

Abstract
Activation Induced Deaminase (AID) is an enzyme responsible for somatic hypermutation (SHM) and immunoglobulin heavy chain class switch recombination (CSR). Because AID causes double-stranded breaks in DNA, its expression is highly regulated and is normally restricted to germinal center B cells. Dysregulated AID expression can lead to cancer as a result of AID-mediated chromosomal translocations. Many transcription factors including Paired box protein 5 (Pax5) have been implicated in regulating the expression of Aicda, the gene encoding AID. In this study, we demonstrate that exogenous expression of Pax5 in a murine plasmacytoma cell line, 558LμM, leads to robust activation of endogenous Aicda transcription. Pax5 is known to initiate transcription both through its N-terminal paired DNA-binding domain (DBD) and C-terminal-activation domain. Through mutational analysis, we demonstrate that Pax5 regulates Aicda transcription through its C-terminal-activation domain. Together, our work describes a novel system that will be useful for determining how Pax5 regulates Aicda transcription.
Keywords: Activation induced cytidine deaminase gene, AID, Pax5, transcriptional regulation
Introduction
Activation Induced Deaminase (AID) is an enzyme required for somatic hypermutation (SHM) and class switch recombination (CSR) in germinal center B cells [1]. Activities of AID are indispensable for immunoglobulin affinity maturation and functional diversification, which are essential for the generation of diverse and effective humoral immune responses. AID is encoded by the Activation-induced cytidine deaminase (Aicda) gene, and is a member of the Apolipoprotein B mRNA editing enzyme catalytic polypeptide (Apobec) family of RNA and DNA editing enzymes [2]. Mechanisms of action of AID are not understood completely. AID modifies single-stranded DNA (ssDNA) by deamination of cytosine to generate uracil in vitro [3]. Similar modifications of bases by AID occur in vivo. The presence of uracil in immunoglobulin variable and switch regions in germinal center B cells is dependent on AID, providing direct evidence for cytosine modifications as part of AID-mediated functions [4]. AID also deaminates 5-methylcytosine (5-meC), resulting in thymine in vitro [5]. Conversion of 5-meC to thymine has been identified as an intermediate step in DNA demethylation that occurs in early embryonic stages of humans and mice [6-8]. In B cells, AID-converted uracils and surrounding bases are substrates for error-prone base excision repair and mismatch repair, which restore the normal sequence of DNA or result in somatic hypermutation (SHM) of DNA encoding Ig variable regions (reviewed in [9]). In the context of switch regions within Ig heavy chain loci, staggered breaks in double-stranded DNA (dsDNA) induced by AID provide ends for deletion of intervening DNA and joining in class switch recombination (CSR). However, despite the extensive characterization of AID's activities in vitro, it is not understood how 1) the expression of AID is regulated in germinal center B cells, 2) it is targeted to DNA sequence ‘hotspots’ [3], and 3) its functions are restricted to these sequences in variable region segments of Ig genes.
Following embryogenesis, AID expression is confined to germinal center B cells. However, promiscuous AID expression is strongly associated with tumorigenesis, including B and T cell lymphomas and leukemias and gastric, lung and colorectal cancers [10-14]. The genesis of these cancers is likely due to dsDNA breaks induced by AID, which can result in chromosomal translocations between Ig and non-Ig gene loci. Robbiani and colleagues found that AID was responsible for the chromosomal breaks in c-myc that promote IgH/c-myc translocations common in Burkitt's lymphomas in humans and plasmacytomas in mice [15, 16]. AID generates dsDNA breaks in a spectrum of non-Ig genes, including genes encoding transcription factors and signaling proteins required for normal B cell functions [17]. The recent detection of chromosomal translocations in normal B cells suggests that the AID-dependent generation of these hybrid loci, which could potentially promote tumorigenesis, is an ongoing process [18]. Thus, it is not surprising that the dosage, cell type-specificity and duration of AID expression is tightly controlled during B cell maturation in the context of germinal centers.
Pax5 is an important B cell lineage commitment factor that functions primarily in the early stages of B cell development. Pax5 has been studied extensively as a driver of early B cell development, where it cooperates with other transcription factors to activate the B cell-specific program of expressed genes [19, 20]. Pax5 is also essential for B cell lineage commitment [21]. Importantly, Pax5 limits the developmental potential of B cells by repressing the transcription of genes of other hematopoietic lineages [22].
Although Pax5 has been demonstrated to be important for transcription of germinal center B cell-specific genes, the role of Pax5 in Aicda transcription is somewhat controversial. Enforced expression of Pax5 in pro-B cell lines activated Aicda transcription [23]. AID expression in chronic lymphocytic leukemia (CLL) is associated with high expression of the Pax5 [24, 25]. Tran and colleagues identified a binding site for Pax5 in the first intron of the Aicda gene [26]. Interestingly, PI3K signaling may regulate Aicda expression by promoting expression of Blimp-1, which in turn represses Pax5 expression [27, 28].
