

Abstract
Dyslipidemia has been frequently observed among individuals infected with human immunodeficiency virus type 1 (HIV-1), and factors related to HIV-1, the host, and antiretroviral therapy (ART) are involved in this phenomenon. This study reviews the roles of genetic polymorphisms, HIV-1 infection, and highly active antiretroviral therapy (HAART) in lipid metabolism. Lipid abnormalities can vary according to the HAART regimen, such as those with protease inhibitors (PIs). However, genetic factors may also be involved in dyslipidemia because not all patients receiving the same HAART regimen and with comparable demographic, virological, and immunological characteristics develop variations in the lipid profile. Polymorphisms in a large number of genes are involved in the synthesis of structural proteins, and enzymes related to lipid metabolism account for variations in the lipid profile of each individual. As some genetic polymorphisms may cause dyslipidemia, these allele variants should be investigated in HIV-1-infected patients to identify individuals with an increased risk of developing dyslipidemia during treatment with HAART, particularly during therapy with PIs. This knowledge may guide individualized treatment decisions and lead to the development of new therapeutic targets for the treatment of dyslipidemia in these patients.
1. Introduction
Serum lipids have a multifactorial etiology that is determined by a large number of environmental and genetic factors [1]. Genetic and dietary factors influence serum cholesterol concentration, but detailed mechanisms of their interactions are not well known. An increase in dietary cholesterol intake raises serum cholesterol concentrations in some but not all subjects.
Human immunodeficiency virus type 1 (HIV-1) infected patients develop dyslipidemia, resulting in a highly atherogenic lipid profile with increased levels of total cholesterol, low-density lipoprotein cholesterol (LDL-C), and triglycerides (TG) and decreased levels of high-density lipoprotein cholesterol (HDL-C) [2]. The pathogenesis of dyslipidemia in HIV-1 infection is complex and involves factors related to the virus, the host, and to the antiretroviral therapy (ART). Moreover, HIV-1 infection and ART are associated with accelerated atherosclerosis and an increased number of cases of myocardial infarction [3].
Highly active antiretroviral therapy (HAART) consists of a combination of drugs that inhibit different stages of viral replication, and it is divided mechanistically into six classes [3] based on whether it targets the viral lifecycle or viral enzymes: nucleoside reverse transcriptase inhibitors (NRTIs), nonnucleoside reverse transcriptase inhibitors (NNRTIs), protease inhibitors (PIs), fusion inhibitor (enfuvirtide or T-20), entry inhibitor chemokine receptor 5 (CCR5) antagonist maraviroc, and HIV-1 integrase strand transfer inhibitor [4, 5].
The introduction of HAART in 1996 dramatically reduced the mortality and morbidity in HIV-1-infected patients, leading to prolonged and improved quality of life and making HIV-1 infection a manageable chronic disease [6]. HAART uses combination formulations containing at least three antiretroviral drugs that are extremely effective in reducing the plasma viral load of HIV-1 RNA to undetectable levels [4, 7, 8].
However, it is increasingly clear that HIV-1-infected patients exhibit an increased risk of developing noninfectious consequences of HIV-1 infection over time. In the last few years, lipodystrophy (characterized by body fat redistribution), insulin resistance, central adiposity, and dyslipidemia have been reported in HIV-1-infected patients, and their relationships with antiretroviral drugs and HIV-1 infection are the subject of global debate and research [9]. Moreover, HAART can induce severe metabolic complications, such as insulin resistance, metabolic syndrome, lipodystrophy, and cardiovascular diseases. The metabolic effects of HAART and the risk of premature and accelerated atherosclerosis in HIV-1-infected patients are well recognized. These clinical conditions have significantly high prevalence in patients infected with HIV-1 that are treated with these drugs [10].
The type and severity of lipid abnormalities vary according to the HAART regimen used. However, genetic factors may be involved in dyslipidemia because not all patients exposed to same HAART regimen and comparable demographic, virological, and immunological characteristics develop lipid profile variations [11–13].
Many polymorphic variants of the genes that regulate lipid metabolism are present in humans, and more than 400 genes are candidate regulators of lipid exchange. Carriers of abnormal alleles exhibit a high risk for obesity and its associated complications, and therefore there is the interest in the association between dyslipidemia, adiposity, and other diseases with different genotypes. The genes involved in the leptin-melanocortin system of regulation of energy metabolism, protein carriers of lipids and cholesterol in the blood, and enzyme-splitting lipids are of particular interest [14].
Genetic variations of enzymes, receptors, and apolipoproteins (apo), which are essential to LDL-C metabolism, are partially involved in the regulation of serum LDL-C and total cholesterol [15]. Recently, the genetic components of dyslipidemia have been intensively investigated. Variations in a large number of genes involved in the synthesis of structural proteins and enzymes associated with lipid metabolism account for variations in the lipid profile of each individual [1].
Genetic variations that occur at a frequency of more than 1% in a study population are called genetic polymorphisms. The genetic basis for these variations can be a single nucleotide change in the DNA sequence, known as single nucleotide polymorphisms (SNPs), insertions or deletions (indels) of one or more base pairs [16], repeats of a large number of nucleotides (variable number of tandem repeats (VNTR) or minisatellite), and repeats of a small number of nucleotides (short tandem repeat (STR) or microsatellite). SNPs are the most common type of sequence variation in the human genome. The 10 to 30 million SNPs in humans represent 90% of all sequence variations [17].
The effect of a polymorphism depends on its interactions with environmental factors that predispose patients to dyslipidemia, such as being overweight, physical inactivity, or smoking [18–20].
There are several factors that can trigger the atherogenic process, including dyslipidemia, smoking, hypertension, diabetes mellitus, physical inactivity, obesity, and a history of premature atherosclerotic disease. However, dyslipidemia is a major risk factor for developing coronary artery disease (CAD) [21].
Among the genetic factors associated with CAD are variations in the genetic loci responsible for the lipoprotein structure and metabolism and the low-density lipoprotein receptor (LDLR), which may contribute to the development of CAD. Some of these genetic variations are associated with increased serum levels of lipids, and therefore, they may be associated with a high risk of CAD [15, 22, 23]. There is a direct relationship between the onset of CAD and high LDL-C because these particles contribute to atherosclerotic plaques [24]. The opposite effect is observed when HDL-C is high. This circulating lipoprotein has the protective effect of reversing cholesterol transport and promotes a set of anti-inflammatory, antioxidant, and anticoagulant actions that inhibit atherosclerosis [25].
CAD is the main cause of mortality in many parts of the industrialized world [26]. In Brazil, CAD is the major cause of mortality and morbidity in women over the age of 40 or 50 years [27]. Hence, the early identification of subjects at risk of developing CAD is an important public health issue. Salazar et al. [28] showed that Brazilian women with CAD had elevated total serum cholesterol, TG, and LDL-C concentrations. These results confirm the well-known association between CAD and high lipid concentration. According to Salazar et al. [23], common DNA polymorphisms in genes associated with lipid metabolism are potentially important genetic markers of variation in the plasma lipid profile and thus susceptibility or resistance to CAD.
Myocardial infarction, angina pectoris, and ischemic stroke resulting from atherosclerosis are the main causes of morbidity and mortality in adults in developed and developing countries [21]. A study showed that 38% of men and 42% of women in Brazil exhibit elevated serum cholesterol [29]. Lipid profile data and the study of polymorphisms in genes encoding structural proteins and enzymes regulating lipid metabolism reveal the prevalence of dyslipidemia in a population, allowing targeted intervention for the control and prevention of atherosclerotic diseases [1, 30].
The considerable improvement in the rates of morbidity and mortality among HIV-1-infected patients due to HAART has progressively transformed the infection into a chronic disease [6, 7, 31, 32]. Given the increased life expectancy of these patients, a systematic evaluation of their risk for early cardiovascular events is important [10].
Considering the importance of determining the contribution of genetic polymorphisms to the multifactorial etiology of dyslipidemia, this study reviews the genetic polymorphisms associated with changes in serum lipids and assesses the role of these polymorphisms in lipid changes in patients with HIV-1.
2. Dyslipidemia in HIV-1-Infected Patients
Dyslipidemia is frequently observed in HIV-1-infected patients. Its pathogenesis is complex and includes factors related to the virus, the host, and the ART. Antiretroviral drugs are associated with a state of accelerated atherosclerosis and an increase in the number of cases of myocardial infarction [3]. Cardiovascular reactions are diverse, due to several factors, such as the HIV-1 infection itself, autoimmunity, immune response against other viral infections, neoplasms, prolonged immunosuppression, malnutrition, drug cardiotoxicity [33, 34], and hormonal changes [35].
2.1. The Role of HIV-1 Infection
HIV-1-associated dyslipidemia was recognized for years before the widespread use of PI-based HAART [36, 37]. Viremia-associated dyslipidemia is characterized by decreased plasma concentrations of total cholesterol, LDL-C, and HDL-C and elevated plasma TG [38–40]. Low HDL-C is correlated with immune activation early in the course of HIV-1 infection [41], the repercussions of which may extend beyond atherosclerosis because of the numerous functions of HDL-C, including antioxidant and anti-inflammatory activities [42–45]. HIV-1 is also associated with an increase in acute phase HDL that lacks the normal atheroprotective functions [46].
Cholesterol is critical for several steps in HIV-1 replication. HIV-1 decreases plasma HDL-C by impairing the cholesterol-dependent efflux transporter ATP-binding cassette protein A1 (ABCA1) in human macrophages, a condition that is highly atherogenic [47]. Additionally, the inflammation stimulates endothelial lipase and certain acute phase proteins, such as serum amyloid A. The plasma level of this enzyme in humans is inversely associated with HDL-C, and the acute phase proteins accelerate the removal of HDL-C by macrophages [45].
The dyslipidemia in HIV-1-infected patients resembles that observed in other chronic infections [48]. The chronic inflammatory processes are characterized by the production of proinflammatory cytokines, such as tumor necrosis factor α (TNFα) and interferon α (IFNα), resulting in the impaired clearance of TG-rich lipoproteins and insulin resistance [49]. Moreover, the nutritional state of HIV-1-infected patients, who may undergo weight loss and protein depletion, might contribute to reduced total plasma cholesterol, HDL-C, and LDL-C levels [38, 50].
Figure 1 illustrates several effects of HIV-1 infection on lipid metabolism and regulation.
Figure 1.
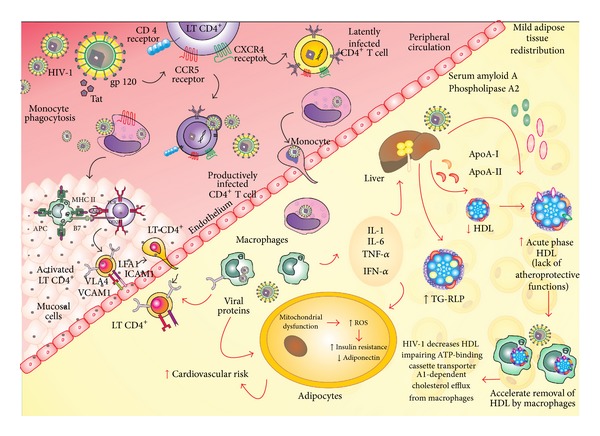
At tissues, human immunodeficiency virus type 1 (HIV-1) infects macrophages using the CD4 as receptor and the CCR5 as coreceptor and induces the local immune response. At peripheral circulation, HIV-1 infects Th1 CD4+ cells, particularly by the coreceptor CXCR4 that persists latently infected or becomes a productively infected cell. The viral proteins induce an proinflammatory response in peripheral circulation and in the tissues and decrease plasma high-density lipoprotein cholesterol (HDL-C) by impairing the cholesterol-dependent efflux transporter ATP-binding cassette protein A1 (ABCA1) in human macrophages, a condition that is highly atherogenic. Additionally, the viral proteins and the proinflammatory cytokines interleukin 1 (IL-1), interleukin 6 (IL-6), tumor necrosis factor α (TN-Fα), and interferon α (IFN-α) stimulate endothelial lipase and certain acute phase proteins, such as serum amyloid A. The viral proteins also exert effects on the adipocytes resulting mitochondrial dysfunction, reactive oxygen species (ROS) production, and insulin resistance and decrease adiponectin. The chronic inflammatory processes increase the production of these proinflammatory cytokines, resulting in the impaired clearance of triglyceride-rich lipoproteins (TG-RLP) and insulin resistance. All these mechanisms increase the risk of cardiovascular diseases in the HIV-1-infected individuals.
