

Abstract
Dental anomalies are common congenital malformations that can occur either as isolated findings or as part of a syndrome. This review focuses on genetic causes of abnormal tooth development and the implications of these abnormalities for clinical care. As an introduction, we describe general insights into the genetics of tooth development obtained from mouse and zebrafish models. This is followed by a discussion of isolated as well as syndromic tooth agenesis, including Van der Woude syndrome, ectodermal dysplasias, oral-facial-digital syndrome type I, Rieger syndrome, holoprosencephaly, and tooth anomalies associated with cleft lip and palate. Next, we review delayed formation and eruption of teeth, as well as abnormalities in tooth size, shape and form. Finally, isolated and syndromic causes of supernumerary teeth are considered, including cleidocranial dysplasia and Gardner syndrome.
Keywords: mouse, zebrafish, teeth, hypodontia, supernumerary teeth, craniofacial, syndrome, tooth
INTRODUCTION
Genetic causes have been identified for both isolated tooth malformations and for the dental anomalies seen in patients with craniofacial developmental abnormalities. Congenitally missing teeth are seen in a host of syndromes, and supernumerary teeth are also central diagnostic findings in a number of syndromes. Additionally, mutations in several genes have been associated with both hypodontia and orofacial clefting in humans and mice, indicating that tooth anomalies and orofacial clefting may share common developmental pathways. Because the study of tooth development is central to understanding the pathogenesis of dental anomalies, this review begins with an overview of recent studies in vertebrate animal models, which is followed by a survey of dental anomalies with known or suspected genetic causes.
LESSONS FROM ANIMAL MODELS
Mouse dentition: the major model system
Most of our knowledge regarding the cellular and genetic basis of mammalian tooth development has come from mouse studies. Although mouse dentition is simpler than that of humans, the developmental mechanisms are thought to be highly conserved between the two. Both humans and rodents have fewer teeth than the unreduced pattern of their mammalian ancestors, in which up to three incisors, one canine, four premolars, and three molars may occur in each dental quadrant. A few species, such as some insectivores, have retained the full pattern of dentition. Humans have two incisors, one canine, two premolars and three molars in the permanent dentition. The adult mouse dentition is much more reduced than in the human, consisting of three molars at the back of the mouth and one incisor at the front, separated by a toothless region called a diastema, in each quadrant (Fig. 1). Another major difference between mouse and human dentition is that mice have only a single set of teeth, whereas in humans the first set of teeth (primary or deciduous teeth) is replaced by a permanent set during childhood. The mouse therefore provides a simplified model for tooth formation in humans.
Figure 1. Comparison of the adult and embryonic tooth pattern in the mouse.

Left: functional dentition in adult mouse. Right: Mouse embryonic tooth pattern. In the upper incisor region, five to six small epithelial prominences are integrated and commonly give rise to the early bud of the upper incisor. In the embryonic mandible, three epithelial prominences predetermine the origin of the prospective functional incisor. During embryonic day (ED) 12.5–13.5, the upper diastema comprises five primordia that do not progress beyond a bud shape (D1–D5), while only a thin epithelial thickening (dashed line) is present in mandible. Two large rudimentary buds develop in the posterior part of the upper (R1, R2) and lower (MS, R2) diastema, and these are the most conspicuous structures in the cheek region until ED 13.5. The upper R1, R2, and MS rudiments cease growth due to epithelial apoptosis and are transformed into epithelial ridges. The lower R2 becomes incorporated into the anterior part of the lower first molar (M1) cap. The buds of the posterior molars (M2, M3) develop at later stages.
Beyond serving as a model for understanding mammalian tooth development in general, studying the development of the reduced dentition in mouse provides two advantages. First, the permanently renewing incisor serves as a model to study the role of stem cells in organ regeneration [Biehs et al., 2013; Feng et al., 2011; Harada et al., 1999; Juuri et al., 2012; Lapthanasupkul et al., 2012; Seidel et al., 2010]. Second, the mouse embryonic jaws contain rudimentary tooth primordia of teeth that were suppressed during evolution [Hovorakova et al., 2011; Peterkova et al., 2002b] (Fig. 1). In the majority of the diastemal tooth primordia, development is arrested [Peterkova et al., 2003], such that the development of the mouse diastema represents a model to study hypodontia [Peterkova et al., 1995]. Some of these rudimentary tooth primordia may be rescued and can give rise to supernumerary teeth [Klein et al., 2006; Peterkova et al., 2002a; Peterkova et al., 2006], which can model controlled tooth regeneration [Cobourne and Sharpe, 2010; Peterkova et al., 2009; Peterkova et al., 2006].
Teeth form through a series of reciprocal interactions between epithelium (derived from oral ectoderm) and mesenchyme (derived from cranial neural crest), which begin at mid-gestation in mouse embryos [Tucker and Sharpe, 2004]. The interactions between oral epithelium and underlying neural-crest derived mesenchyme are mediated by secreted signaling molecules from the major signaling families (FGF, TGF-β, WNT and HH), which lead to various intracellular events, including expression of transcription factors (e.g., members of the Msx, Pax, and Runx families, discussed below) [Jheon et al., 2013; Jussila and Thesleff, 2012].
As the epithelium and mesenchyme interact, the developing tooth (tooth germ) progresses through several stages (Fig. 2). The first morphological sign of tooth development is a localized thickening of the oral epithelium. Next, the thickened (dental) epithelium invaginates into the underlying mesenchyme, forming a dental lamina and tooth buds, while the adjacent dental mesenchyme condenses around the forming tooth buds. Subsequently, the epithelium around the bud tip extends farther into the mesenchyme, forming a cap and then a bell stage tooth germ. The dental epithelium (enamel organ) is surrounded by a layer of dental mesenchyme (dental sac), and the enamel organ encloses the mesenchymal papilla. The dental papilla arises from a small population of highly proliferative mesenchymal cells in close proximity to the inner dental epithelium and the primary enamel knot [Rothova et al., 2012]. Further epithelial morphogenesis and expansion of the dental mesenchyme results in the formation of cusps. During the bell stage, cusp morphogenesis continues and cytodifferentiation begins, as the epithelial cells closest to the dental mesenchyme become enamel-producing ameloblasts, and the adjacent mesenchymal cells become dentin-producing odontoblasts [Ruch, 1995].
Figure 2. Stages of development of the lower first molar in mouse.
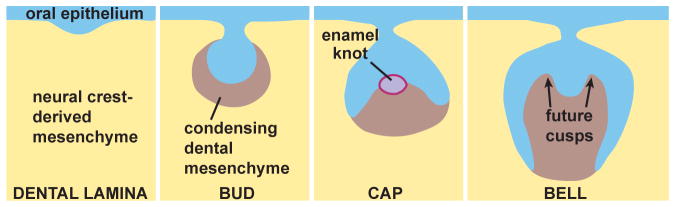
The oral epithelium thickens and then invaginates into the neural crest-derived mesenchyme. Mesenchymal condensation occurs at the bud stage. The enamel knot appears and acts as a signaling center during tooth development at the cap stage. During the bell stage, tooth morphogenesis is accompanied by the differential growth of the interface between the dental epithelium and papilla mesenchyme, which predetermines the form (cusps) of the prospective tooth crown. The matrix will eventually mineralize, forming the tooth crown, and this is followed by root development and tooth eruption.
Epithelial morphogenesis and growth of the dental mesenchyme during the cap and bell stages are thought to be controlled and coordinated by signals produced by the enamel knot, a morphologically distinct region of the epithelium containing densely-packed, non-proliferating cells. The primary enamel knot forms at the center of the tooth germ at the onset of the cap stage [Jernvall et al., 1994] and is subsequently eliminated by apoptosis [Lesot et al., 1996; Vaahtokari et al., 1996]. Secondary enamel knots form at the cusp tips, and signals from them control later aspects of cusp morphogenesis [Jernvall et al., 1998].
In mouse and human, the upper incisors arise largely from the medial nasal process with a minor contribution of the maxillary facial process [Hovorakova et al., 2006; Peterkova et al., 1995]. The upper and lower molars arise from the first pharyngeal arch, which appears to be molecularly patterned in terms of tooth location and identity before any morphological signs of tooth development are evident. The mesenchyme of the first pharyngeal arch initially has ubiquitous odontogenic potential, and the odontogenic mesenchyme is specified by its proximity to the oral epithelium, which is the source of the inductive signal. It is thought that FGF8 from the lateral oral epithelium and BMP4 from the medial oral epithelium differentially regulate the expression of transcription factors (e.g., Dlx1, Dlx2, and Barx1 are expressed laterally, whereas Msx1 and Msx2 are expressed medially) [Bei and Maas, 1998; Keranen et al., 1999; Neubuser et al., 1997; St Amand et al., 2000; Thomas et al., 2000]. These expression patterns have been proposed to represent an “odontogenic homeobox code” that specifies tooth identity, analogous to homeobox codes found in other developmental systems [Sharpe, 1995].
The earliest marker for the location of presumptive teeth is expression of Pax9, which results from antagonistic interactions of FGF and BMP signaling [32]. Fgf8 induces Pax9 expression in first pharyngeal arch mesenchyme, whereas Bmp2 and Bmp4 inhibit this induction [Neubuser et al., 1997]. Therefore, Pax9 is expressed only in regions where Fgf8 is present but Bmp2 and Bmp4 are absent. Interestingly, although Pax9 marks the sites of future tooth development, in mouse studies Pax9 itself appears not to be necessary to position teeth or initiate odontogenesis. Thus, in the mouse Pax9 mutant, teeth develop normally up to the bud stage (E13.5) before arresting, indicating that this gene is critical for bud development but not for tooth initiation [Peters et al., 1998]. The role of Pax9 in human hypodontia is discussed below.
The expression of other genes indicates that, at the earliest stages of tooth development, the instructive information resides in the epithelium. Sonic hedgehog (Shh) expression is restricted to the emerging tooth primordia. The restriction of Shh appears to be due to repression by Wnt7b in the non-dental epithelium [Sarkar et al., 2000]. At the bud stage, the instructive role shifts from the epithelium to the mesenchyme; transcription factors such as Msx1, Pax9, and Runx2 are expressed in the condensed dental mesenchyme [Thesleff, 2006]. These factors, all of which are important in human tooth development as well, promote the expression of secreted signaling molecules including Bmp4, Fgf3, and Wnt5a, which act upon the epithelium and induce the enamel knot.
Zebrafish dentition: an up-and-coming model
In recent years, animal models other than the mouse have emerged for the investigation of early development, organogenesis and regeneration. Zebrafish in particular have become a favorite lab animal, as they are inexpensive to maintain, reproduce easily and abundantly, and have the vertebrate body plan. A vast array of genetic and molecular tools has been developed for zebrafish, which have now been used to model nearly every class of human disease.
The zebrafish has no teeth on its oral jaws, but it has maintained sets of teeth on the rearmost pharyngeal arch as a remnant of the once widespread oral tooth coverage in its remote ancestors. These pharyngeal teeth are continuously replaced throughout life and have been well characterized in terms of patterning, structure and morphodifferentiation [Huysseune et al., 1998; Van Der Heyden and Huysseune, 2000; Van Der Heyden et al., 2000] (Fig. 3). A number of studies have addressed the genetic and molecular underpinnings of tooth development and replacement (reviewed by [Stock, 2007]). Tooth formation and replacement start early, well before many mutations become lethal (at 48 hours and 80 hours post-fertilization, resp.) [Borday-Birraux et al., 2006]. This circumvents the lethality encountered when modeling craniofacial anomalies and dental diseases in mouse models. Additionally, because mice do not replace their dentition, dissecting the mechanism of natural lifelong replacement in zebrafish represents a strategy for understanding tooth replacement in mammals that is not possible with the mouse model.
Figure 3. Zebrafish tooth development.

(A–B) Schematic representation of the pharyngeal dentition of a zebrafish at 6 days post-fertilization (dpf) (A) and one month old (B). Ventral tooth row, yellow, mediodorsal tooth row, ochre, dorsal tooth row, green; replacement teeth not shown in B. At 6 dpf, primary teeth 3V1, 4V1 and 5V1 are attached, and the first replacement tooth (4V2) is mineralizing. (C) Alizarin stained and cleared preparation of the dentition of an 8 dpf zebrafish. Tooth 4V2 (arrowhead) is more advanced compared to the scheme shown in (A). Note keratinized pad (asterisk) opposing the teeth. (D) SEM view of ventral teeth 2V–5V in a juvenile zebrafish (anterior to left). (E–F) One μm plastic cross sections through the pharyngeal dentition of a 5 dpf wild type (WT) zebrafish (E) and 5 dpf edar−/− mutant (F). Teeth 3V1, 4V1, 5V1 and replacement tooth 4V2 are present in the WT; only 4V1 is present in the mutant. (G) One μm plastic cross section through an attaching tooth in a one month zebrafish; note odontoblasts (od) sending processes (arrowhead) into the dentin (d). Scale bars C, E, F = 25 μm, D = 100 μm, G = 50 μm.
