

Abstract
Aims
The first aim was to critically evaluate the extent to which familial hypercholesterolaemia (FH) is underdiagnosed and undertreated. The second aim was to provide guidance for screening and treatment of FH, in order to prevent coronary heart disease (CHD).
Methods and results
Of the theoretical estimated prevalence of 1/500 for heterozygous FH, <1% are diagnosed in most countries. Recently, direct screening in a Northern European general population diagnosed approximately 1/200 with heterozygous FH. All reported studies document failure to achieve recommended LDL cholesterol targets in a large proportion of individuals with FH, and up to 13-fold increased risk of CHD. Based on prevalences between 1/500 and 1/200, between 14 and 34 million individuals worldwide have FH. We recommend that children, adults, and families should be screened for FH if a person or family member presents with FH, a plasma cholesterol level in an adult ≥8 mmol/L(≥310 mg/dL) or a child ≥6 mmol/L(≥230 mg/dL), premature CHD, tendon xanthomas, or sudden premature cardiac death. In FH, low-density lipoprotein cholesterol targets are <3.5 mmol/L(<135 mg/dL) for children, <2.5 mmol/L(<100 mg/dL) for adults, and <1.8 mmol/L(<70 mg/dL) for adults with known CHD or diabetes. In addition to lifestyle and dietary counselling, treatment priorities are (i) in children, statins, ezetimibe, and bile acid binding resins, and (ii) in adults, maximal potent statin dose, ezetimibe, and bile acid binding resins. Lipoprotein apheresis can be offered in homozygotes and in treatment-resistant heterozygotes with CHD.
Conclusion
Owing to severe underdiagnosis and undertreatment of FH, there is an urgent worldwide need for diagnostic screening together with early and aggressive treatment of this extremely high-risk condition.
Keywords: Cholesterol, Low-density lipoprotein, Atherosclerosis, Coronary heart disease, Cardiovascular disease
Introduction
Familial hypercholesterolaemia (FH) is a common genetic cause of premature coronary heart disease (CHD) (i.e. ischaemic heart disease), namely myocardial infarction and angina pectoris, due to lifelong elevated plasma low-density lipoprotein (LDL) cholesterol levels.1,2 If left untreated, men and women with heterozygous FH (later referred to simply as FH, unless specified as heterozygous or homozygous FH) with total cholesterol levels of 8–15 mmol/L (310–580 mg/dL) typically develop CHD before age 55 and 60, respectively, while homozygotes with total cholesterol levels of 12–30 mmol/L (460–1160 mg/dL) typically develop CHD very early in life and if untreated die before age 20. However, once diagnosed, heterozygotes can readily be treated with cholesterol-lowering medication to attenuate development of atherosclerosis and to prevent CHD.3
The extent of underdiagnosis and undertreatment of individuals in the general population with FH is largely unknown. It is generally believed that among whites, 1/500 are heterozygous for FH and 1/1 000 000 are homozygous1,2; however, even these individuals are not diagnosed in most countries.4 Furthermore, these prevalences likely represent underestimates and as cardiovascular disease is the leading cause of death worldwide.5 Indeed, many individuals and families with FH may simply be overlooked among the huge number of individuals with any CHD caused by more common risk factors, and as a consequence be underdiagnosed and undertreated for genetically elevated cholesterol levels.
The aim of the present consensus paper is to critically evaluate the extent to which FH is underdiagnosed and undertreated worldwide. Based on a consensus of the opinions of the experts in this panel and/or on small studies, retrospective studies, and registries (level of evidence C6), the EAS Consensus Panel proposes recommendations on (i) how better to diagnose individuals and families with FH and (ii) therapeutic strategies for best practice aimed to prevent CHD in these extremely high-risk individuals and families. Importantly, the effect of LDL cholesterol lowering on reduction in CHD and all-cause mortality in individuals without FH is based on multiple randomized clinical trials and meta-analyses7 (level of evidence A6). Details of the levels of evidence specifically derived from FH studies can be found elsewhere.8 This Consensus Statement is aimed at cardiologists, endocrinologists, internists, paediatricians, general practitioners, clinical biochemists, public health practitioners, health service planners, other health professionals, and healthcare providers worldwide.
Underdiagnosis
FH was not attributed an independent code in the World Health Organisation International Classification of Diseases, making reliable estimates of the number of individuals diagnosed with this condition difficult. Of the roughly 200 countries/territories in the world, we have therefore only been able to obtain estimates of the number of individuals diagnosed with FH for the 22 countries/territories shown in Figure 1. Upon inclusion of the ∼180 countries/territories not listed in Figure 1, <1% are diagnosed in most countries. The few exceptions are 71% diagnosed in the Netherlands, 43% in Norway, 19% in Iceland, 13% in Switzerland, 12% in the UK, and 6% in Spain. Even these numbers are somewhat unreliable; for example, it has been estimated in Norway that roughly 1/300 have FH,9 and applying this prevalence, instead of 1/500, would mean that 26% rather than 43% of individuals with FH are diagnosed in Norway.
Figure 1.

Estimated per cent of individuals diagnosed with familial hypercholesterolaemia in different countries/territories, as a fraction of those theoretically predicted based on a frequency of 1/500 in the general population. As most countries do not have valid nationwide registries for familial hypercholesterolaemia, several values in this figure represent informed estimates from clinicians/scientists with recognized expertise in and knowledge of familial hypercholesterolaemia in their respective countries. Numbers were provided by Michael Livingston, Steve E. Humphries (UK), Olivier S. Descamps (Belgium).
To date, the prevalence of FH has not been assessed directly in an unselected sample from the general population. Using the Copenhagen General Population Study,10 an unselected European general population sample comprising 69 016 participants with heterozygous FH was diagnosed using the Dutch Lipid Clinic Network (DLCN) criteria (Table 1). The prevalence of individuals classified with definite or probable FH combined (DLCN criteria, >5 points) was ∼1/200 (Figure 2).10 Interestingly, prevalences for definite or probable FH were similar for women and men below age 60; in contrast, above age 60, more women than men were in this category. These findings suggest that many men with FH had died at an earlier age, as was also observed in a UK prevalence study.11
Table 1.
Dutch Lipid Clinic Network criteria for diagnosis of heterozygous familial hypercholesterolaemia in adults
| Group 1: family history | Points |
| (i) First-degree relative with known premature (<55 years, men; <60 years, women) coronary heart disease (CHD) OR | 1 |
| (ii) First-degree relative with known LDL cholesterol >95th percentile by age and gender for country | 1 |
| (iii) First-degree relative with tendon xanthoma and/or corneal arcus OR | 2 |
| (iv) Child(ren) <18 years with LDL cholesterol >95th percentile by age and gender for country | 2 |
| Group 2: clinical history | |
| (i) Subject has premature (<55 years, men; <60 years, women) CHD | 2 |
| (ii) Subject has premature (<55 years, men; <60 years, women) cerebral or peripheral vascular disease | 1 |
| Group 3: physical examination | |
| (i) Tendon xanthoma | 6 |
| (ii) Corneal arcus in a person <45 years | 4 |
| Group 4: biochemical results (LDL cholesterol) | |
| >8.5 mmol/L (>325 mg/dL) | 8 |
| 6.5–8.4 mmol/L (251–325 mg/dL) | 5 |
| 5.0–6.4 mmol/L (191–250 mg/dL) | 3 |
| 4.0–4.9 mmol/L (155–190 mg/dL) | 1 |
| Group 5: molecular genetic testing (DNA analysis) | |
| (i) Causative mutation shown in the LDLR, APOB, or PCSK9 genes | 8 |
A ‘definite FH’ diagnosis can be made if the subject scores >8 points. A ‘probable FH’ diagnosis can be made if the subject scores 6 to 8 points. A ‘possible FH’ diagnosis can be made if the subject scores 3 to 5 points. An ‘unlikely FH’ diagnosis can be made if the subject scores 0 to 2 points. Use of the diagnostic algorithm: per group only one score, the highest applicable, can be chosen. For example, when coronary heart disease and tendon xanthoma as well as dyslipidaemia are present in a family, the highest score for family history is 2. However, if persons with elevated LDL cholesterol levels as well as premature coronary heart disease are present in a family, but no xanthoma or children with elevated LDL cholesterol levels or a causative mutation are found, then the highest score for family history remains 1.
