

Abstract
Transcriptional enhancers are DNA elements capable of regulating gene expression in-cis over great distances. With the recent availability of genomic approaches to define epigenetic marks and RNA levels, these previously difficult to study elements are now being extensively examined for their critical role in lineage-specific transcriptional regulation. This review sets out to highlight the use of embryonic stem cells (ESCs) in the study of enhancers, emphasizing that ESC have become an ideal model system for questions regarding mammalian transcriptional regulation. This review highlights the epigenetic “signature” of enhancers, their mechanism of action, and the role of non-coding RNAs (ncRNAs) on enhancer function. We briefly review insulators, a sub-type of enhancers, and a novel model system for studying enhancer function in vivo. We conclude with some ongoing questions within the field.
Keywords: Embryonic stem cells, Transcriptional enhancers
Introduction
The transcriptional regulation of gene expression in eukaryotes is governed by two distinct classes of genetic elements: trans-acting factors are typically proteins or non-coding RNAs (ncRNAs) which subsequently bind to cis-acting DNA elements such as promoters or enhancers, which must be on the same DNA segment as the gene they regulate. Promoters are short (≈1–2kb) regions of DNA required to be immediately upstream of a gene in the correct orientation to ensure proper transcription. Enhancers, in contrast, are cis-acting elements that act independent of distance from their target gene(s), and are orientation independent. In mammals, given the size of the genome, enhancers have been identified to act upon their target gene up to 100 kb away, and in rare circumstances up to 1 MB away [1].
Enhancers have been a challenge to study because of the vast distances they can act over, and the requirement that they be defined exclusively through functional assays. This has limited their identification on a genome-wide basis. However, recent work utilizing new, genome-wide techniques allows the identification of enhancer based upon epigenetic marks, protein binding events, and/or tissue-specific DNase hypersensitivity sites (DNase HS) [2]. Collectively, these techniques allow for a non-functional definition of enhancers. The purpose of this review is to lay-out the important role that Embryonic Stem Cells (ESCs) have played in recent work to identify transcriptional enhancers and study their biology. For a broader discussion of enhancers in general, the author would like to suggest two excellent recent reviews [3,4]. Given the explosion of literature within this field, an encyclopedic cataloguing of all the work is impossible, and the author would like to apologize if a discussion of some work is omitted for the sake of clarity and/or brevity.
Importance of Enhancers
With the advent of the complete genomic sequence of a number of organisms, a surprising and important discovery was noted, as illustrated in Table 1. While the number of cells varies by many orders of magnitude, the length of the genome from a single-celled eukaryote (S. Cerevisae) to a human varies by slightly more than two orders of magnitude. In contrast, the number of protein coding loci varies by far less with virtually no difference between humans and C. Elegans. This implies that organismal complexity is not directly a function of the number of protein-coding genes, and therefore must be determined by a different mechanism. The most likely explanation for increasing organismal complexity from essentially the same number of protein-coding loci is through more intricate transcriptional regulatory mechanisms. In this way, the combinatorial expression pattern of different factors can be varied to a much greater degree, allowing the same number of protein coding genes to be expressed in a far more complex manner.
Table 1.
Number of cells varying by the length of the genome and number of genes.
| S. Cerevisae | C. Elegan | H. Sapiens | |
|---|---|---|---|
| Cell Number | 1 × 100 | 1 × 103 | 1 × 1012 |
| Genomic Length (bp) | 1 × 107 | 1 × 108 | 3 × 109 |
| Number of Genes | 6 × 103 | 2 × 105 | 2 × 105 |
Complex gene regulation in a multi-cellular organism is akin to tissue-specific regulation. In this situation, specific combinations of both ubiquitous and tissue-specific factors are co-expressed to allow lineage-restricted expression of many genes. While promoters of tissue specific genes often contain binding sites for lineage-restricted transcription factors (TFs), in general promoters are too small to allow for the proper combinatorial binding of all the factors required for lineage-restricted tissue expression. In contrast, enhancers, which can act over great distances, can be utilized much more easily to allow lineage-restricted factors to regulate gene expression. Thus, the increased size of the genome of higher metazoans, at least in part, is needed to encompass the larger number of cis-acting elements required for tissue specific transcription. Multiple groups have demonstrated that enhancers are highly tissue-specific, and likely responsible for most lineage-restricted gene expression [5–9].
Barriers to Studying Enhancers
In a post-genomic world, it is trivial to identify genes and then study the surrounding regions to identify regulatory sites. Promoters have been far easier to study because of their close proximity to the transcriptional start site (TSS) of a gene.
Enhancers have been far more challenging, since they have up until recently been functionally defined. In other cellular contexts, enhancers have most often been identified based upon their (relative) proximity to a locus of interest, and other criteria such as conserved linear DNA sequence across species, because it is assumed that non-coding DNA sequences without regulatory functions will quickly diverge through evolution. However, because enhancers are often lineage specific, linear DNA sequence conservation is a highly stringent criteria, which will miss some possible enhancers [10,11]. If sequence conservation does exist at a DNA element far from a known TSS, the linear DNA sequence can provide hints, based upon the presence of consensus binding sites for transcription factors. In addition, the presence of DNase hypersensitivity sites (DNase HS) within a DNA region, which indicates that a specific DNA sequence is occupied by a protein within the cell, can further substantiate that a region of non-coding DNA may have regulatory potential [12]. While previously a laborious, and time consuming process, DNase HS mapping is now possible on a genome-wide basis by identifying the hypersensitive sites either with microarrays [13] or next-generation sequencing [14]. Nonetheless, the validation of the presence of an enhancer required a functional assay-typically the putative enhancer was cloned into a reporter vector, and the tissue specific expression expressed by transient transfection into a variety of cell lines. While this method remains the classically accepted approach in mammalian cells to assess whether a given DNA sequence contains enhancer activity, it clearly has limitations. In addition to the substantial amount of “wet-bench” work required to validate a region of DNA as possessing enhancer potential, by removing the enhancer from its normal cellular context, a broader, in vivo view of the role of these elements is lost. Lastly, given the very fact that these approaches can be applied essentially only to single sites within the genome, a global view of the role of enhancers on transcriptional regulation was impossible.