Many cell-based systems have been used to study the regulation of AID expression, including the human Burkitt's lymphoma cell lines Ramos and Raji [29, 30] and the murine Ly1+ B cell lymphoma CH12 [31]. However, these cell lines are inefficient models for studies of Aicda regulation because the gene is constitutively transcribed. Here, we demonstrate the efficacy of a novel in vitro system based on the 558LμM murine plasmacytoma cell line [32], which was used previously to identify the Ig-α protein as a component of the B cell receptor (reviewed in [33]). Previously, we employed 558LμM cells to determine requirements for transcriptional activation of the mb-1/Cd79a gene in B cells [34-37]. 558LμM cells do not express key regulators of the early B cell-specific transcriptional program, including Early B cell Factor 1 (EBF1) and Pax5, which activate mb-1 transcription synergistically. In this study, we observed that enforced expression of Pax5 by itself is sufficient to induce transcriptional activation of endogenous Aicda gene in multiple subclones of 558LμM cells. Activation of Aicda transcription requires the previously identified activation domain of Pax5. We conclude that Pax5 is a limiting factor for activation of Aicda transcription in plasmacytoma cells.
Methods
Plasmids
Expression vectors for full length human Pax5 (1-391) or Pax5 (1-149) were described previously [34]. To produce expression vectors for other truncations of Pax5, BSPax5.S1 was amplified using PCR and primer pairs 5’-CTCATCATGGATTTAGAGAAAAATTATCC-3’ (5’ end of all clones) and (CΔ1) 5’-TTACCAGGAGTCGTTGTACGAGGAATA-3’, (CΔ2) 5’-CTATGTCACAATGGGGTAGGACTGCGG-3’, (CΔ3) 5’-CTAGTCTCCCCGCATCTGCTTCCGGAG-3’, or (CΔ4) 5’-CTACGACGAGCCGGCCGAATCCGTGCT-3’[38]. Each amplified fragment was ligated at the filled in (Klenow) XhoI site of MSCV-IRES-GFPαStu.
Cell lines, Transfection, Retroviral Infection, and Flow Cytometry
μM.2 cells stably expressing EBF1:ER, μM.3, and μM.10 cell lines were described previously [34, 37]. The generation of retroviruses and transduction of cells were described previously [34]. GFP+YFP+ cells and YFP+ cells were sorted 72 hours after transduction using a MoFlo XDP sorter (Beckman Coulter, Brea, CA, USA)
Analysis of mRNA transcripts
Isolation of total RNA and preparation of cDNA were described previously [39]. mRNA transcripts were detected by quantitative real time PCR (qPCR) with TaqMan® probes (Invitrogen) specific for murine Hprt (Mm00446968_m1) and Aicda (Mm00507774_m1). Statistical significance was assessed using Student's t test (GraphPad Prism software).
Western Blot
Whole cell extracts were prepared from 2.5×106 GFP+ cells. Cells were washed twice in ice cold 1X PBS. The cells were lysed in RIPA buffer [25mM Tris-HCl (pH 7.6), 150mM NaCl, 1% NP-40, 1% sodium deoxycholate and 0.1% SDS]. Thirty μg of each extract was fractionated on a 4-20% Tris-glycine gel (BioRad). Proteins were transferred to a nitrocellulose membrane and detection of proteins was performed using rabbit polyclonal anti-Pax5 primary antibody (Abcam); HDAC2 was detected using rabbit polyclonal anti-HDAC2 primary antibody (Invitrogen) and IRDye™-700DX–conjugated donkey–anti-rabbit IgG [H and L] secondary antibody (Rockland Immunochemicals, Inc.). Image acquisition and analysis were performed using an Odyssey™ infrared imaging system (LI-COR).
Results
We investigated whether Aicda transcription is activated by EBF1 and Pax5 through enforced expression of these factors in the μM.2 subclone of the 558LμM murine plasmacytoma cell line [34]. Full-length human Pax5 (1-391) was expressed in μM.2 cells using retroviral transduction of MSCV-Pax5-YFP, which co-expresses Pax5 and YFP as separate proteins. Retroviral expression of Pax5 was examined in the absence or presence of a fusion protein comprising EBF1 and the estrogen receptor ligand-binding domain (EBF1:ER) [37], which imparts regulation by 4-hydroxytamoxifen (4-OHT). All cells were treated with 4-OHT 48 hours after transduction to induce EBF1:ER translocation to the nucleus. YFP+ cells were sorted 72 hours after transduction for isolation of mRNA and cDNA synthesis. We performed qPCR using Taqman® probes to assess relative levels of Aicda transcripts in cDNA from sorted cells (Fig 1). All qPCR data were normalized to transcripts of the Hypoxanthine guanine phosphoribosyltransferase (Hprt) gene. In the absence of EBF1:ER and Pax5, μM.2 cells did not express Aicda transcripts detectably, while EBF1:ER by itself induced very low levels of Aicda transcripts. In contrast, exogenous expression of Pax5 resulted in a potent >3000-fold increase in Aicda transcript abundance compared to cells expressing EBF1:ER. However, Pax5 and EBF1:ER did not activate Aicda transcription synergistically.