Figure 2.

Prevalence of definite or probable familial hypercholesterolaemia according to Dutch Lipid Clinic Network Criteria in the Copenhagen General Population Study by 20-year age groups and by gender. Based on 69 016 individuals. This was originally reported as 1/137 but recalculation suggested that the prevalence of definite or probable familial hypercholesterolaemia combined is closer to 1/200 (personal communication Børge G Nordestgaard). FH, familial hypercholesterolaemia. Adapted from Benn et al.10
Based on extrapolations from these estimated 1/500–1/200 prevalences, there are between 14 and 34 million individuals with FH worldwide (Figure 3). Furthermore, even higher prevalences are observed in subpopulations with founder effects.2 Taken together, these data strongly suggest that FH is vastly underdiagnosed in most countries.
Figure 3.

Estimated millions of individuals worldwide with familial hypercholesterolaemia by WHO regions and by income groups. Estimates are shown for the theoretical frequency of heterozygous familial hypercholesterolaemia of 1/500 in the general population,1 as well as for the directly detected frequency of ∼1/200 in the Danish population,10 a typical country in Northern Europe.
Undertreatment
Hitherto, data have not been reported either on the risk of CHD, or on frequency of statin treatment, in FH individuals in a large sample from the general population not subject to ascertainment bias. Using the Copenhagen General Population Study,10 the prevalence of CHD among definite/probable FH participants was 33%; only 48% of subjects with FH received statins. The risk of CHD was increased 13-fold (95% CI: 10- to 17-fold) among individuals with definite/probable FH not receiving statins (Figure 4); similar findings have been reported in FH cohort studies.12 Furthermore, the corresponding increase in risk for CHD among individuals with FH on statin was ten-fold (8- to 14-fold), suggesting that the dose of statin therapy provided resulted in insufficient cholesterol-lowering medication, and was introduced too late in life, when severe atherosclerosis had already developed. Other studies also support massive undertreatment of individuals with FH.3,12,13
Figure 4.
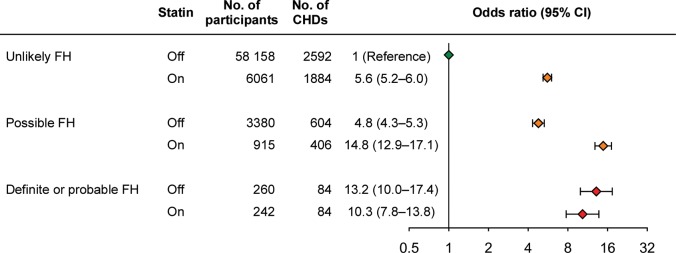
Risk of coronary heart disease as a function of the Dutch Lipid Clinic Network Criteria for a diagnosis of familial hypercholesterolaemia in individuals on or off statin from the general population. Data are based on 69 016 individuals from the Copenhagen General Population Study. CI, confidence interval; FH, familial hypercholesterolaemia; CHD, coronary heart disease = ischaemic heart disease. Adapted from Benn et al.10
Pathophysiology and genetics
FH is caused by mutations in genes encoding key proteins involved in the LDL receptor endocytic and recycling pathways, leading to decreased cellular uptake of LDL and increased plasma LDL cholesterol concentrations1 (Figure 5). Within hepatocytes, cholesterol is recycled or synthesized de novo, with 3-hydroxy-3-methylglutaryl coenzyme A reductase being rate-limiting; statins block the activity of this enzyme. Cholesterol is packaged into apolipoprotein B-containing very low-density lipoproteins (VLDL), the intravascular precursors of LDL, which in turn transports most cholesterol from the liver to peripheral tissues. Regulated endocytosis of LDL via apolipoprotein B by peripheral cells and hepatocytes occurs through the LDL receptor and an adaptor protein (LDLRAP, alias ARH).14 Most LDL receptors recycle, although when proprotein convertase subtilisin/kexin type 9 (PCSK9) is complexed to the LDL receptor, it short-circuits its intracellular recycling from the endosome, thereby reducing receptor numbers.
Figure 5.

Pathophysiology of heterozygous familial hypercholesterolaemia. LDL, low-density lipoprotein; PCSK9, proprotein convertase subtilisin/kexin type 9.
The life-threatening effects of both heterozygous and homozygous FH are related to the resulting elevation in plasma LDL cholesterol, with consequent cholesterol retention in the arterial wall and foam cell formation within the intima of arteries; such early lesions typically progress to occlusive atherosclerosis with angina pectoris and/or plaque rupture with CHD (i.e. myocardial infarction).
Heterozygous FH is caused either by heterozygous loss-of-function mutations in LDLR, heterozygous mutations in APOB that affect the LDL receptor-binding domain of apolipoprotein B, or heterozygous gain-of-function mutations in PCSK9.14 Currently, >1200 mutations have been documented worldwide in LDLR15; these affect all functional domains of the LDL receptor protein and include single-nucleotide mutations, copy number variations, and splicing mutations throughout the LDLR gene. A single mutation, Arg3500Gln, is the only common FH-related mutation in APOB, while >20 different mutations have been detected in PCSK9. Heterozygous LDLR, APOB, and PCSK9 mutations are found in >90, ∼5, and ∼1%, respectively, of heterozygous FH subjects with a causative mutation.2 The prevalence varies geographically.
Homozygous FH results from homozygous, or more often, from compound heterozygous mutations in either the LDLR or ARH genes.16 Some rare subjects are ‘double heterozygotes’, which means they carry mutations in two of the above-mentioned four genes, usually leading to a phenotype that is intermediate between heterozygous and homozygous FH.
Clinical vs. mutation diagnosis
Historically, heterozygous FH was diagnosed clinically and the phenotypically most severe cases were detected, that is, those with severe LDL cholesterol elevations, premature and familial CHD, and tendon xanthomas.1 However, with the increased understanding of the genetic causes of this disease, direct detection of mutations in the LDLR, APOB, PCSK9, and LDLRAP genes is now available in many countries. Such progress has led to the understanding that some 10–40%, depending on referral criteria, of those with a clinical diagnosis will not have a detectable causal mutation; rather, they have a clinical diagnosis of FH, but not a mutation diagnosis (Figure 6).17,18 There may therefore be yet other key genes implicated in this disease; alternatively, these individuals may present a polygenic basis for their LDL cholesterol elevation without contributions from any of the classical FH genes.
Figure 6.
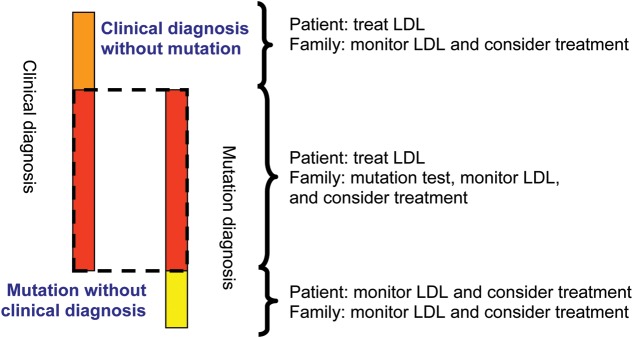
Overlap of clinical and mutation diagnosis of heterozygous familial hypercholesterolaemia. This figure illustrates the fractions of three different clinical scenarios in a study from Spain,18 and therefore not necessarily the exact proportions for these three groups in other countries. ‘Mutation without clinical diagnosis’ means definite, probable, or possible familial hypercholesterolaemia with a familial hypercholesterolaemia-causing mutation but with less severely elevated LDL cholesterol (i.e. below the diagnostic threshold). LDL, low-density lipoprotein cholesterol; FH, familial hypercholesterolaemia.
Conversely, genetic cascade testing from FH subjects with a detected causative mutation has shown that, while on average, relatives who carry the causative mutation have two-fold higher mean LDL cholesterol levels compared with non-carrier relatives, a significant proportion are below the clinical diagnostic cut-off18–21 and thus they have a mutation diagnosis but not a clinical diagnosis of FH (Figure 6).18 Such individuals may possess other favourable genes and/or a lifestyle that reduces the impact of the mutation, but because of their lifetime LDL cholesterol exposure, they should still be offered appropriate lipid-lowering therapy according to the LDL cholesterol targets given later.
