

Abstract
The failure of the remyelination processes in multiple sclerosis contributes to the formation of chronic demyelinated plaques that lead to severe neurological deficits. Long-term cuprizone treatment of C57BL/6 mice resulted in pronounced white matter pathology characterized by oligodendrocyte depletion, irreversible demyelination and persistent functional deficits after cuprizone withdrawal. The use of a combination of in vivo diffusion tensor magnetic resonance imaging (DT-MRI) and histological analyses allowed for an accurate longitudinal assessment of demyelination. Injection of triiodothyronine (T3) hormone over a 3 week interval after cuprizone withdrawal progressively restored the normal DT-MRI phenotype accompanied by an improvement of clinical signs and remyelination. The effects of T3 were not restricted to the later stages of remyelination but increased the expression of sonic hedgehog and the numbers of Olig2+ and PSA-NCAM+ precursors and proliferative cells. Our findings establish a role for T3 as an inducer of oligodendrocyte progenitor cells in adult mouse brain following chronic demyelination.
Keywords: myelin, oligodendrocytes, demyelination therapy, gliotoxic agent, DT-MRI, myelin repair
Introduction
Multiple sclerosis (MS) is an example of chronic inflammatory demyelinating disease of the CNS, with a large heterogeneity in clinical course, MRI patterns and responses to therapy (Kornek and Lassmann, 2003). Several causes have been proposed for the failure of long-term remyelination in MS and for each, therapeutic strategies have been developed (Pluchino et al., 2004). A critical step in preclinical studies is, however, the development of animal models that can be examined in vivo in acute and chronic phases of the MS pathology to characterize the effects of the applied therapy. In mice, the cuprizone-diet model (Ludwin, 1978; Blakemore, 1984; Matsushima and Morell, 2001) is of particular interest because it allows the progression of demyelinated lesions to a chronic state, depending on the duration of cuprizone administration. Effective spontaneous recovery does not occur in brains of long-term cuprizone-treated mice and the model allows the testing of therapeutic strategies for remyelination.
It is likely that factors playing a role in the normal myelination processes participate in the remyelination of the injured CNS. Particularly, molecules implicated in oligodendrocyte differentiation and maturation may act in the generation of positive signals for recovery. Thyroid hormones (THs) are necessary for normal axonal myelination acting at multiple steps during oligodendrocytes development and myelination, via nuclear hormone receptors (Baas et al., 1997; Rodríguez-Peña, 1999; Jones et al., 2003; Sarliève et al., 2004; Schoonover et al., 2004; Kang et al., 2007), but no information are available about the role of TH in the induction of oligodendrocyte lineage in chronic demyelination. We therefore, explored the possibility of stimulating endogenous repair by triiodothyronine (T3) administration in long-term cuprizone-treated mice. Reparative responses were followed in vivo by diffusion tensor magnetic resonance imaging (DT-MRI).
Highly sensitive to the water molecule motion, DT-MRI enables tissue structure to be probed and imaged on a microscopic scale, providing details of the cytoarchitecture of the neural tissue and identifying changes related to a pathological condition (LeBihan, 2003). In the white matter, the hydrophobic nature of the myelin membrane provides barriers for water diffusion and changes in the permeability of these barriers, which are formed during normal and pathologic development, generates modifications in DT-MRI derived parameters. For example, examination of directional diffusivity perpendicular (D⊥) and parallel (D‖) to the fiber tracts allows in vivo assessment of mouse brain dysmyelination and spontaneous recovery (Song et al., 2003, 2005; Harsan et al., 2006, Mori and Zhang, 2006; Harsan et al., 2007).
While it is clear that T3 contributes to the differentiation and maturation of oligodendrocytes (Billon et al., 2001; Schoonover et al., 2004) its role in the regulation of the oligodendrocyte lineage in vivo and particularly in adult brain is unclear. The present findings establish a role for T3 as a potential inducer of oligodendrocyte precursor cells (OPCs) in adult mouse brain in chronic demyelination caused by cuprizone treatment. Moreover, we provide an accurate assessment of demyelination and recovery in vivo, in a longitudinal study that combined DT-MRI and histological analyses of T3-based therapy. We show clearly that T3 acts as a potent inducer of oligodendrogenesis through the expression of sonic hedgehog (Shh) and Olig2 transcription factors and a promoter of myelin regeneration in the chronic demyelination of adult mouse brain.
Materials and Methods
Animals and treatments
Three groups of 8-week-old C57BL/6 female mice, purchased from Janvier Breeding Center were used for DT-MRI exam at different time points as presented in the Figure 1. Duplicates for each group were kept in the same conditions of housing and treatment and used for the histopathological examination.