ESCs to the Rescue: An Ideal Model System to Study Transcriptional Regulation
For the above reasons, the biology of enhancers was limited by both their identification and the need to confirm them with a functional assay. Chromatin Immunoprecipitation (ChIP) coupled with microarray hybridization (ChIP-Chip) or next-generation sequencing (ChIP-Seq [15]), collectively termed genome wide location analysis (GWLA), allows the comprehensive determination of the DNA sites enriched for specific epigenetic marks and/or bound by specific transcription factors (TFs). This technology was early on applied to embryonic stem cells [16–19]. Embryonic stem cells are derived from early mammalian embryos, and in the case of mouse ESC the inner-cell mass (ICM) of day 3.5 dpc embryos [20]. The ICM contains all the cells that eventually go on to form the embryo. Both human and mouse ESCs are similar in that they share two canonical properties of stem cells, self-renewal and pluripotency.
Self-renewal is the ability to continually propagate in an undifferentiated state. Pluripotency is the ability to differentiate into all three primitive germ layers (mesoderm, endoderm, and ectoderm). Given their ability to differentiate into virtually any cell type, pluripotent cells have become an active area of research for regenerative medicine strategies. In addition, because they represent a cell-line that can recapitulate the earliest steps of lineage commitment in mammals, they represent a potent tool for understanding the regulation of early developmental processes.
The importance of transcription regulation on ESCs was explored through a variety of techniques initially. Both of the canonical properties of ESCs, self-renewal and pluripotency, are critically regulated at the level of transcription [20–22]. Collectively, the importance of transcriptional regulation on ESC is highlighted by seminal work showing that a handful (four to six) transcription factors, when expressed together, can reprogram somatic cells into a pluripotent state (iPSC or induced pluripotent stem cells) [23–26]. Defined factor reprogramming has transformed regenerative medicine, with the promise of the generation of patient-derived tissues, thereby bypassing the need for both cadaveric or living donors and the issues with rejection and/or graft-versus host disease.
The central role that pluripotent cells play in regenerative medicine strategies and the importance of transcriptional regulation on their canonical properties has created enormous interest in the pathways utilized by these cells. Better understanding of the gene expression pathways utilized by pluripotent cells will allow these same pathways to be manipulated to increase both the efficiency of iPS generation but also their differentiation into other tissue types. For this reason, a wide range of genome wide approaches such as GWLA, transcriptome analysis (typically microarray based gene expression measurements), and proteomics have been used on ESCs, which are more easily studied than primary cell types because they can be grown in relatively large quantities. An unintended consequence of these studies is that a large range of datasets now exists for ESCs, making them one of the most widely studied mammalian cell systems. In the GEO omnibus alone, there are over 2000 datasets deposited that involve embryonic stem cells in some fashion. These datasets, when integrated, can provide unparalleled understanding behind the interplay between DNA binding proteins such as transcription factors, epigenetic marks such as histone modifications, and their effects on gene expression. Thus, ESCs have become an attractive model system to explore questions regarding mammalian transcriptional regulation. The creation of a novel dataset can be layered onto other datasets to better understand how the complex interplay of a variety of processes influence gene expression on a global scale [27]. In addition, the ability to perturb specific pathways, either by deleting specific DNA elements or depleting trans-acting factors through RNAi strategies, which can be easily utilized in ESCs, allows mechanistic studies to be undertaken. Given the ability of ESCs to differentiate into all three germ-layers, this allows fundamental questions about lineage commitment to be tackled. Collectively, these facts have made ESCs an exceptionally powerful system to understand the role of enhancers on transcriptional regulation, but especially on cell-type specific issues, since ESC can be differentiated into a wide variety of cell types.
Epigenetic marks, TF binding and Enhancers
Epigenetics, in its strictest definition, are heritable traits not encoded within the linear DNA sequence. Mechanistically, this can be explained by the fact that within a cell, there is essentially no isolated DNA, it is almost entirely contained within a complex mixture of proteins, predominantly histones, termed chromatin. There are a wide variety of modifications to DNA (methylation, 5-hydroxy methylation, and others), histones (including methylation, acetylation, and phosphorylation), and nucleosomes positioning which all have important roles in regulating transcriptional expression [28]. Collectively, these epigenetic marks can be a mark of gene expression, but also directly influence it.
The initial work on understanding the effects, histone marks on transcriptional regulation was primarily focused at promoters, since their epigenetic profile could be directly linked to the expression of nearby genes [7,19,29,30]. In ESCs, seminal work(s) by multiple labs highlighted that specific histone methylation marks within promoters could correlate directly with the transcriptional activity of the locus. The most commonly discussed marks near promoters include Histone 3 Lysine 4 trimethylation (H3K4me3), which tends to mark transcriptionally active genes, and H3K27me3 which tends to mark transcriptionally active genes (Figure 1). Most surprisingly, originally observed in ESCs but later extended to other cell types, is that both H3K4me3 and H3k27me3 mark a subset of promoters. These promoters are termed “bivalent” due to the presence of two opposing histone marks, and are typically thought to represent “poised genes”, which can be quickly activated by removal of the repressive H3K27me3 marks [30]. Similar studies, based upon GWLA, determined that the regions immediately adjacent to the TSS of well-annotated genes in ESCs tend to be bound by known pluripotency-associated transcription factors such as Nanog, Oct4, and Sox2 [17,18], with actively transcribed genes required for ESC self-renewal and/or pluripotency bound by two or more of these important TFs. While these studies initially pointed to these highly-occupied TF units as being within the promoter of nearby genes, given the size ranges examined (up to 8–10 kb away from TSS), many of these binding events likely occurred outside of the promoter. These regions are likely enhancers, but in general have been examined in a limited fashion. One of the best-characterized example is a clear enhancer element approximately 5kb upstream of the Nanog TSS [31]. This region has been shown to be highly-occupied by ESC-critical TFs such as Nanog, Oct4, Sox2, and Sall4, all of which contribute to the proper expression of the Nanog locus [17,32].