Whom to screen: how do we recognize index cases?
Probands (index cases) should be identified according to the following criteria:
plasma total cholesterol ≥8 mmol/L (≥310 mg/dL) in an adult or adult family member(s) (or >95th percentile by age and gender for country),
premature CHD in the subject or family member(s),
tendon xanthomas in the subject or family member(s),
sudden premature cardiac death in a family member.
For child probands, criteria #(ii)–(iv) are identical to those of adults, but criterion #(i) should be a plasma total cholesterol ≥6 mmol/L (≥230 mg/dL) in a child or child family member(s) (or >95th percentile by age and gender for country). The highest likelihood of detecting FH is in those with very high LDL cholesterol levels, tendon xanthomas, and/or premature CHD in a family member.17
Drawing a family pedigree (Figure 7) is essential to evaluate the likelihood of FH (Table 1). In cases of probable or definite FH, cascade screening using LDL cholesterol measurement in the family should be conducted and the subject referred for genetic testing if available, with subsequent cascade testing in the family if a causative mutation is found. Initial family members to be tested are biological first-degree relatives, namely parents, siblings, and children. Biological second-degree relatives including grandparents, grandchildren, uncles, aunts, nephews, nieces, and half-siblings should also be considered. ‘Premature CHD’ signifies CHD before age 55 in males and before age 60 in female first-degree relatives, while in second-degree relatives, the corresponding ages are 50 and 55.
Figure 7.
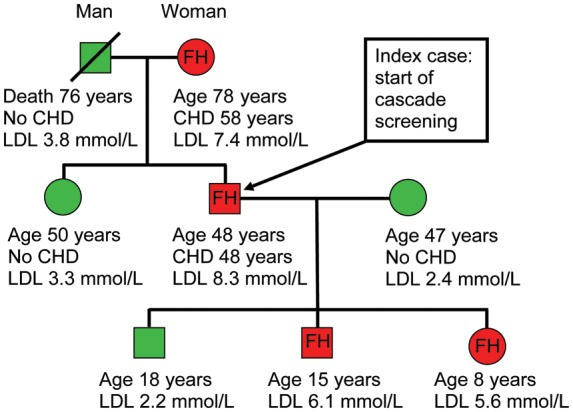
Pedigree of a family with familial hypercholesterolaemia. Red and green colours indicate family members with and without heterozygous familial hypercholesterolaemia. CHD, coronary heart disease; LDL, low-density lipoprotein; FH, familial hypercholesterolaemia.
Diagnosis
Diagnosis of FH relies on five criteria: family history, clinical history of premature CHD, physical examination for xanthomas and corneal arcus, very high LDL cholesterol on repeated measurements, and/or a causative mutation detected by molecular genetics22 (Table 1). Secondary causes of hyperlipidaemia must be excluded by determining that liver enzymes, renal function, and thyroid hormones are normal and that there is no hyperglycaemia or albuminuria.
In addition to drawing a family pedigree (Figure 7), a systematic physical examination for the presence of tendon and tuberous xanthomas and corneal arcus must be performed. Sonographic evaluation of the Achilles tendons increases the rate of xanthoma detection.23
Although total cholesterol levels are ≥8 mmol/L (≥6 mmol/L in children), triglyceride and HDL cholesterol levels are generally unremarkable. The presence of hypertriglyceridaemia does not exclude the FH diagnosis; other reasons for hypertriglyceridaemia should however be evaluated and treated if necessary.24
The DLCN criteria are recommended in order to establish the clinical diagnosis of FH (Table 1). Among individuals with a definite or probable diagnosis of FH (DLCN > 5), and particularly those with an obvious clinical diagnosis with xanthoma and/or high cholesterol plus a family history of premature CHD, molecular genetic testing is strongly recommended. When a causative mutation is found in the index case, a genetic test should be offered to all first-degree relatives (Figure 7).
Lifetime risk assessment and risk factors
It should be stressed that risk calculators such as the European SCORE or the US Framingham Risk Score are not appropriate for FH subjects, as such individuals are at considerably higher risk due to lifelong elevated LDL cholesterol levels (Figure 8). Nevertheless, whether diagnosed clinically or through a causative mutation, not all individuals with FH develop atherosclerosis and CHD to the same extent. Thus, as observed for the development of any CHD,25 other risk factors besides elevated LDL cholesterol act to determine the threshold for CHD (Figure 8), and risk factor counting is critical to assess CHD risk.26 Importantly, as elevated LDL cholesterol is the major problem in FH, this condition is dominated by CHD, whereas cerebrovascular disease is more common in individuals with hypertension and atherosclerosis in the lower limbs is more common among smokers.
Figure 8.
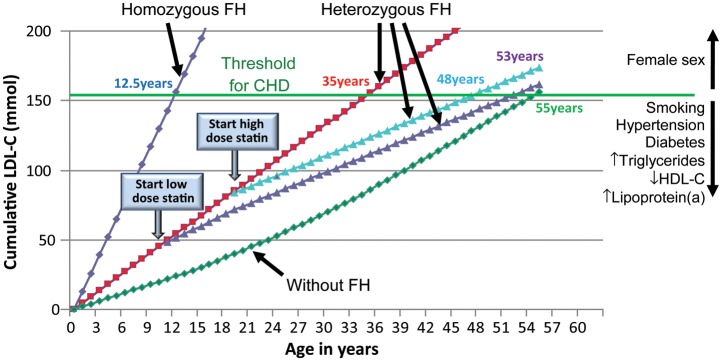
LDL cholesterol burden in individuals with or without familial hypercholesterolaemia as a function of the age of initiation of statin therapy. Data derived from Huijgen et al.20 and Starr et al.21 LDL, low-density lipoprotein; LDL-C, LDL cholesterol; HDL-C, high-density lipoprotein cholesterol; CHD, coronary heart disease; FH, familial hypercholesterolaemia.
The concept of a cumulative LDL cholesterol burden (Figure 8) illustrates the importance of early treatment. The cumulative LDL cholesterol burden of a 55-year-old person without FH is typically 160 mmol, a burden sufficient for CHD to develop (Figure 8; data derived from20,21). For an individual with heterozygous FH, this LDL cholesterol burden is reached by age 35 if untreated, by age 48 if treated since age 18, and by age 53 if treated since age 10. An untreated subject with homozygous FH will reach this level at age 12.5.
Men develop CHD before women; furthermore, hypertension, smoking, diabetes, and high triglycerides/low HDL cholesterol24 are all well-established additional risk factors in FH. In addition, lipoprotein(a) [Lp(a)] may be particularly elevated in clinically diagnosed heterozygous or homozygous FH.27,28 Indeed, elevated Lp(a) is now a well-established causal risk factor for cardiovascular disease irrespective of LDL cholesterol concentration.29,30 Elevated Lp(a) is also an important cardiovascular risk factor in FH.31 Because elevated Lp(a) significantly enhances risk of premature cardiovascular disease in those already at extremely high risk due to FH, additional, aggressive LDL lowering with statins and other drugs should be initiated in FH individuals with high levels of Lp(a). In such individuals, and in extreme cases with severe atherosclerosis and/or CHD, lipoprotein apheresis should be considered.32,33
Asymptomatic atherosclerosis
To improve risk assessment, imaging techniques are recommended to detect asymptomatic atherosclerosis in individuals at intermediate risk in the 2012 European Society of Cardiology (ESC) Guidelines for Cardiovascular Prevention.25 Although subjects with FH on average are at much higher risk, risk within FH is sufficiently variable that assessment of atherosclerosis should also be considered in asymptomatic FH subjects or in those whose family history is unclear. Imaging techniques are also useful in detecting aortic valve calcification and stenosis, which is particularly relevant in homozygous FH and in individuals with elevated Lp(a) levels.34
Techniques available to identify asymptomatic coronary atherosclerosis include exercise electro- and echocardiography, coronary calcium score, and angiography by computed tomography. Some,22,26 but not all,35 guidelines underscore the value of non-invasive imaging of atherosclerosis in assessing and managing asymptomatic FH subjects.
