlate with disease stage, tumor burden, and poor prognosis[9]. Whereas high levels of β2M are associated with an increased release of pro-inflammatory cytokines, including tumor necrosis factor α (TNF-α), interleukin 1 (IL-1), IL-6, and IL-8, β2M diminishes the ability of dendritic cells to enact a T-cell response[10]. Intracellular pro-inflammatory signaling pathways are activated in CLL cells, providing the cells with proliferative and survival advantages and inducing the production of inflammatory cytokines. Novel agents designed to block those pathways induce a dramatic reduction in disease burden and partial restoration of the humeral immune-response in patients with relapsed/refractory disease.
We review here the unique features of the inflammatory response in CLL patients and discuss the effects of established and novel anti-inflammatory agents used to treat this disease.
3. The inflammatory response in CLL
3.1 Soluble inflammatory signals
The role of cytokines and chemokines in the pathogenesis, maintenance, and progression of CLL has been the subject of intense research over the past two decades. In a recent comprehensive analysis of 23 cytokines in the sera of 84 patients with CLL and 49 age-matched healthy individuals, the levels of 17 cytokines, mostly pro-inflammatory cytokines, were significantly higher in the sera of the patients with CLL[11] (Fig 1). More than a 14-fold increase in INF-γ was found in the sera of untreated CLL patients[12]. Similarly, plasma levels of interleukin 6 (IL-6), IL-10[13], IL-8, and TNF-α[14] were also typically increased over levels in controls. Whether produced by CLL cells or other cells, these cytokines contribute, both directly and indirectly, to the survival of CLL cells.
Figure 1.
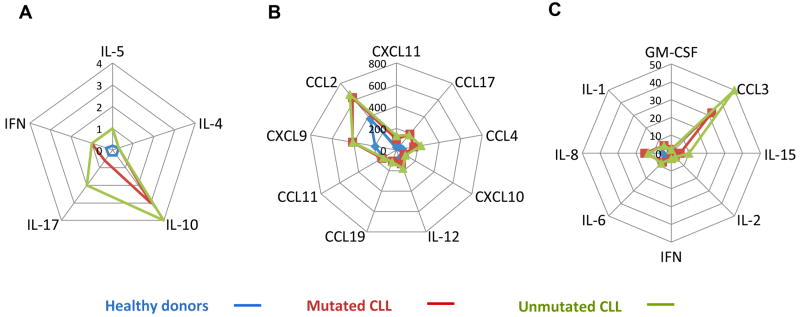
Serum cytokine and chemokine levels in patients with CLL and healthy individuals (Yan et al. (2011). Serum cytokine and chemokine levels were measured in 84 CLL patients and 49 age-matched healthy individuals. Shown are cytokine and chemokine levels that were high, compared to healthy controls, in patients with both IgHV-mutated and –unmutated CLL. Data are depicted according to cytokine levels in serum. A. Low levels: IL-5, IL-4, IL-10, IFN-γ, and IL-17. B. Medium levels: granulocyte macrophage colony-stimulating factor receptor (GM-CSF), IL-1β, IL-8, IL-6, Ifα, IL-2, IL-15, and CCL3. C. High levels: CXCL11, CCL17, CCL4, CXCL10, IL-12, CCL19, CCL11, CXCL9, and CCL2 [11].
In culture, CLL cells undergo spontaneous apoptosis[15]. However, co-culture with T lymphocytes, mesenchymal stromal cells, nurse-like cells (NLCs), or endothelial cells significantly reduces apoptosis rates of CLL cells[16, 17], suggesting that soluble factors and cell-to-cell interactions provide CLL cells with survival signals. Various cytokines whose levels are not increased in CLL also play a role in this process. For example, IL-4 activates the Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathway that protects CLL cells from chemotherapy-induced apoptosis[18]. Although IL-4 levels are not elevated in the seraof patients with CLL[11], IL-4 receptor levels are constitutively high in CLL cells[19]. Similarly, B-cell activating factor (BAFF), a member of the TNF superfamily, is thought to provide CLL cells with a survival advantage [20].
3.2 The cellular arm of chronic inflammation
The presence of CLL cells induces qualitative and quantitative alterations in T-cell populations. However, except in rare cases of spontaneous remission [21], the immune response fails to contain or eradicate the CLL clone. The immune response to CLL is determined by opposing forces: an avid immune reaction triggered by the presence of the malignant clone and impaired immune activity resulting from a defect in effector cell function and CLL cell features that allow evasion of an immune attack.
Large numbers of CD4, CD8, and NK cells are usually detected at the time of CLL diagnosis. The expansion of T cells and natural killer (NK) cells is associated with a prolonged time to first treatment [22], and the relative number of CD8 and CD4 cells is an independent predictor of survival [23], which suggests that these cells have anti-tumor activity. Remarkably, the CD4/CD8 ratio is inverted in nearly half of CLL patients because of an expansion of the cytotoxic T cells [23]. Functionally, these T cells are skewed toward a memory cell phenotype, and the inverted ratio is associated with shorter lymphocyte doubling time and worse prognosis [24]. In addition to quantitative changes, the function of T cells and NK cells may be impaired in patients with CLL. For example, CD4 cells from CLL patients showed increased susceptibility to FAS ligand (CD95)-induced apoptosis[25] and reduced expression of CD40 ligand and CD28[26, 27].
Other cellular components of the immune system also contribute directly to the survival of CLL cells by induction of inflammatory signals. Specifically, monocyte-derived NLCs cells attract, bind, and protect CLL cells from spontaneous and drug-induced apoptosis in a contact-dependent manner [28]. Co-culture with NLCs upregulates the activity of nuclear factor (NF)-κB and the production of pro-inflammatory chemokines, including chemokine ligand 3 (CCL3), CCL4, and CCL22 in CLL cells[29]. Thus, the cross-talk between CLL cells and NLCs activates inflammatory pathways and provides a survival advantage to CLL cells.
3.3 B-cell receptor (BCR) and intracellular pro-inflammatory signaling pathways
The trans-membranal BCR encompasses cell surface immunoglobulin (sIg) bound to a dimer of Ig-α (CD79a) and Ig-β (CD79b) chains [30, 31]. In a normal B cell, antigen binding induces the formation of a complex structure comprised of various kinases and scaffold proteins, termed signalome, tethered at sites of sIg activation[32]. Signalosome activation by kinases, such as Lyn kinase, results in activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and the nuclear factor of activated T cells (NF-AT)[33]. BCR’s two crucial function, signal transduction and antigen presentation to helper T cells, are initiated upon exposure to an antigen. In CLL, stimulation of the BCR induces expansion of the malignant clone [34, 35]. In about 20% of patients with mutated IgHV, an almost identical sequence—the complementarity determining region 3 (CDR3) in both heavy and light chains [36-38] has been identified as a limited set of antigens is recognized by the CLL cell sIg. BCR signaling in CLL is heterogeneous. CLL cells from some patients appear to be unresponsive to antigen engagement when IgM is used for BCR stimulation, whereas cells from other patients retain their signaling capacity [39]. Unlike normal B cells that undergo apoptosis unless they differentiate into plasma or memory cells, CLL cells are sustained by a constitutive tonic BCR activation that provides the substrate for activation of NF-κB and NF-κB-regulated genes [40], induction of pro-survival signals, and production of pro-inflammatory cytokines (Fig. 2).
Figure 2.