While some developmental genes that are expressed early in mammalian tooth development, such as pax9, are not expressed during zebrafish tooth development [Jackman et al., 2004], many parallels with mammalian teeth exist. The importance of Fgf signaling is similar to that in the mouse. Overexpression of Fgf ligands in zebrafish embryos results in supernumerary primary teeth [Jackman et al., 2013], whereas blocking Fgf signaling results in arrest of primary tooth formation [Jackman et al., 2004]. Downregulation of Bmp signaling likewise results in supernumerary teeth [Jackman et al., 2013]. Mutations affecting ectodysplasin (eda) or its receptor (edar) lead to hypodontia, as discussed below for humans [Harris et al., 2008] (Fig. 3, 4).
Figure 4. Ectodermal dysplasia.
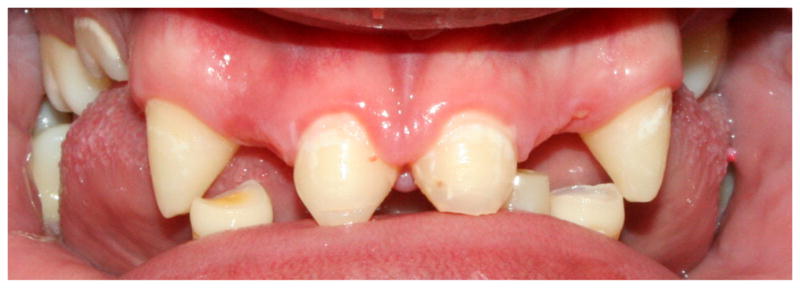
Peg shaped incisors and multiple missing teeth in a 13-year-old male with X-linked hypohidrotic ectodermal dysplasia.
Wnt signaling is a key event in replacement and renewal of ectodermal appendages, and thus potentially also in the replacement of primary by permanent teeth. Although several mutations in components of the canonical Wnt signaling pathway do not affect tooth number in zebrafish (AH personal observations and [Wiweger et al., 2012]), Lef1 mutants display oligodontia [Mcgraw et al., 2011]. Whether the Wnt pathway plays a role at the level of initiation of primary teeth or defective tooth replacement needs to be clarified.
In addition to signaling molecules and transcription factors, there are structural similarities in the tissues and matrices that constitute mammalian and zebrafish teeth. Thus, current studies aim at understanding gene function in cytodifferentiation or mineralization of teeth [Go and Korzh, 2013; Verstraeten et al., 2013], or at elucidating the role of particular genes in rare diseases associated with dental dysplasia [Bloch-Zupan et al., 2011].
Initially, large-scale forward genetic screens were used to identify genes relevant to craniofacial and tooth development, but new technologies are emerging. These include the rapid and targeted introduction of mutations via engineered endonucleases such as ZFNs (zinc finger nucleases) and TALENs (transcription activator-like effector nucleases) (reviewed in [Huang et al., 2012]). These techniques of reverse genetics hold great promise and will continue to increase the relevance of zebrafish as a model for craniofacial and dental diseases.
HUMAN TOOTH DEVELOPMENTAL ANOMALIES
Genetic tooth anomalies can be divided in three main ways. First, the type of anomaly, whether of number, shape, or both, must be determined. These anomalies can include too many teeth (hyperdontia), too few teeth (tooth agenesis), or abnormalities of shape such as taurodontism (enlargement of the body and pulp of the tooth). Second, it is important to know if the anomaly is syndromic, that is, part of a condition with other features, or whether it is isolated. Third, the mode of inheritance must be determined. Sporadic occurrences of genetic anomalies are presumed to be caused by recessive or multifactorial inheritance, by new mutations, or by stochastic occurrences. For the remainder of this review, we will focus on genetic causes of abnormal tooth development and the manifestations of these abnormalities in terms of clinical care; disorders of tooth mineralization are not discussed in this review.
Tooth agenesis: hypodontia, oligodontia and anodontia
Hypodontia refers to the absence of one to six teeth, excluding third molars, whereas oligodontia refers to the absence of more than six teeth, excluding third molars. Third molars are excluded, as these are missing in up to 20% of patients, making this a very common finding. Anodontia is the complete absence of teeth in one or both dentitions. Together, these are referred to as tooth agenesis.
Hypodontia can occur as a sporadic finding, as part of a syndrome, or as a non-syndromic familial form. There are over 80 syndromes that include hypodontia (see Online Mendelian Inheritance in Man, http://www.ncbi.nlm.nih.gov/omim), and some representative syndromes are discussed below. Non-syndromic familial hypodontia may be inherited as an autosomal dominant [Alvesalo and Portin, 1969; Goldenberg et al., 2000; Vastardis et al., 1996], autosomal recessive [Ahmad et al., 1998; Pirinen et al., 2001], or sex-linked trait [De Coster et al., 2009; Erpenstein and Pfeiffer, 1967].
Missing teeth are more common in the permanent dentition than in the primary dentition, but there is a strong correlation between hypodontia in the primary and permanent dentition [Matalova et al., 2008]. In the primary dentition, the prevalence varies from 0.4 to 0.9% in Europe [Jarvinen and Lehtinen, 1981; Ravn, 1971] and is 2.4% in Japan [Yonezu et al., 1997]. In the permanent dentition, the most commonly missing teeth in Caucasians are the mandibular second premolars (4.2%), maxillary lateral incisors (2.3%), and maxillary second premolars (2.2%) [Polder et al., 2004]. Several researchers have reported a higher prevalence of hypodontia among females, with a female to male ratio of 3:2 [Brook, 1975], but the reasons for this are not known.
In individuals with congenitally missing teeth in one region but crowding in another, autotransplantation has good long term prognosis if the transplanted tooth has completed half of its root formation [Paulsen et al., 1995]. Endosseous implant replacement of the missing teeth is another popular and viable option.
Sporadic hypodontia
Sporadic anodontia and oligodontia are rare, but sporadic hypodontia is a relatively common finding. As a general rule, if only one or a few teeth are missing, the missing tooth will be the most distal tooth of any given type. For example, if a molar is missing it is usually the third molar, if an incisor it is the lateral incisor and if a premolar it is usually the second premolar.
Both genetic and environmental factors may contribute to sporadic hypodontia [Schalk-Van Der Weide et al., 1993; Vastardis, 2000]. In terms of environmental influences, development of the permanent teeth may be affected by various factors such as trauma to the jaws, surgical procedures on the jaws, early extraction of the primary teeth, chemotherapy and radiation therapy [Nasman et al., 1997; Schalk-Van Der Weide et al., 1993]. Currently, little is known about the genetic etiologies of sporadic hypodontia, although these may be similar to those that cause familial non-syndromic hypodontia. Mutation in PAX9 has been associated with both sporadic (or low-penetrance familial) hypodontia and oligodontia [Pawlowska et al., 2010].
Familial, non-syndromic hypodontia
In familial hypodontia, the inheritance in the majority of families is autosomal dominant with incomplete penetrance and variable expressivity. Mutations in several genes have been found to cause familial hypodontia. It is also thought that many cases of familial hypodontia may represent a complex, multifactorial condition.
A missense mutation in MSX1 on chromosome 4 was the first mutation found to be associated with non-syndromic hypodontia. The mutation was found in all affected members of a family with missing second premolars and third molars. Some also had missing maxillary first premolars, mandibular first molars, one or both upper lateral incisors or a single lower central incisor. All had normal primary dentitions [Vastardis et al., 1996].
Subsequently, a second gene—PAX9 on chromosome 14—was found to be involved in hypodontia. A frame shift mutation in PAX9 was identified in a family with autosomal dominant hypodontia that had missing permanent molars [Stockton et al., 2000]. Some individuals were missing the maxillary and/or mandibular second premolars as well as central incisors. Since then a number of mutations and polymorphisms have been identified in the human PAX9 region with variable forms of oligodontia that mainly affect the molars [Das et al., 2003; Frazier-Bowers et al., 2002; Mostowska et al., 2006; Mostowska et al., 2003a; Mostowska et al., 2003b; Nieminen et al., 2001].
More recently, hypodontia associated with AXIN2 mutations has been identified to affect a wider range of tooth types. In a four-generation Finnish family, 11 members were found to be missing at least eight permanent teeth along with an increased risk of developing colorectal neoplasia [Lammi et al., 2004]. AXIN2 is a component of the WNT signaling pathway.
Mutations in two genes that can cause ectodermal dysplasia, EDA and WNT10A, can also cause isolated hypodontia; the syndromic effects of mutations in these genes are discussed later in this review. EDA mutations have recently been linked to non-syndromic hypodontia, which typically includes missing mandibular and/or upper incisors and canine [Yang et al., 2013]. A recent study found that 56% of the patients with isolated hypodontia had a mutation in WNT10A, which is strongly expressed in the dental epithelium at the tooth initiation stage and is required for normal tooth development beyond the bud stage [van den Boogaard et al., 2012].
Syndromic hypodontia
Van der Woude syndrome
Van der Woude syndrome (VWS; OMIM #119300) is characterized by paramedian lip pits and sinuses, conical elevations of the lower lip, cleft lip and/or cleft palate, and hypodontia. Adhesions between maxilla and mandible (syngnathia) have been reported [Leck and Aird, 1984]. At times, VWS can be identified solely based on lip pits [Soni et al., 2012]. VWS is the most common clefting syndrome and occurs in approximately 2% of the population with facial clefts [Rintala and Ranta, 1981; Schutte et al., 1996]. The prevalence of VWS is up to 1 in 40,000 still born or live births [Burdick, 1986].
VWS is inherited in an autosomal dominant fashion and is caused by mutations in the interferon regulatory factor 6 (IRF-6) gene [Kondo et al., 2002]. However, there is some genetic heterogeneity in VWS [Wong et al., 2001]. IRF6 mutations also cause popliteal pterygium syndrome (PPS), which in addition to the craniofacial findings of VWS consists of genital abnormalities, webbing of fingers, toes, and behind knees, and other occasional features [Lees et al., 1999].
Lip pits are the most common manifestation of VWS. The occurrence has been reported in up to 88% of the affected individuals [Janku et al., 1980]. VWS is underdiagnosed because lower lip pits are often missed, leading to undetected submucous cleft palate (CP) [Lam et al., 2010]. In CP patients with lower lip sinuses, the incidence of hypodontia was 77.8% [Ranta and Rintala, 1982].
Hypodontia is frequently seen in VWS, and a close association between VWS and congenital absence of second premolars has been shown [Calzavara Pinton et al., 1989; Oberoi and Vargervik, 2005a; Schneider, 1973]. There is a tendency toward greater maxillary hypoplasia in VWS, particularly in the most severe cleft type (bilateral CLP). In addition, the highest incidence of missing teeth is also seen in VWS with the more severe cleft type [Oberoi and Vargervik, 2005a].
Ectodermal dysplasia
There are more than 150 clinically distinct inherited syndromes in which ectodermal dysplasia (ED) is present. ED consists of variable defects in the morphogenesis of ectodermal derivatives including skin, sweat glands, hair, nails and teeth. Many of the ED syndromes have non-ectodermal manifestations, which are not discussed here in detail. Patients with ED can have hypodontia or anodontia, with the anterior teeth usually conical or peg-shaped (Fig. 4); the alveolar ridge is deficient and patients tend to have hypoplastic maxillae with anterior crossbite and low face height with overclosure.
The ED syndromes can be inherited in an autosomal dominant, autosomal recessive, or X-linked form. The most common form of ED is X-linked hypohidrotic ED, or XLHED (OMIM 305100) and is caused by mutations in the gene encoding ectodysplasin-A (EDA), which is a member of the TNF signaling pathway. TNF signaling through EDA activates NFKB1, which is known to play an important role in odontogenesis [Ohazama and Sharpe, 2004]. Affected males show severe oligodontia or anodontia in both primary and permanent dentition. An average of nine permanent teeth develop in individuals with classic XLHED, typically the canines and first molars [Lexner et al., 2007]. Teeth are often smaller than average and have an altered morphology. Anterior teeth tend to be conical in shape. Dental radiographs are helpful in determining the extent of hypodontia. Taurodontism is more common in the molars of individuals with XLHED.
Female carriers have variable, milder phenotypic expressions resulting from X chromosome inactivation. They may have hypodontia or anodontia and abnormally shaped teeth. 60% to 80% of carriers have some degree of hypodontia [Cambiaghi et al., 2000]. In XLHED, both primary and permanent dentition are affected [Clauss et al., 2008].