Figure 1.

Histone Modifications at different classes of cis-regulatory elements. Active promoters tend to be marked by H3K4me3 in the absence of H3K27me3. Inactive promoters tend to be marked by H3K27me3, with the additional presence of H3K4me3 termed the “bivalent” mark, and are thought to represent promoters “poised” for activation after removal of the repressive H3K27me3 mark. Enhancers tend to be marked differently, with active enhancers marked by H3K4me1, H3K27Ac, and Cyclic AMP-responsive element binding protein (CBP)/p300 binding. Poised enhancers tend to be marked by H3K4me1 alone, CBP/p300, and in human ESCs H3K27me3. The combination of histone marks and their implications for gene expression remain an open question. Insulators are defined by the binding of CCCTC-binding factor (CTCF). Transcriptional start sites (TSS) are indicated with arrows.
In the initial era of GWLA in ESC the work focused on promoters, given the ease with which they can be identified simply based upon their proximity to the TSS. However, the first clues as an epigenetic “signature” for enhancers came from the work of multiple labs, predominantly in non-ESC cell types. Cyclic AMP-responsive element binding (CREB) protein (CBP) and p300 are highly similar proteins with histone acetyltransferase activity that can interact with transcription factors as well as directly binding specific histone modifications [33]. These factors are critical in mediating the transcriptional response to multiple cell signaling cascades. Seminal studies done by the lab of Bing Ren in non-ESC cell types demonstrated that CBP/p300 binding events were interspersed throughout genome and identified novel enhancer elements that could recreate lineage specific gene expression patterns in transgenic mouse models [6,7]. This initial work suggested that the binding of specific factors, namely CBP/p300, could be utilized to identify possible enhancers. Within a relatively short amount of time, a number of labs published work based upon ChIP-seq to identify “signature” to describe enhancers. In mouse ESCs, the most commonly accepted marks are H3K4me1, H3k27Ac, and binding by CBP/p300 [5–9]. All three marks have been shown to be lineage specific in a variety of circumstances. In general, these enhancers have been distinguished by their “activity”, with two groups publishing that H3K27Ac marked active enhancers [8,9], whereas H3K4me1 alone tended to indicate enhancers which were developmentally poised. It should be noted that CBP/p300 is known to bind to genomic regions rich in H3K4me1 and acetylate H3K27Ac, and may indicate why putative enhancer which are H3K4me1 rich and co-occupied by CBP/p300 seem to correlate better with enhancer activity [8,9,34,35]. These results, importantly, needed to be put into the broader context of how enhancer “activity” is defined. Typically, this is done by a nearest neighbor analysis, whereby the transcriptional activity of the well-annotated gene most proximal to a given enhancer is assessed. Controls are often included to try and correct for the fact that any one locus may be acted upon by multiple enhancers and vice a versa, but can be a challenge and may muddle the picture. In addition, given that there is a fair degree of overlap between these epigenetic marks, teasing out whether there are important functional differences between them remains an important area of research. Also complicating the picture is that many histone modification(s) that mark enhancers also mark promoters. For example, H3K27Ac is often found at the promoters of actively transcribed genes [11], and thus may define un-annotated genes and/or pseudogenes as opposed to enhancers. Lastly, there may be currently unidentified protein binding events and/or histone modifications that mark enhancers, and therefore a complete annotation of all possible enhancers remains elusive.
One important point is that at least in murine ESCs, the repressive H3K27me3 tends to occur exclusively at promoters. In contrast, recent work [9] has shown that in human ESCs, the distinction between poised and active enhancers is slightly different than murine ESCs. In this study, they characterize active enhancers as occupied by p300, H3K4me1 rich, and also rich in H3K27Ac. In contrast, they noted that poised enhancers are occupied by p300, H3K4me1, and absent H3K27Ac, but in contrast to mouse ESCs also were relatively H3K27me3 high, a polycomb-related mark typically seen close to repressed promoters. The difference between mice and human ESCs represents whether species differences, differences between the developmental origins of the two pluripotent cell types, or simply technical differences between the two groups methodologies remains to be seen. Nonetheless, the discrepancies between these two studies illustrates that, while histone marks can be a powerful method to define enhancers, no “one-size-fits-all” enhancers definition is currently possible. Further studies are needed to determine how enhancers marked by different histone modifications (such as H3K4me1+, H3K27Ac+ versus H3K4me1+ CBP/p300+ enhancers) differ into their ability/role regulate gene expression.
While histone marks such as methylation and acetylation have been the predominant epigenetic mark to be studied in defining signatures for ESCs, other types of epigenetic marks have also been explored. Nucleosome repositioning is the basis for DNase HS mapping, with the displacement of nucleosomes away from areas bound by proteins, thereby providing increased access to DNA unprotected by histones, thereby making them hypersensitive to cleavage by DNase I. The repositioning may be a specific process to mammalian enhancers, with one study showing that the nucleosome remodeler, CHD7 which contains an SNF2 like ATPase domain, localizes to active enhancers in ESCs, where it acts to modulate gene expression [36]. One important consideration is that nucleosome positioning may be a secondary effects of the various epigenetic marks and/or protein binding events by CBP/p300, and therefore required but not sufficient for enhancer function.