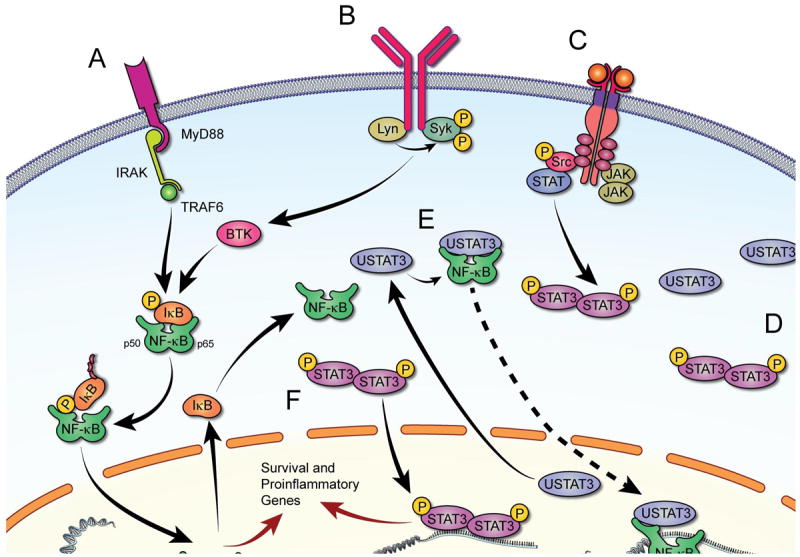
Signaling pathways activated in CLL cells. Upon attachment to their corresponding receptors (A, B and C) various extracellular factors induce signal transduction and activate transcription of inflammatory cytokines and chemokines. A. Ligands binding to TLR induce MYD88-mediated activation of NF-κB via phosphorylation and ubiquitination of IκB, enabling nuclear localization and DNA binding of NF-κB. B. BCR activation recruits BTK, which in turn phosphorylates IκB and activates NF-κB. C. Several ligands phosphorylate membrane-bound tyrosine kinase and non-tyrosine kinase receptors or recruit JAKs that transduce signaling by activating STAT3, ERK, and/or AKT (not shown). Phosphorylated (p) STAT3 forms heterodimers, shuttles to the nucleus, and activates transcription. D. In CLL, STAT3 is constitutively phosphorylated. Because STAT3 activates STAT3, high levels of unphosphorylated STAT3 (USTAT3) are present in CLL cells. E. USTAT3 binds NF-κB in competition with IκB; USTAT3 that shuttles freely in and out of the nucleus “carries” into the nucleus NF-κB that binds to DNA and activates transcription. F. Both pSTAT3 and NF-κB induce production of inflammatory cytokines that provide CLL cells with survival advantage.
In addition to activation of the BCR, cytokine receptors such as toll-like receptor (TLR) and CD40 bind their corresponding ligands and initiate signal transduction of pro-inflammatory pathways. Furthermore, similar to constitutively activated BCR, STAT3 is constitutively activated in CLL [41], providing CLL cells with a survival advantage, activating NF-κB [42] and enhancing the production of inflammatory chemokines and cytokines (Fig. 2).
3.4 Summary
Activation of chronic inflammatory processes, characteristically found in CLL, provide CLL cells with survival advantage and stimulate the neoplastic clone Constitutive activation of pro-inflammatory intracellular master regulators, such as NF-kB and STAT3, contribute to chronic inflammation by the induction of inflammatory cytokines such as IL-6 and IL-1, whose activities are enhanced by cross-talk between CLL cells and their microenvironment, resulting in quantitative and qualitative abnormalities in the effector arm of the immune response. Strategies to target one or more pro-inflammatory pathways are discussed in chapter 5.
4. CLL cell somatic mutations activating pro-inflammatory pathways
Two large-scale DNA deep-sequencing studies detected somatic mutations in CLL cells. In one study, deep sequencing of 105 CLL samples detected 1,246 mutations affecting 1,100 protein-coding genes with a mean of 20 mutations per person [43]. In another study, parallel whole genome sequencing of CLL cells and germ-line DNA from 91 CLL patients’ samples detected 1,838 nonsynonymous mutations in 1608 protein-coding genes and 45 mutations per person [44]. Surprisingly, only 186 recurrent and non-recurrent mutations were identified simultaneously in both data sets. Despite the limited overlap in mutation detection, the mutated genes were clustered in similar pathways in the two data sets, with an overwhelming representation of pro-inflammatory pathways (Table 1).
Table 1.
Pro-inflammatory pathways in which mutated genes were detected in two large whole-genome DNA sequencing studies
| Canonical Pathways [43] | P value | Canonical Pathways[44] | P value |
|---|---|---|---|
| NF-κB signaling | 0.01 | ATM signaling | 0.00006 |
| Toll like receptor signaling | 0.02 | LPS-stimulated MAPK signaling | 0.0007 |
| CD28 signaling in T helper cells | 0.04 | CC receptor 3 signaling in eosinophils | 0.0001 |
| Leukocyte extravasation signaling | 0.04 | Chemokine signaling | 0.001 |
| CXCR4 signaling | 0.05 | T-cell receptor signaling | 0.01 |
| Chemokine signaling | 0.06 | Toll-like receptor signaling | 0.02 |
| B-cell receptor signaling | 0.06 | B-cell receptor signaling | 0.02 |
| ATM signaling | 0.08 | IL-12 signaling and production in macrophages | 0.03 |
| IL-4 signaling | 0.09 | IL-3 signaling | 0.03 |
| IL-12 signaling and production in macrophages | 0.09 | IL-1 signaling | 0.04 |
| IL-15 signaling | 0.1 | CXCR4 signaling | 0.04 |
| Role of NFAT in the regulation of the immune response | 0.1 | MIF regulation of innate immunity | 0.05 |
| IL-8 signaling | 0.05 | ||
| Acute phase response signaling | 0.06 | ||
| NF-κB signaling | 0.06 | ||
| IL-17 signaling | 0.07 | ||
| PI3K signaling in B lymphocytes | 0.1 | ||
| B-cell activating factor signaling | 0.1 | ||
| Role of NFAT in the regulation of the immune response | 0.1 | ||
NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells, ATM, ataxia telangiectasia-mutated; CD, cluster of differentiation; CXCR4, chemokine receptor type 4; IL, interleukin; NFAT, nuclear factor of activated T-cells; MIF, migration inhibitory factor; NK, natural killer; Fcγ, Fc gamma; PI3K, phosphatidylinositide 3-kinases; FcγRIIB, Fcγ receptor IIB; Wnt, Int and Wg gene; MAPK, mitogen-activated protein kinase; BAFF, B-cell activating factor;
The functional analysis presented in this table was generated through the use of IPA (Ingenuity Systems (www.ingenuity.com). Fisher’s exact test was used to calculate a P-value determining the probability that the association between the mutated genes and the canonical pathway could be explained by chance. Pro-inflammatory pathways in which mutated genes were detected in the study of Quesada et al. [43] are depicted in columns 1 and 2, and in the study of Wang et al. [44], in columns 3 and 4. Both studies detected mutations in genes crucial for an inflammatory response, including cytokine signaling pathways (eg: IL-6), innate immunity activation pathways (eg: TLR signaling), chemokine signaling (eg: CXCR4) and pro-inflammatory transcription factors (eg: NF-κB signaling).
A few mutations detected in a significant number of patients significantly affect the activation of pro-inflammatory signaling pathways. The myeloid differentiation factor (MYD88) gene was found to be mutated in 3% to 10% of patients, most of whom had IgHV mutation [44, 45]. The most common MYD88 mutation detected in CLL is an activating mutation in p.L265P [45]. MYD88 encodes the protein MYD88 known to synchronize the assembly of a multi-subunit signaling complex consisting of various members of the interleukin-1 receptor-associated kinase (IRAK) serine-threonine kinase family known to activate NF-κB, STAT3, and induce the secretion of pro-inflammatory cytokines [46, 47]. The NOTCH1 gene was found to be mutated in 4% to 12% of patients with CLL [43, 44], most commonly in a selected group of patients with trisomy 12, unmutated immunoglobulin heavy chain variable (IgVH), and aggressive disease. In CLL patients with those features, 28 of 62 (41.9%) harbored a mutation in NOTCH1 [48]. Similar to the MYD88 activating mutation, NOTCH1 mutations activate components of the NF-κB pathway [49].
Unlike activating mutations in MYD88 and NOTCH1, mutations in the ataxia telangiectasia-mutated (ATM) gene are usually inactivating mutations. ATM is a serine-threonine kinase that activates the checkpoint response upon exposure to stressors that induce DNA damage [50]. Complex links tie ATM to inflammation and cancer; ATM is an immune modulator, and both activated and inactivated ATM promotes inflammation (Fig. 3). The ATM gene is located at the long arm of chromosome 11. Deletion of 11q, found in 2% to 10% of CLL patients, is associated with a lower remission rate [51, 52] and a patient’s prognosis worsens if the mutation occurs in both alleles [53]. The incidence of ATM mutation in patients lacking the 11q abnormality depends on the screening methods. Whole-genome sequencing studies identified ATM mutations in 4% to 10% of patients with CLL [43, 44]. However, gene dosage analysis performed by multiplex ligation probe amplification detected ATM alterations in about 25% of patients at diagnosis. Identifying ATM mutation is of clinical significance because patients harboring those mutations have a shorter disease-free survival [54].
Figure 3.