Odonto-onycho-dermal dysplasia (OMIM 257980) is an autosomal recessive ED syndrome caused by mutations in WNT10A [Adaimy et al., 2007]. These patients present with dry hair, severe hypodontia, smooth tongue, nail dysplasia, hyperhidrosis of palms and soles, and hyperkeratosis. As mentioned above, WNT10A mutations are also a common cause of isolated hypodontia.
Oral-facial-digital syndrome type I
Oral-facial-digital syndrome (OFD) type 1 (OMIM 311200) is a developmental disorder characterized by malformations of the face, oral cavity, digits, central nervous system, and kidneys. The prevalence of OFD1 is 1 in 50,000 to 1 in 250,000 live births. OFD1 is an X-linked disorder caused by mutations in the gene OFD1. This gene is important for formation of a cellular organelle known as the primary cilium. OFD1 affects only females, as this condition is lethal in males. Although clinical features overlap with other types of OFD (of which there are at least 9), X-linked dominant inheritance and polycystic kidney disease are specific to OFD1.
The typical oral manifestations of OFD1 are seen in the tongue, palate, and teeth. The tongue is lobed and is bifid or trifid with nodules (hamartomas or lipomas); this is seen in at least a third of patients with OFD1. Ankyloglossia due to a short lingual frenulum is common. Cleft hard or soft palate, submucous cleft palate, or highly arched palate occur in more than 50% of affected patients. Alveolar clefts and accessory gingival frenulae are common. These fibrous bands are hyperplastic frenulae extending from the buccal mucous membrane to the alveolar ridge, resulting in notching of the alveolar ridges. Dental abnormalities include missing teeth, extra teeth, enamel dysplasia, and malocclusion [Al-Qattan, 1998; Toriello and Franco, 2007]. The lower lateral incisors are missing in 50% of individuals, and this is associated with fibrous bands in the region.
Rieger syndrome
Rieger syndrome (OMIM 601542) is an autosomal dominant disorder characterized by malformations in the anterior chamber of the eye, umbilical anomalies, and hypodontia. Its prevalence is 1 in 200,000. When the ocular abnormality is combined with other craniofacial, dental, and developmental somatic anomalies, it is given the name Axenfeld-Rieger syndrome (ARS; OMIM 180500). Glaucoma is found in 50% of the cases [Shields et al., 1985]. Craniofacial, dental, and umbilical anomalies are also regularly reported in connection with ARS [Childers and Wright, 1986; Dressler and Gramer, 2006]. Characteristic craniofacial features are maxillary hypoplasia, hypertelorism, and telecanthus. Other systemic features like anomalies of the pituitary gland, middle ear deafness, heart defects, hypospadias, short stature, and mental retardation were diagnosed in several ARS patients [Ozeki et al., 1999; Shields et al., 1985].
Three genetic loci have been associated with ARS so far. FOXC1 and PITX2 encode transcription factors and are located on chromosomes 6p25 and 4q25, respectively [Tumer and Bach-Holm, 2009]. A third locus for ARS was mapped to chromosome 13q14 but the gene has not yet been identified. Therefore ARS is considered as a morphologically and genetically heterogeneous disorder.
Dental features include hypodontia/oligodontia of primary and permanent dentition. The most commonly missing teeth are lower second premolars and subsequently the central incisors and upper second premolars [Dressler et al., 2010]. The missing teeth in the anterior maxilla are thought to cause underdevelopment of the premaxilla. Other dental abnormalities include hyperplastic upper labial frenulum, peg-shaped front teeth, and small teeth, enamel hypoplasia, conical-shaped teeth, shortened roots, taurodontism, and delayed eruption.
Holoprosencephaly
Holoprosencephaly (HPE) (OMIM # 236100), which occurs with a frequency of 1 in 16,000 live births and 1 in every 200 spontaneous abortions, is a an etiologically heterogenous condition with teratogenic and genetic factors [Hall et al., 1997]. HPE is caused by impaired midline cleavage of the embryonic forebrain. HPE is the most common defect of the forebrain and mid-face in human [Wallis and Muenke, 2000]. The most severe form is cyclopia, and the mildest phenotype is a single upper central incisor. Several loci for HPE have been mapped. HPE3 is caused by mutations in the Sonic hedgehog (SHH) gene, which was described above in the context of tooth development [Lami et al., 2013]. Both recessive and dominant inheritance of HPE has been reported [Cohen and Gorlin, 1969].
The HPE spectrum is commonly associated with solitary median maxillary control incisor (SMMCI), a rare dental anomaly that can occur in either a primary or permanent dentition. It can be an isolated dental finding or occur in association with other recognized syndromes or specific chromosomal abnormalities [Nanni et al., 2001]. The spectrum of defects is extremely variable, as some individuals can present with the full HPE spectrum, some may have only mild symptoms such as SMMCI, and others may have no symptoms at all [El-Jaick et al., 2007]. Sometimes SMMCI is the most easily recognizable anomaly associated with HPE [Hall et al., 1997]. All HPE patients have SMMCI, but not all SMMCI patients have been diagnosed with HPE [Kopp, 1967]. Several syndromes have been associated with SMMCI, including ED, Duane retraction syndrome, velocardiofacial syndrome, CHARGE syndrome, VACTERL association, and HPE [Oberoi and Vargervik, 2005b].
Tooth anomalies associated with cleft lip and palate
It has long been recognized that hypodontia is associated with clefts of the lip and palate. Studies have found that hypodontia is present in approximately 80% of children with non-syndromic clefts [Shapira et al., 1999], and the prevalence of hypodontia increases markedly with the severity of the cleft [Ranta 1986]. The teeth most frequently missing on the cleft side were the upper permanent lateral incisors; these were absent in 74% of all cleft patients, followed by maxillary and mandibular second premolars. The teeth most often missing on the non-cleft side were the maxillary second premolars, followed by the maxillary lateral incisors and mandibular second premolars. Interestingly, congenital absence of both the maxillary lateral incisors and second premolars was found more frequently in siblings of patients with clefts as well [Eerens et al., 2001].
Hypodontia outside the cleft region is more frequent in cleft individuals than in non-cleft individuals [Shapira et al., 1999], and, in general, hypodontia occurs about 10 times more frequently on the cleft side, with left predominance. This is consistent with the side preference for unilateral cleft lip and palate. Additionally, the prevalence of hypodontia (excluding third molars) has been reported as 50% in Pierre Robin sequence, which comprises a triad of micrognathia, glossoptosis, and cleft palate. In these cases, there is an increased frequency of hypodontia in the lower jaw compared to individuals with isolated cleft lip and palate [Ranta 1986].
In addition, these individuals can have other anomalies related to the shape and size of individual teeth, or the presence of additional teeth. A recent meta-analysis concluded that patients with cleft lip and palate experience not only more tooth agenesis, but also supernumerary teeth and anomalous tooth morphology in comparison to non-cleft patients [Tannure et al., 2012]. The most frequently undersized teeth are upper lateral incisors, and while there is evidence that other teeth in cleft patients have higher levels of agenesis and dysmorphology than in non-cleft patients, the data are conflicting. In cleft patients, a high occurrence of a supernumerary lateral incisor (40%–73%) has been detected in the primary dentition [Bohn, 1950; Hansen and Mehdinia, 2002].
From an embryological point of view, it has been shown using 3D reconstructions that the location of the fusion of the medial nasal and maxillary facial outgrowths transiently appears as a furrow on the mesenchymal aspect of the developing lateral incisor germ in humans until prenatal week 8. This implies that the upper incisor germ takes its origin partially from the maxillary outgrowth (Fig. 5) [Hovorakova et al., 2006]. This complex origin at a critical place of fusion of facial processes can explain the developmental vulnerability and resulting anomalies of the upper lateral incisor [Hovorakova et al., 2006]. Complete orofacial clefts of the lip and alveolus result from the failure of the fusion of the medial nasal and maxillary processes. Consequently, the fusion of the dental epithelia is also absent and the two incisor subcomponents remain separate, such that a duplication of the lateral incisor occurs in the cleft area (Fig. 5). It has been proposed that the number of lateral incisors, even in normal individuals, depends on whether both, one, or no incisor subcomponent is able to give rise to a functional tooth [Hovorakova et al., 2006].
Figure 5. Dual developmental origin and duplication of the upper lateral incisor.
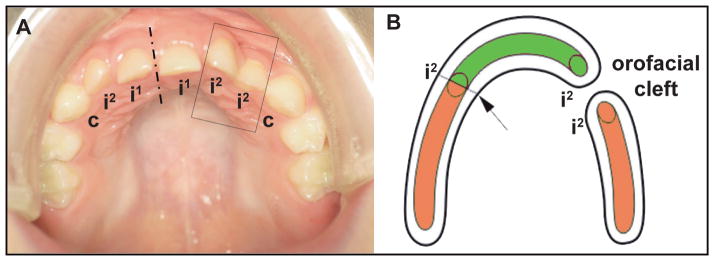
(A) The supernumerary lateral incisor in a patient after the operation of a left-sided cleft lip and palate. (B) Scheme showing the dual developmental origin of the upper lateral incisor and its disturbance associated with a jaw cleft. The upper lateral incisor develops by a physiological fusion (arrow) of two parts, one originating from the dental epithelium of the medial nasal (green) and one originating from the dental epithelium of the maxillary (orange) facial outgrowth. In cleft patients (A), the lateral incisor duplication adjacent to the cleft area has been explained by the non-fusion of both incisor subcomponents as a result of the non-fusion of the medial nasal and maxillary processes (A, B). The increasing extent of hypoplasia of the facial outgrowth tissues determines the formation of two, one, or no lateral incisor, respectively. The lowest level of tissue insufficiency of the facial processes might result in the lateral incisor anomalies even in an intact jaw (without cleft). Dash-and-dotted line represents midline. i1, i2 and c: upper deciduous central incisor, lateral incisor and canine, respectively.
Delayed formation and eruption of teeth
The permanent teeth usually replace the primary teeth between the ages of 6–12 years. However, eruption times for the permanent teeth can vary considerably. The lower incisor shows the least variability and the lower second premolar the greatest in timing of eruption. When two primary teeth are fused, it is linked with the absence of permanent teeth.
Several syndromes have delayed formation and eruption of teeth, including Apert syndrome [Kaloust et al., 1997], cleidocranial dysplasia, Dubowitz syndrome, Goltz syndrome, progeria, Menke syndrome and oculofaciocardiodental syndrome (OFCD) [Oberoi et al., 2005]. Two of these syndromes are discussed below as examples.
In Apert syndrome, there are delays in both development and eruption, and there can also be ectopic eruption and abnormalities in incisor and molar shape [Kaloust et al., 1997]. Erupting teeth remain buried in thickened gingival tissues for long periods of time. The alveolar swellings of the maxillary arch are characteristic of the syndrome and have been shown to contain excessive mucopolysaccharides, predominantly hyaluronic acid [Peterson and Pruzansky, 1974]. Activating mutations in genes encoding receptors for Fibroblast Growth Factors, which were discussed above in the context of tooth development, cause Apert syndrome. However, it is not clear how these mutations contribute to the characteristic dental and gingival findings in Apert syndrome [Kaloust et al., 1997].
Oculofaciocardiodental (OFCD) syndrome (OMIM 300166) is an X-linked condition with characteristic ocular, facial, cardiac and dental findings in affected females and presumed lethality in affected males. The most typical dental anomaly is canine radiculomegaly (enlarged roots). Other findings include delayed dental development and eruption, oligodontia, retained primary teeth and variable root length. Mutations in the BCOR gene have been found in this condition [Ng et al., 2004].
Delayed tooth eruption has also been found in the upper jaw of patients with orofacial clefts. Eruption of the permanent upper lateral incisor, which is also sometimes absent in patients with cleft lip and palate, and the permanent second molar is retarded in patients with cleft lip and palate. In contrast, earlier eruption has been found in the permanent and deciduous maxillary canine, first and second premolars. The delay or acceleration of tooth eruption in cleft patients might be a consequence of the affected bones and teeth [Peterka et al., 1996].
Abnormalities in tooth size, shape and form
Abnormalities in tooth size and shape are thought to result from disturbances in the morphodifferentiation (cap-bell) stage of development. About 5% of the population has a significant “tooth size discrepancy” due to disproportion in the size of upper and lower teeth. The most common abnormality is a variation in size of the upper lateral incisors and second premolars. In patients with hypodontia, the most common abnormality is a peg-shaped upper lateral incisor.
There is very often a discrepancy between tooth size and jaw size. The combination of microdontia and normal jaw size is accompanied by the presence of tremata (free spaces between teeth). Similarly, a shorter jaw in the mesio-distal direction with teeth of normal size results in orthodontic anomalies. This disproportion is most pronounced in the upper jaw of cleft patients, where the permanent teeth have normal mesio-distal dimension, while the upper jaw arch is significantly shorter [Peterka et al., 1996].