Figure 1.
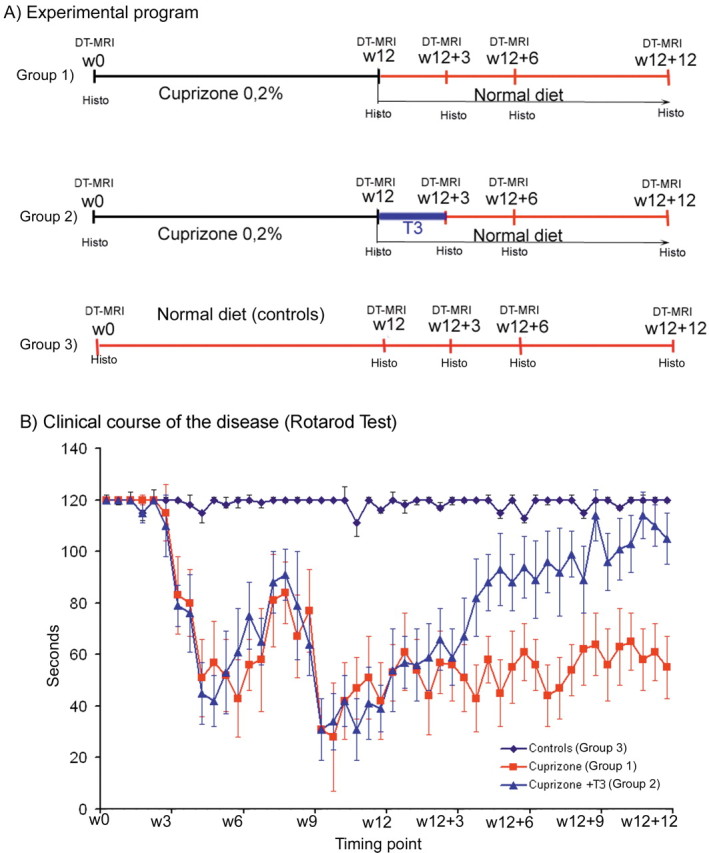
Experimental groups and summary of DT-MRI and histological examinations. A, The longitudinal examination was conducted starting at w0 time point (week before any treatment) and continuing at w12 (week 12 of cuprizone treatment), w12 + 3, w12 + 6, and w12 + 12 (at 3, 6, and 12 weeks after cuprizone removal). B, Results of rotarod test, performed twice a week in each experimental group. The mice were considered as unaffected when performing minimum 2 min (120 s) on the rotarod. The figure shows normal scores for the mice of control group, while the mice subjected to cuprizone diet show clinical signs from the fourth week of regimen, with a relapsing remitting pattern. The mice receiving T3 injections after cuprizone withdrawal (Cuprizone +T3 group) improve considerably their clinical score during the next 12 weeks of observation. On the contrary, only weak improvements were quantified in this period for the mice allowed to recover spontaneously, without any stimulation (Cuprizone group).
Group 1.
Eight female mice were placed on a diet containing 0.2% cuprizone (bis-cyclohexanone-oxaldihydrazone, Sigma) mixed into milled chow and available ad libitum, for 12 weeks (Fig. 1). A cuprizone-free diet followed during the next 12 weeks before death.
Group 2.
Eight females (Fig. 1, group 2) were subjected to cuprizone diet as above for 12 weeks followed by normal diet until death. Immediately after cuprizone-treatment arrest the mice received daily IP injections of T3 hormone (0.3 μg/g body weight) during the following 3 weeks. The mice were further housed under normal diet for other 9 weeks before death.
Group 3.
Eight females (Fig. 1) serving as controls, were bred for 24 weeks in the same house conditions, free of any treatment and normally nourished.
DT-MRI exams and the histology were conducted at 0 and 12 weeks of cuprizone diet (w0 and w12), as well as at 3, 6, and 12 weeks after cuprizone removal (w12 + 3, w12 + 6, and w12 + 12, respectively). Figure 1 presents the treatment groups and the timing of DT-MRI and histological examinations.