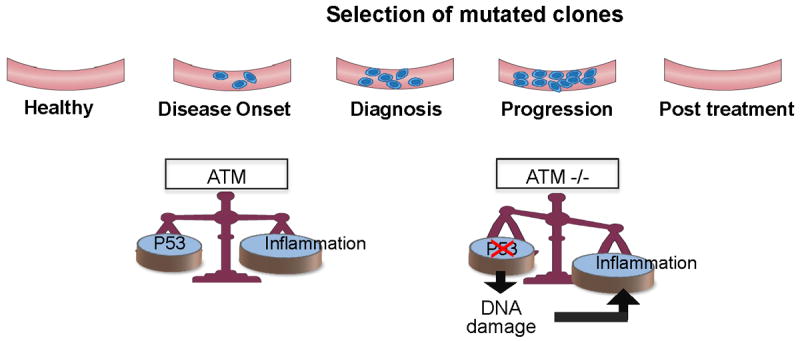
Chronic inflammation is induced in cells with wild-type or deleted ATM at different stages of the disease: At an early stage, wild-type ATM promotes TP53 DNA-damage repair and induction of chronic inflammation and production of inflammatory cytokines such as IL-6. When ATM is mutated, in the absence or presence of 11q deletion, less TP53 is produced and accumulating damaged DNA induces an inflammatory response. This effect is more prominent when the disease burden is high or during disease progression.
Whole genome sequencing studies did not detect a “common” CLL mutation. Rather, they identified pro-inflammatory pathway mutations, providing a rationale for the development of agents that target these pathways. In the next chapter we discuss the effects of well-established anti-inflammatory drugs and novel pro-inflammatory pathway signal transduction inhibitors that are currently investigated in clinical trials.
5. Agents
a. Anti-inflammatory agents
It has been well established that anti-inflammatory agents are active in CLL (Table 2), further supporting the hypothesis that inflammation is a driving force in this disease.
Table 2.
Selected agents that has anti-inflammatory properties used in CLL
| Agents | Primary mechanism of action | Potential benefits in CLL | Clinical efficacy – selected studies |
|---|---|---|---|
| Old agents | |||
| Glucocorticoids | Transactivation of anti-inflammatory genes and transrepression of pro-inflammatory genes | Lympholytic | Partial response in one third of patients. Mobilization from lymph nodes and spleen [55] High dose GC induced responses in 50% if patients with 17p deletion [58] |
| NSAID | Inhibition of (Cox-1) and Cox-2 | Inhibition of Cox-2 induces apoptosis of CLL cells | Concurrent use of aspirin and statins improved OS and PFS in patients with relapsed/refractory disease treated with chemo- immunotherapy [65] |
| Statins | HMG-CoA reductase inhibitors | Decrease synthesis of inflammatory cytokines, induces apoptosis of CLL cells | |
| Novel agents | |||
| Tyrosine Kinase Inhibitors | |||
| Dasatinib (Sprycel) | c-SRC and c-ABL kinase inhibitor | Targets also Lyn and BTK, impairs chemotaxis and adehesion of neutrophils | ORR of 20% in refractory/relapsed CLL [80] |
| Fostamatinib (R788) | Syk kinase inhibitor | Targets also ZAP70 | Limited data in patients with refractory/relapsed CLL [83] |
| GS1101 (CAL-101) | PI3Kδ inhibitor | Reduces secretion of inflammatory cytokines and chemokines | 37 patients with relapsed/refractory CLL enrolled in a Phase 1 study. > 50% reduction in lymphadenopathy in 60% of patients [106] |
| Ibrutinib (PCI-32765) | Irreversible BTK inhibitor | Inhibits BCR, TLR, BAFF, CD40 signaling pathways, reduces levels of CCL3 and CCL4 | ORR of 71% independent of genomic risk factors in 85 patients enrolled in a Phase 1b/2 study [86] |
| Immunomodulatory agents | |||
| Lenalidomide | Suppression of TNF-α levels | Immunomodulatory effects on proliferation of T cells, activation of NK cells and improvement in immunological synapse formation | ORR ranging from 32% to 65%. Elderly patients and patients with 17p benefit [97, 99-101] |
NSAID non-steroidal anti-inflammatory drugs; COX cyclooxygenase; BCR B-cell receptor; TLR toll-like receptor; B-cell activating factor; GC glucocorticoids; OS overall survival; PFS progression free survival; ORR Overall response rate.
i. Glucocorticoids (GCs)
When given as a single agent to CLL patients, GC treatment results in a partial response, with shrinkage of lymph nodes and reduction in splenic size in one-third of patients[55]. An increase in peripheral blood lymphocytes is often seen in the first weeks of treatment and is attributed to the redistribution of lymphocytes that shift from lymphoid organs and bone marrow into the peripheral blood. The anti-tumor effect is at least partially ascribed to the lympholytic effect of these drugs. Steroids induce apoptosis of lymphocytes by different mechanisms. Those include direct activation of transcription of death-specific genes and negative modulation of pro-inflammatory cytokines [56]. Steroid-induced apoptosis occurs without an inflammatory response because cells with intact membranes are removed by phagocytosis, without leaking their cytotoxic cellular contents [56].
High-dose corticosteroids are still used in patients with refractory or relapsed disease, with some patients achieving good, occasionally complete, responses [57]. Because steroids are thought to induce apoptosis in lymphoid cells independent of the p53 pathway, this treatment might be useful in patients with 17p deletion or with p53 mutations. Indeed, Thornton et al. (2003) reported a 50% response rate in patients with p53 abnormalities treated with high-dose methyl prednisolone in a small single-institution retrospective study[58]. Nowadays, the major indication for steroid is autoimmune hemolytic anemia or immune thrombocytopenia [7, 58]. Major side effects include susceptibility to infections and metabolic, psychiatric, dermatologic, musculoskeletal, and gastrointestinal toxicities, which limit its widespread clinical use.
ii. Non-steroidal anti-inflammatory drugs (NSAIDs)
Aspirin is known to decrease CLL cell viability in a dose- and time-dependent manner and to induce apoptosis of CLL cells [59]. More than decades of intense research on the anti-inflammatory effects of NSAID revealed many pathways that are directly or indirectly targeted by this diverse group of agents. However, cyclooxygenases (Cox) 1 and 2 are the principal enzymes targeted by most NSAIDs. Inhibition of Cox reduces prostaglandin production and ameliorates inflammation. Cox-2 is constitutively expressed in CLL cells, and higher Cox-2 levels were detected in patients with poor prognostic factors [60]. Inhibition of Cox-2 induces apoptosis of CLL cells via activation of caspase cascade [59]. Cox-2 inhibitors suppress the anti-oxidant glutathione-inducing oxidative stress and cellular apoptosis [61].
A recent report of 686 newly diagnosed early-stage CLL patients, treated at the Mayo Clinic between 1995 and 2008, did not benefit from NSAID use, as time to first therapy was similar in users and non-users[62]. However, NSAID might have a role in potentiating the effect of chemotherapy in CLL patients. Several in-vitro studies demonstrated that NSAIDs enhance the effects of purine nucleoside analog- and rituximab-based treatments in CLL[63, 64], and a retrospective analysis of 280 patients with relapsed/refractory CLL who received salvage chemoimmunotherapy with fludarabine, cyclophosphamide, and rituximab demonstrated an improved OS and PFS with concurrent use of aspirin [65].
b. Statins
Statins, the broadly used cholesterol-lowering agents, also possess anti-angiogenic [66, 67] and anti-inflammatory properties [68]. Statins decrease the synthesis of inflammatory cytokines and adhesion molecules [69] and reduce the levels of endothelial nitric oxide synthase, thereby reducing oxidative stress and vascular inflammation [69-72]. In vitro studies demonstrated that simvastatin induces apoptosis of CLL cells by activating caspase 9 [73]. Statins can also prevent the homing of lymphocytes to lymph nodes [74, 75], possibly providing additional benefits when combined with chemotherapy. Statins also reduce the cell surface CD20 expression, an effect that might reduce the efficacy of rituximab [76]. Two retrospective studies evaluated the effect of statins on the clinical course of patients with CLL. In both cohorts, statin use did not affect treatment-free survival [62, 77]. Friedman et al. found that the use of statins at the time of diagnosis of CLL is associated with a reduced need for initial therapy [77]. Prospective studies to evaluate the clinical benefits of statins on CLL are warranted.
c. Novel agents
i. Kinase inhibitors
The success of tyrosine kinase inhibitors in the treatment of chronic myelogenous leukemia has inspired many of the current ongoing clinical trials for CLL. These trials use different types of orally administrated kinase inhibitors that prevent phosphorylation of key molecules along signaling pathways that promote the survival and proliferation of the CLL cell.