Sometimes, tooth germs may fuse or germinate during development [Guttal et al., 2010], resulting in teeth with separate pulp chambers joined at the dentin or teeth with a common pulp chamber, respectively. It is often difficult to differentiate between the two, but if a lateral incisor is missing it is most likely because of fusion of the central and lateral incisor primordia.
Taurodontism (OMIM 272700) is characterized by a large pulp chamber and is most commonly seen in molars. The word “taurodontism” was first used to describe the teeth of Neanderthals and another group of prehistoric humans, the Heidelbergs [Keith, 1913]. Taurodontism causes constriction of the cementoenamel junction, thus elongating the pulp chambers vertically creating an apically displaced pulp. There is significant variation among modern day populations. The prevalence has been reported as 0.5% in Japanese [Daito, 1971], 0.57–3.2% in white Americans [Blumberg et al., 1971; Witkop, 1976], 4.3% in African Americans [Jorgenson et al., 1982], 8% in Jordanians [Darwazeh et al., 1998], 33–41% in certain African populations [Shaw, 1928], and 46.4% in young adult Chinese [Macdonald-Jankowski and Li, 1993]. Taurodontism is thought to be a polygenic trait, and both autosomal dominant and recessive inheritance have been suggested.
A taurodontic tooth is thought to result from a disturbance in growth of Hertwig’s epithelial root sheath. Based on this hypothesis, there may be an association between taurodontism and hypodontia, as both conditions may be attributed to defects in the growth of dental epithelium [Hu and Simmer, 2007]. Taurodontism has been described together with isolated [Schalk-Van Der Weide et al., 1993; Seow and Lai, 1989; Stenvik et al., 1972] and syndromic hypodontia in many syndromes, including 18p11.3 deletion [Kantaputra et al., 2006], Smith-Magenis syndrome [Tomona et al., 2006], tricho-dento-osseous syndrome [Hart et al., 1997; Islam et al., 2005; Price et al., 1999; Wright et al., 1997], Klinefelter’s syndrome [Hillebrand et al., 1990; Komatz et al., 1978; Yeh and Hsu, 1999], Williams syndrome [Axelsson et al., 2003], McCune-Albright syndrome [Akintoye et al., 2003], Down syndrome [Alpoz and Eronat, 1997] and Ellis van Creveld syndrome [Hunter and Roberts, 1998]. It has also been described in individuals with cleft lip and palate. An autosomal dominant hypoplastic/hypomature amelogenesis imperfecta (AI) associated with taurodontism (OMIM 104510) has been mapped to the distal-less homeobox (DLX3) locus [Dong et al., 2005]. More recently, taurodontism has been linked with Laurence-Moon/Bardet-Biedl syndrome (LM/BBS) and therefore should be included as a minor criterion in diagnosing LM/BSS [Andersson et al., 2013]. Lastly, there may be a common genetic etiology between VWS, hypodontia, and taurodontism [Nawa et al., 2008]. The frequency of taurodontism in VWS subjects was almost 50% [Nawa et al., 2008].
Supernumerary teeth
A supernumerary tooth is an additional tooth that can be found in any region of the dental arch. Supernumerary teeth result from disturbances during the initiation and proliferation stages of dental development. The most common supernumerary tooth that appears is in the maxillary midline and is called mesiodens. The prevalence of supernumerary teeth is between 0.3% to 0.8% in primary dentition and 1.5% to 3.5% in permanent dentition [Brook, 1974]. A male to female ratio of 2:1 is found in populations with single supernumerary teeth [Kantor et al., 1988] which increases to 3:1 for multiple supernumerary teeth [Gibson, 1979]. Although inherited forms are rare, a familial inheritance has been reported that can be autosomal dominant with incomplete penetrance, autosomal recessive [Cassia et al., 2004] or sex linked pattern of inheritance [Burzynski and Escobar, 1983].
Greater than 90% of supernumeraries will occur in the upper jaw, and approximately 25% of the maxillary anterior supernumerary teeth erupt, but more commonly they are impacted and require extraction. Supernumerary teeth may be unilateral or bilateral, single or multiple, and may be found in one or both jaws. Multiple supernumerary teeth are rare in non-syndromic individuals, so such a finding should prompt a referral to a medical geneticist. An increased prevalence of supernumerary teeth can be found in cleft lip and palate patients, and there are over 20 syndromes with supernumerary teeth, the most common being cleidocranial dysplasia and Gardner syndrome [Moore et al., 2002]. The frequency of supernumerary teeth in individuals with unilateral cleft lip and/or palate was found to be 22.2% [Scheiner and Sampson, 1997], with males affected twice as often as females in the permanent dentition.
There are various hypotheses regarding the etiology of supernumerary teeth. According to one, a supernumerary tooth is created as a result of dichotomy of the tooth bud [Liu, 1995]. Another theory is that supernumeraries are formed as a result of local, independent conditioned hyperactivity of the dental lamina [Liu, 1995; Scheiner and Sampson, 1997]. Sometimes, supernumerary teeth can be interpreted as atavisms, if they appear at locations where teeth were suppressed during evolution [Peterkova et al., 2006; Smith, 1969].
The diagnosis of a supernumerary tooth is confirmed by radiographic examination if abnormal clinical signs are found. Periapical and occlusal radiographs are commonly used in the incisor region. Three-dimensional cone beam CT is valuable as it shows the position of the supernumerary in all 3 dimensions, thereby enabling the correct buccal or palatal approach during removal and direction of force application for orthodontic alignment [Liu et al., 2007].
Some of the problems associated with supernumerary teeth include failure of eruption, displacement [Howard, 1967], crowding, and formation of dentigerous cysts [Awang and Siar, 1989]. Root resorption of adjacent teeth is a rare occurrence as well [Hogstrom and Andersson, 1987].
Cleidocranial dysplasia
The best-known syndrome associated with supernumerary teeth is cleidocranial dysplasia (CCD). CCD (OMIM #119600) is an autosomal dominant skeletal dysplasia associated with clavicle hypoplasia and dental abnormalities and has a prevalence of 1 in 1,000,000. It is caused by mutations in RUNX2, which encodes a transcription factor that activates osteoblast differentiation. One third of CCD cases are sporadic and represent new mutations [Otto et al., 2002].
Individuals with CCD have growth retardation with moderate short stature, delayed fontanelle closure with parietal and frontal bossing, midface hypoplasia, hypertelorism, low nasal bridge, brachydactyly and hearing loss. They may have cleft palate or a narrow, high palate.
Dental manifestations are found in more than 90% of individuals with CCD and include delayed eruption of deciduous and permanent teeth, supernumerary teeth, retention cysts and enamel hypoplasia (Fig. 6) [Golan et al., 2003]. Formation and eruption of the deciduous teeth are usually normal. One suggested explanation for the delayed or non-eruption of many permanent and supernumerary teeth is the lack of cellular cementum in the apical region of impacted teeth [Manjunath et al., 2008]. While CCD patients at a younger age display relatively normal jaw proportions and morphology of the mandible, CCD patients at an older age display short lower face height, acute gonial angle, anterior inclination of the mandible, and mandibular prognathism; due to maxillary hypoplasia, individuals with CCD tend to have a Class III malocclusion [Ishii et al., 1998].
Figure 6. Cleidocranial dysplasia (CCD).
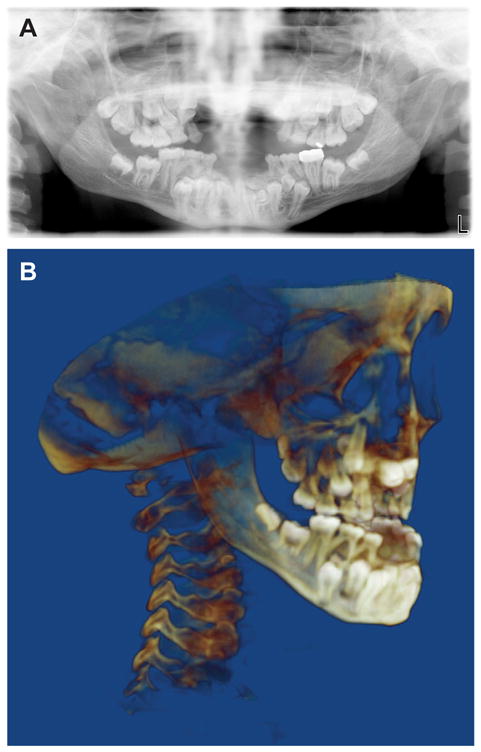
(A) Panoramic radiograph and (B) cone beam CT images of a 14-year-old boy with CCD showing the multiple retained primary teeth and multiple impacted permanent teeth and supernumerary teeth.
Because of challenges in dental management of these individuals with CCD, comprehensive orthodontic and surgical treatment is required. Additionally, there is a wide variation in supernumerary tooth development, including asymmetrical development of supernumerary teeth in the upper and lower jaw [Soni et al., 2012]. Delay of physiologic root resorption results in prolonged retention of the primary teeth, and the eruption of the permanent teeth is delayed and many fail to erupt [Shaikh and Shusterman, 1998]. The presence of supernumerary teeth is not pathognomic for CCD; in fact, some patients may have no supernumerary teeth or even missing teeth [Richardson and Deussen, 1994].
Recent advances in dentistry, such as dental implants, allow better treatment options and outcomes for individuals with CCD. As an example, dental implants can be used not only for orthodontic tooth movement with reduced side effects, but also for replacing teeth that cannot be brought into the arch orthodontically. With the advancements in three-dimensional imaging, Cone Beam Computed Tomography (CBCT) is now routinely used in diagnosis and treatment planning [Liu et al., 2007].
Gardner syndrome
Gardner syndrome, a variant of familial adenomatous polyposis (FAP) (OMIM #175100), is a rare autosomal dominant condition characterized by gastrointestinal polyps, multiple osteomas, and skin and soft tissue tumors including a characteristic retinal lesion. Approximately 10% of FAP individuals are affected by Gardner syndrome [Ramaglia et al., 2007]. Gardner syndrome and FAP are caused by mutations in APC at 5q21 [Groden et al., 1991; Kinzler et al., 1991]. APC is a multidomain protein that plays a major role in tumor suppression by antagonizing the WNT signaling pathway [Barth et al., 1997].
Dental anomalies are present in 30% to 75% of patients with Gardner syndrome, and may include impacted or unerupted teeth, hypodontia, abnormal tooth morphology, supernumerary teeth, hypercementosis, compound odontomas, dentigerous cysts, fused molar roots, long and tapered molar roots, and multiple caries [Basaran and Erkan, 2008; Butler et al., 2005; Madani and Madani, 2007]. Osteomas occur in 68–82% and are generally located in the paranasal sinuses and mandible [Dawlatly et al., 1997; Madani and Madani, 2007]. They can also affect the skull and long bones [Cankaya et al., 2012]. In the mandible, central or lobulated osteomas can be observed; central osteomas are characterisctically near the roots of the teeth, and lobulated types arise form the cortex and most commonly at the mandibular angle [Wesley et al., 1987]. Often the general dentists or orthodontists are the first health care professionals to suspect the diagnosis and refer to the oromaxillofacial surgeon [Cankaya et al., 2012].
CONCLUSION
Dental anomalies are seen as either isolated findings or in individuals with craniofacial developmental abnormalities, and in both cases they can profoundly affect the life of the affected individual. Studies of tooth development in animal models together with genetic studies of patients are improving our understanding of the causes of dental anomalies, and are laying the foundation for future biologically-based treatments.
Acknowledgments
This work was supported by the NIH (DP2-OD00719 and R01-DE021420 to O.D.K.), the California Institute of Regenerative Medicine (RN2-00933 to O.D.K.), and by the Grant Agency of the Czech Republic; CZ:GA ČR:GAP305/12/1766.
Biographies
Ophir D. Klein, M.D., Ph.D. is Associate Professor in the Departments of Orofacial Sciences and Pediatrics, Chair of the Division of Craniofacial Anomalies, and Director of the Program in Craniofacial and Mesenchymal Biology at UCSF. Dr. Klein’s research focuses in large part on understanding the processes underlying craniofacial and dental development. His lab uses mouse models to study the mechanisms responsible for the normal and abnormal development of teeth, facial skeleton and other organs, as well as the regeneration of these organs.
Snehlata Oberoi, D.D.S., M.D.S., is an Associate Professor of Orthodontics with the UCSF Center for Craniofacial Anomalies, where she provides assessment and treatment for children with craniofacial disorders. Her research focuses on developing new methods to assess the outcomes of treatment for cleft lip, cleft palate and other craniofacial anomalies. She also collaborates with the Center’s medical geneticists on research seeking to identify genetic mutations and how they affect dental and facial defects in various craniofacial anomalies.
Ann Huysseune, Ph.D., is Professor and Head of the biology department at Ghent University. Her research interests are focused on the development, structure and evolution of the skeleton, with particular attention to teeth. She uses various teleost fish and other non-mammalian species to study evo-devo aspects of skeletal tissues and skeletal elements. Current studies in her group focus on the dermal skeleton (including the teeth) and on the vertebral column.