Exercise electro- or echocardiography should be considered for risk assessment in adults with FH at very high risk; symptomatic patients should be referred urgently for cardiac specialist review. Coronary artery calcification is a surrogate marker for atherosclerosis, with the calcium score being proportional to atherosclerotic plaque burden and cardiovascular disease risk.25 With the latest techniques, radiation exposure is as low as 1 mSV. Coronary artery calcification and the presence and severity of atherosclerosis detected by computed tomography can identify FH subjects with increased cardiovascular risk who may need more intensive cholesterol-lowering therapy; however, absence should not preclude cholesterol-lowering treatment, because there would likely be diffuse, non-calcified plaques in such individuals. Importantly, presence of coronary calcium is not identical with presence of relevant coronary lesions, because its specificity regarding the potential presence of ≥50% stenosis is only 50%.25 Angiography by computed tomography is at present not recommended for risk assessment.36
Cascade, opportunistic, and universal screening
The most cost-effective approach for identification of new FH subjects is cascade screening of family members of known index cases (Figure 7). Index cases can be detected by opportunistic or targeted systematic screening in primary care guided by a family history of premature CHD and hypercholesterolaemia, and among patients aged <55/60 in men/women with CHD in hospital settings; the DLCN criteria should be used to establish the clinical diagnosis (Table 1). Universal screening of children has often been suggested but has so far only been implemented in Slovenia and at the age of 5.37
Cascade screening using the protocol outlined in Table 2 has been found to be feasible and acceptable to subjects with FH and to physicians.8,26,35 To be maximally cost-effective, cascade screening should be systematic, centrally co-ordinated in a specialized centre and carried out using a combination of plasma lipid profiles and genetic testing. However, if the causative mutation is not known or genetic testing is not available, screening can be performed using the plasma lipid profile alone. Cascade testing in families with a known causative mutation has been carried out very successfully in the Netherlands over the last 15 years using trained genetic field workers (Figure 1).
Table 2.
Cascade testing issues in familial hypercholesterolaemia
| Notification of relatives at risk of familial hypercholesterolaemia should generally not be instituted without the consent of the index case. |
| National and local healthcare service protocols concerning disclosure of medical information without consent should be consulted. |
| A proactive approach that respects privacy, justice, and autonomy is required. |
| All material communicated to relatives and the telephone approach should be comprehensible and not cause alarm. |
| Pre-testing counselling should be offered to at risk family members of an index case prior to phenotypic or genetic testing. |
| If genetic testing detects a causative mutation, a definitive diagnosis of familial hypercholesterolaemia can be made in the tested individual particularly when the phenotype also suggests familial hypercholesterolaemia (Table 1; Figure 6: clinical diagnosis and mutation diagnosis). |
| If genetic testing does not detect a causative mutation, the diagnosis of familial hypercholesterolaemia can be excluded, except when the clinical phenotype is highly suggestive of familial hypercholesterolaemia (Figure 6: clinical diagnosis without mutation). |
| If genetic testing detects a causative mutation but the phenotype does not suggest familial hypercholesterolaemia, then a definitive diagnosis of familial hypercholesterolaemia should not be made; however, the person and family should be monitored every 2–5 years for LDL cholesterol levels (Figure 6: mutation without clinical diagnosis). |
| Genetic testing may have implications for insurance cover in certain countries. |
Another promising but untested targeted approach would be to screen all children for hypercholesterolaemia,37 for example at a time of infant immunization, and then, for children with total cholesterol >6 mmol/L (or >95th percentile), to perform ‘reverse’ cascade screening by testing their parents. This approach is based on the fact that LDL cholesterol values differentiate much better between mutation-negative and mutation-positive FH in children as compared with adults.38 However, as parents may not always be the blood relatives, DNA testing of children presents an ethical dilemma.39 Also, given the relatively small fraction of the population with FH, it is unclear whether such an approach would be feasible; moreover, it would likely be associated with prohibitively high cost and possibly with a high false positive rate.
LDL cholesterol targets
We recommend the following LDL cholesterol targets in FH, in accordance with recent ESC/EAS guidelines:
children <3.5 mmol/L (<135 mg/dL),
adults <2.5 mmol/L (<100 mg/dL),6
adults with CHD or diabetes <1.8 mmol/L (<70 mg/dL).6
These targets are for both heterozygous and homozygous FH regardless of age. However, in children and adults with homozygous FH, these values are extremely difficult to achieve with current treatments.
Due to ethical reasons, no randomized trial has been conducted documenting the benefit of lipid-lowering drug therapy specifically in FH subjects; however, treatment targets are based on large outcome lipid-lowering trials in persons without FH.7 LDL cholesterol is the primary target of therapy and the reduction in both cardiovascular and total mortality is proportional to the degree of LDL cholesterol reduction, with every 1 mmol/L reduction being associated with a corresponding 22% reduction in cardiovascular mortality and a 12% reduction in total mortality over 5 years.7 All untreated individuals with FH above age 40 should be considered to be at very high cardiovascular risk, as they have been exposed to elevated LDL cholesterol levels since birth40 (Figure 8).
Treatment
All subjects with FH and their families should undergo intensive education targeting lifestyle management,41 including intervention on smoking, diet, and physical activity. It is imperative that smokers quit smoking, and such individuals should be referred to a specialized tobacco unit/programme when necessary. Advice to children and young adults not to start smoking is especially important.
A certified dietitian/nutritionist should support implementation of a healthy diet with the involvement of the whole family. A complete record of dietary habits must be obtained, and recommendations for a healthy diet should be individualized. Functional foods known to lower LDL cholesterol, such as plant sterols and stanols, may be considered. The main objective of the nutritional advice is to avoid overweight and to reduce the amount of food and beverages with high cholesterol, saturated fat, and transfat content. Regular physical exercise must be implemented. In adults with FH, assessment of cardiovascular function is advisable before starting any significant exercise programme.
Cholesterol-lowering drugs should be initiated immediately at diagnosis in adults and strongly considered starting at age 8–10 in childhood, along with lifestyle management. The priority for pharmacotherapy should be as follows:
Children:
Statin,
Ezetimibe,
Bile acid-binding resin,
Lipoprotein apheresis in homozygotes.
Statins for children should only be those that have been shown to be safe in this group.
Adults:
Maximal potent statin dose,
Ezetimibe,
Bile acid-binding resins,
Lipoprotein apheresis in homozygotes and in treatment-resistant heterozygotes with CHD.
Maximal potent statin dose should be started at first consultation in adults and could be either atorvastatin 80 mg, rosuvastatin 40 mg, or pitavastatin 4 mg; simvastatin 80 mg should not be used, as this dose is associated with elevated risk of myositis and rhabdomyolysis. We recommend initiation with maximal potent statin dose in adults with FH because among subjects with FH:
<1/20 achieve the recommended LDL cholesterol targets;
most need to decrease LDL cholesterol by at least 50%;
many receive statin doses insufficient to attain LDL cholesterol targets;
many physicians do not uptitrate statin doses despite suboptimal treatment.
Clinical assessment of efficacy and safety is advisable 4–6 weeks after initiating treatment.
Statins are the drug of first choice because of the huge and robust body of evidence for statin-mediated reduction in major cardiovascular events.6,7,25 The introduction of statins has also reduced CHD events in individuals with FH in observational studies,3,4 such that with treatment before onset of CHD, survival without CHD can be similar to that in the general population (Figure 9).
Figure 9.