Tyrosine kinase inhibitors have demonstrated impressive responses in patients with relapsed and refractory CLL. Because of the pivotal role of BCR signaling in CLL, these agents are sometimes collectively referred to as BCR inhibitors, however this name is misleading, and these molecules inhibit converging intra-cellular pathways that deliver signals not only from the BCR, but from various other sources, including TLRs, cytokines, chemokines, and integrins CD40 and BAFF[78]. The final common pathways of all kinase inhibitors are inhibition of key transcription factors, most importantly NF-κB, but also nuclear factor of activated T-cells (NFAT) and STAT3, resulting in the inhibition of pro-survival and pro-inflammatory pathways. Currently, four major kinase inhibitors are actively tested in phase 2 or phase 3 clinical trials. All are associated with strong inhibition of inflammatory pathways.
Dasatinib (Sprycel) is an oral kinase inhibitor targeting SRC and ABL kinases and is approved for treatment in chronic myeloycytic leukemia. The primary target in CLL is considered the non-receptor tyrosine kinase LYN, which initiates the BCR signaling. However, it has been shown that its effect is not specific for LYN, but that BTK and perhaps other kinases are also inhibited [79]. A phase 2 study of patients with refractory/relapsed disease reported overall response rate of 20%, with progression-free survival of 7.5 months and a reduction of > 50% in lymphadenopathy in 7 of 15 patients enrolled[80]. When tested in vitro on human and mouse neutrophils, Dasatinib showed a strong anti-inflammatory effect. Dasatinib completely blocked integrin and Fc receptor-mediated neutrophil functions and significantly impaired chemotaxis and adhesion functions of neutrophils when tested in concentrations that correspond to serum levels in treated patients[81]. Taken together, these studies suggest that the clinical efficacy of Dasatinib is at least partially mediated by its inhibition of pro-inflammatory pathways within the CLL cell and anti-inflammatory signals from the microenvironment.
Fostamatinib (R788) inhibits the spleen tyrosine kinase (SYK) and a number of other kinases, including ZAP70, which is aberrantly expressed in some cases of CLL and correlates with more aggressive disease [78]. SYK inhibitors were initially developed as anti-inflammatory drugs[82], but have shown a promising effect on phase 1/2 studies in patients with refractory/relapsed non-Hodgkin’s lymphoma. Of 11 patients with CLL who participated in this study, 6 (55%) achieved partial response [83].
GS-1101 (CAL-101) is a highly selective inhibitor of the PI3Kδ kinase. This is one of three isoforms of the P13K complex that is responsible for phosphorylation of phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2 [PIP2]) to generate PI(3,4,5)P3 (PIP3). PIP3, in turn, is the docking site of several cytoplasmatic kinases that integrate signals from several pathways, including the BCR, TLR pathways, CD40, and BAFF[78]. In a phase 1 study that included 54 patients with CLL, 80% had a reduction in lymphadenopathy by ≥ 50% and PFS was not reached at 11 months. The clinical effect was independent of traditional prognostic factors, most notably IGVH mutation status [78]. Because of a transient increase in the absolute lymphocyte counts, it was difficult to categorize these patients as responders according to the known International Workshop on Chronic Lymphocytic Leukemia criteria. Subsequently, these criteria have been modified and a Lymphoma Response Foundation-sponsored workshop has suggested adding the term “nodal response” to describe this phenomena (currently only for patients in clinical trials)[84]. Similar to other kinase inhibitors, along the BCR signaling pathways, GS-1101 showed strong inhibition of inflammatory response with markedly reduced secretion of pro-inflammatory cytokines (such as IL-6 and TNF) and chemokines (such as CCL3 and CCL4) mediated by BCR stimulation or NLCs [85].
Ibrutinib (PCI-37265) is an irreversible inhibitor of the BTK kinase. Similar to other kinase inhibitors tested in patients with CLL, it inhibits several signaling pathways, including BCR, TLR, and BAFF CD40, and also disrupts the protective effect of stromal cells[78]. In two phase 1b/2 clinical studies Ibrutinib as monotherapy was tested on patients with relapsed/refractory disease including elderly patients, and induced durable remissions across all groups tested, including elderly patients and patients with high-risk disease. Interestingly, similar to GS-1101, transient lymphocytosis was associated with better response [86, 87]. In addition, partial restoration of the humoral immune deficiency that is found with advanced disease was also reported with sustained increase in levels of IgA [88]. Treatment with Ibrutinib resulted in a rapid decrease in serum concentration of the inflammatory chemokines CCL3 and CCL4 [89]. Furthermore, Ibrutinib was shown to block the secretion of cytokines from activated T cells, while not affecting their survival [78].
ii. Immunomodulatory drugs (IMiDs)
IMiDs include thalidomide and its derivatives lenalidomide (CC-5013) and pomalidomide (CC-4047). These oral medications are synthetic glutamic acid derivatives that are either approved for clinical use or for evaluation in clinical trials in hematologic and non-hematologic malignancies. During the late 1990s and early 2000s, several clinical trials established the efficacy of thalidomide in the treatment of multiple myeloma [90, 91]. Lenalidomide is a 4-amino tultaramide derivative of thalidomide with enhanced immune-modulatory potency, reduced neurosedative toxicity, and a reduced risk of venous thromboembolism [92]. Leprosy was the first indication for thalidomide in the modern era, and its efficacy in treatment patient with leprosy is attributed to its strong anti-inflammatory effect, mainly through the suppression of TNF-α levels [93, 94]. The anti-leukemic effect of this drug is more complex, and in addition to inhibition of cytokine release it may also enhance the activity of the cellular arm of the immune response. It has been shown that lenalidomide stimulates T-cell proliferation and activates NK cells [95]. Furthermore, CLL recognition by T cells is inherently impaired. This is attributed to impaired immunological synapse formation that is partially corrected with lenalidomide [96]. Several phase 2 clinical trials for CLL demonstrated the efficacy of lenalidomide alone or in combination anti-CD20 antibodies, with an overall response rate ranging from 32% to 65%[97-100]. Patients with unfavorable cytogenetic abnormalities, particularly elderly patients, also seem to benefit [100]. Tumor flare reaction is an adverse effect of lenalidomide that is observed uniquely in patients with CLL. It is characterized by a painful swelling of the lymph nodes and/or splenomegaly, accompanied by rash, low-grade fever, and rarely, a rise in the peripheral blood white cell counts [101]. NSAID and low-dose steroid therapy alleviates the symptoms but not the frequency of tumor flare reaction. Whether the appearance of tumor flare is associated with long-term clinical response is not yet clear [101].
6. Conclusions and future perspectives
The introductions of effective chemotherapy agents combined with anti-CD20 antibodies revolutionized the treatment of CLL. A number of patients attain long-term remission and a few patients might have been cured. Nevertheless, for most patients, CLL is still an incurable disease. As in other chronic diseases whose incidences increase with age[102], chronic inflammation contributes to the pathobiology and symptomatology of CLL. Inflammatory cytokine are a result of and contribute to the activation of pro-inflammatory pathways within the malignant clone. Recent whole genome sequencing of CLL cells identified mutations in genes that activate pro-inflammatory signaling pathways, suggesting that those pathways are potential targets for therapeutic intervention. The interaction of CLL cells with cellular and extracellular components of the microenvironment cells of the immune system, such as TH Cells, NK cells, and NLC, contributes to CLL cells’ pro-inflammatory microenvironment.
Recognizing the role of chronic inflammation in CLL expands the arsenal of effective therapeutic modalities. More than 40 years ago, Ezdini et al. reported that some patients with CLL respond to steroid treatment and that this response is accompanied by transient lymphocytosis and shrinkage of lymph nodes[55]. Remarkably, an identical clinical response has been observed in patients treated with novel kinase inhibitors. Clinical response to kinase inhibitors and IMIDs is accompanied by a dramatic reduction in the levels of inflammatory cytokines and chemokines, suggesting other agents targeting pro-inflammatory pathways such as JAK inhibitors[103, 104], recently reported to induce apoptosis of CLL cells[105], might prove to be effective therapeutic agents for CLL.