DT-MRI acquisition and processing
The gaseous anesthesia was achieved in mice by using a mixture of 4% isoflurane (Forene) and oxygen for induction during 3 min and 1.5% for maintenance. The mask placed on the animal nose was connected to the inflow line but also to the outflow line, to remove excess isoflurane from the magnet bore. The animals were placed in a stereotaxic device to immobilize the head, with integrated heating facility to maintain the body temperature at 37°C. An anatomically shaped 1H surface coil for small animals (Rapid Biomedical) was placed on the head to serve as a receiver for the magnetic resonance (MR) signal. The system was then positioned into a 1H resonator (Rapid Biomedical) for rats and mice, which entered into a 20 cm 4.7 T MR magnet equipped with self-shield gradient coils from Magnex Scientific. The MS spectrometer is from S.M.I.S. (now M.R.R.S.). Before the acquisition of images set required for computing the diffusion tensor, a fast spin-echo sequence was used to obtain T2-weighted images as sagittal multislices that cover all the brain. The acquisition parameters were as following (for 4 echoes in the echo train): repetition time (TR) = 3.8 s; spin-echo time (TE) = 40 ms; field of view (FOV) = 20 × 20 mm2, with data matrix 256 × 256 (zero-filled to 512 × 512) and an average of 4. The slice's thickness was 0.5 mm.
For diffusion-weighted images (DWI) acquisition, a conventional spin-echo imaging sequence modified by adding the Stejskal-Tanner diffusion gradient pair was used. Brain sagittal slices were acquired over 2.5 h for each mouse, with a TR of 1.5 s, TE of 35 ms, time (Δ) between the application of diffusion gradient pulses of 21.7 ms, diffusion gradient duration (δ) of 5.6 ms, ramp time of 400 μs and gradient amplitude (G) of 0.135 T/m. The slice thickness was 1 mm, FOV 20 × 20 mm2 and data matrix 256 × 256 (zero-filled to 512 × 512). Diffusion-sensitizing gradients of the same amplitude were applied along six different directions defined by the six unit vectors: (gx, gy, gz) = (1, 0, 0); (0, 1, 0); (0, 0, 1); (1, 1, 0)/; (0, 1, 1)/; (1, 0, 1)/. The used b factor (Le Bihan et al., 1986) values were 0 and 865 s/mm2.
Data processing was performed using software written in Matlab. From the diffusion tensor computation for each voxel of the image (Basser et al., 1994) the 3 eigenvalues λ1, λ2, λ3 (λ1 > λ2 > λ3) and their corresponding eigenvectors (e 1, e 2, e 3) were derived. It is generally assumed that in the CNS environment the main diffusivity (λ1) represents the diffusion parallel to the fiber tracts (axial diffusivity, D‖=λ1) and the mean of the other 2 eigenvalues express the diffusion perpendicular to the tracts, radial diffusion (D⊥=(λ2 + λ3)/2). By using these eigenvalues, the mean diffusivity <D> was also calculated and maps were generated. Fractional anisotropy (FA), which is related to the presence of oriented structures, giving rise to preferred diffusion orientations, was also calculated to yield values from 0 to 1. For an isotropic medium (λ1≅λ2≅λ3), FA = 0, while for an anisotropic medium (for example λ1≫λ2=λ3), FA = 1. Generally brain FA maps show the CSF as isotropic medium (low values of FA) and the highly oriented white matter tracts as anisotropic structures (high FA values). Regions of interest (ROIs) were manually defined (Fig. 2) using a mouse brain atlas as reference (Paxinos and Franklin, 2001).
Figure 2.

A, B, Representative sagittal brain T2-weighted images of individuals from groups 1 (A) and 2 (B) examined at different time points. At w0, corresponding to the exam prior to the cuprizone ingestion, the myelinated areas, corpus callosum (arrows) and cerebellum showed hypointense signals. Enlarged ventricles corresponding to the hyperintense signal are evident after 12 weeks of cuprizone diet in both groups (w12). No significant changes of the T2 images pattern are observed during the w12 to w12 + 12 period in mice from group 1 (A). Partial regain in the hypointensities of corpus callosum and cerebellum are noticed in the group receiving T3 injections (group 2, B) during w12 to w12 + 12 time interval, with spectacular decrease in the ventricle volumes. The T2-weighted images pattern in control mice is identical with that observed at w0 time point in groups 1 and 2 and is not presented. C, Investigated ROIs overlaid on a sagittal DWI of a normal mouse brain at w0.
Statistical analysis was performed using ANOVA, followed by Bonferroni corrections for multiple testing, to quantify the effects of cuprizone diet and age on DT-MRI indices. The analysis showed a significant effect of the cuprizone intoxication at p < 0.05. The same tests were used to quantify the TH effects for remyelination, as expressed by changes of DT-MRI parameter values. The results for each ROI were expressed as mean ± SD. Difference was considered statistically significant at p < 0.05.
Histological analysis
Immunohistofluorescence.