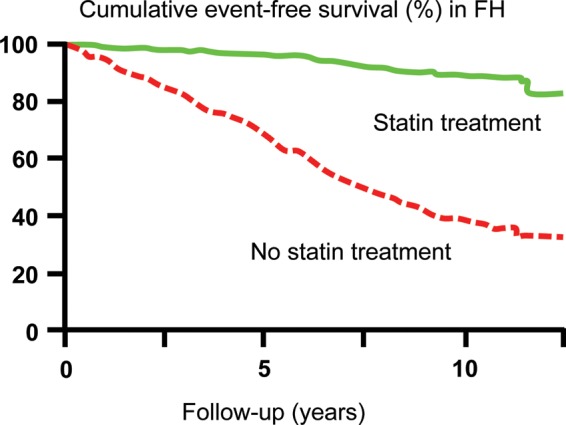
Kaplan–Meier curve estimates of cumulative CHD-free survival among individuals with familial hypercholesterolaemia according to statin treatment (P < 0.001 for difference). Based on 413 and 1537 Dutch subjects with heterozygous familial hypercholesterolaemia on or off statin treatment. CHD, coronary heart disease; FH, familial hypercholesterolaemia. Adapted from Versmissen et al.3
Despite use of the highest doses of potent statins, many subjects with FH will not achieve the LDL cholesterol target with monotherapy alone. Under these conditions, and despite lack of demonstrated clinical benefit in FH of coadministration of the cholesterol absorption inhibitor, ezetimibe, we recommend this agent as an add-on to statin therapy in view of few side effects and high compliance. The statin–ezetimibe combination will decrease LDL cholesterol by 60–70%. For subjects at very high risk with established CHD or type 2 diabetes and with LDL cholesterol >1.8 mmol/L (>70 mg/dL), a bile acid-binding resin (cholestyramine, colestipol, or colesevelam) as a third drug is advised. In some FH patients and in some countries, use of pure niacin (up to 3 g/day) in association with a statin, ezetimibe, or a bile acid-binding resin may be an option for additional reduction of LDL and/or Lp(a). However, niacin in the form of Tredaptive is no longer available.
In FH subjects with elevated triglycerides and low HDL cholesterol or with triglycerides >5.7 mmol/L (>500 mg/dL), maximal potent statin dose combined with fibrates can be considered, in particular fenofibrate given its satisfactory drug–drug interaction profile24 and effect on LDL cholesterol reduction in FH.42 Fenofibrate can also lower LDL cholesterol when triglycerides are normal and may be used if other drugs cannot be tolerated or are unavailable. Details of the efficacy, safety, and management of lipid-lowering drugs are described elsewhere.43
FH individuals with statin intolerance require specialized management to ensure that several different statins have been tested when possible and to combine (depending on the individual case) low dose of statin, ezetimibe, and resins.
In individuals with FH in extreme cases at very high cardiovascular risk with CHD, and with very high LDL cholesterol levels despite drug therapy or because of statin intolerance, adjunctive treatment with lipoprotein apheresis should be considered; this is particularly relevant for children with homozygous FH. Weekly or bi-weekly lipoprotein apheresis can decrease LDL cholesterol and Lp(a) by 50–75% and has clinical benefits in individuals with severe FH.32,33,44–48 Lipoprotein apheresis can be appropriately conducted at specialized lipid clinics, and at centres for haemodialysis and for blood transfusion, and elsewhere. Clinical thresholds for initiation of lipoprotein apheresis may vary between countries.
Children with familial hypercholesterolaemia
In FH, elevated cholesterol is already present at birth and results in early atherosclerotic lesions. Our recommendations in children with this condition rely on intervention trials in children showing good tolerance and efficacy of statins in terms of reduction in LDL cholesterol,49,50 together with reduced progression of subclinical atherosclerosis.51,52
The optimal age range for screening is between 2 and 10 years, as determined by optimal discrimination using cholesterol measurement between children with and without FH. Currently, it is considered unreasonable to start a low-fat diet before age 2, and there are no safety data on the use of statins before age 8–10. On the other hand, the earlier screening and treatment are initiated, the greater the benefit and compliance in the future.52
If total or LDL cholesterol is high, a second lipid profile after 2 or 3 months of dietary guidance, along with other biochemical analyses to exclude secondary hyperlipidaemia (see above) and other risk factors such as Lp(a), should be performed. Once hypercholesterolaemia has been detected in the child, it is important to establish its vertical transmission through a family pedigree (Figure 7), as awareness of the genetic nature may improve the compliance to treatment of both the parents and the child.
For diagnosis in children with one parent with FH, an LDL cholesterol level >3.5 mmol/L (>135 mg/dL) is strongly suggestive. Genetic tests should be performed in all children of parents with FH and a causative mutation, irrespective of whether or not they have high LDL cholesterol; the ethical issues involved in genetic testing of children must also be borne in mind.39 The absence of a positive genetic test in the parents does not exclude FH in a child with high cholesterol (Figure 6), and a clinical diagnosis is likely if a child with LDL cholesterol >3.5 mmol/L has one parent with a DLCN score >5 (Table 1). Importantly, many children in FH families are on a healthy diet and thus have lower LDL cholesterol than expected. In children, xanthomas and corneal arcus are not reliable clinical criteria as they only appear later; however, if present, they are suggestive of homozygous FH. When lifelong drug treatment is under consideration, then genetic demonstration of a causative mutation in LDLR, PCSK9 or APOB is optimal for the diagnosis of FH.
Dietary advice from a certified dietitian/nutritionist given to the parents should start after age 2 of the child. Dietary recommendations are similar to those given to adults with FH; particular caution is however needed to avoid caloric restriction (if weight is normal) and to monitor the growth curve.
Priorities for cholesterol-lowering drugs in children are given earlier. However, it is unknown at what age atherosclerotic lesions become irreversible, only short-term follow-up data are available that low dosages of statins are safe in children, and there is no long-term study evaluating the cardiovascular benefit of cholesterol-lowering drugs in children. Therefore, as is common in paediatrics, the therapeutic decision is based on extrapolation from adult studies and on short-term paediatric studies evaluating the safety and the efficacy of pharmacotherapy on LDL cholesterol lowering or intermediate endpoints.52 Thus, a registry of statin-treated children to collect meaningful follow-up data is urgently needed. Studies in children have shown this medication to be safe when started from age 8 to 10 and, based on the graphical assumption in Figure 8, we therefore recommend initiation of statin therapy at age 8–10, that is, when the diagnosis of FH is supported by a genetic test or by strong clinical arguments including LDL cholesterol >3.5 mmol/L (>135 mg/dL). As mentioned earlier, the LDL cholesterol target in children is <3.5 mmol/L (<135 mg/dL); however, the presence of very high LDL cholesterol or additional cardiovascular risk factors (Figure 8) may lower this target or the age at initiation of statin therapy. Importantly, lipoprotein apheresis should be offered in children with homozygous FH.32,33,44–47 Despite initial enthusiasm, the therapeutic potential of double heart–liver transplantation in children with homozygous FH should be considered with caution.
Cost-effectiveness
Individuals with FH will incur costs to the healthcare system over their lifetime; if unidentified, these may include the cost of the premature CHD they are likely to suffer (Figure 9). If treated however, such costs will include the budget for cholesterol-lowering therapies and for the healthcare professionals that diagnose and treat them. Healthcare economic modelling has demonstrated that there are considerable overall savings in identifying and appropriately treating subjects with FH.53 For individuals in whom the causative mutation has been found, cascade testing of their relatives using genetic testing is highly cost-effective, as roughly 50% will have inherited the mutation. Because of their lifelong burden of LDL cholesterol accumulation (Figure 8), subjects with FH warrant intensive cholesterol-lowering therapy and, even if more expensive agents are used, it remains cost-effective.54
The cost per Life Year Gained for genetic cascade testing and intensive statin therapy in FH is €3–4000, which compares very favourably with mammography for breast cancer screening.55 High-intensity lipid-lowering statin therapy would lead to 101 fewer cardiovascular deaths per 1000 FH individuals treated, and when extrapolating to the 500 million population of the EU (with an estimated 1 000 000 FH subjects), roughly €4700 million could be saved from avoidance of cardiovascular events if all relatives of index cases were identified and treated optimally over a 55-year period, equating to an economy of €86 million per year.56
Novel therapies
Attainment of LDL cholesterol targets over time is imperative in FH subjects, to reduce cumulative lifetime risk40 (Figures 8 and 9). Statin therapy is often inadequate for this goal.10 Novel, well-tolerated therapeutic strategies as add-ons to statin therapy or as sole drugs in case of statin intolerance are therefore essential in FH. New classes of efficacious, LDL- and Lp(a)-lowering agents are currently at advanced stages of development, including therapies targeting PCSK9, anti-sense oligonucleotides targeting APOB, microsomal triglyceride transfer protein inhibitors, and cholesteryl ester transfer protein inhibitors. Additional studies on their long-term safety and efficacy, together with tolerability over time are, however, needed.