Recognition of the central role inflammation plays in the pathobiology of CLL raised the hope that inhibition of inflammatory pathways may pave the road for curing CLL in the near future. Understanding the details of the inflammatory response has already resulted in the development of several agents that are currently under clinical investigations, and has expanded the range of other potential candidate drugs to be considered in future studies.
Acknowledgments
We thank Luanne Jorewicz for editing our manuscript.
This study is supported by a grant from the CLL Global Research Foundation and the National Institutes of Health through MD Anderson’s Cancer Center Support Grant CA016672.
Biography
Dr. Zeev Estrov
Zeev Estrov, M.D., is a Professor of Medicine and Director of Hematopoiesis Research at the Department of Leukemia, The University of Texas MD Anderson Cancer Center. Dr. Estrov received his M.D. degree (with honors) from the Tel-Aviv University School of Medicine in 1973. In 1989 Dr. Estrov joined the MD Anderson Cancer Center where he completed training in Medical Oncology, and appointed clinical faculty member and attending physician at the Department of Leukemia. Dr. Estrov’s research has been focused on the biology of normal and leukemic hematopoietic stem and progenitor cells. His laboratory’s long range objective is to identify the mechanisms that provide leukemia cells with survival advantage and growth advantage and to develop means for inhibiting them. In recent years Dr. Estrov’s laboratory concentrated on the role of signaling pathways and their inducible transcription factors on the growth, survival, plasticity, and apoptosis of normal and leukemia cells. Dr. Estrov’s team is currently investigating novel agents capable of modulating those pathways with the aim of developing new strategies for the treatment of chronic lymphocytic leukemia, myeloproliferative disorders and acute and chronic myeloid leukemias.
Footnotes
Conflict of Interest Disclosure: The authors have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rai KR, K MJ. In: Staging and prognosis of chronic lymphcoytie leukemia. R BD, editor. Waltham, MA: Uptodate. 20052012. [Google Scholar]
- 2.Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Dohner H, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008;111:5446–56. doi: 10.1182/blood-2007-06-093906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Duhrsen U, Augener W, Zwingers T, Brittinger G. Spectrum and frequency of autoimmune derangements in lymphoproliferative disorders: analysis of 637 cases and comparison with myeloproliferative diseases. Br J Haematol. 1987;67:235–9. doi: 10.1111/j.1365-2141.1987.tb02333.x. [DOI] [PubMed] [Google Scholar]
- 4.Weed RI. Exaggerated Delayed Hypersensitivity to Mosquito Bites in Chronic Lymphocytic Leukemia. Blood. 1965;26:257–68. [PubMed] [Google Scholar]
- 5.Davis MD, Perniciaro C, Dahl PR, Randle HW, McEvoy MT, Leiferman KM. Exaggerated arthropod-bite lesions in patients with chronic lymphocytic leukemia: a clinical, histopathologic, and immunopathologic study of eight patients. J Am Acad Dermatol. 1998;39:27–35. doi: 10.1016/s0190-9622(98)70398-6. [DOI] [PubMed] [Google Scholar]
- 6.Tsiodras S, Samonis G, Keating MJ, Kontoyiannis DP. Infection and immunity in chronic lymphocytic leukemia. Mayo Clinic proceedings Mayo Clinic. 2000;75:1039–54. doi: 10.4065/75.10.1039. [DOI] [PubMed] [Google Scholar]
- 7.Dearden C. Disease-specific complications of chronic lymphocytic leukemia. Hematology Am Soc Hematol Educ Program. 2008:450–6. doi: 10.1182/asheducation-2008.1.450. [DOI] [PubMed] [Google Scholar]
- 8.Pavlidis AN, Kalef-Ezra J, Bourantas LC, Lambrou A, Mavridis A. Serum tumor markers in non-Hodgkin’s lymphomas and chronic lymphocytic leukemia. Int J Biol Markers. 1993;8:14–20. doi: 10.1177/172460089300800103. [DOI] [PubMed] [Google Scholar]
- 9.Wierda WG, O’Brien S, Wang X, Faderl S, Ferrajoli A, Do KA, et al. Characteristics associated with important clinical end points in patients with chronic lymphocytic leukemia at initial treatment. J Clin Oncol. 2009;27:1637–43. doi: 10.1200/JCO.2008.18.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xie J, Yi Q. Beta2-microglobulin as a potential initiator of inflammatory responses. Trends Immunol. 2003;24:228–9. doi: 10.1016/s1471-4906(03)00076-0. [DOI] [PubMed] [Google Scholar]
- 11.Yan XJ, Dozmorov I, Li W, Yancopoulos S, Sison C, Centola M, et al. Identification of outcome-correlated cytokine clusters in chronic lymphocytic leukemia. Blood. 2011;118:5201–10. doi: 10.1182/blood-2011-03-342436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mahadevan D, Choi J, Cooke L, Simons B, Riley C, Klinkhammer T, et al. Gene Expression and Serum Cytokine Profiling of Low Stage CLL Identify WNT/PCP, Flt-3L/Flt-3 and CXCL9/CXCR3 as Regulators of Cell Proliferation, Survival and Migration. Hum Genomics Proteomics. 2009;2009:453634. doi: 10.4061/2009/453634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fayad L, Keating MJ, Reuben JM, O’Brien S, Lee BN, Lerner S, et al. Interleukin-6 and interleukin-10 levels in chronic lymphocytic leukemia: correlation with phenotypic characteristics and outcome. Blood. 2001;97:256–63. doi: 10.1182/blood.v97.1.256. [DOI] [PubMed] [Google Scholar]
- 14.Yoon JY, Lafarge S, Dawe D, Lakhi S, Kumar R, Morales C, et al. Association of Interleukin-6 and Interleukin-8 with Poor Prognosis in Elderly Chronic Lymphocytic Leukemia Patients. Leuk Lymphoma. 2012 doi: 10.3109/10428194.2012.666662. [DOI] [PubMed] [Google Scholar]
- 15.Collins RJ, Verschuer LA, Harmon BV, Prentice RL, Pope JH, Kerr JF. Spontaneous programmed death (apoptosis) of B-chronic lymphocytic leukaemia cells following their culture in vitro. Br J Haematol. 1989;71:343–50. doi: 10.1111/j.1365-2141.1989.tb04290.x. [DOI] [PubMed] [Google Scholar]
- 16.Badoux X, Bueso-Ramos C, Harris D, Li P, Liu Z, Burger J, et al. Cross-talk between chronic lymphocytic leukemia cells and bone marrow endothelial cells: role of signal transducer and activator of transcription 3. Hum Pathol. 2011;42:1989–2000. doi: 10.1016/j.humpath.2011.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burger JA. Nurture versus nature: the microenvironment in chronic lymphocytic leukemia. Hematology Am Soc Hematol Educ Program. 2011;2011:96–103. doi: 10.1182/asheducation-2011.1.96. [DOI] [PubMed] [Google Scholar]
- 18.Dietrich S, Kramer OH, Hahn E, Schafer C, Giese T, Hess M, et al. Leflunomide induces apoptosis in fludarabine-resistant and clinically refractory CLL cells. Clin Cancer Res. 2012;18:417–31. doi: 10.1158/1078-0432.CCR-11-1049. [DOI] [PubMed] [Google Scholar]
- 19.Douglas RS, Capocasale RJ, Lamb RJ, Nowell PC, Moore JS. Chronic lymphocytic leukemia B cells are resistant to the apoptotic effects of transforming growth factor-beta. Blood. 1997;89:941–7. [PubMed] [Google Scholar]
- 20.Kern C, Cornuel JF, Billard C, Tang R, Rouillard D, Stenou V, et al. Involvement of BAFF and APRIL in the resistance to apoptosis of B-CLL through an autocrine pathway. Blood. 2004;103:679–88. doi: 10.1182/blood-2003-02-0540. [DOI] [PubMed] [Google Scholar]
- 21.Del Giudice I, Chiaretti S, Tavolaro S, De Propris MS, Maggio R, Mancini F, et al. Spontaneous regression of chronic lymphocytic leukemia: clinical and biologic features of 9 cases. Blood. 2009;114:638–46. doi: 10.1182/blood-2008-12-196568. [DOI] [PubMed] [Google Scholar]