Fig 1.
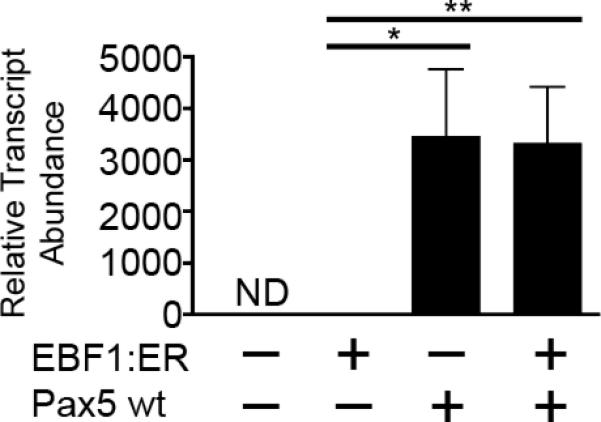
Exogenous expression of human Pax5 wild type (wt) activates transcription of endogenous Aicda in the μM.2 subclone of the 558LμM plasmacytoma cell line. Quantitative PCR of Aicda transcripts in sorted GFP+YFP+ μM.2 cells. All data were normalized to Hprt, n=4 or 5. μM.2 cells + EBF1:ER was set to 1. *p<0.01, **p<0.001.
Previous studies of 558LμM cells revealed their heterogeneity. The μM.2, μM.3, and μM.10 subclones each display different patterns of CpG methylation at their mb-1 (encodes Igα) promoters ([34] and unpublished data). mb-1 promoters of the μM.2 subclone are hypermethylated, which interferes with their transcriptional activation by Pax5. In contrast, mb-1 promoters in the μM.3 and μM.10 subclones are hypomethylated. mb-1 promoters are silent, but activated by Pax5 in μM.10 cells. Curiously, mb-1 promoters in μM.3 cells (originally called 558LμM.3;[32]) are constitutively active in the absence of EBF1 and Pax5 (unpublished data). To determine whether Pax5 activates Aicda transcription in the subclones, we expressed Pax5 using MSCV-Pax5-YFP in μM.2, μM.3, and μM.10 cell lines. qPCR analysis revealed a robust increase in endogenous Aicda transcript abundance in all of the subclones in response to Pax5 (Fig 2). μM.3 cells had low basal levels of Aicda transcripts, but also showed a potent (>1000 fold) elevation in Aicda transcripts in response to Pax5. However, Aicda transcript abundance in response to Pax5 was moderately decreased in μM.3 compared to μM.2 cells. Similar to μM.3 cells, the μM.10 clone also had low basal levels of Aicda transcripts that were markedly upregulated (>3000 fold) in response to Pax5. The upregulation of Aicda transcript levels in μM.10 cells was similar to that seen in μM.2 cells.
Fig 2.
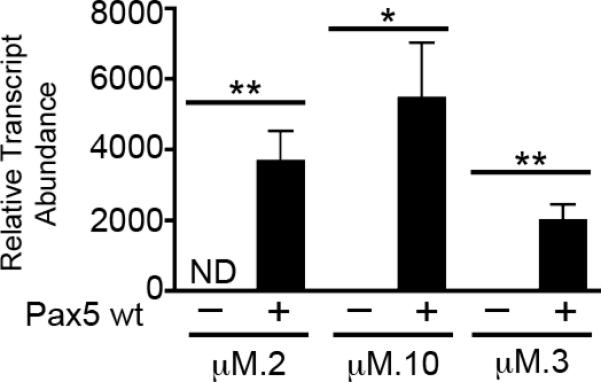
Exogenous expression of Pax5 wild type (wt) activates transcription of endogenous Aicda in three subclones of the 558LμM plasmacytoma cell line to varying degrees. Quantitative PCR of Aicda transcripts in sorted YFP+ μM.2, μM.10, and μM.3 cells. All data were normalized to Hprt, n=3. μM.3 cells with empty YFP vector was set to 1. *p<0.01, **p<0.001.