2.2. The Role of ART
HAART reduces the frequency of opportunistic infections and the number of AIDS-related deaths [6]. However, despite the improvements in quality of life and increased life expectancy gained with the continuous use of HAART, metabolic disorders characterized by hyperglycemia, dyslipidemia, and changes in the distribution of body fat (lipodystrophy) have been observed in HIV-1 seropositive patients [51].
The pathogenesis of HAART-related dyslipidemia is multifactorial and involves various drug-induced effects, chronic inflammatory status, hormonal influences, genetic predisposition, and HIV-1 infection itself [52].
The dyslipidemia associated with HAART is characterized by decreased plasma HDL-C and increased total cholesterol, TG, and LDL-C, which together constitute a highly atherogenic lipid profile [53].
HAART-related dyslipidemia appears mainly with the use of PIs. PIs may increase the hepatic synthesis of TG, VLDL-C, and to a lesser extent, cholesterol. Additionally, these drugs impair the hydrolysis of TG-rich lipoproteins by lipase, reduce free fatty acid trapping, and interfere with normal postprandial free fatty acid metabolism [54].
The treatment of HIV-1-infected patients is related to lipodystrophy, and dyslipidemia primarily affects those who use PIs. According to Carr et al. [55] and Chi et al. [56], over 60% of patients who are treated with PIs develop metabolic changes, such as hyperlipidemia, endothelial dysfunction, hyperglycemia, and central obesity. Persistent dyslipidemia in HIV-1-infected patients appears to be associated with increased cardiovascular risk, with a relative rate of myocardial infarction of 1.2 per year of PI exposure [57, 58].
One proposed mechanism of PI-induced dyslipidemia is based on the structural similarity between the catalytic region of HIV-1 protease and the LDL-receptor-related protein (LRP). This receptor is a member of the LDLR superfamily and participates in lipid metabolism. LRP normally binds to lipoprotein lipase (LPL) on the capillary endothelium, which hydrolyzes fatty acids from TG to promote free fatty acid storage in adipocytes. PIs bind to LRP due to this structural similarity and interfere with LRP-LPL complex formation; as a result, they reduce the adipose storage capacity and increase plasma TG-rich lipoproteins [59].
PI-induced dyslipidemia is also based upon the structural similarity with the amino acid sequence of the C-terminal region of cytoplasmic retinoic acid-binding protein type 1 (CRABP-1). During normal lipid metabolism, CRABP-1 converts retinoic acid to cis-9-retinoic acid, which binds the retinoid X receptor-peroxisome proliferator-activated receptor γ (RXR-PPARγ) heterodimer found in adipocyte nuclei, inhibiting adipocyte apoptosis and stimulating adipocyte proliferation and differentiation. PIs likely bind to CRABP-1, increasing apoptosis and diminishing the proliferation of peripheral adipocytes [59, 60].
PIs also suppress the proteasome-mediated degradation of sterol regulatory element binding proteins (nSREBPs) in the liver and adipocytes. These transcription factors stimulate fatty acid and TG synthesis in the liver and adipose tissue and control several steps of cholesterol synthesis. The hepatic accumulation of nSREBPs increases TG and cholesterol biosynthesis, whereas accumulation in adipose tissue causes insulin resistance and reduced leptin expression and lipodystrophy [61].
In vitro, PIs and NRTIs increase the expression and secretion of proinflammatory cytokines, such as TNF-α, interleukin 6 (IL-6), and interleukin 1β (IL-1β), that are involved in altered adipocyte functions and decreased adiponectin. These alterations are also observed in fat and serum from HIV-1-patients with lipodystrophy that are treated with these drugs [62]. Upon entry into the cell, NRTIs are metabolized to the active triphosphorylated form and can be utilized as substrates by the mitochondrial DNA polymerase γ. Subsequently, they may inhibit mitochondrial DNA (mtDNA) replication and/or increase the number of mutations in mtDNA. This can lead to mtDNA depletion, the disruption of oxidative phosphorylation, decreases in ATP production, increases in reactive oxygen species, and, ultimately, inappropriate mitochondrial and cellular toxicity.
HAART-related dyslipidemia may involve genetic predisposition, as not all patients taking HAART develop comparable metabolic disturbances [48]. In a study of 745 HIV-infected participants, Rotger et al. [30] demonstrated that 42 SNPs of genome-wide contribute to the development of dyslipidemia independent of other genetic variables, HAART, underlying conditions, sex, age, ethnicity, and HIV disease parameters. The genetic background alone explained up to 7.6% of lipid variation in HIV-infected patients (7.6% non-HDL cholesterol, 6.2% HDL-C, and 6.8% TG), and HAART alone explained up to 6.2% of lipid variation (3.9% non-HDL cholesterol, 1.5% HDL-C, and 6.2% TG). An individual with the most dyslipidemic antiretroviral and genetic background risk factors exhibits three- to fivefold increased risk of sustained dyslipidemia compared with an individual with the fewest dyslipidemic therapy and genetic background risk factors.
Figures 2 and 3 illustrate the main mechanisms involved in dyslipidemia associated with the PI and NRTI ART regimens, respectively.
Figure 2.
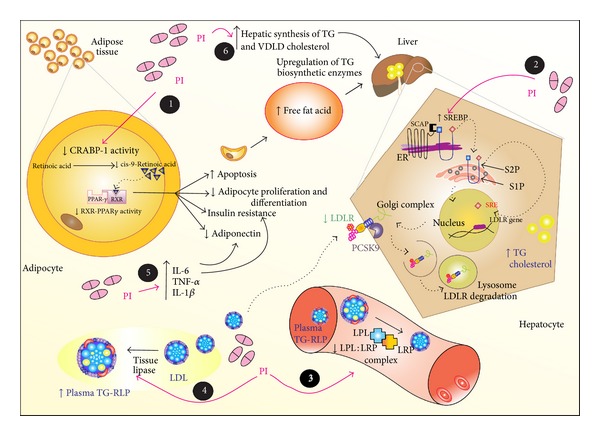
The dyslipidemia associated with protease inhibitor (PI) is characterized by decreased plasma high-density lipoprotein cholesterol (HDL-C) and increased total cholesterol, triglyceride (TG), and low-density lipoprotein cholesterol (LDL-C), which together constitute a highly atherogenic lipid profile. Several mechanisms are proposed such that: (1) the PI-induced dyslipidemia is based upon the structural similarity with the amino acid sequence of the C-terminal region of cytoplasmic retinoic acid-binding protein type 1 (CRABP-1). The PI likely binds to CRABP-1, increasing apoptosis and diminishing the proliferation of peripheral adipocytes; (2) PI suppresses the proteasome-mediated degradation of sterol regulatory element binding proteins (nSREBP) in the liver and adipocytes. These transcription factors stimulate fatty acid and TG synthesis in the liver and adipose tissue and control several steps of cholesterol synthesis. The hepatic accumulation of nSREBP increases TG and cholesterol biosynthesis, whereas accumulation in adipose tissue causes insulin resistance reduced leptin expression and lipodystrophy; (3 and 4) PI-induced dyslipidemia is also based on the structural similarity between the catalytic region of HIV-1 protease and the LDL-receptor-related protein (LRP) and interferes with LRP-LPL complex formation, as a result it reduces the adipose storage capacity and increases plasma TG-rich lipoproteins; (5) PI also increases the expression and secretion of proinflammatory cytokines, such as tumor necrosis factor alpha (TNF-α), interleukin 6 (IL-6), and interleukin 1β (IL-1β), which are involved in altered adipocyte functions and decreased adiponectin; and (6) PI increases the hepatic synthesis of TG, very-low density lipoprotein cholesterol (VLDL-C), and to a lesser extent, cholesterol.
Figure 3.
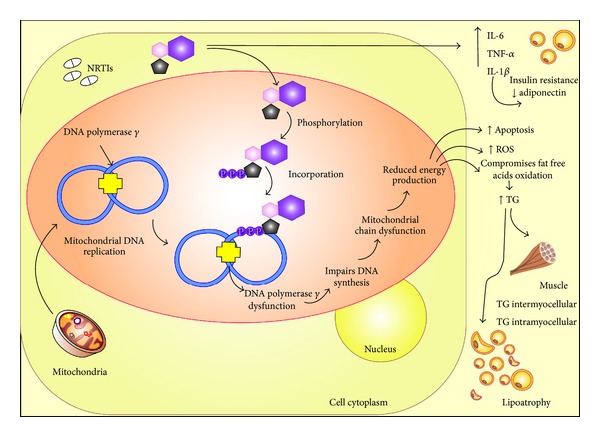
Some mechanisms are proposed to explain the effects of nucleoside reverse transcriptase inhibitors (NRTIs) in the lipid profile of human immunodeficiency virus type 1- (HIV-1-) infected individuals treated with this class of antiretroviral. (1) NRTIs increase the expression and secretion of proinflammatory cytokines, such as tumor necrosis factor alpha (TNF-α), interleukin 6 (IL-6), and interleukin 1β (IL-1β), that are involved in altered adipocyte function, insulin resistance, and adiponectin expression; (2) Upon entry into the cell, NRTIs are metabolized to the active triphosphorylated form and can be used as substrates by the mitochondrial DNA polymerase γ. Subsequently, they may inhibit mitochondrial DNA (mtDNA) replication and/or increase the number of mutations in mtDNA. This effect can lead to mtDNA depletion, the disruption of oxidative phosphorylation, decrease in ATP production, increase in reactive oxygen species (ROS), and, ultimately, inappropriate mitochondrial and cellular toxicity.
3. Genetic Polymorphisms Associated with Dyslipidemia
Polymorphisms in genes associated with dyslipidemia in patients with HIV-1 infection, either treated with ART or untreated, are reviewed.
3.1. Polymorphisms in the LDLR Gene
The LDLR plays a major role in the removal of LDL-C particles from the blood, which, in turn, regulates cholesterol homeostasis. The LDLR modulates plasma levels of LDL-C by regulating LDL-C particle uptake by the liver. It also delivers cholesterol to the adrenal gland and gonads for steroid hormone synthesis and to the liver for bile acid synthesis [63].
Many mutations in the LDLR gene have been identified in patients with familial hypercholesterolemia (FH) [64–66]. Individuals with these mutations exhibit plasma cholesterol concentrations that are elevated twofold or more above normal concentrations and have an increased risk of developing atherosclerosis and CAD [63]. Considering the crucial role of LDLR in cholesterol homeostasis, SNPs in the LDLR gene may also contribute to the variation in plasma cholesterol levels in the general population [23].
Located on chromosome 19p13.2, the LDLR gene comprises 18 exons and 17 introns and encodes a protein of 839 amino acids [67]. More than 1,288 different variants in the LDLR gene have been reported in FH patients as follows: 55% exonic substitutions, 22% exonic small rearrangements (<100 bp), 11% large rearrangements (>100 bp), 2% promoter variants, 10% intronic variants, and 1 variant in the 3′ untranslated sequence [68].
The polymorphic nature of the LDLR gene has been demonstrated by its restriction fragment length polymorphisms (RFLPs) [35, 69]. The AvaII (T20001C, rs5925), HincII (C16730T, rs688) [23], and PvuII (C>T, intron 15) polymorphisms in LDLR are associated with differences in serum lipid concentrations in Brazilian subjects with high risk for CAD [15].