Mice for histological analysis were killed under pentobarbital deep anesthesia and perfused through the left ventricle with freshly prepared solution of 4% paraformaldehyde (PFA) in phosphate buffer (0.1 m, pH 7.5, PBS). Further fixation was achieved by maintaining the brains overnight in the same fixative. The tissues were next embedded in paraffin wax and 5 μm thick sagittal sections were made using the microtome Leica (Leica Instruments).
Double immunolabeling with a rabbit antibody against carbonic anhydrase II (CA II at 1:200 dilution) and a mouse monoclonal antibody against guinea pig myelin basic protein (MBP at 1:10 dilution) (both prepared in our laboratory) was performed according to the procedure previously described (Harsan et al., 2004). The oligodendrocytes marked by CA II antibody were counted in the total length of corpus callosum (genu, body, and splenium) in the different groups of mice. A minimum of six sagittal sections (at the levels 0.25, 0.50, and 0.75 mm laterally, in both hemispheres) from each animal (n = 4 for each time point in each experimental group) were captured at a 40× magnification using a BX60 microscope, equipped with DP70 digital camera (Olympus). The images were analyzed and the oligodendrocytes counted using the NIH ImageJ software.
To estimate the number of proliferating oligodendrocyte, we performed double labeling with a mouse antibody against proliferation cell nuclear antigen (PCNA, 1:100 dilution) (Santa Cruz Biotechnology) and the rabbit CA II antibody, on paraffin sections. The secondary antibodies were Alexa Fluor 488-conjugated anti-rabbit IgG (1:200) and Alexa Fluor 546-conjugated anti-mouse IgG (1:200) (Invitrogen). We also used a double immunolabeling procedure to label the proliferating cells using anti-bromodeoxyuridine (BrdU at 1:50 dilution) antibody after BrdU incorporation. CA II antibody was used for the double labeling as described in the study by Jalabi et al. (2005). Mice received daily injections of 100 μg/g body weight BrdU, during the 4 d previous to perfusion.
The mouse monoclonal anti-polysialic acid (PSA-NCAM, 1:400) as well as the rabbit polyclonal anti-NG2 chondroitin sulfate proteoglycan (1:100, Millipore) antibodies were used to detect the migrating cells and the oligodendrocytes precursors respectively.
A rat anti-mouse CD45 (leukocyte common antigen) at 1:50 dilution was also used to detect the reactive gliosis, while the mouse monoclonal glial fibrillary acidic protein antibody (GFAP, 1:300, Dako) was used for the astrocytosis detection. The sections were examined with BX60 microscope equipped with DP70 digital camera (Olympus).
Immunohistochemistry.
Paraffin-embedded sections from fixed brains or cut as frozen sections were used. Sections were subject to antigen retrieval using 10 mm sodium citrate buffer containing 0.05% Tween 20 at pH 6.0 for 10 min at 98°C then removed from heat and allowed to cool in buffer at room temperature (RT) for 20 min. Sections were rinsed for 10 min in TBS at pH 6.0, followed by serial rinses in 1×PBS, pH 7.4. All sections were preblocked in PBS containing 5–10% normal goat serum and 0.1–0.3% Triton X-100 for 1–2 h at RT. They were then incubated in block containing one of the following primary antibodies at 4°C overnight: Olig-2 at 1:500 (AB9610 Millipore); MBP at 1:500 (Clone SMI-99 Covance); Shh at 1:100 (SC-9024 Santa Cruz) and GFAP at 1:500 (Z0334 Dako). The next day, sections were rinsed in 1× PBS then incubated in the appropriate biotinylated secondary for 1 h at RT: goat anti-mouse IgG at 1:200 (55587 ICN) or goat anti-rabbit IgG at 1:250 (AP132 B Millipore). Signal was amplified using an Avidin-Biotin Complex (PK-6100 Vector) for 30 min at RT and visualized by immunoperoxidase reaction using DAB (SK-4100 Vector). Olig2 immunohistochemistry by using Elite Vecta Stain rabbit ABC kit (Vector Laboratories) was also combined with CA II immunohistofluorescence, as described above for vibratomed brain slices. Sections were then dehydrated and mounted in Permount and photographed on a Leica DM5000 B microscope using a DFC-500 camera with Leica Application Software or with BX60 microscope equipped with DP70 digital camera (Olympus).
Electron microscopy.
At each experimental time point and for each experimental group of animals, mice were specially perfused with 2% PFA and 6% glutaraldehyde (Sigma) for electronic microscopy examination of the brain. Sagittal brain sections (100 μm) were prepared and afterward kept overnight in the same fixative. Tissue slices were embedded in epoxy resin. Five to ten 1 μm semithin sections from brains were stained with toluidine blue, analyzed under ×100 objective and photographs taken. Myelinated axons were counted all over the corpus callosum.