Maria Hovorakova, Ph.D., is a researcher in the the Laboratory of Odontogenesis, Department of Teratology at the Institute of Experimental Medicine at the Academy of Sciences CR, Prague, Czech Republic. She is currently working on tooth development in wild-type and mutant mice, with a focus on the role of rudiments in tooth development.
Miroslav Peterka, M.D., Ph.D., is Head of the Department of Teratology at the Institute of Experimental Medicine at the Academy of Sciences CR, Prague, Czech Republic. He is an Associate Professor at the 1st Medical Faculty, Charles University in Prague and a clinical teratologist involved in prevention of inborn defects at the Clinic of Plastic Surgery, 3rd Medical Faculty in Prague. His research interests are experimental and clinical teratology, as well as pathogenesis and epidemiology of developmental anomalies.
Renata Peterkova, M.D., Ph.D., is Head of the Laboratory of Odontogenesis, Department of Teratology at the Institute of Experimental Medicine at the Academy of Sciences CR, Prague, Czech Republic. Her focus is morphology, including embryology, histology, and anatomy, and her field of interest is the normal and pathological development of teeth and adjacent structures. During her research career, she has studied rudimentary structures during orofacial development. She is interested in their role during normal ontogeny, their involvement in the origin of developmental anomalies, and their evolutionary significance.
References
- Adaimy L, Chouery E, Megarbane H, Mroueh S, Delague V, Nicolas E, Belguith H, De Mazancourt P, Megarbane A. Mutation in wnt10a is associated with an autosomal recessive ectodermal dysplasia: The odonto-onycho-dermal dysplasia. Am J Hum Genet. 2007;81:821–828. doi: 10.1086/520064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad W, Brancolini V, Ul Faiyaz MF, Lam H, Ul Haque S, Haider M, Maimon A, Aita VM, Owen J, Brown D, Zegarelli DJ, Ahmad M, Ott J, Christiano AM. A locus for autosomal recessive hypodontia with associated dental anomalies maps to chromosome 16q12.1. Am J Hum Genet. 1998;62:987–991. doi: 10.1086/301799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akintoye SO, Lee JS, Feimster T, Booher S, Brahim J, Kingman A, Riminucci M, Robey PG, Collins MT. Dental characteristics of fibrous dysplasia and mccune-albright syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2003;96:275–282. doi: 10.1016/s1079-2104(03)00225-7. [DOI] [PubMed] [Google Scholar]
- Al-Qattan MM. Cone-shaped epiphyses in the toes and trifurcation of the soft palate in oral-facial-digital syndrome type-i. Br J Plast Surg. 1998;51:476–479. doi: 10.1054/bjps.1997.0297. [DOI] [PubMed] [Google Scholar]
- Alpoz AR, Eronat C. Taurodontism in children associated with trisomy 21 syndrome. J Clin Pediatr Dent. 1997;22:37–39. [PubMed] [Google Scholar]
- Alvesalo L, Portin P. The inheritance pattern of missing, peg-shaped, and strongly mesio-distally reduced upper lateral incisors. Acta Odontol Scand. 1969;27:563–575. doi: 10.3109/00016356909026309. [DOI] [PubMed] [Google Scholar]
- Andersson E, Axelsson S, Gjolstad L, Storhaug K. Taurodontism: A minor diagnostic criterion in laurence-moon/bardet-biedl syndromes. Acta Odontologica Scandinavica. 2013:1–4. doi: 10.3109/00016357.2013.794389. [DOI] [PubMed] [Google Scholar]
- Awang MN, Siar CH. Dentigerous cyst due to mesiodens: Report of two cases. J Ir Dent Assoc. 1989;35:117–118. [PubMed] [Google Scholar]
- Axelsson S, Bjornland T, Kjaer I, Heiberg A, Storhaug K. Dental characteristics in williams syndrome: A clinical and radiographic evaluation. Acta Odontol Scand. 2003;61:129–136. doi: 10.1080/00016350310001451. [DOI] [PubMed] [Google Scholar]
- Barth AI, Nathke IS, Nelson WJ. Cadherins, catenins and apc protein: Interplay between cytoskeletal complexes and signaling pathways. Curr Opin Cell Biol. 1997;9:683–690. doi: 10.1016/s0955-0674(97)80122-6. [DOI] [PubMed] [Google Scholar]
- Basaran G, Erkan M. One of the rarest syndromes in dentistry: Gardner syndrome. Eur J Dent. 2008;2:208–212. [PMC free article] [PubMed] [Google Scholar]
- Bei M, Maas R. Fgfs and bmp4 induce both msx1-independent and msx1-dependent signaling pathways in early tooth development. Development. 1998;125:4325–4333. doi: 10.1242/dev.125.21.4325. [DOI] [PubMed] [Google Scholar]
- Biehs B, Hu JK, Strauli NB, Sangiorgi E, Jung H, Heber RP, Ho S, Goodwin AF, Dasen JS, Capecchi MR, Klein OD. Bmi1 represses ink4a/arf and hox genes to regulate stem cells in the rodent incisor. Nat Cell Biol. 2013 doi: 10.1038/ncb2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloch-Zupan A, Jamet X, Etard C, Laugel V, Muller J, Geoffroy V, Strauss JP, Pelletier V, Marion V, Poch O, Strahle U, Stoetzel C, Dollfus H. Homozygosity mapping and candidate prioritization identify mutations, missed by whole-exome sequencing, in smoc2, causing major dental developmental defects. Am J Hum Genet. 2011;89:773–781. doi: 10.1016/j.ajhg.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumberg JE, Hylander WL, Goepp RA. Taurodontism: A biometric study. Am J Phys Anthropol. 1971;34:243–255. doi: 10.1002/ajpa.1330340208. [DOI] [PubMed] [Google Scholar]
- Bohn A. Anomalies of the lateral incisor in cases of harelip and cleft palate. Acta Odontol Scand. 1950;9:41–59. doi: 10.3109/00016355009087225. [DOI] [PubMed] [Google Scholar]
- Borday-Birraux V, Van Der Heyden C, Debiais-Thibaud M, Verreijdt L, Stock DW, Huysseune A, Sire JY. Expression of dlx genes during the development of the zebrafish pharyngeal dentition: Evolutionary implications. Evol Dev. 2006;8:130–141. doi: 10.1111/j.1525-142X.2006.00084.x. [DOI] [PubMed] [Google Scholar]
- Brook AH. Dental anomalies of number, form and size: Their prevalence in british schoolchildren. J Int Assoc Dent Child. 1974;5:37–53. [PubMed] [Google Scholar]
- Brook AH. Variables and criteria in prevalence studies of dental anomalies of number, form and size. Community Dent Oral Epidemiol. 1975;3:288–293. doi: 10.1111/j.1600-0528.1975.tb00326.x. [DOI] [PubMed] [Google Scholar]
- Burdick AB. Genetic epidemiology and control of genetic expression in van der woude syndrome. J Craniofac Genet Dev Biol Suppl. 1986;2:99–105. [PubMed] [Google Scholar]
- Burzynski NJ, Escobar VH. Classification and genetics of numeric anomalies of dentition. Birth Defects Orig Artic Ser. 1983;19:95–106. [PubMed] [Google Scholar]
- Butler J, Haealy C, Toner M, Flint S. Gardener’s syndrome-review and report of a case. Oral oncology Extra. 2005;41:89–92. [Google Scholar]
- Calzavara Pinton PG, Gavazzoni R, Carlino A, Leali C. van der woude syndrome. G Ital Dermatol Venereol. 1989;124:171–173. [PubMed] [Google Scholar]
- Cambiaghi S, Restano L, Paakkonen K, Caputo R, Kere J. Clinical findings in mosaic carriers of hypohidrotic ectodermal dysplasia. Arch Dermatol. 2000;136:217–224. doi: 10.1001/archderm.136.2.217. [DOI] [PubMed] [Google Scholar]
- Cankaya AB, Erdem MA, Isler SC, Cifter M, Olgac V, Kasapoglu C, Oral CK. Oral and maxillofacial considerations in gardner’s syndrome. Int J Med Sci. 2012;9:137–141. doi: 10.7150/ijms.3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassia A, El-Toum S, Feki A, Megarbane A. Five mandibular incisors: An autosomal recessive trait? Br Dent J. 2004;197:307–309. doi: 10.1038/sj.bdj.4811648. [DOI] [PubMed] [Google Scholar]
- Childers NK, Wright JT. Dental and craniofacial anomalies of axenfeld-rieger syndrome. J Oral Pathol. 1986;15:534–539. doi: 10.1111/j.1600-0714.1986.tb00572.x. [DOI] [PubMed] [Google Scholar]
- Clauss F, Maniere MC, Obry F, Waltmann E, Hadj-Rabia S, Bodemer C, Alembik Y, Lesot H, Schmittbuhl M. Dento-craniofacial phenotypes and underlying molecular mechanisms in hypohidrotic ectodermal dysplasia (hed): A review. J Dent Res. 2008;87:1089–1099. doi: 10.1177/154405910808701205. [DOI] [PubMed] [Google Scholar]
- Cobourne MT, Sharpe PT. Making up the numbers: The molecular control of mammalian dental formula. Semin Cell Dev Biol. 2010;21:314–324. doi: 10.1016/j.semcdb.2010.01.007. [DOI] [PubMed] [Google Scholar]
- Cohen MMJ, Gorlin RJ. Genetic consideration in a sibship of cyclopia and clefts. Birth Defects Orig Art Ser. 1969;5:113–118. [Google Scholar]
- Daito K. surfaces of stone models made in elastic material impression. I. Stone model surfaces and treatment of alginate impressions. Kokubyo Gakkai Zasshi. 1971;38:266–274. doi: 10.5357/koubyou.38.266. [DOI] [PubMed] [Google Scholar]
- Darwazeh AM, Hamasha AA, Pillai K. Prevalence of taurodontism in jordanian dental patients. Dentomaxillofac Radiol. 1998;27:163–165. doi: 10.1038/sj/dmfr/4600342. [DOI] [PubMed] [Google Scholar]
- Das P, Hai M, Elcock C, Leal SM, Brown DT, Brook AH, Patel PI. Novel missense mutations and a 288-bp exonic insertion in pax9 in families with autosomal dominant hypodontia. Am J Med Genet A. 2003;118:35–42. doi: 10.1002/ajmg.a.10011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawlatly ED, Al-Qurain AA, Salih-Mahmud M. Gardner’s syndrome presenting with a giant osteoma. Ann Saudi Med. 1997;17:542–544. doi: 10.5144/0256-4947.1997.542. [DOI] [PubMed] [Google Scholar]
- De Coster PJ, Marks LA, Martens LC, Huysseune A. Dental agenesis: Genetic and clinical perspectives. J Oral Pathol Med. 2009;38:1–17. doi: 10.1111/j.1600-0714.2008.00699.x. [DOI] [PubMed] [Google Scholar]
- Dong J, Amor D, Aldred MJ, Gu T, Escamilla M, Macdougall M. Dlx3 mutation associated with autosomal dominant amelogenesis imperfecta with taurodontism. Am J Med Genet A. 2005;133A:138–141. doi: 10.1002/ajmg.a.30521. [DOI] [PubMed] [Google Scholar]
- Dressler P, Gramer E. morphology, family history, and age at diagnosis of 26 patients with axenfeld-rieger syndrome and glaucoma or ocular hypertension. Ophthalmologe. 2006;103:393–400. doi: 10.1007/s00347-006-1335-6. [DOI] [PubMed] [Google Scholar]
- Dressler S, Meyer-Marcotty P, Weisschuh N, Jablonski-Momeni A, Pieper K, Gramer G, Gramer E. Dental and craniofacial anomalies associated with axenfeld-rieger syndrome with pitx2 mutation. Case Rep Med. 2010;2010:621984. doi: 10.1155/2010/621984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eerens K, Vlietinck R, Heidbuchel K, Van Olmen A, Derom C, Willems G, Carels C. Hypodontia and tooth formation in groups of children with cleft, siblings without cleft, and nonrelated controls. Cleft Palate Craniofac J. 2001 Jul;38(4):374–378. doi: 10.1597/1545-1569_2001_038_0374_hatfig_2.0.co_2. [DOI] [PubMed] [Google Scholar]
- El-Jaick KB, Fonseca RF, Moreira MA, Ribeiro MG, Bolognese AM, Dias SO, Pereira ET, Castilla EE, Orioli IM. Single median maxillary central incisor: New data and mutation review. Birth Defects Res A Clin Mol Teratol. 2007;79:573–580. doi: 10.1002/bdra.20380. [DOI] [PubMed] [Google Scholar]