Salazar et al. [23] investigated the effects of LDLR gene polymorphisms at the AvaII site in exon 13 (T20001C, rs5925) and the HincII site in exon 12 (C16730T, rs688) on circulating lipids of 170 unrelated white individuals presenting a lipid profile with high risk for coronary heart disease (HRG) and 130 controls. CHD subjects showed a higher frequency of the AvaII (A+) and HincII (H+) alleles compared with controls, and the frequency of the A+A+ (AvaII) and H+H+ (HincII) genotypes was greater in the HRG group than in the control group (32 versus 16% and 32 versus 18%, resp.). Moreover, in the HRG group, the A+A+ and H+H+ genotypes were associated with high concentrations of total serum cholesterol and LDL-C (P = 0.0001). Interestingly, neither the AvaII (rs5925) nor HincII (rs688) polymorphism was observed to affect serum lipid profiles in control individuals [23]. The strong association between A+A+ (AvaII) and H+H+ (HincII) genotypes with high total cholesterol and circulating LDL-C levels shows that LDLR genetic polymorphisms affect cholesterol levels in individuals with a high risk of CAD. Additionally, common polymorphisms in the LDLR gene are associated with inter-individual differences in plasma LDL-C levels in normal and hypercholesterolemic subjects [70–73].
The PvuII intron 15 polymorphism is linked to other variations in LDLR that structurally alter the receptor activity or alter its function in a regulatory manner [73]. A PvuII intron 15 polymorphism of the LDLR gene is associated with differences in LDL-C concentration in normal and hypercholesterolemic individuals from different countries [74, 75]. Salazar et al. [15] demonstrated the influence of PvuII intron 15 polymorphisms of LDLR on serum lipid profiles in individuals with low or high risk for CAD (HRG). The authors analyzed 128 white subjects with lipid profiles suggesting HRG and 100 white normolipidemic individuals (controls). The P1P1 genotype frequency for the PvuII intron 15 polymorphism (homozygous for the absence of a restriction site) was greater in HRG-affected individuals than in control subjects (57%versus 38%, P < 0.05). Moreover, this genotype was strongly associated with high total cholesterol, TG, LDL-C, and VLDL-C and low HDL-C in HRG patients. Similarly, the control individuals with the P1P1 genotype presented higher concentrations of total cholesterol and LDL-C compared to those with other genotypes (P1P2 and P2P2) [15].
In a study of Brazilian Caucasian women with CAD, Salazar et al. [28] showed that the A+A+ and P1P1 homozygous genotypes (AvaII and PvuII polymorphisms in the LDLR gene, resp.) were significantly higher in women with CAD than in the control group (44% versus 16%, P < 0.001 and 64% versus 39%, P < 0.05, resp.). Similarly, the frequency of the A+ and P1 alleles observed among women with CAD was higher than in controls (62% versus 44%, P < 0.05 and 78% versus 65%, P < 0.05, resp.). For the HincII polymorphism in LDLR, no significant difference in genotype distribution or in relative allele frequencies was observed between patients and controls.
Salazar et al. [76] also evaluated the AvaII (exon 13), HincII (exon 12), and PvuII intron 15 polymorphisms in 50 unrelated Brazilian individuals clinically diagnosed as FH heterozygotes and in 130 normolipidemic controls. The FH subjects showed higher frequencies of A+A+ (AvaII), H+H+ (HincII), and P1P1 (PvuII) homozygous genotypes compared with the control group (P < 0.05). In addition, FH subjects presented higher frequencies of A+ (58%), H+ (61%), and P1 (78%) alleles compared with normolipidemic individuals (45%, 45%, and 64%, resp.). The strong association observed between these alleles and FH suggests that AvaII, HincII, and PvuII polymorphisms could be useful for monitoring FH inheritance in Brazilian families.
3.2. Apo E Gene Polymorphism
The apo E protein is incorporated in the structure of HDLs-C, very low-density lipoproteins cholesterol (VLDLs-C), chylomicrons, and lipolytic degradation products (i.e., the remnants of chylomicrons and intermediate density lipoprotein cholesterol (IDL-C)). This plasma protein binds to cellular receptors. Furthermore, it is important for the transport of cholesterol and other lipids from peripheral tissues to the liver, where they are metabolized [77, 78].
Apo E is also important for the catabolism of TG-rich lipoproteins and reverse cholesterol transport in various tissues [79], which involves its binding to LDLR and the apo E hepatic receptor, the activation of enzymes including hepatic lipase, and hepatic production of VLDL-C [80, 81]. The LDLR in the liver can clear both LDL- and apo E-containing lipoproteins, but the LRP-mediated clearance of remnants is absolutely dependent on apo E [82]. Moreover, apo E influences enteral cholesterol absorption, immunoregulation, and neurobiological events such as neuronal repair, remodeling, and protection [83, 84].
Apo E is synthesized primarily in the liver (>90%) and also in the gut, brain, lungs, kidneys, and macrophages, and it is secreted as a glycosylated protein [83]. In addition to its important effects on lipid metabolism, vascular disease, and cholesterol modulation, apo E also regulates the growth of smooth muscle cells in the arterial wall, which impacts the progression or regression of atherosclerotic lesions [85].
The apo E gene is located on the long arm of chromosome 19 and encodes a protein of 299 amino acids [79]. According to Andrade and Hutz [1], the apo E gene exerts a strong influence on the serum levels of LDL-C.
The apo E gene has a common polymorphism, HhaI (T112C, rs429358 and C158T, rs7412), which is located in exon 4 and generates three alleles, ε2, ε3, and ε4; these alleles determine the six genotypes (ε2/ε2, ε2/ε3, ε2/ε4, ε3/ε3, ε3/ε4, and ε4/ε4) [79, 83]. The allele frequencies differ significantly between ethnic groups [86, 87], but ε3 is the most common allele in several populations [88].
According to Schwanke et al. [83], the apo E polymorphisms modify the protein structure and function. Apo E isoforms interact differently with lipoprotein receptors, altering their metabolism and consequently the plasma level of the circulating lipids [89].
According to Davignon et al. [90], in industrialized societies, individuals carrying the ε4 allele exhibit high serum levels of total cholesterol and LDL-C, while individuals carrying the ε3 allele exhibit intermediate levels, and those carrying the ε2 allele present the lowest levels. Hallman et al. [86] reported that associations between the ε4 allele and increased total and LDL-C levels and between the ε2 allele and low levels of these lipids have been documented in many studies, independently of ethnic group.
The association between apo E polymorphisms and CAD has been studied with regard to cardiology, as apo E affects lipoprotein metabolism and cholesterol transport [80, 81, 91]. The apo Eε4 allele is consistently associated with an increased risk of CAD, although its impact seems to vary according to other factors, such as gender, ethnic origin, and lifestyle [90, 92, 93].
Salazar et al. [28] demonstrated that the HhaI polymorphism in the apo E gene is strongly associated with CAD. Brazilian women with CAD present a higher frequency of the ε3/ε4 genotype compared with controls (40% versus 14%, P < 0.001). In addition, women with CAD present a higher frequency of the ε4 allele compared with controls (23% versus 11%, P < 0.05), suggesting that this allele promotes premature CAD. However, in a study of 184 Afro-Brazilian individuals, the HhaI polymorphism in apo E was not associated with hypertension or variations in serum lipid concentrations [94].
3.3. Apo B Gene Polymorphisms
Apo B is the major protein in human LDL-C and VLDL-C, and it is synthesized in the liver and intestine. This protein is essential for the assembly, secretion, and metabolism of lipoprotein particles and for the removal of LDL-C from the circulation by LDLR on cell surfaces [63, 95].
Structural and genetic alterations in apo B are associated with defective binding to LDLR and lead to hypercholesterolemia, an important risk factor for atherosclerosis and premature CAD [96–98].
The apo B gene is located on chromosome 2p23-p24, and several mutations and SNPs are associated with either variations in plasma lipid concentrations [79] or with CAD and myocardial infarction [99–101]. The SNPs in apo B include the XbaI at exon 26 (C7673T, rs693), EcoRI at exon 29 (G12669A, rs1042031), MspI at exon 26 (rs676210), an indel at exon 1 within the signal peptide (rs17240441), and a hypervariable region at the 3′ end (3′HVR) [102, 103].
Polymorphisms in the apo B gene, as evaluated by RFLP using the restriction enzymes XbaI (rs693), EcoRI (rs1042031), and MspI (rs67210), are also associated with variability in serum cholesterol levels and coronary atherosclerosis [22, 104–106].
The indel, MspI (rs676210), XbaI (rs693), and 3′HVR polymorphisms may be associated with variations in lipid levels, CAD, and myocardial infarction [104, 107–111], but these findings are controversial [112, 113].
The XbaI polymorphism in exon 26 of the apo B gene is associated with increased total cholesterol, altered postprandial lipoprotein metabolism, and increased CAD [114–117]. The EcoRI polymorphism in exon 29 is associated with variations in total cholesterol and TG levels, obesity, and CAD [22, 110, 118, 119]. Furthermore, the signal peptide indel polymorphism is associated with increased serum TG, total cholesterol, and LDL-C [120, 121].
Salazar et al. [28] reported that women with CAD present a higher frequency of the X-X genotype for the XbaI polymorphism compared with controls (42% versus 12%, P < 0.0001). The frequency of the X allele is also higher in women with CAD compared with controls (0.66 versus 0.39, P < 0.0001). The XbaI polymorphism is associated with increased total cholesterol, LDL-C, and CAD in Brazilian Caucasian women.
In a study of the genotypes at three polymorphic sites of ApoB (the indelat the signal peptide, XbaI at exon 26, and EcoRI at exon 29), Machado et al. [122] reported the simultaneous presence of the rare X+ and Del alleles (X+Del haplotype) in males with CHD was associated with significantly high serum levels of total cholesterol (P < 0.01), TG (P < 0.05), and LDL-cholesterol (P < 0.05) and with a high total cholesterol/HDL-C ratio (P < 0.05). These data indicate that a single haplotype, X+Del, within the apo B gene impacts lipid metabolism and may contribute to CHD susceptibility in Brazilian males.
Cavalli et al. [123] investigated four apo B gene polymorphisms, MspI, (rs676210), XbaI (C7673T, rs693), the indel, and 3′HVR, in 177 white hypercholesterolemic Brazilian subjects and 100 control individuals. The genotype distribution and allele frequency of the MspI, XbaI, and indelpolymorphisms were similar between hypercholesterolemic and control individuals, and the frequency of the alleles with ≤43 repeats in the 3′HVR was higher in the hypercholesterolemic group than in the control group (16.4 versus 8.5%, P < 0.05). Moreover, these alleles were associated with higher serum total cholesterol hypercholesterolemic individuals (P < 0.05). On the other hand, hypercholesterolemic individuals carrying at least one allele with ≤43 repeats presented higher total serum cholesterol compared with the individuals carrying both alleles with >43 repeats. In addition, an association between the indel and 3′HVR polymorphisms was observed. The alleles with ≤43 repeats and the Del allele were more frequent in the hypercholesterolemic individuals (P < 0.05). Taken together, these findings show that the apo B 3′HVR polymorphism may be an important genetic marker to evaluate the risk of atherosclerotic disease.
3.4. Apo AI-CIII-AV Gene Cluster Polymorphisms
Apo A-I, apo C-III, and apo A-V are mainly synthesized in the liver [124, 125]. Apo A-I is the major protein found in HDL cholesterol and is a cofactor for lecithin cholesterol acyltransferase (LCAT), the enzyme required for reverse cholesterol transport metabolism [126, 127]. The MspI polymorphism in the promoter region of apo AI is associated with differences in the plasma levels of apo AI and HDL-C [128].
ApoC-III is the major apolipoprotein of hepatic VLDL-C and; due to the role in the transport and metabolism of cholesterol, it is a candidate for determining genetic associations with serum lipid or lipoprotein levels and dyslipidemia. In vitro studies show that apo C-III is a noncompetitive inhibitor of LPL activity, which suggests that it plays an important role in TG-rich lipoprotein catabolism [129]. There are several polymorphisms in the apo C-III gene, [130]. Genetic variations in the 3′ untranslated region of apo C-III (SstI polymorphism, rs10892152) are more frequent in hypertriglyceridemic individuals [108, 131].