Sixty-nanometer ultrathin sections were also examined in a transmission electron microscope (Siemens Elmiskop 102) at 60 kV. The calibers of 1500 axons from similar regions of the corpus callosum were measured in electron micrographs for each examined brain, by using NIH ImageJ plugins.
Thyroid hormone assays
Four hundred microliters of blood was collected from each mouse in heparin-treated tubes at the end of cuprizone treatment (w12) and from nontreated control mice. Plasma samples were obtained after blood centrifugation and then stored at −20°C. Total T4, total T3, and thyroid stimulating hormone (TSH) determinations were performed as previously described (Streckfuss et al., 2005) with one modification in the TSH assay: 125I-labeled rat TSH was obtained from BioCode-Hycel (Belgium).
All the experimental procedures were performed in accordance with the guidelines of Animal Care Committee of Louis Pasteur University, Strasbourg, France.
Results
To assess the effects of T3-based therapy, we induced severe white matter pathology in C57BL/6 mice, by 12 weeks of cuprizone diet. The cuprizone is a well described gliotoxic agent that alters the functions of oligodendrocyte mitochondria resulting in cell death. Importantly, our results showed that long-term cuprizone treatment does not affect significantly the thyroid hormones levels, T3 and T4, nor the TSH in the present model of chronic demyelination in adult mice (Table 1). The death by apoptosis of oligodendrocytes results in myelin breakdown and underlies the progression of the disease toward a chronic state. The experimental program and the established animal groups examined in DT-MRI and histology are presented in Figure 1 A (see also Materials and Methods).
Table 1.
T3, T4, and TSH levels measured in blood plasma collected from control and cuprizone-treated mice at the end of intoxication period
| w12 cuprizone-treated and control mice | T3 ng/ml |
T4 ng/ml |
TSH ng/ml |
|||
|---|---|---|---|---|---|---|
| Control | Cuprizone | Control | Cuprizone | Control | Cuprizone | |
| Mean | 0.80 | 0.79 | 41.45 | 35.85 | 152 | 190 |
| SD | 0.17 | 0.17 | 9.5 | 5.85 | 38.26 | 55.14 |
| No. of animals | 10 | 11 | 6 | 6 | 6 | 6 |
t test was not significant for any group.
Clinical course of the disease
During the period of cuprizone ingestion mice in groups 1 and 2 (Fig. 1 B) showed clinical signs of demyelination (loss of body weight, seizures, loss of coordinate movements and locomotors disabilities assessed on the rotarod) starting at the fourth week (w4) of cuprizone administration, with a relapsing remitting pattern and gradually increasing severity till the end of the treatment. No significant improvements of the clinical score were detected in group 1, after returning to the normal diet (Fig. 1 B), while the mice that received daily T3 injections at the end of the cuprizone diet showed a gradual significant improvement of clinical scores during the w12 to w12 + 12 period (Fig. 1 B). The locomotor disabilities were assessed by performing the rotarod test twice a week over a 24 week interval (Fig. 1 B).
T2-weighted images
Acquired T2-weighted images (T2-transverse relaxation time constant) revealed dramatic effects of the cuprizone intoxication on mouse brains at w12 (Fig. 2 A,B). Corpora callosa showed a hypointense signal at w0, corresponding to a well myelinated structure (because T2 has short values in myelin) before cuprizone treatment (Fig. 2 A,B). This pattern was maintained in all examined ages of mice receiving normal diet (data not shown).
The first pathologic signs were clearly observed at w12 in T2-weighted images of mice brains receiving cuprizone diet (groups 1 and 2). CSF was observed as a hyperintense signal in the enlarged ventricular regions and infiltrating the surrounding structures (Fig. 2 A,B, w12). Simultaneously, the hypointense signal in T2-weighted images was lost in the corpus callosum after 12 weeks of cuprizone treatment, corresponding to the myelin breakdown.
Restoring a normal diet had no effect on the appearance of brain T2 images of group 1 (Fig. 2 A). The hyperintense signal, denoting major vasogenic edema and inflammation combined with an enlargement of the ventricles, was visible at all MRI time-points while corpus callosum and neighboring structures were not clearly identified.
In contrast, brain images of T3-treated mice of group 2, showed normal ventricle size as early as w12 + 3 and progressively, a hypointense signal was regained in the corpora callosa and cerebella (Fig. 2 B). However, a quantitative assessment of the myelination state from the T2-weighted images was not possible and further DT-MRI was performed.