Pax5 has a well characterized C-terminal transcriptional activation (transactivation) domain [40]. However, this domain is not required for transcriptional activation of genes including mb-1 and Lef1 gene [38, 41]. To determine requirements for Aicda transcription in plasmacytoma cells, we transduced μM.2 cells with a series of retroviruses (as MSCV-Pax5-GFP) that express Pax5 with progressively larger C-terminal truncations (Fig 3A). GFP+ cells were sorted 72 hours after infection. qPCR was used to assess levels of Aicda transcription in μM.2 cells in the absence or presence of full length wild type or truncated Pax5 mutants. Wild type Pax5 demonstrated the greatest activation of Aicda transcription (>3000 fold) (Fig 3B). The CΔ1 mutant, which interacts with Groucho, or Grg4 [42], also activated Aicda transcription at somewhat decreased levels relative to full length Pax5 (>1000 fold). Transcription of Aicda was almost completely ablated after removal of the entire transactivation domain (CΔ2). Other Pax5 truncation mutants also failed to activate Aicda transcription. These results indicate that C-terminal residues between 304 and 391 of Pax5 are required for Aicda transcriptional activation in plasmacytoma cells. We verified human Pax5 expression in μM.2 by Western Blot (Fig 3C). The μM.2 cells with exogenous human Pax5 had similar levels of Pax5 protein expression, while the empty GFP control did not express Pax5. This verifies that activation of Aicda transcription was not due to varying amounts of Pax5, but due to the truncations of the proteins.
Fig 3.
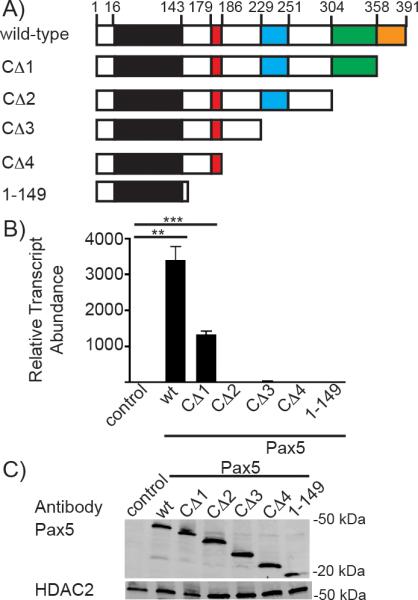
The C-terminal-transactivation domain of Pax5 is required for endogenous Aicda transcription in μM.2 cells. A) Schematic of the Pax5 wild-type (wt) and deletion mutants used in this study. B) Quantitative PCR of Aicda transcripts in sorted GFP+ μM.2 cells. All data were normalized to Hprt, n=3. μM.2 cells transduced with empty GFP vector was set to 1. **p<0.001, ***p<0.0001. C) Western detection of wild type and truncated Pax5 proteins in sorted GFP+ μM.2 cells. HDAC2 was detected as a loading control.
Discussion
In this study we demonstrate that Pax5 induces endogenous Aicda transcription in transduced plasmacytoma cells. Our results strongly suggest that Pax5 is important for the expression of endogenous Aicda. However, it remains unclear whether factors synergize with Pax5 to activate endogenous Aicda transcription. Our data also demonstrate that the C-terminal-transactivation domain of Pax5 is required to activate endogenous Aicda transcription. This indicates that Pax5 may target the Aicda gene locus in a manner similar to its activation of the Cd19 gene through interaction with the C-terminal-activation domain versus the N-terminal-paired DNA-binding domain [43]. In contrast, Pax5 activates transcription of other genes such as mb-1 and Lef1 through binding of the paired DNA-binding domain located in the N-terminus [38, 44]. In the case of the mb-1 promoter, direct interactions between Pax5 and Ets family transcription factors activate mb-1 transcription [38]. We did not obtain evidence for similar interactions between Pax5 and Ets proteins on the Aicda promoter (data not shown). We also observed little or no enhancement of Aicda gene activation by EBF1, although low levels of Aicda transcripts were observed in the presence of EBF1 by itself.
The levels of Aicda transcript abundance observed in μM.2 cells expressing exogenous Pax5 are similar to those in an unstimulated IgA+ subclone of the CH12 murine Ly1+ B cell lymphoma cell line, which mimics a germinal center B cell. CH12 cells differ from the 558LμM cell lines in that they express endogenous EBF1 and Pax5. Upon stimulation with IL-4, TGF-β and CD40-specific antibodies, CH12 cells increase AID expression dramatically due to activation of the NF-κB pathway and class switch to IgA [31, 45]. The 558LμM cell lines have constitutive NF-κB expression (data not shown) and therefore activate Aicda transcription without additional stimuli once wild type Pax5 is introduced.