Apo A-V is observed at lower concentrations than other apolipoproteins; however, studies have shown that it participates in TG metabolism. Apo A-V deficiency is associated with severe hypertriglyceridemia in humans because this apolipoprotein reduces plasma TG by reducing hepatic VLDL-TG production and by enhancing the lipolytic conversion of TG-rich lipoproteins [125, 132]. Three mutations in the Apo A-V gene have been described, at positions 148, 139, and 97 (Q148X, Q138X, and Q97X, resp.). These mutations produce three different glutamine nonsense mutations that result in Apo A-V deficiencies.
3.5. PCSK9 Gene Polymorphisms
Another protein related to dyslipidemia is proprotein convertase subtilisin/kexin type 9 (PCSK9). The PCSK9 gene is located on chromosome 1p32, has 12 exons, and encodes a 692 amino acid protein. There are several mutations in PCSK9, including c.G1120T (p.Asp374Tyr), c.T381A (p.Ser127Arg), c.T646A (p.Phe216Leu), c.A654T (p.Arg218Ser), R46L (rs11591147), and rs11206510. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia (ADH) [133]. The overexpression of PCSK9 in HepG2 cells accelerates the degradation of cell-surface LDLR through a nonproteasomal mechanism in a postendoplasmic reticulum compartment and leads to increased total cholesterol and LDL-C [134, 135].
3.6. Cholesteryl Ester Transfer Protein Gene Polymorphisms
Cholesteryl ester transfer protein (CETP) is an enzyme with a key role in HDL-C metabolism. CETP promotes the exchange of TG and cholesterol between lipoproteins, and it transfers cholesteryl esters from HDL-C to other lipoproteins for subsequent absorption of cholesterol by hepatocytes. Cholesteryl esters are transferred to LDL-Cs and VLDL-Cs in exchange for TG [136–138]. By increasing the amount of cholesteryl esters in LDL-Cs and VLDL-Cs, CETP increases the atherogenicity of these lipoproteins. High plasma CETP concentration is associated with reduced HDL-C, a strong and independent risk factor for atherosclerosis [139, 140].
The CETP gene is located on chromosome 16 and contains 16 exons [141, 142]. The protein is expressed primarily in the liver, spleen, and adipose tissue, but low levels have been detected in the small intestine, adrenal glands, heart, kidney, and skeletal muscle [143]. CETP-deficient patients exhibit elevated plasma HDL-C levels and low plasma LDL-C levels [144].
The relationship between plasma CETP, HDL-C, and atherosclerosis is complex, and CETP gene polymorphisms have been studied to better define this relationship [145]. Polymorphisms at the CETP gene locus are associated with the progression of coronary atherosclerosis independently of plasma lipase activity and HDL-C concentration.
The TaqIB (rs708272) polymorphism affects lipid transfer activity and HDL-C. TaqIB (rs708272) is one of the best studied polymorphisms in CETP; it consists of a silent guanine-to-adenine nucleotide substitution in intron 1. The less common allele, B2, is associated with decreased CETP activity, and in normolipemic individuals, this allele is associated with an increase in HDL-C due to decreased CETP activity [18, 146–148].
3.7. Lipoprotein Lipase Gene Polymorphisms
Lipoprotein lipase (LPL) is linked to the vascular endothelium and plays a crucial role in plasma lipoprotein processing. LPL catalyzes TG hydrolysis, which is the limiting step in the removal of TG-rich lipoproteins such as chylomicrons, VLDL-C, and LDL-C from the circulation [149]. LPL acts as a ligand for LDLR-related protein and for the uptake of VLDL-C and LDL-C [150].
The LPL gene is located on chromosome 8 (8p22), and it is composed of 10 exons [151, 152]. The known polymorphisms result in three functional variants: D9N (G28A, rs1801177), S291N (A1127G, rs268), and S447X or MnlI (rs328) and two SNPs located on introns: HindIII at intron 8 (T381G, rs320) and PvuII at intron 6 (rs285). Generally, these variants are associated with increased TG, but the S447X mutation, which truncates the last two amino acids of the polypeptide chain, decreases TG [153–155].
The HindIII (T381G, rs320) and PvuII (rs285) polymorphisms, located on introns 8 and 6 of the LPL gene, respectively, are associated with angiographic CAD. However, Anderson et al. [156] demonstrated that HindIII(+) allele is moderately associated with CAD, and the PvuII(−) allele is only modestly associated with CAD.
4. Genetic Polymorphisms Associated with Dyslipidemia in HIV-1 Infected Patients
There have been few studies of the effects of the LDLR gene on plasma cholesterol in HIV-1-infected patients. Tran et al. [157] showed that HIV-1 patients receiving PIs such as nelfinavir have decreased LDLR and LRP mRNA and protein levels, resulting in the reduced functional activity of these two receptors, which are involved in cholesterol metabolism. Moreover, individuals receiving nelfinavir have reduced levels of active SREBP in the nucleus.
Plasma LDL-C levels may be influenced through the regulation of hepatic LDLR expression. The expression of LDLR is under metabolic and hormonal control. Insulin, dehydroepiandrosterone (DHEA), and growth hormone (GH) may stimulate LDLR expression and reduce plasma LDL cholesterol levels [158–160]. Petit et al. [35] evaluated the LDLR expression in HIV-patients with or without lipodystrophy. These authors found that HIV-lipodystrophy was associated with low expression of LDLR and that this decreased LDLR expression was independent of DHEA or insulin secretion.
A study of 60 HIV-1-infected patients receiving PI therapy showed an association between apo C-III polymorphisms and a genetic predisposition to develop high TG and low HDL-C levels [161]; these authors suggested that apo C-III polymorphism genotyping could identify patients who are at risk for both hypertriglyceridemia and lipoatrophy [162]. Foulkes et al. [163] showed that there are associations between ethnic differences, apo C-III variants, and the development of hypertriglyceridemia in HIV-1- infected patients treated with PIs. These authors also demonstrated that Hispanics carrying the variant alleles at apo C-III exhibited smaller TG increases after receiving PIs compared with those carrying the wild-type genotype. According to Aragonès et al. [164], the apo C-III rs10892152 polymorphism predisposes HIV-1-infected patients, especially those treated with PIs, to an unfavorable lipid profile. Apo A-V polymorphisms also enhance PI-associated hyperlipidemia [52], and variations in this gene are risk factors for extreme hypertriglyceridemia [165].
Tarr et al. [166] evaluated the influence of apo C-III, apo E, and TNF polymorphisms on the risk of ART-associated lipid disorders. No association between TNF and lipoatrophy was observed, whereas apo C-III and apo E contributed to an unfavorable lipid profile in ART-treated HIV-1 infected patients. In another study, 20 SNPs of 13 genes involved in lipid transport and metabolism were evaluated in 438 HIV-infected individuals receiving ART, and the results showed that SNPs in the ABCA1, apo A-V, and apo C-III genes contributed to hypertriglyceridemia, whereas SNPs in the apo A-V and CETP genes contributed to low HDL-C [11]. In a recent report by Egaña-Gorroño et al. [13], 192 SNPs in 87 genes from the lipid metabolism pathway were assessed in 727 HIV-1-infected patients starting ART. The results of this study showed that one SNP in the apo B gene (rs10495712) was associated with high LDL-C levels.
5. Conclusion
Dyslipidemia leads to atherosclerosis and CAD; thus, understanding the etiology of changes in the lipid profile is extremely important. Dyslipidemia is a complex and multifactorial condition caused by polymorphisms in genes involved in lipid metabolism and regulation and by environmental factors such as smoking, sedentary lifestyle, stress, and diet. The main genes studied in relation to dyslipidemia are those that encode proteins, receptors, and enzymes related to lipid metabolism and regulation. Polymorphisms in the LDLR, apoE, apo B, apo A-I, apo C-III, apo A-V, PCSK9, CETP, and LPL genes are associated with changes in lipid profile.
Moreover, HIV-1-infected patients often have lipid disorders. The pathogenesis of these disorders is complex and multifactorial, involving viral and host factors and ART. By itself, HIV-1 causes lipid disorders, and it acts synergistically with ART to generate dyslipidemia, insulin resistance, and lipodystrophy syndrome, especially in patients who are treated with PIs.
The genetic causes of dyslipidemia in HIV-1-infected patients have been investigated because not all patients who use HAART exhibit metabolic disorders. Some polymorphisms in these patients are associated with lipid profile changes. Moreover, the genetic contribution to dyslipidemia alone explains up to 7.6% of the variation in HIV-1-infected patients, and HAART explains up to 6.2% of the variation. The combination of genotype and ART increases the risk of sustained dyslipidemia in HIV-1-infected individuals by up to 5-fold, with increased plasma concentrations of total cholesterol, LDL-C, and TG and decreased plasma HDL-C.
The genetic contribution to dyslipidemia is similar to or greater than the contribution of HAART. Thus, clinicians should consider genetics and the effects of ART when selecting an antiretroviral regimen for HIV-1 patients. Because gene polymorphisms cause dyslipidemia, they should be investigated in HIV-1-infected patients to identify individuals with an increased risk of developing dyslipidemia when treated with ART, especially those containing PIs. This knowledge could guide individualized treatment decisions and lead to new therapeutic targets for the treatment of dyslipidemia.
References
- 1.Andrade FM, Hutz MH. O componente genético da determinação dos lipídeos séricos. Ciência & Saúde Coletiva. 2002;7(1):175–182. [Google Scholar]
- 2.Valente AM, Reis AF, Machado DM, Succi RC, Chacra AR. Alterações metabólicas da síndrome lipodistrofia do HIV. Arquivos Brasileiros de Endocrinologia & Metabologia. 2005;49(6):10–17. doi: 10.1590/s0004-27302005000600004. [DOI] [PubMed] [Google Scholar]
- 3.Estrada V, Portilla J. Dyslipidemia related to antiretroviral therapy. AIDS Reviews. 2011;13(1):49–56. [PubMed] [Google Scholar]
- 4.Wlodawer A, Vondrasek J. Terapia anti-aids. Annual Review of Biophysics and Biomolecular Structure. 1998;27(249):10–16. doi: 10.1146/annurev.biophys.27.1.249. [DOI] [PubMed] [Google Scholar]
- 5.Menéndez-Arias L. Molecular basis of human immunodeficiency virus type 1 drug resistance: overview and recent developments. Antiviral Research. 2013;98(1):93–120. doi: 10.1016/j.antiviral.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 6.UNAIDS. World AIDS Day Report. 2012, http://www.unaids.org/en/media/unaids/contentassets/documents/epidemiology/2012/gr2012/JC2434_WorldAIDSday_results_en.pdf.