In addition, to nucleosome position, the third common epigenetic mark is direct alterations to DNA, with methylation on cytosine being the most widely studied. Bisulfite sequencing, the preferred method for identifying regions of methylated DNA was technically challenging on a genome wide scale until recently. A novel technique, whole genome bisulfite sequencing (BiSeq), was recently applied to ESCs and neural progenitors (NPs) [37]. Local regions of quantitative hypomethylation, termed low-methylated regions (LMRs) appear to be a unique pattern of methylation, occur distal to promoters of well-annotated genes, and are distinct from classic CpG islands. Based upon a variety of criteria, these regions of methylation overlap substantially with distal regulatory regions, either enhancers or insulators, bound by CCCTC-binding factor (CTCF, discussed further below). These LMRs are shaped by the binding of specific factors, with CTCF and RE1-silencing transcription factor (REST) both being able to change a region from hypermethylation into an LMR by binding to the site. The ability to modulate the epigenetic marks at a specific site through DNA binding is important, because often it has been a challenge to determine how much influence any given epigenetic change is the result of binding by a protein, versus the other way around. One important caveat of this study is its limited study of only two cell types, ESCs and neural progenitors, and therefore the conclusion may not be broadly applicable to other tissues/lineages. In addition to methylation on cytosine, a more recent DNA modification, 5′ hydroxylmethylation on cytosine (5′ hmC) has been described, with the conversion being catalyzed by the Tet families of proteins [38]. Tet1 and Tet2 have been shown to be required for ESC biology [39–41]. Genome wide-location analysis of Tet1 in mouse ESCs or direct identification of 5′hmC in DNA all revealed that 5′hmC accumulated near the promoters of genes, perhaps more frequently within CpG islands and genes which exhibit the bivalent histone mark (H3K4me3/H3K27me3) [40,42–44]. Similar results have been obtained in human ESCs, but there also seems to be enrichment for 5′ hmC within enhancers defined by a variety of conditions [45,46]. However, the differences between human and mouse ESCs may reflect the underlying biological differences between the two, or perhaps simply represent technical differences in the approaches and analysis models used by the different groups. These differences highlight the importance of using, if possible, a single bioinformatics “pipeline” to analyze published datasets, thereby eliminating disparities that may arise simply from the use of competing algorithms. Importantly, the role of 5′ hmC in gene regulation remains to be determined, including whether it is simply an intermediate DNA base pair, prior to conversion to another species.
Importantly, the distinction between these different types of enhancers remains unclear. Many active enhancers, i.e. those actively engaged with promoters in any given cell-type, likely contain a group of epigenetic marks, perhaps H3K27Ac and H3K4me1, etc. While the presence of these marks seems to be better defined, the mechanism by which they are laid down and/or removed remains undetermined. One recent paper indicates that the histone demethylase LSD1 is recruited to ESC specific enhancers, where doing the process of differentiation it removes the H3K4me1 mark, thereby “decommissioning” the enhancer [47]. This important finding is one of the first examples of how an enhancer can be “turned off” during a developmental process. However, the recruitment and selective activity of different epigenetic modifiers to make a specific DNA sequence either an active, poised, or inactive enhancer, remains an important questions within the field.
Mechanism of Enhancer Action
Given the great distances that enhancers can act over, the obvious mechanistic question is, how does this occur? Multiple models have been generated to explain their effects but virtually all are variations on the so-called “looping” model [3,4,48] (Figure 2). This model postulates that for enhancers to act they must be brought into close physical contact with a promoter, thereby creating a DNA loop with the intervening segment. While most labs agree that some type of DNA loop is formed, further details of the mechanism remain contentious [49–51]. How the actual enhancer: promoter interaction occurs, whether simply by diffusion or some type of active scanning mechanism remains to be definitively determined [51]. In addition, some have postulated that RNA Pol II is initially nucleated at the enhancer element, and then transferred to the promoter, which remains controversial [52].
Figure 2.
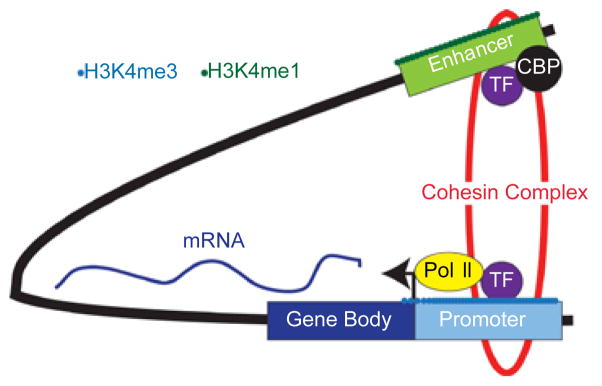
Illustrating of the “looping” model of enhancer function. In this model, the promoter and enhancer, which are separated by a large distance, are brought into close physical proximity by “looping” out the intervening DNA segment. This allows direct, physical contact between the two DNA elements and their associated epigenetic marks and bound proteins. In this example, the enhancer is marked by H3K4me1, and co-occupied by a transcription factor (TF) and Cyclic AMP-responsive element binding protein (CBP). The promoter is marked by H3K4me3, indicating it is actively transcribed by RNA Pol II (Pol II). The DNA loop is stabilized by the cohesin complex.