Diagnostic and treatment summary
We recommend that most individuals with FH should be treated in primary care, preferably in a family context, while complex cases including children should be referred to specialized lipid or FH clinics. However, as FH management in primary care poses major challenges such as appropriate use of genetic testing, frequent requirement for polypharmacy, specialist knowledge of non-invasive testing, and complex organizational requirements for family cascade screening, ‘shared care’ between primary care and specialized lipid or FH clinics is another attractive option. In most countries, there is an urgent need for education of physicians to deal with FH, to establish networks of Lipid/FH clinics, and to establish laboratories for genetic screening and testing, preferably in a concerted manner (Figure 10).
Figure 10.

Summary of diagnostic and treatment strategies.
Authors’ contributions
EAS Consensus Panel members were nominated by the Co-chairs M.J.C. and H.N.G. to represent expertise across FH clinical management and research from across the World. The Panel met twice, organized and chaired by M.J.C. and H.N.G. The first meeting critically reviewed the literature while the second meeting reviewed additional literature and scrutinized the first draft of the consensus paper. All panel members agreed to conception and design; contributed to interpretation of available data; suggested revisions for this document; and approved the final document before submission.
Funding
Supported by unrestricted educational grants to EAS from Amgen, Aegerion, AstraZeneca, Genzyme, Hoffman-La Roche, Kowa Europe, Novartis, and Sanofi-Aventis/Regeneron. These companies were not present at the Consensus Panel meetings, had no role in the design or content of the Consensus Statement, and had no right to approve or disapprove the final document.
Conflict of interest: Consensus Panel members have received lecture honoraria, consultancy fees and/or research funding from Aegerion (M.J.C., R.D.S., M.A., A.L.C., K.G.P., E.B.), Amgen (M.J.C., H.N.G., F.J.R., L.M., K.G.P.), Pfizer (B.G.N., M.J.C., H.N.G., J.B., G.W., O.S.D., R.D.S., M.A., G.K.H., K.K.R., O.W.), Astra Zeneca (B.G.N., M.J.C., H.N.G., J.B., G.W., O.S.D., L.M., R.D.S., K.K.R., K.G.P., O.W., E.B.), Danone (M.J.C.. E.B., L.M., E.B.), Genzyme (M.J.C., H.N.G., S.H.E., R.D.S., M.A., G.K.H., K.G.P., E.B.), Bristol Myers Squibb (H.N.G., R.D.S., K.G.P.), Merck/Shering Plough (B.G.N., M.J.C., H.N.G., J.B., G.W., O.S.D., A.L.C., M.R.T., L.M., R.D.S., M.A., G.K.H., K.K.R., K.G.P., O.W., E.B.), Abbott (H.N.G., K.R., G.W., L.T., K.G.P.), Boehringer Ingelheim (H.N.G., F.A., M.R.T., K.G.P.), Sanofi-Aventis/Regeneron (B.G.N., M.J.C., H.N.G., J.B., G.W, L.T., O.S.D., M.R.T., F.J.R., L.M., M.A., C.B., K.K.R., K.G.P., O.W., E.B.), Lilly (M.R.T., K.G.P.), Solvay (O.D.S., K.K.R.), Novartis (H.N.G., M.R.T., L.M., K.K.R., K.G.P.), Novo-Nordisk (R.D.S., K.K.R.), Kowa (H.N.G., A.L.C., M.J.C., L.M., M.R.T., K.K.R.), Hoffman-La Roche (M.J.C., M.A., E.B.), Unilever (E.B.), Genfit (E.B.), Kraft (E.B.), and ISIS Pharmaceuticals (B.G.N., F.J.R., K.G.P.).
Acknowledgements
We thank Ms Jane Stock for co-ordination and Michael Livingston for providing numbers for Figure 1.
References
- 1.Goldstein JK, Hobbs HH, Brown MS. Familial hypercholesterolemia. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic & Molecular Bases of Inherited Disease. 8th ed. New York: McGraw-Hill; 2001. pp. 2863–2913. [Google Scholar]
- 2.Austin MA, Hutter CM, Zimmern RL, Humphries SE. Genetic causes of monogenic heterozygous familial hypercholesterolemia: a HuGE prevalence review. Am J Epidemiol. 2004;160:407–420. doi: 10.1093/aje/kwh236. [DOI] [PubMed] [Google Scholar]
- 3.Versmissen J, Oosterveer DM, Yazdanpanah M, Defesche JC, Basart DC, Liem AH, Heeringa J, Witteman JC, Lansberg PJ, Kastelein JJ, Sijbrands EJ. Efficacy of statins in familial hypercholesterolaemia: a long term cohort study. BMJ. 2008;337:a2423. doi: 10.1136/bmj.a2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marks D, Thorogood M, Neil HA, Humphries SE. A review on the diagnosis, natural history, and treatment of familial hypercholesterolaemia. Atherosclerosis. 2003;168:1–14. doi: 10.1016/s0021-9150(02)00330-1. [DOI] [PubMed] [Google Scholar]
- 5.World Health Organization. World Healths Statistics 2012. Internet http://www.who.int/gho/publications/world_health_statistics/2012/en/ 9 October 2012.
- 6.Reiner Z, Catapano AL, De BG, Graham I, Taskinen MR, Wiklund O, Agewall S, Alegria E, Chapman MJ, Durrington P, Erdine S, Halcox J, Hobbs R, Kjekshus J, Filardi PP, Riccardi G, Storey RF, Wood D. ESC/EAS Guidelines for the management of dyslipidaemias: the Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS) Eur Heart J. 2011;32:1769–1818. doi: 10.1093/eurheartj/ehr158. [DOI] [PubMed] [Google Scholar]
- 7.Baigent C, Blackwell L, Emberson J, Holland LE, Reith C, Bhala N, Peto R, Barnes EH, Keech A, Simes J, Collins R. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376:1670–1681. doi: 10.1016/S0140-6736(10)61350-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Minhas R, Humphries SE, Davies D, Lee P, McDowell I, Neil A, Qureshi N, Rowlands P, Seed M, Stracey H, Thorogood M, Watson M, Barbir M, Lucassen A, Parke A, Wierzbicki A, Williams H, Wray R, Shaw E, Turnbull N, DeMott K, Kathoria M, Nunes V, Nherera L, Ritchie G. UK NICE Guideline on Identification and Management of FH (CG71) Internet http://www.nice.org.uk/CG071. 30 March 2013.