- 7.Palella FJ, Jr., Delaney KM, Moorman AC, et al. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. The New England Journal of Medicine. 1998;338(13):853–860. doi: 10.1056/NEJM199803263381301. [DOI] [PubMed] [Google Scholar]
- 8.Detels R, Muñoz A, McFarlane G, et al. Effectiveness of potent antiretroviral therapy on time to AIDS and death in men with known HIV infection duration. Journal of the American Medical Association. 1998;280(17):1497–1503. doi: 10.1001/jama.280.17.1497. [DOI] [PubMed] [Google Scholar]
- 9.Sudano I, Spieker LE, Noll G, Corti R, Weber R, Lüscher TF. Cardiovascular disease in HIV infection. American Heart Journal. 2006;151(6):1147–1155. doi: 10.1016/j.ahj.2005.07.030. [DOI] [PubMed] [Google Scholar]
- 10.Guimarães MMM, Greco DB, Júnior ARO, Penido MG, Machado LJC. Distribuição da gordura corporal e perfis lipídico e glicêmico de pacientes infectados pelo HIV. Arquivos Brasileiros de Endocrinologia & Metabologia. 2007;51(1):42–51. doi: 10.1590/s0004-27302007000100008. [DOI] [PubMed] [Google Scholar]
- 11.Arnedo M, Taffé P, Sahli R, et al. Contribution of 20 single nucleotide polymorphisms of 13 genes to dyslipidemia associated with antiretroviral therapy. Pharmacogenetics and Genomics. 2007;17(9):755–764. doi: 10.1097/FPC.0b013e32814db8b7. [DOI] [PubMed] [Google Scholar]
- 12.Tarr PE, Rotger M, Telenti A. Dyslipidemia in HIV-infected individuals: from pharmacogenetics to pharmacogenomics. Pharmacogenomics. 2010;11(4):587–594. doi: 10.2217/pgs.10.35. [DOI] [PubMed] [Google Scholar]
- 13.Egaña-Gorroño L, Martínez E, Cormand B, Escribà T, Gatell J, Arnedo M. Impact of genetic factors on dyslipidemia in HIV-infected patients starting antiretroviral therapy. AIDS. 2013;27:529–538. doi: 10.1097/QAD.0b013e32835d0da1. [DOI] [PubMed] [Google Scholar]
- 14.Kirillova OO. Modern concepts of gene polymorphisms which regulate lipid metabolism. Vopr Pitan. 2012;81(4):48–58. [PubMed] [Google Scholar]
- 15.Salazar LA, Hirata MH, Forti N, et al. Pvu II intron 15 polymorphism at the LDL receptor gene is associated with differences in serum lipid concentrations in subjects with low and high risk for coronary artery disease from Brazil. Clinica Chimica Acta. 2000;293(1-2):75–88. doi: 10.1016/s0009-8981(99)00218-1. [DOI] [PubMed] [Google Scholar]
- 16.Lander ES, Whitehead Institute for Biomedical Research Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 17.Collins FS, Brooks LD, Chakravarti A. A DNA polymorphism discovery resource for research on human genetic variation. Genome Research. 1998;8(12):1229–1231. doi: 10.1101/gr.8.12.1229. [DOI] [PubMed] [Google Scholar]
- 18.Freeman DJ, Griffin BA, Holmes AP, et al. Regulation of plasma HDL cholesterol and subfraction distribution by genetic and environmental factors: associations between the TaqI B RFLP in the CETP gene and smoking and obesity. Arteriosclerosis and Thrombosis. 1994;14(3):336–344. doi: 10.1161/01.atv.14.3.336. [DOI] [PubMed] [Google Scholar]
- 19.Vohl M-C, Lamarche B, Pascot A, et al. Contribution of the cholesteryl ester transfer protein gene TaqlB polymorphism to the reduced plasma HDL-cholesterol levels found in abdominal obese men with the features of the insulin resistance syndrome. International Journal of Obesity. 1999;23(9):918–925. doi: 10.1038/sj.ijo.0800972. [DOI] [PubMed] [Google Scholar]
- 20.Fiegenbaum M. Estudo da variabilidade em genes de apolipoproteínas sobre níveis lipídicos e parâmetros de massa e gordura corporal na população de Porto Alegre [Dissertação de Mestrado] Porto Alegre, Brazil: Programa de Pós-Graduação em Genética e Biologia Molecular, UFRGS; 2001. [Google Scholar]
- 21.Santos RD. Sociedade Brasileira de Cardiologia. III Diretrizes Brasileiras sobre Dislipidemias e Diretrizes de Prevenção da Aterosclerose do Departamento de Aterosclerose da Sociedade Brasileira de Dislipidemias. Arquivos Brasileiros de Cardiologia. 2001;77(supplement 3):1–48. [PubMed] [Google Scholar]
- 22.Stepanov VA, Puzyrev VP, Karpov RS, Kutmin AI. Genetic markers in coronary artery disease in a Russian population. Human Biology. 1998;70(1):47–57. [PubMed] [Google Scholar]
- 23.Salazar LA, Hirata MH, Giannini SD, et al. Effects of AvaII and HincII polymorphisms at the LDL-receptor gene on serum lipid levels of Brazilian individuals with high risk for coronary heart disease. Journal of Clinical Laboratory Analysis. 1999;13:251–258. doi: 10.1002/(SICI)1098-2825(1999)13:6<251::AID-1>3.0.CO;2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Siqueira AFA, Abdalla DSP, Ferreira SRG. LDL: da síndrome metabólica à instabilização da placa aterosclerótica. Arquivos Brasileiros de Endocrinologia & Metabologia. 2006;50(2):334–343. doi: 10.1590/s0004-27302006000200020. [DOI] [PubMed] [Google Scholar]
- 25.Lima ES, Couto RD. Estrutura, metabolismo e funções fisiológicas da lipoproteína de alta densidade. Jornal Brasileiro de Patologia e Medicina Laboratorial. 2006;42(3):169–178. [Google Scholar]
- 26.Simons LA. Interrelations of lipids and lipoproteins with coronary artery disease mortality in 19 countries. American Journal of Cardiology. 1986;57(14):5G–10G. doi: 10.1016/0002-9149(86)90659-4. [DOI] [PubMed] [Google Scholar]
- 27.Faria EC, Moraes VSC, Oliveira MLPS, Varriano AA, Silva CAM, Castilho LN. Risk factors for coronary artery disease in women. A study in a Brazilian population. Atherosclerosis. 1999;144(article 101) [Google Scholar]
- 28.Salazar LA, Hirata MH, Giannini SD, et al. Seven DNA polymorphisms at the candidate genes of atherosclerosis in Brazilian women with angiographically documented coronary artery disease. Clinica Chimica Acta. 2000;300(1-2):139–149. doi: 10.1016/s0009-8981(00)00308-9. [DOI] [PubMed] [Google Scholar]
- 29.Sposito AC. Sociedade Brasileira de Cardiologia. IV Diretrizes Brasileiras sobre Dislipidemias e Prevenção da Aterosclerose do Departamento de Aterosclerose da Sociedade Brasileira de Dislipidemias. Arquivos Brasileiros de Cardiologia. 2007;88(supplement 1):1–19. [PubMed] [Google Scholar]
- 30.Rotger M, Bayard C, Taffé P, et al. Contribution of genome-wide significant single-nucleotide polymorphisms and antiretroviral therapy to dyslipidemia in HIV-infected individuals: a longitudinal study. Circulation. 2009;2(6):621–628. doi: 10.1161/CIRCGENETICS.109.874412. [DOI] [PubMed] [Google Scholar]
- 31.Hogg RS, Yip B, Kully C, et al. Improved survival among HIV-infected patients after initiation of triple-drug antiretroviral regimens. Canadian Medical Association Journal. 1999;160(5):659–665. [PMC free article] [PubMed] [Google Scholar]
- 32.Marins JRP, Jamal LF, Chen SY, et al. Dramatic improvement in survival among adult Brazilian AIDS patients. AIDS. 2003;17(11):1675–1682. doi: 10.1097/00002030-200307250-00012. [DOI] [PubMed] [Google Scholar]
- 33.Arshad A, Bansal A, Patel RC, Frishman WH. Cardiac complications of human immunodeficiency virus infection: diagnostic and therapeutic considerations. Heart Disease. 2000;2(2):133–145. [PubMed] [Google Scholar]
- 34.Barbaro G. Cardiovascular manifestations of HIV infection. Circulation. 2002;106(11):1420–1425. doi: 10.1161/01.cir.0000031704.78200.59. [DOI] [PubMed] [Google Scholar]
- 35.Petit JM, Duong M, Duvillard L, et al. LDL-receptors expression in HIV-infected patients: relations to antiretroviral therapy, hormonal status, and presence of lipodystrophy. European Journal of Clinical Investigation. 2002;32(5):354–359. doi: 10.1046/j.1365-2362.2002.00989.x. [DOI] [PubMed] [Google Scholar]
- 36.Constans J, Pellegrin JL, Peuchant E, et al. Plasma lipids in HIV-infected patients: a prospective study in 95 patients. European Journal of Clinical Investigation. 1994;24(6):416–420. doi: 10.1111/j.1365-2362.1994.tb02185.x. [DOI] [PubMed] [Google Scholar]
- 37.Hellerstein MK, Grunfeld C, Wu K, et al. Increased de novo hepatic lipogenesis in human immunodeficiency virus infection. Journal of Clinical Endocrinology and Metabolism. 1993;76(3):559–565. doi: 10.1210/jcem.76.3.8445011. [DOI] [PubMed] [Google Scholar]
- 38.Grunfeld C, Kotler DP, Hamadeh R, Tierney A, Wang J, Pierson RN., Jr. Hypertriglyceridemia in acquired immunodeficiency syndrome. American Journal of Medicine. 1989;86(1):27–31. doi: 10.1016/0002-9343(89)90225-8. [DOI] [PubMed] [Google Scholar]
- 39.Penzak SR, Chuck SK. Hyperlipidemia associated with HIV protease inhibitor use: pathophysiology, prevalence, risk factors and treatment. Scandinavian Journal of Infectious Diseases. 2000;32(2):111–123. doi: 10.1080/003655400750045196. [DOI] [PubMed] [Google Scholar]
- 40.Rose H, Woolley I, Hoy J, et al. HIV infection and high-density lipoprotein: the effect of the disease vs the effect of treatment. Metabolism. 2006;55(1):90–95. doi: 10.1016/j.metabol.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 41.Zangerle R, Sarcletti M, Gallati H, Reibnegger G, Wachter H, Fuchs D. Decreased plasma concentrations of HDL cholesterol in HIV-infected individuals are associated with immune activation. Journal of Acquired Immune Deficiency Syndromes. 1994;7(11):1149–1156. [PubMed] [Google Scholar]
- 42.Pirich C, Efthimiou Y, O’Grady J, Zielinski C, Sinzinger H. Apolipoprotein A and biological half-life of prostaglandin I2 in HIV-1 infection. Thrombosis Research. 1996;81(2):213–218. doi: 10.1016/0049-3848(95)00238-3. [DOI] [PubMed] [Google Scholar]
- 43.Khovidhunkit W, Memon RA, Shigenaga JK, et al. Plasma platelet-activating factor acetylhydrolase activity in human immunodeficiency virus infection and the acquired immunodeficiency syndrome. Metabolism. 1999;48(12):1524–1531. doi: 10.1016/s0026-0495(99)90240-8. [DOI] [PubMed] [Google Scholar]
- 44.Coll B, van Wijk JPH, Parra S, et al. Effects of rosiglitazone and metformin on postprandial paraoxonase-1 and monocyte chemoattractant protein-1 in human immunodeficiency virus-infected patients with lipodystrophy. European Journal of Pharmacology. 2006;544(1–3):104–110. doi: 10.1016/j.ejphar.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 45.Rader DJ. Molecular regulation of HDL metabolism and function: implications for novel therapies. Journal of Clinical Investigation. 2006;116(12):3090–3100. doi: 10.1172/JCI30163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sala F, Catapano AL, Norata GD. High-density lipoproteins and atherosclerosis: emerging aspects. Journal of Geriatric Cardiology. 2012;9:401–407. doi: 10.3724/SP.J.1263.2011.12282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mujawar Z, Rose H, Morrow MP, et al. Human immunodeficiency virus impairs reverse cholesterol transport from macrophages. PLoS Biology. 2006;4(11) doi: 10.1371/journal.pbio.0040365.e365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oh J, Hegele RA. HIV-associated dyslipidaemia: pathogenesis and treatment. The Lancet Infectious Diseases. 2007;7(12):787–796. doi: 10.1016/S1473-3099(07)70287-6. [DOI] [PubMed] [Google Scholar]