Nonetheless, the presence of a DNA loop between a promoter and enhancer is considered in vivo evidence of an enhancer’s engagement and regulation of a specific locus. These DNA loops can be assessed by an assay originally developed in 2002 termed chromosomal conformational capture (3C), in which DNA from nuclei is isolated and interactions between genomic regions are determined by crosslinking, endonuclease digestion, intermolecular ligation, and PCR or quantitative PCR analysis of ligated products [48]. Using this assay, interactions can be mapped and quantified, allowing an in vivo assessment of not just enhancer activity, but their developmental regulation. Genome wide approaches based upon this approach include coupling 3C to next-generation sequencing in approaches such as 4C, 5C, or Hi-C, [53]. Collectively, these assays allow the identification and characterization of the DNA loops formed during enhancer engagement with a promoter.
While the formation of these loops has been studied for some time, a group recently focused on the proteins critical for DNA loop formation/stabilization using the power of ESCs. Kagey et al. [54] first performed a short-hairpin RNA interference (shRNA) screen to identify factors required for pluripotency. Common ESC-critical TFs such as Nanog, Sall4, and Tcf1 were recovered, but surprisingly, two important protein complexes were also uncovered, the cohesin complex and the mediator complex. The mediator complex is a large, ill-defined group of proteins conserved through evolution and are known to mediate transcriptional activation by enhancers [55]. The cohesin complex was surprising. This is a ubiquitously expressed group of proteins, which collectively assist in maintaining sister-chromatid cohesin through mitosis [56,57]. Recently, they have been shown to be critical in transcriptional regulation through a direct interaction with the insulator protein CTCF [58]. Thus, the fact that a widely expressed group of proteins could be critical for an ESC-specific property, pluripotency, was surprising. Through a combination of GWLA and 3C analysis, the authors were able to demonstrate that the cohesin complex mediates DNA loops to facilitate the engagement of enhancer elements into promoters of critical pluripotency loci such as Nanog, Oct4, and others. These DNA loops were distinct from the elements where the cohesin complex mediates looping of insulator elements through a direct interaction with CTCF. Thus, a single group of proteins, the cohesin complex, are responsible for mediating DNA loops between different types of cis-acting elements. Collectively, this work highlights not just the importance of enhancers in ESC biology, but also raises important questions, about how a ubiquitously expressed group of proteins can mediate a cell-type specific phenotype, pluripotency, when depleted.
Insulators Versus Enhancers
While enhancers are classically defined as cis-acting elements that can act regulate transcription over great distances, this relatively broad definition covers a variety of DNA elements. Two broad classes that are discussed predominantly in ESCs are enhancers, which are usually meant to indicate DNA elements that cause transcriptional activation in specific cells types. Insulators, in contrast, are thought to be a distinct subclass of distal cis-acting elements, which are typically bound by CTCF [59]. While insulators have pleiotropic effects in mammalian cells, in general they are thought to repress transcription. This may be done either by the insulator creating a buffer between the chromatin structure and epigenetic marks of nearby elements, or by preventing enhancers from being brought into close proximity of promoters. The latter is termed the “enhancer blocking” model, and is illustrated in Figure 3. In contrast to enhancers, insulator tend to be relatively invariant across different cell types, making it unclear precisely how these elements work. One group recently attempted to identify on a genome wide basis all the binding sites of CTCF in mouse ESCs and all the possible DNA loops generated by a combination analysis termed chromatin interaction analysis-paired end tag sequencing (ChIA-PET) [60]. In this pioneering work, the authors identified 39,371 CTCF binding sites, and a total of 1,816 DNA sites that were distal to each other but clearly interacted in this assay. Surprisingly, of these interactions, 1,480 were intrachromosomal and 336 (19%) were interchromosomal. Many of these interchromosomal interactions could be verified by 4C methodologies or fuorescent in-situ hybridization (FISH). This opens the intriguing and counter-intuitive idea that insulators, and possibly other types of enhancer(s) could act in trans. In addition, it may indicate that insulators, and possibly other types of distal cis-acting elements could play an important role in regulating the global architecture of chromatin within the nucleus, perhaps by organizing specific DNA segments into actively transcribed regions (enhancesomes) and repressed, heterochromatin regions. This may be a global phenomenon, in which repressed regions of chromatin are targets to specific areas of the nucleus, such as the nuclear lamina in a sequence specific manner [61].
Figure 3.
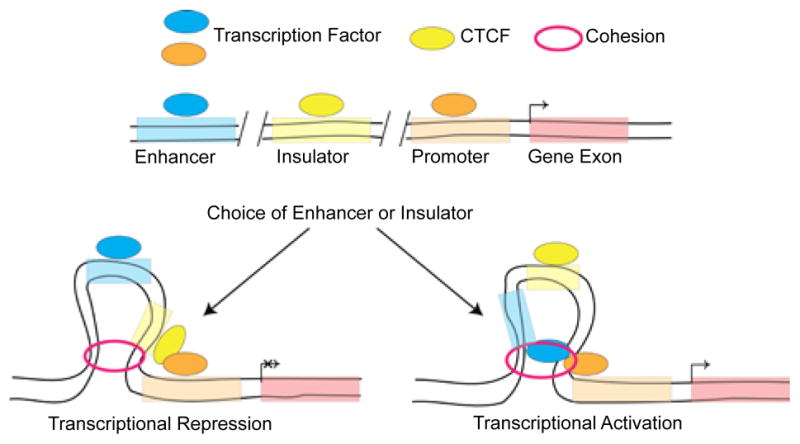
Model of transcriptional looping. The cohesion complex can loop either insulator, defined by CCCTC-binding factor (CTCF) binding, or enhancer elements bound by transcription factors. As can be seen, the intervening DNA sequence is “looped-out” to allow the enhancer/insulator and promoters to be brought into close physical proximity. In addition, the ability of an insulator to “block” the enhancer from interacting with its target promoter is illustrated. How the choice between which type of element, enhancer versus insulator, is utilized remains unknown.