- 9.Heiberg A, Berg K. The inheritance of hyperlipoproteinaemia with xanthomatosis. A study of 132 kindreds. Clin Genet. 1976;9:203–233. doi: 10.1111/j.1399-0004.1976.tb01569.x. [DOI] [PubMed] [Google Scholar]
- 10.Benn M, Watts GF, Tybjaerg-Hansen A, Nordestgaard BG. Familial hypercholesterolemia in the Danish general population: prevalence, coronary artery disease, and cholesterol-lowering medication. J Clin Endocrinol Metab. 2012;97:3956–3964. doi: 10.1210/jc.2012-1563. [DOI] [PubMed] [Google Scholar]
- 11.Neil HA, Hammond T, Huxley R, Matthews DR, Humphries SE. Extent of underdiagnosis of familial hypercholesterolaemia in routine practice: prospective registry study. BMJ. 2000;321:148. doi: 10.1136/bmj.321.7254.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scientific Steering Committee on behalf of the Simon Broome Register Group. Mortality in treated heterozygous familial hypercholesterolaemia: implications for clinical management. Atherosclerosis. 1999;142:105–112. [PubMed] [Google Scholar]
- 13.Huijgen R, Vissers MN, Defesche JC, Lansberg PJ, Kastelein JJ, Hutten BA. Familial hypercholesterolemia: current treatment and advances in management. Expert Rev Cardiovasc Ther. 2008;6:567–581. doi: 10.1586/14779072.6.4.567. [DOI] [PubMed] [Google Scholar]
- 14.Rader DJ, Cohen J, Hobbs HH. Monogenic hypercholesterolemia: new insights in pathogenesis and treatment. J Clin Invest. 2003;111:1795–1803. doi: 10.1172/JCI18925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Usifo E, Leigh SE, Whittall RA, Lench N, Taylor A, Yeats C, Orengo CA, Martin AC, Celli J, Humphries SE. Low-density lipoprotein receptor gene familial hypercholesterolemia variant database: update and pathological assessment. Ann Hum Genet. 2012;76:387–401. doi: 10.1111/j.1469-1809.2012.00724.x. [DOI] [PubMed] [Google Scholar]
- 16.Raal FJ, Santos RD. Homozygous familial hypercholesterolemia: current perspectives on diagnosis and treatment. Atherosclerosis. 2012;223:262–268. doi: 10.1016/j.atherosclerosis.2012.02.019. [DOI] [PubMed] [Google Scholar]
- 17.Civeira F, Ros E, Jarauta E, Plana N, Zambon D, Puzo J, Martinez de Esteban JP, Ferrando J, Zabala S, Almagro F, Gimeno JA, Masana L, Pocovi M. Comparison of genetic versus clinical diagnosis in familial hypercholesterolemia. Am J Cardiol. 2008;102:1187–1193. doi: 10.1016/j.amjcard.2008.06.056. 1193. [DOI] [PubMed] [Google Scholar]
- 18.Palacios L, Grandoso L, Cuevas N, Olano-Martin E, Martinez A, Tejedor D, Stef M. Molecular characterization of familial hypercholesterolemia in Spain. Atherosclerosis. 2012;221:137–142. doi: 10.1016/j.atherosclerosis.2011.12.021. [DOI] [PubMed] [Google Scholar]
- 19.Thorsson B, Sigurdsson G, Gudnason V. Systematic family screening for familial hypercholesterolemia in Iceland. Arterioscler Thromb Vasc Biol. 2003;23:335–338. doi: 10.1161/01.atv.0000051874.51341.8c. [DOI] [PubMed] [Google Scholar]
- 20.Huijgen R, Hutten BA, Kindt I, Vissers MN, Kastelein JJ. Discriminative ability of LDL-cholesterol to identify patients with familial hypercholesterolemia: a cross-sectional study in 26,406 individuals tested for genetic FH. Circ Cardiovasc Genet. 2012;5:354–359. doi: 10.1161/CIRCGENETICS.111.962456. [DOI] [PubMed] [Google Scholar]
- 21.Starr B, Hadfield SG, Hutten BA, Lansberg PJ, Leren TP, Damgaard D, Neil HA, Humphries SE. Development of sensitive and specific age- and gender-specific low-density lipoprotein cholesterol cutoffs for diagnosis of first-degree relatives with familial hypercholesterolaemia in cascade testing. Clin Chem Lab Med. 2008;46:791–803. doi: 10.1515/CCLM.2008.135. [DOI] [PubMed] [Google Scholar]
- 22.Civeira F. Guidelines for the diagnosis and management of heterozygous familial hypercholesterolemia. Atherosclerosis. 2004;173:55–68. doi: 10.1016/j.atherosclerosis.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 23.Jarauta E, Junyent M, Gilabert R, Plana N, Mateo-Gallego R, de GE, Cenarro A, Nunez I, Coll B, Masana L, Ros E, Civeira F. Sonographic evaluation of Achilles tendons and carotid atherosclerosis in familial hypercholesterolemia. Atherosclerosis. 2009;204:345–347. doi: 10.1016/j.atherosclerosis.2008.09.040. [DOI] [PubMed] [Google Scholar]
- 24.Chapman MJ, Ginsberg HN, Amarenco P, Andreotti F, Boren J, Catapano AL, Descamps OS, Fisher E, Kovanen PT, Kuivenhoven JA, Lesnik P, Masana L, Nordestgaard BG, Ray KK, Reiner Z, Taskinen MR, Tokgozoglu L, Tybjaerg-Hansen A, Watts GF. Triglyceride-rich lipoproteins and high-density lipoprotein cholesterol in patients at high risk of cardiovascular disease: evidence and guidance for management. Eur Heart J. 2011;32:1345–1361. doi: 10.1093/eurheartj/ehr112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perk J, De BG, Gohlke H, Graham I, Reiner Z, Verschuren WM, Albus C, Benlian P, Boysen G, Cifkova R, Deaton C, Ebrahim S, Fisher M, Germano G, Hobbs R, Hoes A, Karadeniz S, Mezzani A, Prescott E, Ryden L, Scherer M, Syvanne M, Scholte Op Reimer WJ, Vrints C, Wood D, Zamorano JL, Zannad F. European Guidelines on cardiovascular disease prevention in clinical practice (version 2012): The Fifth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of nine societies and by invited experts) Atherosclerosis. 2012;223:1–68. doi: 10.1016/j.atherosclerosis.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 26.Watts GF, Sullivan DR, Poplawski N, van BF, Hamilton-Craig I, Clifton PM, O'Brien R, Bishop W, George P, Barter PJ, Bates T, Burnett JR, Coakley J, Davidson P, Emery J, Martin A, Farid W, Freeman L, Geelhoed E, Juniper A, Kidd A, Kostner K, Krass I, Livingston M, Maxwell S, O'Leary P, Owaimrin A, Redgrave TG, Reid N, Southwell L, Suthers G, Tonkin A, Towler S, Trent R. Familial hypercholesterolaemia: a model of care for Australasia. Atheroscler Suppl. 2011;12:221–263. doi: 10.1016/j.atherosclerosissup.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 27.Wiklund O, Angelin B, Olofsson SO, Eriksson M, Fager G, Berglund L, Bondjers G. Apolipoprotein(a) and ischaemic heart disease in familial hypercholesterolaemia. Lancet. 1990;335:1360–1363. doi: 10.1016/0140-6736(90)91242-3. [DOI] [PubMed] [Google Scholar]
- 28.Kraft HG, Lingenhel A, Raal FJ, Hohenegger M, Utermann G. Lipoprotein(a) in homozygous familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2000;20:522–528. doi: 10.1161/01.atv.20.2.522. [DOI] [PubMed] [Google Scholar]
- 29.Kamstrup PR, Tybjaerg-Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301:2331–2339. doi: 10.1001/jama.2009.801. [DOI] [PubMed] [Google Scholar]
- 30.Nordestgaard BG, Chapman MJ, Ray K, Boren J, Andreotti F, Watts GF, Ginsberg H, Amarenco P, Catapano A, Descamps OS, Fisher E, Kovanen PT, Kuivenhoven JA, Lesnik P, Masana L, Reiner Z, Taskinen MR, Tokgozoglu L, Tybjaerg-Hansen A. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31:2844–2853. doi: 10.1093/eurheartj/ehq386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jansen AC, van Aalst-Cohen ES, Tanck MW, Trip MD, Lansberg PJ, Liem AH, van Lennep HW, Sijbrands EJ, Kastelein JJ. The contribution of classical risk factors to cardiovascular disease in familial hypercholesterolaemia: data in 2400 patients. J Intern Med. 2004;256:482–490. doi: 10.1111/j.1365-2796.2004.01405.x. [DOI] [PubMed] [Google Scholar]
- 32.Stefanutti C. Treatment of severe genetic dyslipidemia: where are we going? Ther Apher Dial. 2013;17:122–123. doi: 10.1111/1744-9987.12028. [DOI] [PubMed] [Google Scholar]
- 33.Stefanutti C, Morozzi C, Di GS. Italian multicenter study on low-density lipoprotein apheresis Working Group 2009 survey. Ther Apher Dial. 2013;17:169–178. doi: 10.1111/j.1744-9987.2012.01142.x. [DOI] [PubMed] [Google Scholar]