- 49.Tape C, Kisilevsky R. Apolipoprotein A-I and apolipoprotein SAA half-lives during acute inflammation and amyloidogenesis. Biochimica et Biophysica Acta. 1990;1043(3):295–300. doi: 10.1016/0005-2760(90)90030-2. [DOI] [PubMed] [Google Scholar]
- 50.Grunfeld C, Kotler DP, Shigenaga JK, et al. Circulating interferon-α levels and hypertriglyceridemia in the acquired immunodeficiency syndrome. American Journal of Medicine. 1991;90(2):154–162. [PubMed] [Google Scholar]
- 51.Farhi L, De Lima DB, Cunha CB. Dyslipidemia in HIV/AIDS patients in antiretroviral therapy in a university hospital, Rio de Janeiro, Brazil. Jornal Brasileiro de Patologia e Medicina Laboratorial. 2008;44(3):175–184. [Google Scholar]
- 52.Guardiola M, Ferré R, Salazar J, et al. Protease inhibitor-associated dyslipidemia in HIV-infected patients is strongly influenced by the APOA5-1131T→C gene variation. Clinical Chemistry. 2006;52(10):1914–1919. doi: 10.1373/clinchem.2006.069583. [DOI] [PubMed] [Google Scholar]
- 53.Riddler SA, Smit E, Cole SR, et al. Impact of HIV Infection and HAART on Serum Lipids in Men. Journal of the American Medical Association. 2003;289(22):2978–2982. doi: 10.1001/jama.289.22.2978. [DOI] [PubMed] [Google Scholar]
- 54.Reeds DN, Yarasheski KE, Fontana L, et al. Alterations in liver, muscle, and adipose tissue insulin sensitivity in men with HIV infection and dyslipidemia. American Journal of Physiology. 2006;290(1):E47–E53. doi: 10.1152/ajpendo.00236.2005. [DOI] [PubMed] [Google Scholar]
- 55.Carr A, Samaras K, Burton S, et al. A syndrome of peripheral lipodystrophy, hyperlipidaemia and insulin resistance in patients receiving HIV protease inhibitors. AIDS. 1998;12(7):F51–F58. doi: 10.1097/00002030-199807000-00003. [DOI] [PubMed] [Google Scholar]
- 56.Chi D, Henry J, Kelley J, Thorpe R, Smith JK, Krishnaswamy G. The effects of HIV infection on endothelial function. Endothelium. 2000;7(4):223–242. doi: 10.3109/10623320009072210. [DOI] [PubMed] [Google Scholar]
- 57.Lundgren JD. Combination antiretroviral therapy and the risk of myocardial infarction: the data collection on adverse events of Anti-HIV Drugs (DAD) Study Group. The New England Journal of Medicine. 2003;349(21):1993–2003. doi: 10.1056/NEJMoa030218. [DOI] [PubMed] [Google Scholar]
- 58.Friis-Møller N, Reiss P, Sabin CA, et al. Class of antiretroviral drugs and the risk of myocardial infarction. The New England Journal of Medicine. 2007;356(17):1723–1735. doi: 10.1056/NEJMoa062744. [DOI] [PubMed] [Google Scholar]
- 59.Carr A, Samaras K, Chisholm DJ, Cooper DA. Pathogenesis of HIV-1-protease inhibitor-associated peripheral lipodystrophy, hyperlipidaemia, and insulin resistance. The Lancet. 1998;351(9119):1881–1883. doi: 10.1016/S0140-6736(98)03391-1. [DOI] [PubMed] [Google Scholar]
- 60.Calza L, Manfredi R, Chiodo F. Dyslipidaemia associated with antiretroviral therapy in HIV-infected patients. Journal of Antimicrobial Chemotherapy. 2004;53(1):10–14. doi: 10.1093/jac/dkh013. [DOI] [PubMed] [Google Scholar]
- 61.Hui DY. Effects of HIV protease inhibitor therapy on lipid metabolism. Progress in Lipid Research. 2003;42(2):81–92. doi: 10.1016/s0163-7827(02)00046-2. [DOI] [PubMed] [Google Scholar]
- 62.Lagathu C, Kim M, Maachi M, et al. HIV antiretroviral treatment alters adipokine expression and insulin sensitivity of adipose tissue in vitro and in vivo. Biochimie. 2005;87(1):65–71. doi: 10.1016/j.biochi.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 63.Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232(4746):34–47. doi: 10.1126/science.3513311. [DOI] [PubMed] [Google Scholar]
- 64.Tolleshaug H, Goldstein JL, Schneider WJ, Brown MS. Posttranslational processing of the LDL receptor and its genetic disruption in familial hypercholesterolemia. Cell. 1982;30(3):715–724. doi: 10.1016/0092-8674(82)90276-8. [DOI] [PubMed] [Google Scholar]
- 65.Goldstein JL, Brown MS. Progress in understanding the LDL receptor and HMG-CoA reductase, two membrane proteins that regulate the plasma cholesterol. Journal of Lipid Research. 1984;25(13):1450–1461. [PubMed] [Google Scholar]
- 66.Hobbs HH, Brown MS, Goldstein JL. Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Human Mutation. 1992;1(6):445–466. doi: 10.1002/humu.1380010602. [DOI] [PubMed] [Google Scholar]
- 67.Sudhof TC, Goldstein JL, Brown MS, Russell DW. The LDL receptor gene: a mosaic of exons shared with different proteins. Science. 1985;228(4701):815–822. doi: 10.1126/science.2988123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Usifo E, Leigh SE, Whittall RA, et al. Low-density lipoprotein receptor gene familial hypercholesterolemia variant database: update and pathological assessment. Annals of Human Genetics. 2012;76(5):387–401. doi: 10.1111/j.1469-1809.2012.00724.x. [DOI] [PubMed] [Google Scholar]
- 69.Pedersen JC, Berg K. Normal DNA polymorphism at the low density lipoprotein receptor (LDLR) locus associated with serum cholesterol level. Clinical Genetics. 1988;34(5):306–312. doi: 10.1111/j.1399-0004.1988.tb02883.x. [DOI] [PubMed] [Google Scholar]
- 70.Myant NB, Gallagher JJ, Knight BL, et al. Clinical signs of familial hypercholesterolemia in patients with familial defective apolipoprotein B-100 and normal low density lipoprotein receptor function. Arteriosclerosis and Thrombosis. 1991;11(3):691–703. doi: 10.1161/01.atv.11.3.691. [DOI] [PubMed] [Google Scholar]
- 71.Wiseman SA, Powell JT, Humphries SE, Press M. The magnitude of the hypercholesterolemia of hypothyroidism is associated with variation in the low density lipoprotein receptor gene. Journal of Clinical Endocrinology and Metabolism. 1993;77(1):108–112. doi: 10.1210/jcem.77.1.8100826. [DOI] [PubMed] [Google Scholar]
- 72.Gylling H, Kontula K, Koivisto U-M, Miettinen HE, Miettinen TA. Polymorphisms of the genes encoding apoproteins A-I, B, C-III, and E and LDL receptor, and cholesterol and LDL metabolism during increased cholesterol intake: common alleles of the apoprotein E gene show the greatest regulatory impact. Arteriosclerosis, Thrombosis, and Vascular Biology. 1997;17(1):38–44. doi: 10.1161/01.atv.17.1.38. [DOI] [PubMed] [Google Scholar]
- 73.Gudnason V, Zhou T, Thormar K, et al. Detection of the low density lipoprotein receptor gene PvuII intron 15 polymorphism using the polymerase chain reaction: association with plasma lipid traits in healthy men and women. Disease Markers. 1998;13(4):209–220. doi: 10.1155/1998/842051. [DOI] [PubMed] [Google Scholar]
- 74.Leitersdorf E, Chakravarti A, Hobbs HH. Polymorphic DNA haplotypes at the LDL receptor locus. American Journal of Human Genetics. 1989;44(3):409–421. [PMC free article] [PubMed] [Google Scholar]
- 75.Chaves FJ, Puig O, García-Sogo M, et al. Seven DNA polymorphisms in the LDL receptor gene: application to the study of familial hypercholesterolemia in Spain. Clinical Genetics. 1996;50(1):28–35. doi: 10.1111/j.1399-0004.1996.tb02342.x. [DOI] [PubMed] [Google Scholar]
- 76.Salazar LA, Cavalli SA, Hirata MH, et al. Polymorphisms of the low-density lipoprotein receptor gene in Brazilian individuals with heterozygous familial hypercholesterolemia. Brazilian Journal of Medical and Biological Research. 2000;33(11):1301–1304. doi: 10.1590/s0100-879x2000001100006. [DOI] [PubMed] [Google Scholar]
- 77.Curtiss LK, Boisvert WA. Apolipoprotein E and atherosclerosis. Current Opinion in Lipidology. 2000;11(3):243–251. doi: 10.1097/00041433-200006000-00004. [DOI] [PubMed] [Google Scholar]
- 78.Brandão AC, Pinheiro S, Jr., Pinhel MA, et al. Polimorfismo genético da apolipoproteína E na doença arterial periférica. Jornal Vascular Brasileiro. 2004;3(4):317–322. [Google Scholar]
- 79.Forti N, Salazar LA, Diament J, Giannini SD, Hirata MH, Hirata RDC. Genetic changes and cholesterolemia: recent Brazilian studies. Arquivos Brasileiros de Cardiologia. 2003;80(5):565–571. doi: 10.1590/s0066-782x2003000500010. [DOI] [PubMed] [Google Scholar]
- 80.Shore VG, Shore B. Heterogeneity of human plasma very low density lipoproteins. Separation of species differing in protein components. Biochemistry. 1973;12(3):502–507. doi: 10.1021/bi00727a022. [DOI] [PubMed] [Google Scholar]
- 81.Ginsberg HN. Lipoprotein physiology. Endocrinology and Metabolism Clinics of North America. 1998;27(3):503–519. doi: 10.1016/s0889-8529(05)70023-2. [DOI] [PubMed] [Google Scholar]
- 82.Linton MF, Hasty AH, Babaev VR, Fazio S. Hepatic apo E expression is required for remnant lipoprotein clearance in the absence of the low density lipoprotein receptor. Journal of Clinical Investigation. 1998;101(8):1726–1736. doi: 10.1172/JCI2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schwanke CHA, Cruz IBM, Leal NF, Scheibe R, Moriguchi Y, Moriguchi EH. Análise da associação entre polimorfismo do gene da apolipoproteína E e fatores de risco cardiovascular em idosos longevos. Arquivos Brasileiros de Cardiologia. 2002;78(6):561–570. doi: 10.1590/s0066-782x2002000600004. [DOI] [PubMed] [Google Scholar]
- 84.Mahley RW, Huang Y. Apoliprotein E: from atherosclerosis to Alzheimer’s disease and beyond. Current Opinion in Lipidology. 1999;10:207–217. doi: 10.1097/00041433-199906000-00003. [DOI] [PubMed] [Google Scholar]
- 85.Mazzone T. Apolipoprotein E secretion by macrophages: its potential physiological functions. Current Opinion in Lipidology. 1996;7(5):303–307. doi: 10.1097/00041433-199610000-00008. [DOI] [PubMed] [Google Scholar]
- 86.Hallman DM, Boerwinkle E, Saha N, et al. The apolipoprotein E polymorphism: a comparison of allele frequencies and effects in nine populations. American Journal of Human Genetics. 1991;49(2):338–349. [PMC free article] [PubMed] [Google Scholar]
- 87.Gerdes LU, Klausen IC, Sihm I, Faergeman O. Apolipoprotein E polymorphism in a Danish population compared to findings in 45 other study populations around the world. Genetic Epidemiology. 1992;9(3):155–167. doi: 10.1002/gepi.1370090302. [DOI] [PubMed] [Google Scholar]
- 88.Eichner JE, Dunn ST, Perveen G, Thompson DM, Stewart KE, Stroehla BC. Apolipoprotein E polymorphism and cardiovascular disease: a HuGE review. American Journal of Epidemiology. 2002;155(6):487–495. doi: 10.1093/aje/155.6.487. [DOI] [PubMed] [Google Scholar]