Non-coding RNAs and Enhancers
One of the most important recent insights from the advent of next-generation sequencing of RNA (RNA-seq) based techniques is that a large fraction of the genome is transcriptionally active, even though a small percentage (approximately 1–2%) encodes for proteins. While many of these transcriptional products remain to be defined, a relatively large number are non-coding (ncRNAs). Their biology is actively being investigated, and the “classification” of ncRNAs remains in flux [62]. While one class of ncRNAs, microRNAs, have well-defined biological functions and mechanisms attached to them, others are poorly understood. Within this group of non-coding RNAs, it has been recently noted that a wide-range of enhancer are bound by RNA Pol II, and produce short, bidirectional transcripts, referred to as eRNAs [63–65]. In all the initial publications (neurons, macrophages, and prostate cancer cells), these transcripts were assessed at extragenic sites enriched in the enhancer specific marks H3K4me1 and co-occupied by p300/CBP. These transcripts appear to be distinct from another class of ncRNAs, termed long intergenic ncRNAs (lincRNAs), which have histone marks (H3K4me3 and H3K36me3) more classically associated with protein coding loci. The bi-directional nature of these transcripts is intriguing. In ESCs, these same transcripts have been identified at a subset of enhancers in ESCs [66] and appear to be more highly correlated with the H3K27Ac mark in murine ESCs [8]. In human ESCs, developmentally poised enhancers see an increase in their eRNA production during differentiation [9]. There are many intriguing hypothesis to explain these transcripts. One is that they are the result of spurious transcription of actively transcribed loci, with Pol II “accidentally” transcribed the enhancer when it is in close proximity to the promoter. This seems unlikely, with at least one example near the β-globin locus where transcription occurs independent of the promoter [67]. Another hypothesis is that these eRNAs play a critical role in enhancer function. This question remains untested, and will be required to determine if enhancer derived transcripts serve a biological function. Nonetheless, these transcripts may eventually allow another method for enhancer identification on a genome-wide scale [66].
Novel Methods for Enhancer Functional Assessment
While identifying enhancers by epigenetic marks has allowed a non-functional approach to their identification, a functional validation scheme remains useful. First, this allows careful, mechanistic studies to be performed upon enhancers. Second, it is not possible to profile histone marks in all possible cell type, making an in vivo technique that could be utilized in animals appealing. Lastly, an approach that could be utilized in a high-throughput fashion, to screen a large number of enhancers important for early development would be invaluable. Transient transfection assays, in which a DNA element with putative enhancer activity are fused to a reporter sequence containing a minimal promoter is the classic approach. The advantage of this approach is it allows for careful, mechanistic question to be addressed. Unfortunately, in this situation, the cellular context, and the complexities of chromatin are fundamentally lost. Similar studies in animals, whereby the effects of deleting/altering the linear DNA sequence of a given enhancer element in vivo are laborious and time-consuming, eliminating the chances of assessing a large number of enhancers. Recently, a new ESC based system has been generated to allow rapid in vivo screening of enhancer activity [68]. Homologous recombination allows the insertion of putative enhancers coupled to β-lactamase (lacZ) into the ubiquitously expressed Rosa26 locus. The ESCs can then be differentiated in vitro or injected into blastocysts for in vivo analysis to study the developmental expression of enhancers. Given the rapid throughput possible with these cells, this approach could also be utilized to perform the kinds of mechanistic studies that have been the purview of transient transfection assays.
Conclusions, pending questions, and Future directions
While the work outlined above in ESCs and other model systems has uncovered new methods to identify and characterize enhancers, a whole host of questions remain. First and foremost, the mechanism by which enhancers operate remains an open question. Do they load RNA Pol II onto promoters, change RNA Pol II conformation/phosphorylation state to enhancer transcription, perhaps by causing it to be released from promoter proximal pausing? Next, how do the different epigenetic marks seen at enhancers, H3K4me1, H3K27Ac, etc. compare? Are they developmentally distinct, do they recruit different proteins/TF to mediate their function, and by what mechanism are these marks laid down and removed? With the intriguing finding that enhancers are bound by Pol II and transcribed into eRNAs, it is important to understand if this small group of enhancers have special biological properties, and if the eRNAs produced themselves perform a specific function. Other types of ncRNAs such as lincRNAs can act as scaffolds to recruit epigenetic modifiers to specific loci in trans. While eRNAs may perform a similar role, until this is proven their biology remains unclear. Perhaps the most fundamental question that remains is how does any given promoter “choose” which enhancer(s) it is engaged with at any given time (Figure 1). Given the sheer size of the genome, and the exciting possibility that enhancers may be able to work in trans [60], in theory a promoter could be activated by virtually any enhancer within the genome, even on different chromosomes. This fundamental question may help elucidate the reasons behind the size, and regulatory complexity of the mammalian genome.
Many of the above questions are complex and will require the use of both currently known and yet undeveloped techniques, ESCs remain well positioned to make substantial contributions to all of the above questions. As transcriptional regulation in ESCs remains a broad, and widely studied model system, the ESC may soon become the prototypical model system for understanding mammalian gene regulation.
Acknowledgments
The author is supported by an NHLBI sponsored Career Development Award (7K08HL087951).