- 22.Palmer S, Hanson CA, Zent CS, Porrata LF, Laplant B, Geyer SM, et al. Prognostic importance of T and NK-cells in a consecutive series of newly diagnosed patients with chronic lymphocytic leukaemia. Br J Haematol. 2008;141:607–14. doi: 10.1111/j.1365-2141.2008.07070.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gonzalez-Rodriguez AP, Contesti J, Huergo-Zapico L, Lopez-Soto A, Fernandez-Guizan A, Acebes-Huerta A, et al. Prognostic significance of CD8 and CD4 T cells in chronic lymphocytic leukemia. Leuk Lymphoma. 2010;51:1829–36. doi: 10.3109/10428194.2010.503820. [DOI] [PubMed] [Google Scholar]
- 24.Nunes C, Wong R, Mason M, Fegan C, Man S, Pepper C. Expansion of a CD8(+)PD-1(+) replicative senescence phenotype in early stage CLL patients is associated with inverted CD4:CD8 ratios and disease progression. Clin Cancer Res. 2012;18:678–87. doi: 10.1158/1078-0432.CCR-11-2630. [DOI] [PubMed] [Google Scholar]
- 25.Tinhofer I, Marschitz I, Kos M, Henn T, Egle A, Villunger A, et al. Differential sensitivity of CD4+ and CD8+ T lymphocytes to the killing efficacy of Fas (Apo-1/CD95) ligand+ tumor cells in B chronic lymphocytic leukemia. Blood. 1998;91:4273–81. [PubMed] [Google Scholar]
- 26.Cantwell M, Hua T, Pappas J, Kipps TJ. Acquired CD40-ligand deficiency in chronic lymphocytic leukemia. Nat Med. 1997;3:984–9. doi: 10.1038/nm0997-984. [DOI] [PubMed] [Google Scholar]
- 27.Rossi E, Matutes E, Morilla R, Owusu-Ankomah K, Heffernan AM, Catovsky D. Zeta chain and CD28 are poorly expressed on T lymphocytes from chronic lymphocytic leukemia. Leukemia. 1996;10:494–7. [PubMed] [Google Scholar]
- 28.Burger JA, Tsukada N, Burger M, Zvaifler NJ, Dell’Aquila M, Kipps TJ. Blood-derived nurse-like cells protect chronic lymphocytic leukemia B cells from spontaneous apoptosis through stromal cell-derived factor-1. Blood. 2000;96:2655–63. [PubMed] [Google Scholar]
- 29.Burger JA, Quiroga MP, Hartmann E, Burkle A, Wierda WG, Keating MJ, et al. High-level expression of the T-cell chemokines CCL3 and CCL4 by chronic lymphocytic leukemia B cells in nurselike cell cocultures and after BCR stimulation. Blood. 2009;113:3050–8. doi: 10.1182/blood-2008-07-170415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dal Porto JM, Gauld SB, Merrell KT, Mills D, Pugh-Bernard AE, Cambier J. B cell antigen receptor signaling 101. Mol Immunol. 2004;41:599–613. doi: 10.1016/j.molimm.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 31.Niiro H, Clark EA. Regulation of B-cell fate by antigen-receptor signals. Nat Rev Immunol. 2002;2:945–56. doi: 10.1038/nri955. [DOI] [PubMed] [Google Scholar]
- 32.Pierce SK, Liu W. The tipping points in the initiation of B cell signalling: how small changes make big differences. Nat Rev Immunol. 2010;10:767–77. doi: 10.1038/nri2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gauld SB, Dal Porto JM, Cambier JC. B cell antigen receptor signaling: roles in cell development and disease. Science. 2002;296:1641–2. doi: 10.1126/science.1071546. [DOI] [PubMed] [Google Scholar]
- 34.Chiorazzi N, Ferrarini M. B cell chronic lymphocytic leukemia: lessons learned from studies of the B cell antigen receptor. Annu Rev Immunol. 2003;21:841–94. doi: 10.1146/annurev.immunol.21.120601.141018. [DOI] [PubMed] [Google Scholar]
- 35.Stevenson FK, Caligaris-Cappio F. Chronic lymphocytic leukemia: revelations from the B-cell receptor. Blood. 2004;103:4389–95. doi: 10.1182/blood-2003-12-4312. [DOI] [PubMed] [Google Scholar]
- 36.Ghiotto F, Fais F, Valetto A, Albesiano E, Hashimoto S, Dono M, et al. Remarkably similar antigen receptors among a subset of patients with chronic lymphocytic leukemia. J Clin Invest. 2004;113:1008–16. doi: 10.1172/JCI19399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stamatopoulos K, Belessi C, Moreno C, Boudjograh M, Guida G, Smilevska T, et al. Over 20% of patients with chronic lymphocytic leukemia carry stereotyped receptors: Pathogenetic implications and clinical correlations. Blood. 2007;109:259–70. doi: 10.1182/blood-2006-03-012948. [DOI] [PubMed] [Google Scholar]
- 38.Widhopf GF, 2nd, Rassenti LZ, Toy TL, Gribben JG, Wierda WG, Kipps TJ. Chronic lymphocytic leukemia B cells of more than 1% of patients express virtually identical immunoglobulins. Blood. 2004;104:2499–504. doi: 10.1182/blood-2004-03-0818. [DOI] [PubMed] [Google Scholar]
- 39.Stevenson FK, Krysov S, Davies AJ, Steele AJ, Packham G. B-cell receptor signaling in chronic lymphocytic leukemia. Blood. 2011;118:4313–20. doi: 10.1182/blood-2011-06-338855. [DOI] [PubMed] [Google Scholar]
- 40.Stadanlick JE, Kaileh M, Karnell FG, Scholz JL, Miller JP, Quinn WJ, 3rd, et al. Tonic B cell antigen receptor signals supply an NF-kappaB substrate for prosurvival BLyS signaling. Nat Immunol. 2008;9:1379–87. doi: 10.1038/ni.1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frank DA, Mahajan S, Ritz J. B lymphocytes from patients with chronic lymphocytic leukemia contain signal transducer and activator of transcription (STAT) 1 and STAT3 constitutively phosphorylated on serine residues. J Clin Invest. 1997;100:3140–8. doi: 10.1172/JCI119869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu Z, Hazan-Halevy I, Harris DM, Li P, Ferrajoli A, Faderl S, et al. STAT-3 activates NF-kappaB in chronic lymphocytic leukemia cells. Mol Cancer Res. 2011;9:507–15. doi: 10.1158/1541-7786.MCR-10-0559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quesada V, Conde L, Villamor N, Ordonez GR, Jares P, Bassaganyas L, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet. 2012;44:47–52. doi: 10.1038/ng.1032. [DOI] [PubMed] [Google Scholar]
- 44.Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med. 2011;365:2497–506. doi: 10.1056/NEJMoa1109016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Puente XS, Pinyol M, Quesada V, Conde L, Ordonez GR, Villamor N, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475:101–5. doi: 10.1038/nature10113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ishii KJ, Akira S. Innate immune recognition of, and regulation by, DNA. Trends Immunol. 2006;27:525–32. doi: 10.1016/j.it.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 47.Iwasaki A, Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science. 2010;327:291–5. doi: 10.1126/science.1183021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Balatti V, Bottoni A, Palamarchuk A, Alder H, Rassenti LZ, Kipps TJ, et al. NOTCH1 mutations in CLL associated with trisomy 12. Blood. 2012;119:329–31. doi: 10.1182/blood-2011-10-386144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Screpanti I, Bellavia D, Campese AF, Frati L, Gulino A. Notch, a unifying target in T-cell acute lymphoblastic leukemia? Trends Mol Med. 2003;9:30–5. doi: 10.1016/s1471-4914(02)00003-5. [DOI] [PubMed] [Google Scholar]
- 50.Khanna KK, Jackson SP. DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet. 2001;27:247–54. doi: 10.1038/85798. [DOI] [PubMed] [Google Scholar]
- 51.Dohner H, Stilgenbauer S, Benner A, Leupolt E, Krober A, Bullinger L, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343:1910–6. doi: 10.1056/NEJM200012283432602. [DOI] [PubMed] [Google Scholar]
- 52.Fegan C, Robinson H, Thompson P, Whittaker JA, White D. Karyotypic evolution in CLL: identification of a new sub-group of patients with deletions of 11q and advanced or progressive disease. Leukemia. 1995;9:2003–8. [PubMed] [Google Scholar]