We hypothesized that differences in mb-1 promoter responsiveness to transcription factors between 558LμM subclones extends to Aicda promoters. This could affect the ability of Pax5 to induce Aicda transcription. Previous work in the laboratory demonstrated that 558LμM cells are a heterogeneous population. Differences in promoter methylation affect endogenous mb-1 transcription and the cells’ response to transcription factors including EBF1, Runx1 and Pax5 [34, 36, 37]. Prior to transduction, μM.2 and μM.10 do not express significant levels of mb-1 transcripts. Expression of Pax5 alone in μM.10, but not μM.2, cells significantly increases endogenous mb-1 transcription. μM.2 cells require the activities of both EBF1 and Pax5 for significant mb-1 expression. In contrast, the μM.3 subclone expresses mb-1 transcripts constitutively. Wild type Pax5 activated endogenous Aicda transcription in three subclones of the 558LμM cells: μM.2, μM.3, and μM.10. The greatest levels of Aicda transcription in response to Pax5 were observed in μM.10 cells. Future studies will assess the status of DNA methylation at Aicda promoters in these cells; however, Pax5-dependent transcriptional efficiency may be due to other differences between the subclones, such as the presence of other factors that regulate Aicda transcription.
Pax5 likely synergizes with other regulators of Aicda transcription in plasmacytoma cells. Multiple B cell-specific and more widely expressed transcription factors have been implicated in the control of Aicda gene transcription. The basal Aicda promoter comprises binding sites for Sp factors including Sp1 and Sp3 [46]. Studies in mice suggest that basic helix-loop-helix proteins (E12 and E47, collectively termed E-proteins) encoded by the Tcfe2a (E2A) gene activate endogenous Aicda transcription. Sayegh et al. (2003) reported that B cells overexpressing the Inhibition of differentiation 3 (Id3) protein, a post-translational inhibitor of E-proteins, failed to activate endogenous Aicda transcription [47]. Kwon et al. (2008) reported that E2A-deficient mice express normal levels of AID, although the generation of germinal center B cells is impaired [48]. Additionally, Tran et al. (2010) identified functionally important E boxes in an intron of the Aicda gene [26]. Thus, E-proteins may regulate Aicda transcription directly. Other factors linked with Aicda transcription include HoxC4 [49], Irf8 [50], STAT6 and NF-κB [45], which may alleviate silencing of Aicda genes by c-Myb and E2F [26]. More recently, the basic leucine zipper transcription factor BATF was shown to activate Aicda transcription directly [51]. We previously detected E proteins and nuclear NF-κB in the μM.2 subclone. These cells also express significant levels of Blimp1 transcripts (A.L. Shaffer, unpublished data). Blimp1 may negatively regulate Aicda expression [27]. Additional studies will focus on whether additional regulatory factors are present in plasmacytoma cells.
558LμM have proven useful for studying activation of endogenous Aicda expression. Future studies will focus on determining additional requirements for endogenous Aicda transcriptional activation. Additionally, these cells can be used to examine the roles that chromatin remodeling complexes may play in transcriptional activation of Aicda.
Acknowledgements
We are indebted to Michael Reth for providing the parent 558LμM and μM.3 subclone and Marian Koshland for the IgA+ CH12 cells. We thank Holly Maier and Julita Ramirez for technical (HM and JR) and editorial (JR) assistance. This work was supported by NIH grants R01 AI054661 and AI081878. CD was in part supported by the Cancer Research Institute Pre-doctoral Emphasis Pathway in Tumor Immunology Fellowship.