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Lettice LA, Heaney SJ, Purdie LA, Li L, de Beer P, et al. A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum Mol Genet. 2003;12:1725–1735. doi: 10.1093/hmg/ddg180. [DOI] [PubMed] [Google Scholar]
- 2.Hardison RC, Taylor J. Genomic approaches towards finding cis-regulatory modules in animals. Nat Rev Genet. 2012;13:469–483. doi: 10.1038/nrg3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ong CT, Corces VG. Enhancer function: new insights into the regulation of tissue-specific gene expression. Nat Rev Genet. 2011;12:283–293. doi: 10.1038/nrg2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bulger M, Groudine M. Functional and Mechanistic Diversity of Distal Transcription Enhancers. Cell. 2011;144:327–339. doi: 10.1016/j.cell.2011.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, et al. ChlP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–858. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 8.Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci USA. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, et al. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470:279–283. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Visel A, Rubin EM, Pennacchio LA. Genomic views of distant-acting enhancers. Nature. 2009;461:199–205. doi: 10.1038/nature08451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiao S, Xie D, Cao X, Yu P, Xing X, et al. Comparative Epigenomic Annotation of Regulatory DNA. Cell. 2012;149:1381–1392. doi: 10.1016/j.cell.2012.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elgin SC. The formation and function of DNase I hypersensitive sites in the process of gene activation. J Biol Chem. 1988;263:19259–19262. [PubMed] [Google Scholar]
- 13.Crawford GE, Davis S, Scacheri PC, Renaud G, Halawi MJ, et al. DNase-chip: a high-resolution method to identify DNase I hypersensitive sites using tiled microarrays. Nat Meth. 2006;3:503–509. doi: 10.1038/NMETH888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Song L, Crawford GE. DNase-seq: A High-Resolution Technique for Mapping Active Gene Regulatory Elements across the Genome from Mammalian Cells. Cold Spring Harb Protoc. 2010 doi: 10.1101/pdb.prot5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet. 2009;10:669–680. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, et al. Core Transcriptional Regulatory Circuitry in Human Embryonic Stem Cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim J, Chu J, Shen X, Wang J, Orkin SH. An extended transcriptional network for pluripotency of embryonic stem cells. Cell. 2008;132:1049–1061. doi: 10.1016/j.cell.2008.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen X, Xu H, Yuan P, Fang F, Huss M, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- 19.Azuara V, Perry P, Sauer S, Spivakov M, Jørgensen HF, et al. Chromatin signatures of pluripotent cell lines. Nat Cell Biol. 2006;8:532–538. doi: 10.1038/ncb1403. [DOI] [PubMed] [Google Scholar]
- 20.Young RA. Control of the Embryonic Stem Cell State. Cell. 2011;144:940–954. doi: 10.1016/j.cell.2011.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rao S, Orkin SH. Unraveling the transcriptional network controlling ES cell pluripotency. Genome Biol. 2006;7:230. doi: 10.1186/gb-2006-7-8-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Orkin SH, Hochedlinger K. Chromatin Connections to Pluripotency and Cellular Reprogramming. Cell. 2011;145:835–850. doi: 10.1016/j.cell.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takahashi K, Yamanaka S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 24.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, et al. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 25.Park IH, Zhao R, West JA, Yabuuchi A, Huo H, et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451:141–146. doi: 10.1038/nature06534. [DOI] [PubMed] [Google Scholar]
- 26.Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 27.Hawkins RD, Hon GC, Ren B. Next-generation genomics: an integrative approach. Nat Rev Genet. 2010;11:476–486. doi: 10.1038/nrg2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet. 2010;12:7–18. doi: 10.1038/nrg2905. [DOI] [PubMed] [Google Scholar]
- 29.Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, et al. A Bivalent Chromatin Structure Marks Key Developmental Genes in Embryonic Stem Cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 31.Wu Q, Chen X, Zhang J, Loh YH, Low TY, et al. Sall4 Interacts with Nanog and Co-occupies Nanog Genomic Sites in Embryonic Stem Cells. J Biol Chem. 2006;281:24090–24094. doi: 10.1074/jbc.C600122200. [DOI] [PubMed] [Google Scholar]
- 32.Rao S, Zhen S, Roumiantsev S, McDonald LT, Yuan GC, et al. Differential Roles of Sall4 Isoforms in Embryonic Stem Cell Pluripotency. Mol Cell Biol. 2010;30:5364–5380. doi: 10.1128/MCB.00419-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bedford DC, Kasper LH, Fukuyama T, Brindle PK. Target gene context influences the transcriptional requirement for the KAT3 family of CBP and p300 histone acetyltransferases. Epigenetics. 2010;5:9–15. doi: 10.4161/epi.5.1.10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tie F, Banerjee R, Stratton CA, Prasad-Sinha J, Stepanik V, et al. CBP-mediated acetylation of histone H3 lysine 27 antagonizes Drosophila Polycomb silencing. Development. 2009;136:3131–3141. doi: 10.1242/dev.037127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schnetz MP, Handoko L, Akhtar-Zaidi B, Bartels CF, Pereira CF, et al. CHD7 Targets Active Gene Enhancer Elements to Modulate ES Cell-Specific Gene Expression. PLoS Genet. 2010;6:e1001023. doi: 10.1371/journal.pgen.1001023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature. 2011;480:490–495. doi: 10.1038/nature10716. [DOI] [PubMed] [Google Scholar]
- 38.Branco MR, Ficz G, Reik W. Uncovering the role of 5-hydroxymethylcytosine in the epigenome. Nat Rev Genet. 2011;13:7–13. doi: 10.1038/nrg3080. [DOI] [PubMed] [Google Scholar]