- 34.Thanassoulis G, Campbell CY, Owens DS, Smith JG, Smith AV, Peloso GM, Kerr KF, Pechlivanis S, Budoff MJ, Harris TB, Malhotra R, O'Brien KD, Kamstrup PR, Nordestgaard BG, Tybjaerg-Hansen A, Allison MA, Aspelund T, Criqui MH, Heckbert SR, Hwang SJ, Liu Y, Sjogren M, van der Pals J, Kalsch H, Muhleisen TW, Nothen MM, Cupples LA, Caslake M, Di AE, Danesh J, Rotter JI, Sigurdsson S, Wong Q, Erbel R, Kathiresan S, Melander O, Gudnason V, O'Donnell CJ, Post WS. Genetic associations with valvular calcification and aortic stenosis. N Engl J Med. 2013;368:503–512. doi: 10.1056/NEJMoa1109034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goldberg AC, Hopkins PN, Toth PP, Ballantyne CM, Rader DJ, Robinson JG, Daniels SR, Gidding SS, de Ferranti SD, Ito MK, McGowan MP, Moriarty PM, Cromwell WC, Ross JL, Ziajka PE. Familial hypercholesterolemia: screening, diagnosis and management of pediatric and adult patients: clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 2011;5:S1–S8. doi: 10.1016/j.jacl.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 36.Greenland P, Alpert JS, Beller GA, Benjamin EJ, Budoff MJ, Fayad ZA, Foster E, Hlatky MA, Hodgson JM, Kushner FG, Lauer MS, Shaw LJ, Smith SC, Jr, Taylor AJ, Weintraub WS, Wenger NK, Jacobs AK, Smith SC, Jr, Anderson JL, Albert N, Buller CE, Creager MA, Ettinger SM, Guyton RA, Halperin JL, Hochman JS, Kushner FG, Nishimura R, Ohman EM, Page RL, Stevenson WG, Tarkington LG, Yancy CW. ACCF/AHA guideline for assessment of cardiovascular risk in asymptomatic adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2010;56:e50–103. doi: 10.1016/j.jacc.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 37.Kusters DM, de BC, Widhalm K, Guardamagna O, Bratina N, Ose L, Wiegman A. Paediatric screening for hypercholesterolaemia in Europe. Arch Dis Child. 2012;97:272–276. doi: 10.1136/archdischild-2011-300081. [DOI] [PubMed] [Google Scholar]
- 38.Vuorio AF, Turtola H, Kontula K. Neonatal diagnosis of familial hypercholesterolemia in newborns born to a parent with a molecularly defined heterozygous familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. 1997;17:3332–3337. doi: 10.1161/01.atv.17.11.3332. [DOI] [PubMed] [Google Scholar]
- 39.Genetic testing in asymptomatic minors: recommendations of the European Society of Human Genetics. Eur J Hum Genet. 2009;17:720–721. doi: 10.1038/ejhg.2009.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Horton JD, Cohen JC, Hobbs HH. PCSK9: a convertase that coordinates LDL catabolism. J Lipid Res. 2009;50(Suppl):S172–S177. doi: 10.1194/jlr.R800091-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Broekhuizen K, Jelsma GJ, van Poppel NM, Koppes LL, Brug J, van MW. Is the process of delivery of an individually tailored lifestyle intervention associated with improvements in LDL cholesterol and multiple lifestyle behaviours in people with Familial Hypercholesterolemia? BMC Public Health. 2012;12:348. doi: 10.1186/1471-2458-12-348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weisweiler P. Low-dose colestipol plus fenofibrate: effects on plasma lipoproteins, lecithin:cholesterol acyltransferase, and postheparin lipases in familial hypercholesterolemia. Metabolism. 1989;38:271–274. doi: 10.1016/0026-0495(89)90086-3. [DOI] [PubMed] [Google Scholar]
- 43.Robinson JG, Goldberg AC. Treatment of adults with familial hypercholesterolemia and evidence for treatment: recommendations from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 2011;5:S18–S29. doi: 10.1016/j.jacl.2011.03.451. [DOI] [PubMed] [Google Scholar]
- 44.Kucukcongar A, Yenicesu I, Tumer L, Kasapkara CS, Ezgu FS, Pasaoglu O, Demirtas C, Celik B, Dilsiz G, Hasanoglu A. Apheresis-inducible cytokine pattern change in children with homozygous familial hypercholesterolemia. Transfus Apher Sci. 2013;48:391–396. doi: 10.1016/j.transci.2013.04.024. [DOI] [PubMed] [Google Scholar]
- 45.Fernandez-Fuertes LF, Tapia MM, Nieves PI, Novoa Mogollon FJ, Diaz CJ. Low-density lipoprotein apheresis using double filtration plasmapheresis: 27-month use in a child with homozygous familial hypercholesterolemia. Ther Apher Dial. 2010;14:484–485. doi: 10.1111/j.1744-9987.2010.00839.x. [DOI] [PubMed] [Google Scholar]
- 46.Stefanutti C, Di GS, Vivenzio A, Colloridi V, Bosco G, Berni A, Rabbone I, Cerutti F, Bertolini S. Low-density lipoprotein apheresis in a patient aged 3.5 years. Acta Paediatr. 2001;90:694–701. doi: 10.1080/080352501750258793. [DOI] [PubMed] [Google Scholar]
- 47.Stefanutti C, Vivenzio A, Ferraro PM, Morozzi C, Belotherkovsky D. Apheresis-inducible cytokine pattern change in severe, genetic dyslipidemias. Cytokine. 2011;56:835–841. doi: 10.1016/j.cyto.2011.08.024. [DOI] [PubMed] [Google Scholar]
- 48.Schuff-Werner P, Fenger S, Kohlschein P. Role of lipid apheresis in changing times. Clin Res Cardiol Suppl. 2012;7:7–14. doi: 10.1007/s11789-012-0049-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Avis HJ, Vissers MN, Stein EA, Wijburg FA, Trip MD, Kastelein JJ, Hutten BA. A systematic review and meta-analysis of statin therapy in children with familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2007;27:1803–1810. doi: 10.1161/ATVBAHA.107.145151. [DOI] [PubMed] [Google Scholar]
- 50.Arambepola C, Farmer AJ, Perera R, Neil HA. Statin treatment for children and adolescents with heterozygous familial hypercholesterolaemia: a systematic review and meta-analysis. Atherosclerosis. 2007;195:339–347. doi: 10.1016/j.atherosclerosis.2006.09.030. [DOI] [PubMed] [Google Scholar]
- 51.Wiegman A, Hutten BA, de GE, Rodenburg J, Bakker HD, Buller HR, Sijbrands EJ, Kastelein JJ. Efficacy and safety of statin therapy in children with familial hypercholesterolemia: a randomized controlled trial. JAMA. 2004;292:331–337. doi: 10.1001/jama.292.3.331. [DOI] [PubMed] [Google Scholar]
- 52.Rodenburg J, Vissers MN, Wiegman A, van Trotsenburg AS, van der Graaf A, de GE, Wijburg FA, Kastelein JJ, Hutten BA. Statin treatment in children with familial hypercholesterolemia: the younger, the better. Circulation. 2007;116:664–668. doi: 10.1161/CIRCULATIONAHA.106.671016. [DOI] [PubMed] [Google Scholar]
- 53.Marks D, Wonderling D, Thorogood M, Lambert H, Humphries SE, Neil HA. Cost effectiveness analysis of different approaches of screening for familial hypercholesterolaemia. BMJ. 2002;324:1303. doi: 10.1136/bmj.324.7349.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nherera L, Calvert NW, Demott K, Humphries SE, Neil HA, Minhas R, Thorogood M. Cost-effectiveness analysis of the use of a high-intensity statin compared to a low-intensity statin in the management of patients with familial hypercholesterolaemia. Curr Med Res Opin. 2010;26:529–536. doi: 10.1185/03007990903494934. [DOI] [PubMed] [Google Scholar]
- 55.Nherera L, Marks D, Minhas R, Thorogood M, Humphries SE. Probabilistic cost-effectiveness analysis of cascade screening for familial hypercholesterolaemia using alternative diagnostic and identification strategies. Heart. 2011;97:1175–1181. doi: 10.1136/hrt.2010.213975. [DOI] [PubMed] [Google Scholar]
- 56.Nherera LM. Saving lives, saving families: the health, social and economic advantages of detecting and treating familial hypercholesterolaemia (FH) Economics Chapter: Estimating the benefits from treatment and increasing the implementation of cascading screening Internet http://heartuk.org.uk/files/uploads/documents/HUK_HealthEconomics_FINAL2012_2702.pdf. 17 December 2012.