- 89.Siest G, Pillot T, Regis-Bailly A, et al. Apolipoprotein E: an important gene and protein to follow in laboratory medicine. Clinical Chemistry. 1995;41(8):1068–1086. [PubMed] [Google Scholar]
- 90.Davignon J, Gregg RE, Sing CF. Apolipoprotein E polymorphism and atherosclerosis. Arteriosclerosis. 1988;8(1):1–21. doi: 10.1161/01.atv.8.1.1. [DOI] [PubMed] [Google Scholar]
- 91.Mansur AP. Análise do componente genético da doença coronariana. Arquivos Brasileiros de Cardiologia. 2000;74:531–533. [Google Scholar]
- 92.Tiret L, De Knijff P, Menzel H-J, Ehnholm C, Nicaud V, Havekes LM. ApoE polymorphism and predisposition to coronary heart disease in youths of different European population: the EARS study. Arteriosclerosis and Thrombosis. 1994;14(10):1617–1624. doi: 10.1161/01.atv.14.10.1617. [DOI] [PubMed] [Google Scholar]
- 93.Bercedo-Sanz A, Gonzalez-Lamuno D, Malaga S, Garcia-Fuentes M. Impact of ApoE4 allele on total cholesterol levels of children in northern Spain. Clinical Genetics. 1999;55(1):69–70. doi: 10.1034/j.1399-0004.1999.550115.x. [DOI] [PubMed] [Google Scholar]
- 94.Sakuma T, Hirata RDC, Hirata MH. Five polymorphisms in gene candidates for cardiovascular disease in Afro-Brazilian individuals. Journal of Clinical Laboratory Analysis. 2004;18(6):309–316. doi: 10.1002/jcla.20044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Young SG. Recent progress in understanding apolipoprotein B. Circulation. 1990;82(5):1574–1594. doi: 10.1161/01.cir.82.5.1574. [DOI] [PubMed] [Google Scholar]
- 96.Avogaro P, Bittolo Bon G, Cazzolato G, Rorai E. Relationship between apolipoproteins and chemical components of lipoproteins in survivors of myocardial infarction. Atherosclerosis. 1980;37(1):69–76. doi: 10.1016/0021-9150(80)90094-5. [DOI] [PubMed] [Google Scholar]
- 97.Levy RI. Cholesterol, lipoproteins, apoproteins, and heart disease: present status and future prospects. Clinical Chemistry. 1981;27(5):653–662. [PubMed] [Google Scholar]
- 98.Lewis B. The lipoproteins: predictors, protectors, and pathogens. British Medical Journal. 1983;287(6400):1161–1164. doi: 10.1136/bmj.287.6400.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wu JH, Wen MS, Lo SK, Chern MS. Increased frequency of apolipoprotein B signal peptide sp24/24 in patients with coronary artery disease. General allele survey in the population of Taiwan and comparison with Caucasians. Clinical Genetics. 1994;45(5):250–254. doi: 10.1111/j.1399-0004.1994.tb04150.x. [DOI] [PubMed] [Google Scholar]
- 100.Turner PR, Talmud PJ, Visvikis S, Ehnholm C, Tiret L. DNA polymorphisms of the apoprotein B gene are associated with altered plasma lipoprotein concentrations but not with perceived risk of cardiovascular disease: European Atherosclerosis Research Study. Atherosclerosis. 1995;116(2):221–234. doi: 10.1016/0021-9150(94)05550-3. [DOI] [PubMed] [Google Scholar]
- 101.Gardemann A, Ohly D, Fink M, et al. Association of the insertion/deletion gene polymorphism of the apolipoprotein B signal peptide with myocardial infarction. Atherosclerosis. 1998;141(1):167–175. doi: 10.1016/s0021-9150(98)00161-0. [DOI] [PubMed] [Google Scholar]
- 102.Ludwig EH, McCarthy BJ. Haplotype analysis of the human apolipoprotein B mutation associated with familial defective apolipoprotein B100. American Journal of Human Genetics. 1990;47(4):712–720. [PMC free article] [PubMed] [Google Scholar]
- 103.Tai D-Y, Pan J-P, Lee-Chen G-J. Identification and haplotype analysis of apolipoprotein B-100 Arg3500 → Trp mutation in hyperlipidemic Chinese. Clinical Chemistry. 1998;44(8):1659–1665. [PubMed] [Google Scholar]
- 104.Berg K. DNA polymorphism at the apolipoprotein B locus is associated with lipoprotein level. Clinical Genetics. 1986;30(6):515–520. doi: 10.1111/j.1399-0004.1986.tb01920.x. [DOI] [PubMed] [Google Scholar]
- 105.Hansen PS, Defesche JC, Kastelein JJP, et al. Phenotypic variation in patients heterozygous for familial defective apolipoprotein B (FDB) in three European countries. Arteriosclerosis, Thrombosis, and Vascular Biology. 1997;17(4):741–747. doi: 10.1161/01.atv.17.4.741. [DOI] [PubMed] [Google Scholar]
- 106.Guzman ECR, Hirata MH, Quintao ECR, Hirata RDC. Association of the apolipoprotein B gene polymorphisms with cholesterol levels and response to fluvastatin in Brazilian individuals with high risk for coronary heart disease. Clinical Chemistry and Laboratory Medicine. 2000;38(8):731–736. doi: 10.1515/CCLM.2000.103. [DOI] [PubMed] [Google Scholar]
- 107.Law A, Wallis SC, Powell LM. Common DNA polymorphism within coding sequence of apolipoprotein B gene associated with altered lipid levels. The Lancet. 1986;1(8493):1301–1302. doi: 10.1016/s0140-6736(86)91222-5. [DOI] [PubMed] [Google Scholar]
- 108.Hegele RA, Huang L-S, Herbert PN. Apolipoprotein B-gene DNA polymorphisms associated with myocardial infarction. The New England Journal of Medicine. 1986;315(24):1509–1515. doi: 10.1056/NEJM198612113152403. [DOI] [PubMed] [Google Scholar]
- 109.Talmud PJ, Barni N, Kessling AM. Apolipoprotein B gene variants are involved in the determination of serum cholesterol levels: a study in normo- and hyperlipidaemic individuals. Atherosclerosis. 1987;67(1):81–89. doi: 10.1016/0021-9150(87)90267-x. [DOI] [PubMed] [Google Scholar]
- 110.Genest JJ, Jr., Ordovas JM, McNamara JR, et al. DNA polymorphisms of the apolipoprotein B gene in patients with premature coronary artery disease. Atherosclerosis. 1990;82(1-2):7–17. doi: 10.1016/0021-9150(90)90138-9. [DOI] [PubMed] [Google Scholar]
- 111.Peacock R, Dunning A, Hamsten A, Tornvall P, Humphries S, Talmud P. Apolipoprotein B gene polymorphisms, lipoproteins and coronary atherosclerosis: a study of young myocardial infarction survivors and healthy population-based individuals. Atherosclerosis. 1992;92(2-3):151–164. doi: 10.1016/0021-9150(92)90274-k. [DOI] [PubMed] [Google Scholar]
- 112.Gaffney D, Freeman DJ, Shepherd J, Packard CJ. The ins/del polymorphism in the signal sequence of apolipoprotein B has no effect on lipid parameters. Clinica Chimica Acta. 1993;218(2):131–138. doi: 10.1016/0009-8981(93)90177-6. [DOI] [PubMed] [Google Scholar]
- 113.Glišić S, Prljić J, Radovanović N, Alavantić D. Study of apoB gene signal peptide insertion/deletion polymorphism in a healthy Serbian population: no association with serum lipid levels. Clinica Chimica Acta. 1997;263(1):57–65. doi: 10.1016/s0009-8981(97)06556-x. [DOI] [PubMed] [Google Scholar]
- 114.Myant NB, Gallagher J, Barbir M, Thompson GR, Wile D, Humphries SE. Restriction fragment length polymorphisms in the apo B gene in relation to coronary artery disease. Atherosclerosis. 1989;77(2-3):193–201. doi: 10.1016/0021-9150(89)90081-6. [DOI] [PubMed] [Google Scholar]
- 115.Bohn M, Bakken A, Erikssen J, Berg K. XbaI polymorphism in DNA at the apolipoprotein B locus is associated with myocardial infarction (MI) Clinical Genetics. 1993;44(5):241–248. doi: 10.1111/j.1399-0004.1993.tb03890.x. [DOI] [PubMed] [Google Scholar]
- 116.Ukkola O, Savolainen MJ, Salmela PI, Von Dickhoff K, Antero Kesaniemi Y. Apolipoprotein B gene DNA polymorphisms are associated with macro- and microangiopathy in non-insulin-dependent diabetes mellitus. Clinical Genetics. 1993;44(4):177–184. doi: 10.1111/j.1399-0004.1993.tb03875.x. [DOI] [PubMed] [Google Scholar]
- 117.Lopez-Miranda J, Ordovas JM, Ostos MA, et al. Dietary fat clearance in normal subjects is modulated by genetic variation at the apolipoprotein B gene locus. Arteriosclerosis, Thrombosis, and Vascular Biology. 1997;17(9):1765–1773. doi: 10.1161/01.atv.17.9.1765. [DOI] [PubMed] [Google Scholar]
- 118.Paulweber B, Friedl W, Krempler F, Humphries SE, Sandhofer F. Association of DNA polymorphism at the apolipoprotein B gene locus with coronary heart disease and serum very low density lipoprotein levels. Arteriosclerosis. 1990;10(1):17–24. doi: 10.1161/01.atv.10.1.17. [DOI] [PubMed] [Google Scholar]
- 119.Pouliot M-C, Despres J-P, Dionne FT, et al. ApoB-100 gene EcoRI polymorphism: relations to plasma lipoprotein changes associated with abdominal visceral obesity. Arteriosclerosis and Thrombosis. 1994;14(4):527–533. doi: 10.1161/01.atv.14.4.527. [DOI] [PubMed] [Google Scholar]
- 120.Bohn M, Bakken A, Erikssen J, Berg K. The apolipoprotein B signal peptide insertion/deletion polymorphism is not associated with myocardial infarction in Norway. Clinical Genetics. 1994;45(5):255–259. doi: 10.1111/j.1399-0004.1994.tb04151.x. [DOI] [PubMed] [Google Scholar]
- 121.Hong SH, Lee CC, Kim JQ. Genetic variation of the apolipoprotein b gene in korean patients with coronary artery disease. Molecules and Cells. 1997;7(4):521–525. [PubMed] [Google Scholar]
- 122.Machado MO, Hirata MH, Bertolami MC, Hirata RDC. Apo B gene haplotype is associated with lipid profile of higher risk for coronary heart disease in Caucasian Brazilian men. Journal of Clinical Laboratory Analysis. 2001;15:19–24. doi: 10.1002/1098-2825(2001)15:1<19::AID-JCLA4>3.0.CO;2-#. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Cavalli SA, Hirata MH, Salazar LA, et al. Apolipoprotein B gene polymorphisms: prevalence and impact on serum lipid concentrations in hypercholesterolemic individuals from Brazil. Clinica Chimica Acta. 2000;302:189–203. doi: 10.1016/s0009-8981(00)00367-3. [DOI] [PubMed] [Google Scholar]
- 124.Bruns GAP, Karathanasis SK, Breslow JL. Human apolipoprotein A-I-C-III gene complex is located on chromosome 11. Arteriosclerosis. 1984;4(2):97–102. doi: 10.1161/01.atv.4.2.97. [DOI] [PubMed] [Google Scholar]
- 125.Schaap FG, Rensen PCN, Voshol PJ, et al. ApoAV reduces plasma triglycerides by inhibiting very low density lipoprotein-triglycerides (VLDL-TG) production and stimulating lipoprotein lipase-mediated VLDL-TG hydrolysis. Journal of Biological Chemistry. 2004;279(27):27941–27947. doi: 10.1074/jbc.M403240200. [DOI] [PubMed] [Google Scholar]
- 126.Fielding CJ, Shore VG, Fielding PE. A protein cofactor of lecithin: cholesterol acyltransferase. Biochemical and Biophysical Research Communications. 1972;46(4):1493–1498. doi: 10.1016/0006-291x(72)90776-0. [DOI] [PubMed] [Google Scholar]
- 127.Jenner JL, Seman LJ, Millar JS, et al. The metabolism of apolipoproteins (a) and B-100 within plasma lipoprotein (a) in human beings. Metabolism. 2005;54(3):361–369. doi: 10.1016/j.metabol.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 128.Peacock RE, Hamsten A, Johansson J, Nilsson-Ehle P, Humphries SE. Associations of genotypes at the apolipoprotein AI-CIII-AIV, apolipoprotein B and lipoprotein lipase gene loci with coronary atherosclerosis and high density lipoprotein subclasses. Clinical Genetics. 1994;46(4):273–282. doi: 10.1111/j.1399-0004.1994.tb04159.x. [DOI] [PubMed] [Google Scholar]