- 39.Koh KP, Yabuuchi A, Rao S, Huang Y, Cunniff K, et al. Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell. 2011;8:200–213. doi: 10.1016/j.stem.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Williams K, Christensen J, Pedersen MT, Johansen JV, Cloos PA, et al. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature. 2011;473:343–348. doi: 10.1038/nature10066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu H, D’Alessio AC, Ito S, Xia K, Wang Z, et al. Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature. 2011;473:389–393. doi: 10.1038/nature09934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ficz G, Branco MR, Seisenberger S, Santos F, Krueger F, et al. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature. 2011;473:398–402. doi: 10.1038/nature10008. [DOI] [PubMed] [Google Scholar]
- 43.Guo JU, Su Y, Zhong C, Ming GL, Song H. Hydroxylation of 5-Methylcytosine by TET1 Promotes Active DNA Demethylation in the Adult Brain. Cell. 2011;145:423–434. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pastor WA, Pape UJ, Huang Y, Henderson HR, Lister R, et al. Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature. 2011;473:394–397. doi: 10.1038/nature10102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stroud H, Feng S, Morey Kinney S, Pradhan S, Jacobsen SE. 5-hydroxymethylcytosine is associated with enhancers and gene bodies in human embryonic stem cells. Genome Biol. 2011;12:R54. doi: 10.1186/gb-2011-12-6-r54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Szulwach KE, Li X, Li Y, Song CX, Han JW, et al. Integrating 5-Hydroxymethylcytosine into the Epigenomic Landscape of Human Embryonic Stem Cells. PLoS Genet. 2011;7:e1002154. doi: 10.1371/journal.pgen.1002154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Whyte WA, Bilodeau S, Orlando DA, Hoke HA, Frampton GM, et al. Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature. 2012;482:221–225. doi: 10.1038/nature10805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation. Science. 2002;295:1306–1311. doi: 10.1126/science.1067799. [DOI] [PubMed] [Google Scholar]
- 49.De Laat W, Klous P, Kooren J, Noordermeer D, Palstra R, et al. Three-Dimensional Organization of Gene Expression in Erythroid Cells. In: Bieker JJ, editor. Current Topics in Developmental Biology. Academic Press; 2008. pp. 117–139. [DOI] [PubMed] [Google Scholar]
- 50.Bulger M, Groudine M. Looping versus linking: toward a model for long-distance gene activation. Genes Dev. 1999;13:2465–2477. doi: 10.1101/gad.13.19.2465. [DOI] [PubMed] [Google Scholar]
- 51.Ptashne M. Gene regulation by proteins acting nearby and at a distance. Nature. 1986;322:697–701. doi: 10.1038/322697a0. [DOI] [PubMed] [Google Scholar]
- 52.Blackwood EM, Kadonaga JT. Going the distance: a current view of enhancer action. Science. 1998;281:60–63. doi: 10.1126/science.281.5373.60. [DOI] [PubMed] [Google Scholar]
- 53.de Wit E, de Laat W. A decade of 3C technologies: insights into nuclear organization. Genes Dev. 2012;26:11–24. doi: 10.1101/gad.179804.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kagey MH, Newman JJ, Bilodeau S, Zhan Y, Orlando DA, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467:430–435. doi: 10.1038/nature09380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Taatjes DJ. The human Mediator complex: a versatile, genome-wide regulator of transcription. Trends Biochem Sci. 2010;35:315–322. doi: 10.1016/j.tibs.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dorsett D. Cohesin: genomic insights into controlling gene transcription and development. Curr Opin Genet Dev. 2011;21:199–206. doi: 10.1016/j.gde.2011.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wood AJ, Severson AF, Meyer BJ. Condensin and cohesin complexity: the expanding repertoire of functions. Nat Rev Genet. 2010;11:391–404. doi: 10.1038/nrg2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wendt KS, Yoshida K, Itoh T, Bando M, Koch B, et al. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature. 2008;451:796–801. doi: 10.1038/nature06634. [DOI] [PubMed] [Google Scholar]
- 59.Phillips JE, Corces VG. CTCF: master weaver of the genome. Cell. 2009;137:1194–1211. doi: 10.1016/j.cell.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Handoko L, Xu H, Li G, Ngan CY, Chew E, et al. CTCF-mediated functional chromatin interactome in pluripotent cells. Nat Genet. 2011;43:630–638. doi: 10.1038/ng.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zullo JM, Demarco IA, Piqué-Regi R, Gaffney DJ, Epstein CB, et al. DNA Sequence-Dependent Compartmentalization and Silencing of Chromatin at the Nuclear Lamina. Cell. 2012;149:1474–1487. doi: 10.1016/j.cell.2012.04.035. [DOI] [PubMed] [Google Scholar]
- 62.Wright MW, Bruford EA. Naming ‘junk’: human non-protein coding RNA (ncRNA) gene nomenclature. Hum Genomics. 2011;5:90–98. doi: 10.1186/1479-7364-5-2-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang D, Garcia-Bassets I, Benner C, Li W, Su X, et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature. 2011;474:390–394. doi: 10.1038/nature10006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, et al. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465:182–187. doi: 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.De Santa F, Barozzi I, Mietton F, Ghisletti S, Polletti S, et al. A Large Fraction of Extragenic RNA Pol II Transcription Sites Overlap Enhancers. Plos Biol. 2010;8:e1000384. doi: 10.1371/journal.pbio.1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Melgar MF, Collins FS, Sethupathy P. Discovery of active enhancers through bidirectional expression of short transcripts. Genome Biol. 2011;12:R113. doi: 10.1186/gb-2011-12-11-r113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ling J, Baibakov B, Pi W, Emerson BM, Tuan D. The HS2 enhancer of the beta-globin locus control region initiates synthesis of non-coding, polyadenylated RNAs independent of a cis-linked globin promoter. J Mol Biol. 2005;350:883–896. doi: 10.1016/j.jmb.2005.05.039. [DOI] [PubMed] [Google Scholar]
- 68.Tsanov KM, Nishi Y, Peterson KA, Liu J, Baetscher M, et al. An embryonic stem cell-based system for rapid analysis of transcriptional enhancers. Genesis. 2012;50:443–450. doi: 10.1002/dvg.20820. [DOI] [PMC free article] [PubMed] [Google Scholar]