- Erpenstein H, Pfeiffer RA. sex-linked-dominant hereditary reduction in number of teeth. Humangenetik. 1967;4:280–293. doi: 10.1007/BF00292201. [DOI] [PubMed] [Google Scholar]
- Feng J, Mantesso A, De Bari C, Nishiyama A, Sharpe PT. Dual origin of mesenchymal stem cells contributing to organ growth and repair. Proc Natl Acad Sci U S A. 2011;108:6503–6508. doi: 10.1073/pnas.1015449108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazier-Bowers SA, Guo DC, Cavender A, Xue L, Evans B, King T, Milewicz D, D’souza RN. A novel mutation in human pax9 causes molar oligodontia. J Dent Res. 2002;81:129–133. [PubMed] [Google Scholar]
- Gibson AC. Concomitant hypo-hyperodontia. Br J Orthod. 1979;6:101–105. doi: 10.1179/bjo.6.2.101. [DOI] [PubMed] [Google Scholar]
- Go W, Korzh V. Plasma membrane ca(2+) atpase atp2b1a regulates bone mineralization in zebrafish. Bone. 2013;54:48–57. doi: 10.1016/j.bone.2013.01.026. [DOI] [PubMed] [Google Scholar]
- Golan I, Baumert U, Hrala BP, Mussig D. Dentomaxillofacial variability of cleidocranial dysplasia: Clinicoradiological presentation and systematic review. Dentomaxillofac Radiol. 2003;32:347–354. doi: 10.1259/dmfr/63490079. [DOI] [PubMed] [Google Scholar]
- Goldenberg M, Das P, Messersmith M, Stockton DW, Patel PI, D’souza RN. Clinical, radiographic, and genetic evaluation of a novel form of autosomal-dominant oligodontia. J Dent Res. 2000;79:1469–1475. doi: 10.1177/00220345000790070701. [DOI] [PubMed] [Google Scholar]
- Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H, Joslyn G, Stevens J, Spirio L, Robertson M, et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66:589–600. doi: 10.1016/0092-8674(81)90021-0. [DOI] [PubMed] [Google Scholar]
- Guttal KS, Naikmasur VG, Bhargava P, Bathi RJ. Frequency of developmental dental anomalies in the indian population. Eur J Dent. 2010;4:263–269. [PMC free article] [PubMed] [Google Scholar]
- Hall RK, Bankier A, Aldred MJ, Kan K, Lucas JO, Perks AG. Solitary median maxillary central incisor, short stature, choanal atresia/midnasal stenosis (smmci) syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1997;84:651–662. doi: 10.1016/s1079-2104(97)90368-1. [DOI] [PubMed] [Google Scholar]
- Hansen K, Mehdinia M. Isolated soft tissue cleft lip: The influence on the nasal cavity and supernumerary laterals. Cleft Palate Craniofac J. 2002;39:322–326. doi: 10.1597/1545-1569_2002_039_0322_istclt_2.0.co_2. [DOI] [PubMed] [Google Scholar]
- Harada H, Kettunen P, Jung HS, Mustonen T, Wang YA, Thesleff I. Localization of putative stem cells in dental epithelium and their association with notch and fgf signaling. J Cell Biol. 1999;147:105–120. doi: 10.1083/jcb.147.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris MP, Rohner N, Schwarz H, Perathoner S, Konstantinidis P, Nusslein-Volhard C. Zebrafish eda and edar mutants reveal conserved and ancestral roles of ectodysplasin signaling in vertebrates. PLoS Genet. 2008;4:e1000206. doi: 10.1371/journal.pgen.1000206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart TC, Bowden DW, Bolyard J, Kula K, Hall K, Wright JT. Genetic linkage of the tricho-dento-osseous syndrome to chromosome 17q21. Hum Mol Genet. 1997;6:2279–2284. doi: 10.1093/hmg/6.13.2279. [DOI] [PubMed] [Google Scholar]
- Hillebrand U, Mohr C, Plewa G. taurodontism in patients with sex chromosome anomalies. Dtsch Z Mund Kiefer Gesichtschir. 1990;14:187–189. [PubMed] [Google Scholar]
- Hogstrom A, Andersson L. Complications related to surgical removal of anterior supernumerary teeth in children. ASDC J Dent Child. 1987;54:341–343. [PubMed] [Google Scholar]
- Hovorakova M, Lesot H, Peterkova R, Peterka M. Origin of the deciduous upper lateral incisor and its clinical aspects. J Dent Res. 2006;85:167–171. doi: 10.1177/154405910608500210. [DOI] [PubMed] [Google Scholar]
- Hovorakova M, Prochazka J, Lesot H, Smrckova L, Churava S, Boran T, Kozmik Z, Klein O, Peterkova R, Peterka M. Shh expression in a rudimentary tooth offers new insights into development of the mouse incisor. J Exp Zool B Mol Dev Evol. 2011;316:347–358. doi: 10.1002/jez.b.21408. [DOI] [PubMed] [Google Scholar]
- Howard RD. The unerupted incisor. A study of the postoperative eruptive history of incisors delayed in their eruption by supernumerary teeth. Dent Pract Dent Rec. 1967;17:332–341. [PubMed] [Google Scholar]
- Hu JC, Simmer JP. Developmental biology and genetics of dental malformations. Orthod Craniofac Res. 2007;10:45–52. doi: 10.1111/j.1601-6343.2007.00384.x. [DOI] [PubMed] [Google Scholar]
- Huang P, Zhu Z, Lin S, Zhang B. Reverse genetic approaches in zebrafish. J Genet Genomics. 2012;39:421–433. doi: 10.1016/j.jgg.2012.07.004. [DOI] [PubMed] [Google Scholar]
- Hunter ML, Roberts GJ. Oral and dental anomalies in ellis van creveld syndrome (chondroectodermal dysplasia): Report of a case. Int J Paediatr Dent. 1998;8:153–157. doi: 10.1046/j.1365-263x.1998.00069.x. [DOI] [PubMed] [Google Scholar]
- Huysseune A, Van Der Heyden C, Sire JY. Early development of the zebrafish (danio rerio) pharyngeal dentition (teleostei, cyprinidae) Anat Embryol (Berl) 1998;198:289–305. doi: 10.1007/s004290050185. [DOI] [PubMed] [Google Scholar]
- Ishii K, Nielsen IL, Vargervik K. Characteristics of jaw growth in cleidocranial dysplasia. Cleft Palate Craniofac J. 1998;35:161–166. doi: 10.1597/1545-1569_1998_035_0161_cojgic_2.3.co_2. [DOI] [PubMed] [Google Scholar]
- Islam M, Lurie AG, Reichenberger E. Clinical features of tricho-dento-osseous syndrome and presentation of three new cases: An addition to clinical heterogeneity. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;100:736–742. doi: 10.1016/j.tripleo.2005.04.017. [DOI] [PubMed] [Google Scholar]
- Jackman WR, Davies SH, Lyons DB, Stauder CK, Denton-Schneider BR, Jowdry A, Aigler SR, Vogel SA, Stock DW. Manipulation of fgf and bmp signaling in teleost fishes suggests potential pathways for the evolutionary origin of multicuspid teeth. Evolution & Development. 2013;15:107–118. doi: 10.1111/ede.12021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman WR, Draper BW, Stock DW. Fgf signaling is required for zebrafish tooth development. Dev Biol. 2004;274:139–157. doi: 10.1016/j.ydbio.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Janku P, Robinow M, Kelly T, Bralley R, Baynes A, Edgerton MT. The van der woude syndrome in a large kindred: Variability, penetrance, genetic risks. Am J Med Genet. 1980;5:117–123. doi: 10.1002/ajmg.1320050203. [DOI] [PubMed] [Google Scholar]
- Jarvinen S, Lehtinen L. Supernumerary and congenitally missing primary teeth in finnish children. An epidemiologic study. Acta Odontol Scand. 1981;39:83–86. doi: 10.3109/00016358109162264. [DOI] [PubMed] [Google Scholar]
- Jernvall J, Aberg T, Kettunen P, Keranen S, Thesleff I. The life history of an embryonic signaling center: Bmp-4 induces p21 and is associated with apoptosis in the mouse tooth enamel knot. Development. 1998;125:161–169. doi: 10.1242/dev.125.2.161. [DOI] [PubMed] [Google Scholar]
- Jernvall J, Kettunen P, Karavanova I, Martin LB, Thesleff I. Evidence for the role of the enamel knot as a control center in mammalian tooth cusp formation: Non-dividing cells express growth stimulating fgf-4 gene. Int J Dev Biol. 1994;38:463–469. [PubMed] [Google Scholar]
- Jheon AH, Seidel K, Biehs B, Klein OD. From molecules to mastication: The development and evolution of teeth. Wiley Interdisciplinary Reviews: Developmental Biology. 2013;2:165–182. doi: 10.1002/wdev.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgenson RJ, Salinas CF, Shapiro SD. The prevalence of taurodontism in a select population. J Craniofac Genet Dev Biol. 1982;2:125–135. [PubMed] [Google Scholar]
- Jussila M, Thesleff I. Signaling networks regulating tooth organogenesis and regeneration, and the specification of dental mesenchymal and epithelial cell lineages. Cold Spring Harb Perspect Biol. 2012;4:a008425. doi: 10.1101/cshperspect.a008425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juuri E, Saito K, Ahtiainen L, Seidel K, Tummers M, Hochedlinger K, Klein OD, Thesleff I, Michon F. Sox2+ stem cells contribute to all epithelial lineages of the tooth via sfrp5+ progenitors. Dev Cell. 2012;23:317–328. doi: 10.1016/j.devcel.2012.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaloust S, Ishii K, Vargervik K. Dental development in apert syndrome. Cleft Palate Craniofac J. 1997;34:117–121. doi: 10.1597/1545-1569_1997_034_0117_ddias_2.3.co_2. [DOI] [PubMed] [Google Scholar]
- Kantaputra PN, Limwongse C, Tochareontanaphol C, Mutirangura A, Mevatee U, Praphanphoj V. Contiguous gene syndrome of holoprosencephaly and hypotrichosis simplex: Association with an 18p11.3 deletion. Am J Med Genet A. 2006;140:2598–2602. doi: 10.1002/ajmg.a.31386. [DOI] [PubMed] [Google Scholar]
- Kantor ML, Bailey CS, Burkes EJ., Jr Duplication of the premolar dentition. Oral Surg Oral Med Oral Pathol. 1988;66:62–64. doi: 10.1016/0030-4220(88)90068-0. [DOI] [PubMed] [Google Scholar]
- Keith A. Abnormal crania-achondroplastic and acrocephalic. J Anat Physiol. 1913;47:189–206. [PMC free article] [PubMed] [Google Scholar]
- Keranen SV, Kettunen P, Aberg T, Thesleff I, Jernvall J. Gene expression patterns associated with suppression of odontogenesis in mouse and vole diastema regions. Dev Genes Evol. 1999;209:495–506. doi: 10.1007/s004270050282. [DOI] [PubMed] [Google Scholar]
- Kinzler KW, Nilbert MC, Su LK, Vogelstein B, Bryan TM, Levy DB, Smith KJ, Preisinger AC, Hedge P, Mckechnie D, et al. Identification of fap locus genes from chromosome 5q21. Science. 1991;253:661–665. doi: 10.1126/science.1651562. [DOI] [PubMed] [Google Scholar]
- Klein OD, Minowada G, Peterkova R, Kangas A, Yu BD, Lesot H, Peterka M, Jernvall J, Martin GR. Sprouty genes control diastema tooth development via bidirectional antagonism of epithelial-mesenchymal fgf signaling. Dev Cell. 2006;11:181–190. doi: 10.1016/j.devcel.2006.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatz Y, Tomoyoshi T, Yoshida O, Fujimoto A, Yoshitake K. Taurodontism and klinefelter’s syndrome. J Med Genet. 1978;15:452–454. doi: 10.1136/jmg.15.6.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo S, Schutte BC, Richardson RJ, Bjork BC, Knight AS, Watanabe Y, Howard E, De Lima RL, Daack-Hirsch S, Sander A, Mcdonald-Mcginn DM, Zackai EH, Lammer EJ, Aylsworth AS, Ardinger HH, Lidral AC, Pober BR, Moreno L, Arcos-Burgos M, Valencia C, Houdayer C, Bahuau M, Moretti-Ferreira D, Richieri-Costa A, Dixon MJ, Murray JC. Mutations in irf6 cause van der woude and popliteal pterygium syndromes. Nat Genet. 2002;32:285–289. doi: 10.1038/ng985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp WK. A herediatry congenitally missig maxillary central incisor. Oral Surg Oral Med Oral Pathol. 1967;24:367. [Google Scholar]