- 53.Austen B, Skowronska A, Baker C, Powell JE, Gardiner A, Oscier D, et al. Mutation status of the residual ATM allele is an important determinant of the cellular response to chemotherapy and survival in patients with chronic lymphocytic leukemia containing an 11q deletion. J Clin Oncol. 2007;25:5448–57. doi: 10.1200/JCO.2007.11.2649. [DOI] [PubMed] [Google Scholar]
- 54.Guarini A, Marinelli M, Tavolaro S, Bellacchio E, Magliozzi M, Chiaretti S, et al. ATM gene alterations in chronic lymphocytic leukemia patients induce a distinct gene expression profile and predict disease progression. Haematologica. 2012;97:47–55. doi: 10.3324/haematol.2011.049270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ezdinli EZ, Stutzman L, Aungst CW, Firat D. Corticosteroid therapy for lymphomas and chronic lymphocytic leukemia. Cancer. 1969;23:900–9. doi: 10.1002/1097-0142(196904)23:4<900::aid-cncr2820230427>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 56.Greenstein S, Ghias K, Krett NL, Rosen ST. Mechanisms of glucocorticoid-mediated apoptosis in hematological malignancies. Clin Cancer Res. 2002;8:1681–94. [PubMed] [Google Scholar]
- 57.Molica S. High-dose dexamethasone in refractory B-cell chronic lymphocytic leukemia patients. Am J Hematol. 1994;47:334. doi: 10.1002/ajh.2830470421. [DOI] [PubMed] [Google Scholar]
- 58.Thornton PD, Matutes E, Bosanquet AG, Lakhani AK, Grech H, Ropner JE, et al. High dose methylprednisolone can induce remissions in CLL patients with p53 abnormalities. Annals of hematology. 2003;82:759–65. doi: 10.1007/s00277-003-0710-5. [DOI] [PubMed] [Google Scholar]
- 59.Bellosillo B, Pique M, Barragan M, Castano E, Villamor N, Colomer D, et al. Aspirin and salicylate induce apoptosis and activation of caspases in B-cell chronic lymphocytic leukemia cells. Blood. 1998;92:1406–14. [PubMed] [Google Scholar]
- 60.Ryan EP, Pollock SJ, Kaur K, Felgar RE, Bernstein SH, Chiorazzi N, et al. Constitutive and activation-inducible cyclooxygenase-2 expression enhances survival of chronic lymphocytic leukemia B cells. Clin Immunol. 2006;120:76–90. doi: 10.1016/j.clim.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 61.Ryan EP, Bushnell TP, Friedman AE, Rahman I, Phipps RP. Cyclooxygenase-2 independent effects of cyclooxygenase-2 inhibitors on oxidative stress and intracellular glutathione content in normal and malignant human B-cells. Cancer Immunol Immunother. 2008;57:347–58. doi: 10.1007/s00262-007-0374-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shanafelt TD, Rabe KG, Kay NE, Zent CS, Call TG, Slager SL, et al. Statin and non-steroidal anti-inflammatory drug use in relation to clinical outcome among patients with Rai stage 0 chronic lymphocytic leukemia. Leuk Lymphoma. 2010;51:1233–40. doi: 10.3109/10428194.2010.486877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Robak P, Linke A, Cebula B, Robak T, Smolewski P. Cytotoxic effect of R-etodolac (SDX-101) in combination with purine analogs or monoclonal antibodies on ex vivo B-cell chronic lymphocytic leukemia cells. Leuk Lymphoma. 2006;47:2625–34. doi: 10.1080/10428190600948147. [DOI] [PubMed] [Google Scholar]
- 64.Lindhagen E, Nissle S, Leoni L, Elliott G, Chao Q, Larsson R, et al. R-etodolac (SDX-101) and the related indole-pyran analogues SDX-308 and SDX-309 potentiate the antileukemic activity of standard cytotoxic agents in primary chronic lymphocytic leukaemia cells. Cancer chemotherapy and pharmacology. 2007;60:545–53. doi: 10.1007/s00280-006-0400-9. [DOI] [PubMed] [Google Scholar]
- 65.Chae Young Kwang, T LX, Keating Michael J, Estrov Zeev. Concurrent Use of Aspirin with the Fludarabine, Cyclophophamide, Rituximab (FCR) Regimen Improves Outcome in Patients with Relapsed/Refractory CLL American Society of Hematology. Atlanta Georgia: 2012. [Google Scholar]
- 66.Filip-Ciubotaru FM, Manciuc C, Foia L. Statins and endothelial dysfunction. Rev Med Chir Soc Med Nat Iasi. 2009;113:975–83. [PubMed] [Google Scholar]
- 67.Larose E, Ganz P. Statins and endothelial dysfunction. Semin Vasc Med. 2004;4:333–46. doi: 10.1055/s-2004-869590. [DOI] [PubMed] [Google Scholar]
- 68.Weitz-Schmidt G. Statins as anti-inflammatory agents. Trends Pharmacol Sci. 2002;23:482–6. doi: 10.1016/s0165-6147(02)02077-1. [DOI] [PubMed] [Google Scholar]
- 69.Davignon J. Beneficial cardiovascular pleiotropic effects of statins. Circulation. 2004;109:III39–43. doi: 10.1161/01.CIR.0000131517.20177.5a. [DOI] [PubMed] [Google Scholar]
- 70.Hernandez-Perera O, Perez-Sala D, Navarro-Antolin J, Sanchez-Pascuala R, Hernandez G, Diaz C, et al. Effects of the 3-hydroxy-3-methylglutaryl-CoA reductase inhibitors, atorvastatin and simvastatin, on the expression of endothelin-1 and endothelial nitric oxide synthase in vascular endothelial cells. J Clin Invest. 1998;101:2711–9. doi: 10.1172/JCI1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wassmann S, Laufs U, Muller K, Konkol C, Ahlbory K, Baumer AT, et al. Cellular antioxidant effects of atorvastatin in vitro and in vivo. Arteriosclerosis, thrombosis, and vascular biology. 2002;22:300–5. doi: 10.1161/hq0202.104081. [DOI] [PubMed] [Google Scholar]
- 72.Zhou MS, Schuman IH, Jaimes EA, Raij L. Renoprotection by statins is linked to a decrease in renal oxidative stress, TGF-beta, and fibronectin with concomitant increase in nitric oxide bioavailability. Am J Physiol Renal Physiol. 2008;295:F53–9. doi: 10.1152/ajprenal.00041.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chapman-Shimshoni D, Yuklea M, Radnay J, Shapiro H, Lishner M. Simvastatin induces apoptosis of B-CLL cells by activation of mitochondrial caspase 9. Exp Hematol. 2003;31:779–83. doi: 10.1016/s0301-472x(03)00192-9. [DOI] [PubMed] [Google Scholar]
- 74.Schramm R, Menger MD, Harder Y, Schmits R, Adam O, Weitz-Schmidt G, et al. Statins inhibit lymphocyte homing to peripheral lymph nodes. Immunology. 2007;120:315–24. doi: 10.1111/j.1365-2567.2006.02505.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ulivieri C, Fanigliulo D, Benati D, Pasini FL, Baldari CT. Simvastatin impairs humoral and cell-mediated immunity in mice by inhibiting lymphocyte homing, T-cell activation and antigen cross-presentation. Eur J Immunol. 2008;38:2832–44. doi: 10.1002/eji.200838278. [DOI] [PubMed] [Google Scholar]
- 76.Winiarska M, Bil J, Wilczek E, Wilczynski GM, Lekka M, Engelberts PJ, et al. Statins impair antitumor effects of rituximab by inducing conformational changes of CD20. PLoS Med. 2008;5:e64. doi: 10.1371/journal.pmed.0050064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Friedman DR, Magura LA, Warren HA, Harrison JD, Diehl LF, Weinberg JB. Statin use and need for therapy in chronic lymphocytic leukemia. Leuk Lymphoma. 2010;51:2295–8. doi: 10.3109/10428194.2010.520050. [DOI] [PubMed] [Google Scholar]