References
- 1.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102(5):553–63. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 2.Muramatsu M, Sankaranand VS, Anant S, Sugai M, Kinoshita K, Davidson NO, et al. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. J Biol Chem. 1999;274(26):18470–6. doi: 10.1074/jbc.274.26.18470. [DOI] [PubMed] [Google Scholar]
- 3.Dickerson SK, Market E, Besmer E, Papavasiliou FN. AID mediates hypermutation by deaminating single stranded DNA. J Exp Med. 2003;197(10):1291–6. doi: 10.1084/jem.20030481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maul RW, Saribasak H, Martomo SA, McClure RL, Yang W, Vaisman A, et al. Uracil residues dependent on the deaminase AID in immunoglobulin gene variable and switch regions. Nat Immunol. 2011;12(1):70–6. doi: 10.1038/ni.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morgan HD, Dean W, Coker HA, Reik W, Petersen-Mahrt SK. Activation-induced cytidine deaminase deaminates 5-methylcytosine in DNA and is expressed in pluripotent tissues: implications for epigenetic reprogramming. J Biol Chem. 2004;279(50):52353–60. doi: 10.1074/jbc.M407695200. [DOI] [PubMed] [Google Scholar]
- 6.Bhutani N, Brady JJ, Damian M, Sacco A, Corbel SY, Blau HM. Reprogramming towards pluripotency requires AID-dependent DNA demethylation. Nature. 2010;463(7284):1042–7. doi: 10.1038/nature08752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Popp C, Dean W, Feng S, Cokus SJ, Andrews S, Pellegrini M, et al. Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature. 2010;463(7284):1101–5. doi: 10.1038/nature08829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rai K, Huggins IJ, James SR, Karpf AR, Jones DA, Cairns BR. DNA demethylation in zebrafish involves the coupling of a deaminase, a glycosylase, and gadd45. Cell. 2008;135(7):1201–12. doi: 10.1016/j.cell.2008.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peled JU, Kuang FL, Iglesias-Ussel MD, Roa S, Kalis SL, Goodman MF, et al. The biochemistry of somatic hypermutation. Annu Rev Immunol. 2008;26:481–511. doi: 10.1146/annurev.immunol.26.021607.090236. [DOI] [PubMed] [Google Scholar]
- 10.Gruber TA, Chang MS, Sposto R, Muschen M. Activation-induced cytidine deaminase accelerates clonal evolution in BCR-ABL1-driven B-cell lineage acute lymphoblastic leukemia. Cancer Res. 2010;70(19):7411–20. doi: 10.1158/0008-5472.CAN-10-1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marantidou F, Dagklis A, Stalika E, Korkolopoulou P, Saetta A, Anagnostopoulos A, et al. Activation-induced cytidine deaminase splicing patterns in chronic lymphocytic leukemia. Blood Cells Mol Dis. 2010;44(4):262–7. doi: 10.1016/j.bcmd.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 12.Matsumoto Y, Marusawa H, Kinoshita K, Niwa Y, Sakai Y, Chiba T. Up-regulation of activation-induced cytidine deaminase causes genetic aberrations at the CDKN2b-CDKN2a in gastric cancer. Gastroenterology. 2010;139(6):1984–94. doi: 10.1053/j.gastro.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 13.Shinmura K, Igarashi H, Goto M, Tao H, Yamada H, Matsuura S, et al. Aberrant expression and mutation-inducing activity of AID in human lung cancer. Ann Surg Oncol. 2011;18(7):2084–92. doi: 10.1245/s10434-011-1568-8. [DOI] [PubMed] [Google Scholar]
- 14.Endo Y, Marusawa H, Chiba T. Involvement of activation-induced cytidine deaminase in the development of colitis-associated colorectal cancers. J Gastroenterol. 2011;46(Suppl 1):6–10. doi: 10.1007/s00535-010-0326-1. [DOI] [PubMed] [Google Scholar]
- 15.Robbiani DF, Bothmer A, Callen E, Reina-San-Martin B, Dorsett Y, Difilippantonio S, et al. AID is required for the chromosomal breaks in c-myc that lead to c-myc/IgH translocations. Cell. 2008;135(6):1028–38. doi: 10.1016/j.cell.2008.09.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robbiani DF, Bunting S, Feldhahn N, Bothmer A, Camps J, Deroubaix S, et al. AID produces DNA double-strand breaks in non-Ig genes and mature B cell lymphomas with reciprocal chromosome translocations. Mol Cell. 2009;36(4):631–41. doi: 10.1016/j.molcel.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu M, Duke JL, Richter DJ, Vinuesa CG, Goodnow CC, Kleinstein SH, et al. Two levels of protection for the B cell genome during somatic hypermutation. Nature. 2008;451(7180):841–5. doi: 10.1038/nature06547. [DOI] [PubMed] [Google Scholar]