- 129.Wang C-S, McConathy W, Kloer HU, Alaupovic P. Modulation of lipoprotein lipase activity by apolipoproteins. Effect of apolipoprotein C-III. Journal of Clinical Investigation. 1985;75(2):384–390. doi: 10.1172/JCI111711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Porkka KVK, Taimela S, Kontula K, et al. Variability gene effects of DNA polymorphisms at the apo B, apo AI/CIII and apo E loci on serum lipids: the Cardiovascular Risk in Young Finns Study. Clinical Genetics. 1994;45(3):113–121. doi: 10.1111/j.1399-0004.1994.tb04007.x. [DOI] [PubMed] [Google Scholar]
- 131.Dallongeville J, Meirhaeghe A, Cottel D, Fruchart J-C, Amouyel P, Helbecque N. Gender related association between genetic variations of APOC-III gene and lipid and lipoprotein variables in northern France. Atherosclerosis. 2000;150(1):149–157. doi: 10.1016/s0021-9150(99)00362-7. [DOI] [PubMed] [Google Scholar]
- 132.Oliva CP, Pisciotta L, Li Volti G, et al. Inherited apolipoprotein A-V deficiency in severe hypertriglyceridemia. Arteriosclerosis, Thrombosis, and Vascular Biology. 2005;25(2):411–417. doi: 10.1161/01.ATV.0000153087.36428.dd. [DOI] [PubMed] [Google Scholar]
- 133.Abifadel M, Varret M, Rabès J-P, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nature Genetics. 2003;34(2):154–156. doi: 10.1038/ng1161. [DOI] [PubMed] [Google Scholar]
- 134.Benjannet S, Rhainds D, Essalmani R, et al. NARC-1/PCSK9 and its natural mutants: zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. Journal of Biological Chemistry. 2004;279(47):48865–48875. doi: 10.1074/jbc.M409699200. [DOI] [PubMed] [Google Scholar]
- 135.Maxwell KN, Fisher EA, Breslow JL. Overexpression of PCSK9 accelerates the degradation of the LDLR in a post-endoplasmic reticulum compartment. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(6):2069–2074. doi: 10.1073/pnas.0409736102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yen FT, Deckelbaum RJ, Mann CJ, Marcel YL, Milne RW, Tall AR. Inhibition of cholesteryl ester transfer protein activity by monoclonal antibody. Effects of cholesteryl ester formation and neutral lipid mass transfer in human plasma. Journal of Clinical Investigation. 1989;83(6):2018–2024. doi: 10.1172/JCI114112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tall A. Plasma lipid transfer proteins. Annual Review of Biochemistry. 1995;64:235–257. doi: 10.1146/annurev.bi.64.070195.001315. [DOI] [PubMed] [Google Scholar]
- 138.Barter PJ. Hugh Sinclair Lecture: the regulation and remodelling of HDL by plasma factors. Atherosclerosis Supplements. 2002;3(4):39–47. doi: 10.1016/s1567-5688(02)00041-7. [DOI] [PubMed] [Google Scholar]
- 139.McPherson R, Mann CJ, Tall AR, et al. Plasma concentrations of cholesteryl ester transfer protein in hyperlipoproteinemia: relation to cholesteryl ester transfer protein activity and other lipoprotein variables. Arteriosclerosis and Thrombosis. 1991;11(4):797–804. doi: 10.1161/01.atv.11.4.797. [DOI] [PubMed] [Google Scholar]
- 140.Tall AR. Plasma cholesteryl ester transfer protein. Journal of Lipid Research. 1993;34(8):1255–1274. [PubMed] [Google Scholar]
- 141.Angelon LB, Quinet EM, Gillete TG, Drayna DT, Brown ML, Tall AR. Organization of the human cholesteryl ester transfer protein gene. Biochemistry. 1990;29(6):1372–1376. doi: 10.1021/bi00458a004. [DOI] [PubMed] [Google Scholar]
- 142.Callen DF, Hildebrand CE, Reeders S. Report of the second international workshop on human chromosome 16 mapping. Cytogenetics and Cell Genetics. 1992;60:158–167. [PubMed] [Google Scholar]
- 143.Ordovas JM, Cupples LA, Corella D, et al. Association of cholesteryl ester transfer protein-TaqIB polymorphism with variations in lipoprotein subclasses and coronary heart disease risk: the Framingham Study. Arteriosclerosis, Thrombosis, and Vascular Biology. 2000;20:1323–1329. doi: 10.1161/01.atv.20.5.1323. [DOI] [PubMed] [Google Scholar]
- 144.Inazu A, Jiang X-C, Haraki T, et al. Genetic cholesteryl ester transfer protein deficiency caused by two prevalent mutations as a major determinant of increased levels of high density lipoprotein cholesterol. Journal of Clinical Investigation. 1994;94(5):1872–1882. doi: 10.1172/JCI117537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kuivenhoven JA, Jukema JW, Zwinderman AH, et al. The role of a common variant of the cholesteryl ester transfer protein gene in the progression of coronary atherosclerosis. The New England Journal of Medicine. 1998;338(2):86–93. doi: 10.1056/NEJM199801083380203. [DOI] [PubMed] [Google Scholar]
- 146.Drayna D, Lawn R. Multiple RFLPs at the human cholesteryl ester transfer protein (CETP) locus. Nucleic Acids Research. 1987;15(11, article 4698) doi: 10.1093/nar/15.11.4698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kondo I, Berg K, Drayna D, Lawn R. DNA polymorphism at the locus for human cholesteryl ester transfer protein (CETP) is associated with high density lipoprotein cholesterol and apolipoprotein levels. Clinical Genetics. 1989;35(1):49–56. doi: 10.1111/j.1399-0004.1989.tb02904.x. [DOI] [PubMed] [Google Scholar]
- 148.Hannuksela ML, Johanna Liinamaa M, Antero Kesaniemi Y, Savolainen MJ. Relation of polymorphisms in the cholesteryl ester transfer protein gene to transfer protein activity and plasma lipoprotein levels in alcohol drinkers. Atherosclerosis. 1994;110(1):35–44. doi: 10.1016/0021-9150(94)90065-5. [DOI] [PubMed] [Google Scholar]
- 149.Eckel RH. Lipoprotein lipase: a multifunctional enzyme relevant to common metabolic diseases. The New England Journal of Medicine. 1989;320(16):1060–1068. doi: 10.1056/NEJM198904203201607. [DOI] [PubMed] [Google Scholar]
- 150.Mulder M, Lombardi P, Jansen H, Van Berkel TJC, Frants RR, Havekes LM. Low density lipoprotein receptor internalizes low density and very low density lipoproteins that are bound to heparan sulfate proteoglycans via lipoprotein lipase. Journal of Biological Chemistry. 1993;268(13):9369–9375. [PubMed] [Google Scholar]
- 151.Deeb SS, Peng R. Structure of the human lipoprotein lipase gene. Biochemistry. 1989;28(10):4131–4135. doi: 10.1021/bi00436a001. [DOI] [PubMed] [Google Scholar]
- 152.Oka K, Tkalcevic GT, Nakano T, Tucker H, Ishimura-Oka K, Brown WV. Structure and polymorphic map of human lipoprotein lipase gene. Biochimica et Biophysica Acta. 1990;1049(1):21–26. doi: 10.1016/0167-4781(90)90079-h. [DOI] [PubMed] [Google Scholar]
- 153.Hokanson JE. Functional variants in the lipoprotein lipase gene and risk of cardiovascular disease. Current Opinion in Lipidology. 1999;10(5):393–399. doi: 10.1097/00041433-199910000-00003. [DOI] [PubMed] [Google Scholar]
- 154.Wittrup HH, Tybjærg-Hansen A, Nordestgaard BG. Lipoprotein lipase mutations, plasma lipids and lipoproteins, and risk of ischemic heart disease: a meta-analysis. Circulation. 1999;99(22):2901–2907. doi: 10.1161/01.cir.99.22.2901. [DOI] [PubMed] [Google Scholar]
- 155.Razzaghi H, Aston CE, Hamman RF, Kamboh MI. Genetic screening of the lipoprotein lipase gene for mutations associated with high triglyceride/low HDL-cholesterol levels. Human Genetics. 2000;107(3):257–267. doi: 10.1007/s004390000367. [DOI] [PubMed] [Google Scholar]
- 156.Anderson JL, King GJ, Bair TL, et al. Association of lipoprotein lipase gene polymorphisms with coronary artery disease. Journal of the American College of Cardiology. 1999;33(4):1013–1020. doi: 10.1016/s0735-1097(98)00677-9. [DOI] [PubMed] [Google Scholar]
- 157.Tran H, Robinson S, Mikhailenko I, Strickland DK. Modulation of the LDL receptor and LRP levels by HIV protease inhibitors. Journal of Lipid Research. 2003;44(10):1859–1869. doi: 10.1194/jlr.M200487-JLR200. [DOI] [PubMed] [Google Scholar]
- 158.Wade DP, Knight BL, Soutar AK. Regulation of low-density-lipoprotein-receptor mRNA by insulin in human hepatoma Hep G2 cells. European Journal of Biochemistry. 1989;181(3):727–731. doi: 10.1111/j.1432-1033.1989.tb14784.x. [DOI] [PubMed] [Google Scholar]
- 159.Pascale RM, Simile MM, De Miglio MR, et al. Inhibition of 3-hydroxy-3-methylglutaryl-CoA reductase activity and gene expression by dehydroepiandrosterone in preneoplastic liver nodules. Carcinogenesis. 1995;16(7):1537–1542. doi: 10.1093/carcin/16.7.1537. [DOI] [PubMed] [Google Scholar]
- 160.Rudling M, Olivecrona H, Eggertsen G, Angelin B. Regulation of rat hepatic low density lipoprotein receptors: in vivo stimulation by growth hormone is not mediated by insulin-like growth factor. Journal of Clinical Investigation. 1996;97(2):292–299. doi: 10.1172/JCI118415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Fauvel J, Bonnet E, Ruidavets J-B, et al. An interaction between apo C-III variants and protease inhibitors contributes to high triglyceride/low HDL levels in treated HIV patients. AIDS. 2001;15(18):2397–2406. doi: 10.1097/00002030-200112070-00007. [DOI] [PubMed] [Google Scholar]
- 162.Bonnet E, Bernard J, Fauvel J, Massip P, Ruidavets J-B, Perret B. Association of APOC3 polymorphisms with both dyslipidemia and lipoatrophy in HAART-receiving patients. AIDS Research and Human Retroviruses. 2008;24(2):169–171. doi: 10.1089/aid.2007.0076. [DOI] [PubMed] [Google Scholar]
- 163.Foulkes AS, Wohl DA, Frank I, et al. Associations among race/ethnicity, apoC-III genotypes, and lipids in HIV-1-infected individuals on antiretroviral therapy. PLoS Medicine. 2006;3(3):337–347. doi: 10.1371/journal.pmed.0030052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Aragonès G, Alonso-Villaverde C, Pardo-Reche P, et al. Antiretroviral treatment-induced dyslipidemia in HIV-infected patients is influenced by the APOC3-related rs10892151 polymorphism. BMC Medical Genetics. 2011;12, article 120 doi: 10.1186/1471-2350-12-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Chang S-Y, Ko W-S, Kao J-T, et al. Association of single-nucleotide polymorphism 3 and c.553G>T of APOA5 with hypertriglyceridemia after treatment with highly active antiretroviral therapy containing protease inhibitors in HIV-infected individuals in Taiwan. Clinical Infectious Diseases. 2009;48(6):832–835. doi: 10.1086/597099. [DOI] [PubMed] [Google Scholar]
- 166.Tarr PE, Taffé P, Bleiber G, et al. Modeling the influence of APOC3, APOE, and TNF polymorphisms on the risk of antiretroviral therapy-associated lipid disorders. Journal of Infectious Diseases. 2005;191(9):1419–1426. doi: 10.1086/429295. [DOI] [PubMed] [Google Scholar]