DT-MRI and histopathological observation
DWI, as well as the FA maps generated after diffusion tensor computation provide good contrast for identifying the brain macrostructures and for choosing the regions of interest (ROIs) (Fig. 2 C). High FA values or high anisotropy are usually found in normal white matter tracts, whereas low FA values are generally measured in isotropic tissues where water diffuses equally in all directions (like in CSF or to a lesser degree in gray matter or abnormal white matter tracts).
Cuprizone intoxication has been demonstrated to differentially induce demyelination in the mouse brain structures, having very significant effects on the corpus callosum (Blakemore, 1984; Komoly, 2005) while other regions are less affected (Tansey et al., 1996). Therefore, two separate areas of corpus callosum, the splenium and the genu were selected for analyses, along with other regions (Fig. 2 C). DT-MRI and histological analyses showed that the consequence of the cuprizone ingestion was anatomically variable throughout the brain, with statistically significant effects quantified by DT-MRI only in the corpus callosum and the cerebellum. Therefore, these regions were selected for detailed data presentation.
Cuprizone intoxication effects
DT-MRI was performed on all animals before the onset of cuprizone treatment, corresponding to w0 time point, the fully myelinated state. FA maps showed high anisotropy of the white matter, especially in the corpus callosum, as well as in the cerebellar folia (Fig. 3 A,B, w0). The regions that showed the greatest anisotropy corresponded to the areas with strong staining intensity of myelin on histological sections (Fig. 4). Staining with an anti-myelin basic protein (MBP) antibody revealed well myelinated normal brains at 8 weeks of age (w0) (Fig. 4 A). At this age, many carbonic anhydase II (CA II)-positive oligodendrocytes were detected throughout the brains and predominantly in the white matter (Fig. 4 A). Few cells that expressed proliferation nuclear antigen (PCNA) and low numbers of BrdU+ cells were present in the corpus callosum. Limited number of PSA-NCAM+ cells was observed and these were restricted to the rostral migratory stream with no staining detected in the corpus callosum. A small number of oligodendrocyte precursors NG2+ cells were detected.
Figure 3.

A, B, Fractional anisotropy maps of representative individuals from experimental groups 1 (A) and 2 (B), followed longitudinally at different time points. Normal brain images at w0 show high anisotropic values in the well formed corpus callosum (white arrows). FA maps resulted from DT-MRI exams at w12 display very low values of this parameter all along the corpus callosum. No significant recovery of the FA is visible during the 12 weeks after the cuprizone removal in the brain images of mouse belonging to group 1 (A). Gradual increase in the FA is observed in the brain white matter of the mouse receiving T3 treatment after returning to normal diet (B, w12 + 3; w12 + 6; w12 + 12). The FA pattern in control mice is identical with that observed at w0 time point in group 1 and 2 and is not presented. FA values are distributed on a scale from 0 (blue) to 1 (red).
Figure 4.
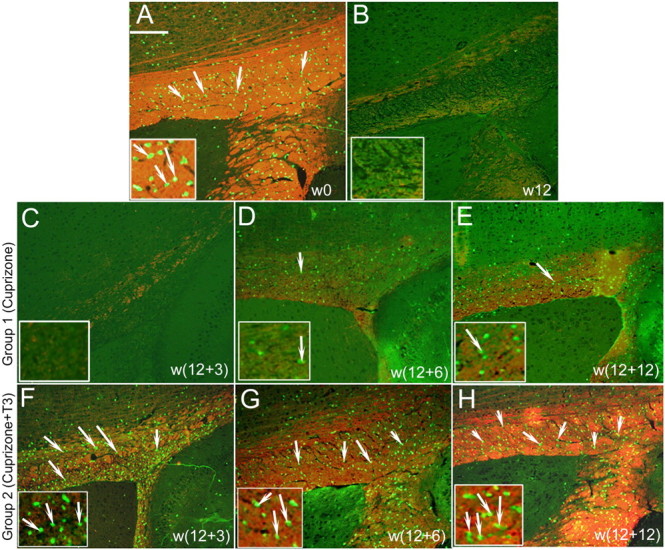
Representative images of double immunostaining (merged) of the mice corpora callosa with anti-MBP and anti-CA II antibodies. The anti-MBP antibody detects the myelin in red, while oligodendrocytes are green for anti-CA II antibody. Higher magnification of regions of corpus callosum is presented in the bottom of each image. A, At w0, before the cuprizone treatment, many green CA II+ oligodendrocytes (arrows) and MBP+ myelinated fibers were detected. The same staining patterns were obtained in the sections of control mice at each time point and are omitted from presentation. B, After 12 weeks of cuprizone insult (w12), very few oligodendrocytes and only scattered myelinated fibers were noticed. C–E, Failure of consistent remyelination is quantified after the cuprizone withdrawal in mice belonging to group 1 at w12 + 3 (C), w12 + 6 (D), w12 + 12 (E). F–H, Important progressive recovery of the myelin (MBP red staining) and repopulation by a great number of oligodendrocytes (CA II green staining) are evident in the corpus callosum of mice from group 2, at w12 + 3 (F), w12 + 6 (G), and w12 + 12 (H). Scale bar, 200 μm.