- 18.Klein IA, Resch W, Jankovic M, Oliveira T, Yamane A, Nakahashi H, et al. Translocation-capture sequencing reveals the extent and nature of chromosomal rearrangements in B lymphocytes. Cell. 2011;147(1):95–106. doi: 10.1016/j.cell.2011.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nutt SL, Kee BL. The transcriptional regulation of B cell lineage commitment. Immunity. 2007;26(6):715–25. doi: 10.1016/j.immuni.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 20.Ramirez J, Lukin K, Hagman J. From hematopoietic progenitors to B cells: mechanisms of lineage restriction and commitment. Curr Opin Immunol. 2010;22(2):177–84. doi: 10.1016/j.coi.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nutt SL, Heavey B, Rolink AG, Busslinger M. Commitment to the B-lymphoid lineage depends on the transcription factor Pax5. Nature. 1999;401(6753):556–62. doi: 10.1038/44076. [DOI] [PubMed] [Google Scholar]
- 22.Delogu A, Schebesta A, Sun Q, Aschenbrenner K, Perlot T, Busslinger M. Gene repression by Pax5 in B cells is essential for blood cell homeostasis and is reversed in plasma cells. Immunity. 2006;24(3):269–81. doi: 10.1016/j.immuni.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 23.Gonda H, Sugai M, Nambu Y, Katakai T, Agata Y, Mori KJ, et al. The balance between Pax5 and Id2 activities is the key to AID gene expression. J Exp Med. 2003;198(9):1427–37. doi: 10.1084/jem.20030802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oppezzo P, Dumas G, Lalanne AI, Payelle-Brogard B, Magnac C, Pritsch O, et al. Different isoforms of BSAP regulate expression of AID in normal and chronic lymphocytic leukemia B cells. Blood. 2005;105(6):2495–503. doi: 10.1182/blood-2004-09-3644. [DOI] [PubMed] [Google Scholar]
- 25.Reiniger L, Bodor C, Bognar A, Balogh Z, Csomor J, Szepesi A, et al. Richter's and prolymphocytic transformation of chronic lymphocytic leukemia are associated with high mRNA expression of activation-induced cytidine deaminase and aberrant somatic hypermutation. Leukemia. 2006;20(6):1089–95. doi: 10.1038/sj.leu.2404183. [DOI] [PubMed] [Google Scholar]
- 26.Tran TH, Nakata M, Suzuki K, Begum NA, Shinkura R, Fagarasan S, et al. B cell-specific and stimulation-responsive enhancers derepress Aicda by overcoming the effects of silencers. Nat Immunol. 2010;11(2):148–54. doi: 10.1038/ni.1829. [DOI] [PubMed] [Google Scholar]
- 27.Omori SA, Cato MH, Anzelon-Mills A, Puri KD, Shapiro-Shelef M, Calame K, et al. Regulation of class-switch recombination and plasma cell differentiation by phosphatidylinositol 3-kinase signaling. Immunity. 2006;25(4):545–57. doi: 10.1016/j.immuni.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 28.Lin KI, Angelin-Duclos C, Kuo TC, Calame K. Blimp-1-dependent repression of Pax-5 is required for differentiation of B cells to immunoglobulin M-secreting plasma cells. Mol Cell Biol. 2002;22(13):4771–80. doi: 10.1128/MCB.22.13.4771-4780.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang W, Bardwell PD, Woo CJ, Poltoratsky V, Scharff MD, Martin A. Clonal instability of V region hypermutation in the Ramos Burkitt's lymphoma cell line. Int Immunol. 2001;13(9):1175–84. doi: 10.1093/intimm/13.9.1175. [DOI] [PubMed] [Google Scholar]
- 30.Ruckerl F, Busse B, Bachl J. Episomal vectors to monitor and induce somatic hypermutation in human Burkitt-Lymphoma cell lines. Mol Immunol. 2006;43(10):1645–52. doi: 10.1016/j.molimm.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 31.Nakamura M, Kondo S, Sugai M, Nazarea M, Imamura S, Honjo T. High frequency class switching of an IgM+ B lymphoma clone CH12F3 to IgA+ cells. Int Immunol. 1996;8(2):193–201. doi: 10.1093/intimm/8.2.193. [DOI] [PubMed] [Google Scholar]
- 32.Hombach J, Tsubata T, Leclercq L, Stappert H, Reth M. Molecular components of the B-cell antigen receptor complex of the IgM class. Nature. 1990;343(6260):760–2. doi: 10.1038/343760a0. [DOI] [PubMed] [Google Scholar]
- 33.Hagman J. Conveying the message: identification of Ig-alpha and Ig-beta as components of the B cell receptor complex. J Immunol. 2009;183(3):1503–4. doi: 10.4049/jimmunol.0990055. [DOI] [PubMed] [Google Scholar]
- 34.Maier H, Colbert J, Fitzsimmons D, Clark DR, Hagman J. Activation of the Early B-Cell-Specific mb-1 (Ig- ) Gene by Pax-5 Is Dependent on an Unmethylated Ets Binding Site. Molecular and Cellular Biology. 2003;23(6):1946–60. doi: 10.1128/MCB.23.6.1946-1960.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maier H, Ostraat R, Parenti S, Fitzsimmons D, Abraham LJ, Garvie CW, et al. Requirements for selective recruitment of Ets proteins and activation of mb-1/Ig-alpha gene transcription by Pax-5 (BSAP). Nucleic Acids Res. 2003;31(19):5483–9. doi: 10.1093/nar/gkg785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maier H, Ostraat R, Gao H, Fields S, Shinton SA, Medina KL, et al. Early B cell factor cooperates with Runx1 and mediates epigenetic changes associated with mb-1 transcription. Nat Immunol. 2004;5(10):1069–77. doi: 10.1038/ni1119. [DOI] [PubMed] [Google Scholar]