- Lam AK, David DJ, Townsend GC, Anderson PJ. Van der woude syndrome: Dentofacial features and implications for clinical practice. Aust Dent J. 2010;55:51–58. doi: 10.1111/j.1834-7819.2009.01178.x. [DOI] [PubMed] [Google Scholar]
- Lami F, Carli D, Ferrari P, Marini M, Alesi V, Iughetti L, Percesepe A. Holoprosencephaly: Report of four cases and genotype-phenotype correlations. J Genet. 2013;92:97–101. doi: 10.1007/s12041-013-0215-5. [DOI] [PubMed] [Google Scholar]
- Lammi L, Arte S, Somer M, Jarvinen H, Lahermo P, Thesleff I, Pirinen S, Nieminen P. Mutations in axin2 cause familial tooth agenesis and predispose to colorectal cancer. Am J Hum Genet. 2004;74:1043–1050. doi: 10.1086/386293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapthanasupkul P, Feng J, Mantesso A, Takada-Horisawa Y, Vidal M, Koseki H, Wang L, An Z, Miletich I, Sharpe PT. Ring1a/b polycomb proteins regulate the mesenchymal stem cell niche in continuously growing incisors. Dev Biol. 2012;367:140–153. doi: 10.1016/j.ydbio.2012.04.029. [DOI] [PubMed] [Google Scholar]
- Leck GD, Aird JC. An incomplete form of the popliteal pterygium syndrome? Br Dent J. 1984;157:318–319. doi: 10.1038/sj.bdj.4805480. [DOI] [PubMed] [Google Scholar]
- Lees MM, Winter RM, Malcolm S, Saal HM, Chitty L. Popliteal pterygium syndrome: A clinical study of three families and report of linkage to the van der woude syndrome locus on 1q32. J Med Genet. 1999;36:888–892. [PMC free article] [PubMed] [Google Scholar]
- Lesot H, Vonesch JL, Peterka M, Tureckova J, Peterkova R, Ruch JV. Mouse molar morphogenesis revisited by three-dimensional reconstruction. Ii. Spatial distribution of mitoses and apoptosis in cap to bell staged first and second upper molar teeth. Int J Dev Biol. 1996;40:1017–1031. [PubMed] [Google Scholar]
- Lexner MO, Bardow A, Bjorn-Jorgensen J, Hertz JM, Almer L, Kreiborg S. Anthropometric and cephalometric measurements in x-linked hypohidrotic ectodermal dysplasia. Orthod Craniofac Res. 2007;10:203–215. doi: 10.1111/j.1601-6343.2007.00402.x. [DOI] [PubMed] [Google Scholar]
- Liu DG, Zhang WL, Zhang ZY, Wu YT, Ma XC. Three-dimensional evaluations of supernumerary teeth using cone-beam computed tomography for 487 cases. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2007;103:403–411. doi: 10.1016/j.tripleo.2006.03.026. [DOI] [PubMed] [Google Scholar]
- Liu JF. Characteristics of premaxillary supernumerary teeth: A survey of 112 cases. ASDC J Dent Child. 1995;62:262–265. [PubMed] [Google Scholar]
- Macdonald-Jankowski DS, Li TT. Taurodontism in a young adult chinese population. Dentomaxillofac Radiol. 1993;22:140–144. doi: 10.1259/dmfr.22.3.8299833. [DOI] [PubMed] [Google Scholar]
- Madani M, Madani F. Gardner’s syndrome presenting with dental complaints. Arch Iran Med. 2007;10:535–539. [PubMed] [Google Scholar]
- Manjunath K, Kavitha B, Saraswathi TR, Sivapathasundharam B, Manikandhan R. Cementum analysis in cleidocranial dysostosis. Indian J Dent Res. 2008;19:253–256. doi: 10.4103/0970-9290.42960. [DOI] [PubMed] [Google Scholar]
- Matalova E, Fleischmannova J, Sharpe PT, Tucker AS. Tooth agenesis: From molecular genetics to molecular dentistry. J Dent Res. 2008;87:617–623. doi: 10.1177/154405910808700715. [DOI] [PubMed] [Google Scholar]
- Mcgraw HF, Drerup CM, Culbertson MD, Linbo T, Raible DW, Nechiporuk AV. Lef1 is required for progenitor cell identity in the zebrafish lateral line primordium. Development. 2011;138:3921–3930. doi: 10.1242/dev.062554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore SR, Wilson DF, Kibble J. Sequential development of multiple supernumerary teeth in the mandibular premolar region -- a radiographic case report. Int J Paediatr Dent. 2002;12:143–145. doi: 10.1046/j.1365-263x.2002.00336.x. [DOI] [PubMed] [Google Scholar]
- Mostowska A, Biedziak B, Trzeciak WH. A novel mutation in pax9 causes familial form of molar oligodontia. Eur J Hum Genet. 2006;14:173–179. doi: 10.1038/sj.ejhg.5201536. [DOI] [PubMed] [Google Scholar]
- Mostowska A, Kobielak A, Biedziak B, Trzeciak WH. Novel mutation in the paired box sequence of pax9 gene in a sporadic form of oligodontia. Eur J Oral Sci. 2003a;111:272–276. doi: 10.1034/j.1600-0722.2003.00036.x. [DOI] [PubMed] [Google Scholar]
- Mostowska A, Kobielak A, Trzeciak WH. Molecular basis of non-syndromic tooth agenesis: Mutations of msx1 and pax9 reflect their role in patterning human dentition. Eur J Oral Sci. 2003b;111:365–370. doi: 10.1034/j.1600-0722.2003.00069.x. [DOI] [PubMed] [Google Scholar]
- Nanni L, Ming JE, Du Y, Hall RK, Aldred M, Bankier A, Muenke M. Shh mutation is associated with solitary median maxillary central incisor: A study of 13 patients and review of the literature. Am J Med Genet. 2001;102:1–10. doi: 10.1002/1096-8628(20010722)102:1<1::aid-ajmg1336>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Nasman M, Forsberg CM, Dahllof G. Long-term dental development in children after treatment for malignant disease. Eur J Orthod. 1997;19:151–159. doi: 10.1093/ejo/19.2.151. [DOI] [PubMed] [Google Scholar]
- Nawa H, Oberoi S, Vargervik K. Taurodontism and van der woude syndrome. Is there an association? Angle Orthod. 2008;78:832–837. doi: 10.2319/081707-384.1. [DOI] [PubMed] [Google Scholar]
- Neubuser A, Peters H, Balling R, Martin GR. Antagonistic interactions between fgf and bmp signaling pathways: A mechanism for positioning the sites of tooth formation. Cell. 1997;90:247–255. doi: 10.1016/s0092-8674(00)80333-5. [DOI] [PubMed] [Google Scholar]
- Ng D, Thakker N, Corcoran CM, Donnai D, Perveen R, Schneider A, Hadley DW, Tifft C, Zhang L, Wilkie AO, Van Der Smagt JJ, Gorlin RJ, Burgess SM, Bardwell VJ, Black GC, Biesecker LG. Oculofaciocardiodental and lenz microphthalmia syndromes result from distinct classes of mutations in bcor. Nat Genet. 2004;36:411–416. doi: 10.1038/ng1321. [DOI] [PubMed] [Google Scholar]
- Nieminen P, Arte S, Tanner D, Paulin L, Alaluusua S, Thesleff I, Pirinen S. Identification of a nonsense mutation in the pax9 gene in molar oligodontia. Eur J Hum Genet. 2001;9:743–746. doi: 10.1038/sj.ejhg.5200715. [DOI] [PubMed] [Google Scholar]
- Oberoi S, Vargervik K. Hypoplasia and hypodontia in van der woude syndrome. Cleft Palate Craniofac J. 2005a;42:459–466. doi: 10.1597/04-028.1. [DOI] [PubMed] [Google Scholar]
- Oberoi S, Vargervik K. Velocardiofacial syndrome with single central incisor. Am J Med Genet A. 2005b;132A:194–197. doi: 10.1002/ajmg.a.30434. [DOI] [PubMed] [Google Scholar]
- Oberoi S, Winder AE, Johnston J, Vargervik K, Slavotinek AM. Case reports of oculofaciocardiodental syndrome with unusual dental findings. Am J Med Genet A. 2005;136:275–277. doi: 10.1002/ajmg.a.30811. [DOI] [PubMed] [Google Scholar]
- Ohazama A, Sharpe PT. Tnf signalling in tooth development. Curr Opin Genet Dev. 2004;14:513–519. doi: 10.1016/j.gde.2004.07.008. [DOI] [PubMed] [Google Scholar]
- Otto F, Kanegane H, Mundlos S. Mutations in the runx2 gene in patients with cleidocranial dysplasia. Hum Mutat. 2002;19:209–216. doi: 10.1002/humu.10043. [DOI] [PubMed] [Google Scholar]
- Ozeki H, Shirai S, Ikeda K, Ogura Y. Anomalies associated with axenfeld-rieger syndrome. Graefes Arch Clin Exp Ophthalmol. 1999;237:730–734. doi: 10.1007/s004170050304. [DOI] [PubMed] [Google Scholar]
- Paulsen HU, Andreasen JO, Schwartz O. Pulp and periodontal healing, root development and root resorption subsequent to transplantation and orthodontic rotation: A long-term study of autotransplanted premolars. Am J Orthod Dentofacial Orthop. 1995;108:630–640. doi: 10.1016/s0889-5406(95)70009-9. [DOI] [PubMed] [Google Scholar]
- Pawlowska E, Janik-Papis K, Poplawski T, Blasiak J, Szczepanska J. Mutations in the pax9 gene in sporadic oligodontia. Orthod Craniofac Res. 2010;13:142–152. doi: 10.1111/j.1601-6343.2010.01488.x. [DOI] [PubMed] [Google Scholar]
- Peterka M, Peterkova R, Likovsky Z. Timing of exchange of the maxillary deciduous and permanent teeth in boys with three types of orofacial clefts. Cleft Palate Craniofac J. 1996;33:318–323. doi: 10.1597/1545-1569_1996_033_0318_toeotm_2.3.co_2. [DOI] [PubMed] [Google Scholar]
- Peterkova R, Churava S, Lesot H, Rothova M, Prochazka J, Peterka M, Klein OD. Revitalization of a diastemal tooth primordium in spry2 null mice results from increased proliferation and decreased apoptosis. J Exp Zool B Mol Dev Evol. 2009;312B:292–308. doi: 10.1002/jez.b.21266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterkova R, Kristenova P, Lesot H, Lisi S, Vonesch JL, Gendrault JL, Peterka M. Different morphotypes of the tabby (eda) dentition in the mouse mandible result from a defect in the mesio-distal segmentation of dental epithelium. Orthod Craniofac Res. 2002a;5:215–226. doi: 10.1034/j.1600-0544.2002.02226.x. [DOI] [PubMed] [Google Scholar]
- Peterkova R, Lesot H, Peterka M. Phylogenetic memory of developing mammalian dentition. J Exp Zool B Mol Dev Evol. 2006;306:234–250. doi: 10.1002/jez.b.21093. [DOI] [PubMed] [Google Scholar]
- Peterkova R, Peterka M, Lesot H. The developing mouse dentition: A new tool for apoptosis study. Ann N Y Acad Sci. 2003;1010:453–466. doi: 10.1196/annals.1299.083. [DOI] [PubMed] [Google Scholar]
- Peterkova R, Peterka M, Viriot L, Lesot H. Development of the vestigial tooth primordia as part of mouse odontogenesis. Connect Tissue Res. 2002b;43:120–128. doi: 10.1080/03008200290000745. [DOI] [PubMed] [Google Scholar]
- Peterkova R, Peterka M, Vonesch JL, Ruch JV. Contribution of 3-d computer-assisted reconstructions to the study of the initial steps of mouse odontogenesis. Int J Dev Biol. 1995;39:239–247. [PubMed] [Google Scholar]
- Peters H, Neubuser A, Kratochwil K, Balling R. Pax9-deficient mice lack pharyngeal pouch derivatives and teeth and exhibit craniofacial and limb abnormalities. Genes Dev. 1998;12:2735–2747. doi: 10.1101/gad.12.17.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson SJ, Pruzansky S. Palatal anomalies in the syndromes of apert and crouzon. Cleft Palate J. 1974;11:394–403. [PubMed] [Google Scholar]
- Pirinen S, Kentala A, Nieminen P, Varilo T, Thesleff I, Arte S. Recessively inherited lower incisor hypodontia. J Med Genet. 2001;38:551–556. doi: 10.1136/jmg.38.8.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polder BJ, Van’t Hof MA, Van Der Linden FP, Kuijpers-Jagtman AM. A meta-analysis of the prevalence of dental agenesis of permanent teeth. Community Dent Oral Epidemiol. 2004;32:217–226. doi: 10.1111/j.1600-0528.2004.00158.x. [DOI] [PubMed] [Google Scholar]