Electron micrographs showed ∼40 myelinated axons/100 μm2 in the corpus callosum, with a mean axonal diameter of 1.12 μm, in all the groups of mice at w0 (Fig. 5 I,J). No significant changes of DT-MRI and histopathology data were observed in control animals from w0 to w12 + 12.
Figure 5.
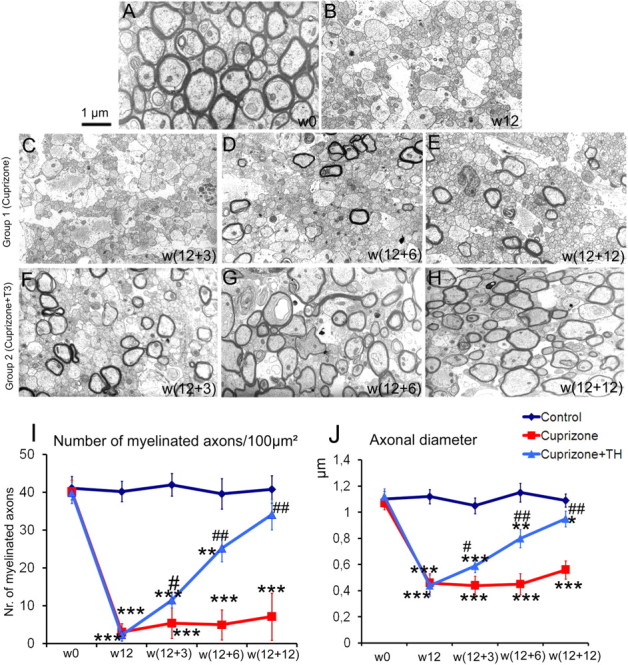
Representative electron micrographs of the mouse corpus callosum. A, At w0, before cuprizone treatment, well myelinated axons are observed. Similar electron micrographs were obtained from sections of control mice at each time point (data not shown). B, After 12 weeks of cuprizone insult (w12), most axons are demyelinated and have small caliber. Scale bar, 1 μm. C–E, Failure of consistent remyelination is quantified after cuprizone withdrawal in mice belonging to group 1 at w12 + 3 (C), w12 + 6 (D), w12 + 12 (E). F–H, Important progressive recovery in both, number of myelinated axons and myelin thickness is evident in the corpora callosa of mice from group 2 (that received T3 hormone), at w12 + 3 (F), w12 + 6 (G), and w12 + 12 (H). I, Number of myelinated axons/100 μm2 counted in semithin sections of the corpus callosum in animals from groups 1 and 2, at each experimental time point. J, Axonal caliber measurements performed on electron micrographs acquired from the corpus callosum region of mice from groups 1 and 2, at each experimental time point. * represents the statistical significance (*p < 0.05, **p < 0.01, ***p < 0.001) versus the control group, while # expresses the statistically significant differences (# p < 0.05, ## p < 0.001) among groups 1 (cuprizone treatment) and 2 (cuprizone plus T3 treatments).
The brain FA maps of all mice subjected to 12 weeks (w12) cuprizone diet (groups 1 and 2) showed abnormally decreased values in the white matter areas, and especially the corpus callosum (Fig. 3 A,B, w12). Cerebellar atrophy and reduced FA values were also observed. Significant increases of the radial diffusion values made this parameter the most efficient for quantifying the intoxication effect in the genu (increased by 53%) and splenium (increased by 50%) of corpus callosum at w12 (Fig. 6 A,C). The elevation of D⊥ was accompanied by a decrease in the axial diffusion values, but the reduction was not statistically significant (Fig. 6 B,D). This may reflect high SD values resulting from interindividual variations in response to cuprizone insult. Eigenvalues or diffusivities analyses in cerebellum showed increased diffusion coefficients by 34% in radial direction while the axial diffusivity did not change significantly (Fig. 6 E,F). Brain histopathological examination at w12 in animals from sibling groups showed a dramatic reduction by 90% (see Fig. 9 K) in oligodendrocyte number at w12 compared with w0 in the corpus callosum and a decrease by 50% in the cerebellum of cuprizone-treated mice. At w12 MBP staining was absent in corpus callosum of cuprizone-treated mice (Fig. 4 B), while cerebellar structures showed also only patches of MBP labels. Large numbers of CD45+ reactive microglial cells were seen in demyelinated white matter, while only very few NG2-positive cells were detected at w12. PSA-NCAM+ cells were absent in the corpus callosum and the subventricular zone at the end of cuprizone treatment. Limited numbers of PCNA+ and BrdU+ cells were detected in the subventricular area of these mice at w12 and these cells were not double stained for CA II.