- 37.Gao H, Lukin K, Ramirez J, Fields S, Lopez D, Hagman J. Opposing effects of SWI/SNF and Mi-2/NuRD chromatin remodeling complexes on epigenetic reprogramming by EBF and Pax5. Proc Natl Acad Sci U S A. 2009;106(27):11258–63. doi: 10.1073/pnas.0809485106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fitzsimmons D, Hodsdon W, Wheat W, Maira SM, Wasylyk B, Hagman J. Pax-5 (BSAP) recruits Ets proto-oncogene family proteins to form functional ternary complexes on a B-cell-specific promoter. Genes Dev. 1996;10(17):2198–211. doi: 10.1101/gad.10.17.2198. [DOI] [PubMed] [Google Scholar]
- 39.Fields S, Ternyak K, Gao H, Ostraat R, Akerlund J, Hagman J. The ‘zinc knuckle’ motif of Early B cell Factor is required for transcriptional activation of B cell-specific genes. Mol Immunol. 2008;45(14):3786–96. doi: 10.1016/j.molimm.2008.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dorfler P, Busslinger M. C-terminal activating and inhibitory domains determine the transactivation potential of BSAP (Pax-5), Pax-2 and Pax-8. EMBO J. 1996;15(8):1971–82. [PMC free article] [PubMed] [Google Scholar]
- 41.Nutt SL, Morrison AM, Dorfler P, Rolink A, Busslinger M. Identification of BSAP (Pax-5) target genes in early B-cell development by loss- and gain-of-function experiments. EMBO J. 1998;17(8):2319–33. doi: 10.1093/emboj/17.8.2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eberhard D, Jimenez G, Heavey B, Busslinger M. Transcriptional repression by Pax5 (BSAP) through interaction with corepressors of the Groucho family. EMBO J. 2000;19(10):2292–303. doi: 10.1093/emboj/19.10.2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kozmik Z, Wang S, Dorfler P, Adams B, Busslinger M. The promoter of the CD19 gene is a target for the B-cell-specific transcription factor BSAP. Mol Cell Biol. 1992;12(6):2662–72. doi: 10.1128/mcb.12.6.2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nutt SL, Urbanek P, Rolink A, Busslinger M. Essential functions of Pax5 (BSAP) in pro-B cell development: difference between fetal and adult B lymphopoiesis and reduced V-to-DJ recombination at the IgH locus. Genes Dev. 1997;11(4):476–91. doi: 10.1101/gad.11.4.476. [DOI] [PubMed] [Google Scholar]
- 45.Dedeoglu F, Horwitz B, Chaudhuri J, Alt FW, Geha RS. Induction of activation-induced cytidine deaminase gene expression by IL-4 and CD40 ligation is dependent on STAT6 and NFkappaB. Int Immunol. 2004;16(3):395–404. doi: 10.1093/intimm/dxh042. [DOI] [PubMed] [Google Scholar]
- 46.Yadav A, Olaru A, Saltis M, Setren A, Cerny J, Livak F. Identification of a ubiquitously active promoter of the murine activation-induced cytidine deaminase (AICDA) gene. Mol Immunol. 2006;43(6):529–41. doi: 10.1016/j.molimm.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 47.Sayegh CE, Quong MW, Agata Y, Murre C. E-proteins directly regulate expression of activation-induced deaminase in mature B cells. Nat Immunol. 2003;4(6):586–93. doi: 10.1038/ni923. [DOI] [PubMed] [Google Scholar]
- 48.Kwon K, Hutter C, Sun Q, Bilic I, Cobaleda C, Malin S, et al. Instructive role of the transcription factor E2A in early B lymphopoiesis and germinal center B cell development. Immunity. 2008;28(6):751–62. doi: 10.1016/j.immuni.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 49.Park SR, Zan H, Pal Z, Zhang J, Al-Qahtani A, Pone EJ, et al. HoxC4 binds to the promoter of the cytidine deaminase AID gene to induce AID expression, class-switch DNA recombination and somatic hypermutation. Nat Immunol. 2009;10(5):540–50. doi: 10.1038/ni.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee CH, Melchers M, Wang H, Torrey TA, Slota R, Qi CF, et al. Regulation of the germinal center gene program by interferon (IFN) regulatory factor 8/IFN consensus sequence-binding protein. J Exp Med. 2006;203(1):63–72. doi: 10.1084/jem.20051450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ise W, Kohyama M, Schraml BU, Zhang T, Schwer B, Basu U, et al. The transcription factor BATF controls the global regulators of class-switch recombination in both B cells and T cells. Nat Immunol. 2011;12(6):536–43. doi: 10.1038/ni.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]