- Price JA, Wright JT, Walker SJ, Crawford PJ, Aldred MJ, Hart TC. Tricho-dento-osseous syndrome and amelogenesis imperfecta with taurodontism are genetically distinct conditions. Clin Genet. 1999;56:35–40. doi: 10.1034/j.1399-0004.1999.550105.x. [DOI] [PubMed] [Google Scholar]
- Ramaglia L, Morgese F, Filippella M, Colao A. Oral and maxillofacial manifestations of gardner’s syndrome associated with growth hormone deficiency: Case report and literature review. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2007;103:e30–34. doi: 10.1016/j.tripleo.2007.01.013. [DOI] [PubMed] [Google Scholar]
- Ranta R. A review of tooth formation in children with cleft lip/palate. Am J Orthod Dentofacial Orthop. 1986 Jul;90(1):11–18. doi: 10.1016/0889-5406(86)90022-3. [DOI] [PubMed] [Google Scholar]
- Ranta R, Rintala A. Tooth anomalies associated with congenital sinuses of the lower lip and cleft lip/palate. Angle Orthod. 1982;52:212–221. doi: 10.1043/0003-3219(1982)052<0212:TAAWCS>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Ravn JJ. Aplasia, supernumerary teeth and fused teeth in the primary dentition. An epidemiologic study. Scand J Dent Res. 1971;79:1–6. doi: 10.1111/j.1600-0722.1971.tb01986.x. [DOI] [PubMed] [Google Scholar]
- Richardson A, Deussen FF. Facial and dental anomalies in cleidocranial dysplasia: A study of 17 cases. Int J Paediatr Dent. 1994;4:225–231. doi: 10.1111/j.1365-263x.1994.tb00139.x. [DOI] [PubMed] [Google Scholar]
- Rintala AE, Ranta R. Lower lip sinuses: I. Epidemiology, microforms and transverse sulci. Br J Plast Surg. 1981;34:26–30. doi: 10.1016/0007-1226(81)90090-4. [DOI] [PubMed] [Google Scholar]
- Rothova M, Peterkova R, Tucker AS. Fate map of the dental mesenchyme: Dynamic development of the dental papilla and follicle. Dev Biol. 2012;366:244–254. doi: 10.1016/j.ydbio.2012.03.018. [DOI] [PubMed] [Google Scholar]
- Ruch JV. Tooth crown morphogenesis and cytodifferentiations: Candid questions and critical comments. Connect Tissue Res. 1995;32:1–8. doi: 10.3109/03008209509013699. [DOI] [PubMed] [Google Scholar]
- Sarkar L, Cobourne M, Naylor S, Smalley M, Dale T, Sharpe PT. Wnt/shh interactions regulate ectodermal boundary formation during mammalian tooth development. Proc Natl Acad Sci U S A. 2000;97:4520–4524. doi: 10.1073/pnas.97.9.4520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schalk-Van Der Weide Y, Steen WH, Bosman F. Taurodontism and length of teeth in patients with oligodontia. J Oral Rehabil. 1993;20:401–412. doi: 10.1111/j.1365-2842.1993.tb01624.x. [DOI] [PubMed] [Google Scholar]
- Scheiner MA, Sampson WJ. Supernumerary teeth: A review of the literature and four case reports. Aust Dent J. 1997;42:160–165. doi: 10.1111/j.1834-7819.1997.tb00114.x. [DOI] [PubMed] [Google Scholar]
- Schneider EL. Lip pits and congenital absence of second premolars: Varied expression of the lip pits syndrome. J Med Genet. 1973;10:346–349. doi: 10.1136/jmg.10.4.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schutte BC, Sander A, Malik M, Murray JC. Refinement of the van der woude gene location and construction of a 3.5-mb yac contig and sts map spanning the critical region in 1q32-q41. Genomics. 1996;36:507–514. doi: 10.1006/geno.1996.0496. [DOI] [PubMed] [Google Scholar]
- Seidel K, Ahn CP, Lyons D, Nee A, Ting K, Brownell I, Cao T, Carano RA, Curran T, Schober M, Fuchs E, Joyner A, Martin GR, De Sauvage FJ, Klein OD. Hedgehog signaling regulates the generation of ameloblast progenitors in the continuously growing mouse incisor. Development. 2010;137:3753–3761. doi: 10.1242/dev.056358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seow WK, Lai PY. Association of taurodontism with hypodontia: A controlled study. Pediatr Dent. 1989;11:214–219. [PubMed] [Google Scholar]
- Shaikh R, Shusterman S. Delayed dental maturation in cleidocranial dysplasia. ASDC J Dent Child. 1998;65:325–329. 355. [PubMed] [Google Scholar]
- Shapira Y, Lubit E, Kuftinec MM. Congenitally missing second premolars in cleft lip and cleft palate children. Am J Orthod Dentofacial Orthop. 1999;115:396–400. doi: 10.1016/s0889-5406(99)70258-1. [DOI] [PubMed] [Google Scholar]
- Sharpe PT. Homeobox genes and orofacial development. Connect Tissue Res. 1995;32:17–25. doi: 10.3109/03008209509013701. [DOI] [PubMed] [Google Scholar]
- Shaw JC. Taurodont teeth in south african races. J Anat. 1928;62:476–498. 471. [PMC free article] [PubMed] [Google Scholar]
- Shields MB, Buckley E, Klintworth GK, Thresher R. Axenfeld-rieger syndrome. A spectrum of developmental disorders. Survey of Ophthalmology. 1985;29:387–409. doi: 10.1016/0039-6257(85)90205-x. [DOI] [PubMed] [Google Scholar]
- Smith JD. Hyperdontia: Report of case. J Am Dent Assoc. 1969;79:1191–1192. doi: 10.14219/jada.archive.1969.0068. [DOI] [PubMed] [Google Scholar]
- Soni R, Vivek R, Srivastava A, Singh A, Srivastava S, Chaturvedi TP. Van der woude syndrome associated with hypodontia: A rare clinical entity. Case Rep Dent. 2012;2012:283946. doi: 10.1155/2012/283946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Amand TR, Zhang Y, Semina EV, Zhao X, Hu Y, Nguyen L, Murray JC, Chen Y. Antagonistic signals between bmp4 and fgf8 define the expression of pitx1 and pitx2 in mouse tooth-forming anlage. Dev Biol. 2000;217:323–332. doi: 10.1006/dbio.1999.9547. [DOI] [PubMed] [Google Scholar]
- Stenvik A, Zachrisson BU, Svatun B. Taurodontism and concomitant hypodontia in siblings. Oral Surg Oral Med Oral Pathol. 1972;33:841–845. doi: 10.1016/0030-4220(72)90454-9. [DOI] [PubMed] [Google Scholar]
- Stock DW. Zebrafish dentition in comparative context. J Exp Zool B Mol Dev Evol. 2007;308:523–549. doi: 10.1002/jez.b.21187. [DOI] [PubMed] [Google Scholar]
- Stockton DW, Das P, Goldenberg M, D’souza RN, Patel PI. Mutation of pax9 is associated with oligodontia. Nat Genet. 2000;24:18–19. doi: 10.1038/71634. [DOI] [PubMed] [Google Scholar]
- Tannure PN, Oliveira CaGR, Maria LC, Vierira AR, Granjeiro JM, Costa MC. Prevalence of dental anomalies in non-syndromic individuals with cleft lip and palate: A systematic review and met-analysis. Cleft Palate Craniofac. 2012;49:194–200. doi: 10.1597/10-043. [DOI] [PubMed] [Google Scholar]
- Thesleff I. The genetic basis of tooth development and dental defects. Am J Med Genet A. 2006;140:2530–2535. doi: 10.1002/ajmg.a.31360. [DOI] [PubMed] [Google Scholar]
- Thomas BL, Liu JK, Rubenstein JL, Sharpe PT. Independent regulation of dlx2 expression in the epithelium and mesenchyme of the first branchial arch. Development. 2000;127:217–224. doi: 10.1242/dev.127.2.217. [DOI] [PubMed] [Google Scholar]
- Tomona N, Smith AC, Guadagnini JP, Hart TC. Craniofacial and dental phenotype of smith-magenis syndrome. Am J Med Genet A. 2006;140:2556–2561. doi: 10.1002/ajmg.a.31371. [DOI] [PubMed] [Google Scholar]
- Toriello HV, Franco B. Oral-facial-digital syndrome type i. Seattle: University of Washington; 2007. GeneReviews. [PubMed] [Google Scholar]
- Tucker A, Sharpe P. The cutting-edge of mammalian development; how the embryo makes teeth. Nat Rev Genet. 2004;5:499–508. doi: 10.1038/nrg1380. [DOI] [PubMed] [Google Scholar]
- Tumer Z, Bach-Holm D. Axenfeld-rieger syndrome and spectrum of pitx2 and foxc1 mutations. Eur J Hum Genet. 2009;17:1527–1539. doi: 10.1038/ejhg.2009.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaahtokari A, Aberg T, Thesleff I. Apoptosis in the developing tooth: Association with an embryonic signaling center and suppression by egf and fgf-4. Development. 1996;122:121–129. doi: 10.1242/dev.122.1.121. [DOI] [PubMed] [Google Scholar]
- Van Der Heyden C, Huysseune A. Dynamics of tooth formation and replacement in the zebrafish (danio rerio) (teleostei, cyprinidae) Dev Dyn. 2000;219:486–496. doi: 10.1002/1097-0177(2000)9999:9999<::AID-DVDY1069>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Van Der Heyden C, Huysseune A, Sire JY. Development and fine structure of pharyngeal replacement teeth in juvenile zebrafish (danio rerio) (teleostei, cyprinidae) Cell Tissue Res. 2000;302:205–219. doi: 10.1007/s004410000180. [DOI] [PubMed] [Google Scholar]
- Vastardis H. The genetics of human tooth agenesis: New discoveries for understanding dental anomalies. Am J Orthod Dentofacial Orthop. 2000;117:650–656. [PubMed] [Google Scholar]
- Vastardis H, Karimbux N, Guthua SW, Seidman JG, Seidman CE. Ahuman msx1 homeodomain missense mutation causes selective tooth agenesis. Nat Genet. 1996;13:417–421. doi: 10.1038/ng0896-417. [DOI] [PubMed] [Google Scholar]
- Verstraeten B, Van Hengel J, Sanders E, Van Roy F, Huysseune A. N-cadherin is required for cytodifferentiation during zebrafish odontogenesis. J Dent Res. 2013;92:365–370. doi: 10.1177/0022034513477424. [DOI] [PubMed] [Google Scholar]
- Wallis D, Muenke M. Mutations in holoprosencephaly. Hum Mutat. 2000;16:99–108. doi: 10.1002/1098-1004(200008)16:2<99::AID-HUMU2>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Wesley RK, Cullen CL, Bloom WS. Gardner’s syndrome with bilateral osteomas of coronoid process resulting in limited opening. Pediatr Dent. 1987;9:53–57. [PubMed] [Google Scholar]
- Witkop CJ. Clinical aspects of dental anomalies. Int Dent J. 1976;26:378–390. [PubMed] [Google Scholar]
- Wiweger MI, Zhao Z, Van Merkesteyn RJ, Roehl HH, Hogendoorn PC. Hspg-deficient zebrafish uncovers dental aspect of multiple osteochondromas. PLoS One. 2012;7:e29734. doi: 10.1371/journal.pone.0029734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong FK, Koillinen H, Rautio J, Teh BT, Ranta R, Karsten A, Larson O, Linder-Aronson S, Huggare J, Larsson C, Kere J. Genetic heterogeneity and exclusion of a modifying locus at 17p11.2-p11.1 in finnish families with van der woude syndrome. J Med Genet. 2001;38:198–202. doi: 10.1136/jmg.38.3.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright JT, Kula K, Hall K, Simmons JH, Hart TC. Analysis of the tricho-dento-osseous syndrome genotype and phenotype. Am J Med Genet. 1997;72:197–204. doi: 10.1002/(sici)1096-8628(19971017)72:2<197::aid-ajmg14>3.3.co;2-0. [DOI] [PubMed] [Google Scholar]
- Yang Y, Luo L, Xu J, Zhu P, Xue W, Wang J, Li W, Wang M, Cheng K, Liu S, Tang Z, Ring BZ, Su L. Novel eda p.Ile260ser mutation linked to non-syndromic hypodontia. J Dent Res. 2013;92:500–506. doi: 10.1177/0022034513487557. [DOI] [PubMed] [Google Scholar]
- Yeh SC, Hsu TY. Endodontic treatment in taurodontism with klinefelter’s syndrome: A case report. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1999;88:612–615. doi: 10.1016/s1079-2104(99)70094-6. [DOI] [PubMed] [Google Scholar]
- Yonezu T, Hayashi Y, Sasaki J, Machida Y. Prevalence of congenital dental anomalies of the deciduous dentition in japanese children. Bull Tokyo Dent Coll. 1997;38:27–32. [PubMed] [Google Scholar]