- 78.Wiestner A. Emerging role of kinase-targeted strategies in chronic lymphocytic leukemia. Blood. 2012;120:4684–91. doi: 10.1182/blood-2012-05-423194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hantschel O, Rix U, Schmidt U, Burckstummer T, Kneidinger M, Schutze G, et al. The Btk tyrosine kinase is a major target of the Bcr-Abl inhibitor dasatinib. Proc Natl Acad Sci U S A. 2007;104:13283–8. doi: 10.1073/pnas.0702654104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Amrein PC, Attar EC, Takvorian T, Hochberg EP, Ballen KK, Leahy KM, et al. Phase II study of dasatinib in relapsed or refractory chronic lymphocytic leukemia. Clin Cancer Res. 2011;17:2977–86. doi: 10.1158/1078-0432.CCR-10-2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Futosi K, Nemeth T, Pick R, Vantus T, Walzog B, Mocsai A. Dasatinib inhibits proinflammatory functions of mature human neutrophils. Blood. 2012;119:4981–91. doi: 10.1182/blood-2011-07-369041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Weinblatt ME, Kavanaugh A, Genovese MC, Musser TK, Grossbard EB, Magilavy DB. An oral spleen tyrosine kinase (Syk) inhibitor for rheumatoid arthritis. N Engl J Med. 2010;363:1303–12. doi: 10.1056/NEJMoa1000500. [DOI] [PubMed] [Google Scholar]
- 83.Friedberg JW, Sharman J, Sweetenham J, Johnston PB, Vose JM, Lacasce A, et al. Inhibition of Syk with fostamatinib disodium has significant clinical activity in non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood. 2010;115:2578–85. doi: 10.1182/blood-2009-08-236471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cheson BD, Byrd JC, Rai KR, Kay NE, O’Brien SM, Flinn IW, et al. Novel targeted agents and the need to refine clinical end points in chronic lymphocytic leukemia. J Clin Oncol. 2012;30:2820–2. doi: 10.1200/JCO.2012.43.3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Herman SE, Johnson AJ. Molecular pathways: targeting phosphoinositide 3-kinase p110-delta in chronic lymphocytic leukemia. Clin Cancer Res. 2012;18:4013–8. doi: 10.1158/1078-0432.CCR-11-1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, et al. Targeting BTK with Ibrutinib in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med. 2013 doi: 10.1056/NEJMoa1215637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Herman SE, Gordon AL, Hertlein E, Ramanunni A, Zhang X, Jaglowski S, et al. Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood. 2011;117:6287–96. doi: 10.1182/blood-2011-01-328484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Byrd John C, F RR, Coutre Steven, Flinn Ian W, Burger Jan A, Blum Kristie A, Sharman Jeff P, Grant Barbara, Jones Jeffrey A, Wierda William G, Zhao Weiqiang, Heerema Nyla A, Johnson Amy J, Tran Anh, Clow Fong, Kunkel Lori, James Danelle F, O’Brien Susan. The Bruton’s Tyrosine Kinase (BTK) Inhibitor Ibrutinib (PCI-32765) Promotes High Response Rate, Durable Remissions, and Is Tolerable in Treatment Naïve (TN) and Relapsed or Refractory (RR) Chronic Lymphocytic Leukemia (CLL) or Small Lymphocytic Lymphoma (SLL) Patients Including Patients with High-Risk (HR) Disease: New and Updated Results of 116 Patients in a Phase Ib/II Study American Society of Hematology. Atlanta, Georgia: 2012. [Google Scholar]
- 89.Ponader S, Chen SS, Buggy JJ, Balakrishnan K, Gandhi V, Wierda WG, et al. The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood. 2012;119:1182–9. doi: 10.1182/blood-2011-10-386417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rajkumar SV, Rosinol L, Hussein M, Catalano J, Jedrzejczak W, Lucy L, et al. Multicenter, randomized, double-blind, placebo-controlled study of thalidomide plus dexamethasone compared with dexamethasone as initial therapy for newly diagnosed multiple myeloma. J Clin Oncol. 2008;26:2171–7. doi: 10.1200/JCO.2007.14.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Singhal S, Mehta J, Desikan R, Ayers D, Roberson P, Eddlemon P, et al. Antitumor activity of thalidomide in refractory multiple myeloma. N Engl J Med. 1999;341:1565–71. doi: 10.1056/NEJM199911183412102. [DOI] [PubMed] [Google Scholar]
- 92.Carballido E, Veliz M, Komrokji R, Pinilla-Ibarz J. Immunomodulatory drugs and active immunotherapy for chronic lymphocytic leukemia. Cancer Control. 2012;19:54–67. doi: 10.1177/107327481201900106. [DOI] [PubMed] [Google Scholar]
- 93.Sampaio EP, Sarno EN, Galilly R, Cohn ZA, Kaplan G. Thalidomide selectively inhibits tumor necrosis factor alpha production by stimulated human monocytes. J Exp Med. 1991;173:699–703. doi: 10.1084/jem.173.3.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sheskin J. Thalidomide in the Treatment of Lepra Reactions. Clin Pharmacol Ther. 1965;6:303–6. doi: 10.1002/cpt196563303. [DOI] [PubMed] [Google Scholar]
- 95.Corral LG, Haslett PA, Muller GW, Chen R, Wong LM, Ocampo CJ, et al. Differential cytokine modulation and T cell activation by two distinct classes of thalidomide analogues that are potent inhibitors of TNF-alpha. J Immunol. 1999;163:380–6. [PubMed] [Google Scholar]
- 96.Ramsay AG, Johnson AJ, Lee AM, Gorgun G, Le Dieu R, Blum W, et al. Chronic lymphocytic leukemia T cells show impaired immunological synapse formation that can be reversed with an immunomodulating drug. J Clin Invest. 2008;118:2427–37. doi: 10.1172/JCI35017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Badoux XC, Keating MJ, Wen S, Lee BN, Sivina M, Reuben J, et al. Lenalidomide as initial therapy of elderly patients with chronic lymphocytic leukemia. Blood. 2011;118:3489–98. doi: 10.1182/blood-2011-03-339077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chanan-Khan A, Miller KC, Musial L, Lawrence D, Padmanabhan S, Takeshita K, et al. Clinical efficacy of lenalidomide in patients with relapsed or refractory chronic lymphocytic leukemia: results of a phase II study. J Clin Oncol. 2006;24:5343–9. doi: 10.1200/JCO.2005.05.0401. [DOI] [PubMed] [Google Scholar]
- 99.Chen CI, Bergsagel PL, Paul H, Xu W, Lau A, Dave N, et al. Single-agent lenalidomide in the treatment of previously untreated chronic lymphocytic leukemia. J Clin Oncol. 2011;29:1175–81. doi: 10.1200/JCO.2010.29.8133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ferrajoli A, Lee BN, Schlette EJ, O’Brien SM, Gao H, Wen S, et al. Lenalidomide induces complete and partial remissions in patients with relapsed and refractory chronic lymphocytic leukemia. Blood. 2008;111:5291–7. doi: 10.1182/blood-2007-12-130120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chanan-Khan AA, Cheson BD. Lenalidomide for the treatment of B-cell malignancies. J Clin Oncol. 2008;26:1544–52. doi: 10.1200/JCO.2007.14.5367. [DOI] [PubMed] [Google Scholar]
- 102.Jenny NS. Inflammation in aging: cause, effect, or both? Discovery medicine. 2012;13:451–60. [PubMed] [Google Scholar]
- 103.Ivanenkov YA, Balakin KV, Tkachenko SE. New approaches to the treatment of inflammatory disease : focus on small-molecule inhibitors of signal transduction pathways. Drugs R D. 2008;9:397–434. doi: 10.2165/0126839-200809060-00005. [DOI] [PubMed] [Google Scholar]
- 104.Vijayakrishnan L, Venkataramanan R, Gulati P. Treating inflammation with the Janus kinase inhibitor CP-690,550. Trends Pharmacol Sci. 2011;32:25–34. doi: 10.1016/j.tips.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 105.Martinez-Lostao L, Briones J, Forne I, Martinez-Gallo M, Ferrer B, Sierra J, et al. Role of the STAT1 pathway in apoptosis induced by fludarabine and JAK kinase inhibitors in B-cell chronic lymphocytic leukemia. Leuk Lymphoma. 2005;46:435–42. doi: 10.1080/10428190400018398. [DOI] [PubMed] [Google Scholar]
- 106.Furman RR, Byrd JC, Brown JR, Coutre SE, Benson DM, Wagner-Johnston ND, et al. CAL-101, An Isoform-Selective Inhibitor of Phosphatidylinositol 3-Kinase P110 delta, Demonstrates Clinical Activity and Pharmacodynamic Effects In patients with Relapsed or Refractory Chronic Lymphocytic Leukemia. Blood. 2010;116:31. [Google Scholar]