Figure 6.

A–F, Time course of D⊥ (A, C, E) and D‖ (B, D, F) in the genu (A, B) and splenium (C, D) of corpus callosum and cerebellum (E, F) in all experimental groups. * represents the statistical significance (*p < 0.05, **p < 0.001) versus the control group, while # expresses the statistically significant differences (# p < 0.05, ## p < 0.001) among groups 1 (cuprizone treatment) and 2 (cuprizone +T3 treatments).
Figure 9.
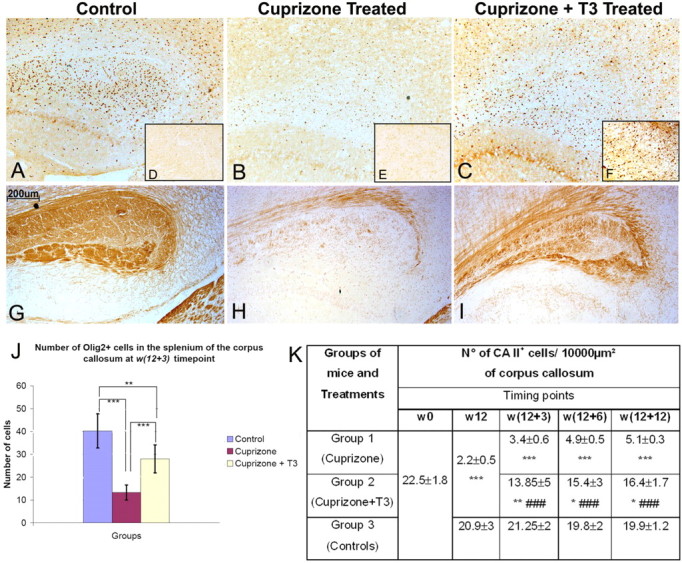
A–H, Olig2 (A–C), Shh (D–F), and MBP (G–I) immunoperoxidase staining in splenium of corpus callosum. In control mice (A) a significant number of Olig2+ cells are present while the number was dramatically reduced after cuprizone treatment (B). A massive reappearance of Olig2+ (C) and Shh+ cells (F) was observed under the influence of T3 treatment (C, F). Shh+ cells were almost absent in control mice (D) and a weak staining of limited number of Shh+ cells were observed in cuprizone-treated mice (E). The induction of Olig2+ and Shh+ cells was accompanied by a massive recovery in MBP immunostaining (I) after a dramatic reduction of MBP immunoreactivity in cuprizone-treated mice (H) compared with the control (G). Magnification ×250. The number of Olig2 cells in splenium of corpus callosum (J) was counted in 4–6 fields per tissue section photographed under microscope using an objective ×10 and two sections per animal. The number of CA II+ cells/10,000 μm2 counted in the corpus callosum (K). The CA II+ cells were counted using the Image J software on micrographs taken with the ×40 objective along the entire corpus callosum. Minimum of 5 slides/animal were analyzed (n = 5/group). * represents the statistical significance (*p < 0.05, **p < 0.01, ***p < 0.001) versus the control group, while # expresses the statistically significant differences (### p < 0.001) among groups 1 (cuprizone treatment) and 2 (cuprizone +T3 treatments).
The electron micrographs revealed a drastic decrease in the number of myelinated axons at w12, from 40 axons/100 μm2 (before treatment) to 2.3 myelinated axons/100 μm2 (Fig. 5 B,I). Furthermore, the mean value of the axonal diameter decreased from 1.12 μm at w0 to 0.44 μm at w12 (Fig. 5 B,J).
Failure of spontaneous recovery in long-term cuprizone-treated mice
After cessation of cuprizone treatment at w12, FA maps in the corpus callosum of the mice from the group 1 displayed constantly low values for as long as w12 + 12 time point (Fig. 3 A). These mice also showed persistent locomotor deficits, with minor improvements after 12 weeks following return to normal diet (Fig. 1 B). Analyses of the radial diffusion in the splenium of the corpus callosum showed only a weak decline of its values from w12 to w12 + 12 (Fig. 6 C). The axial diffusion remained under control values at all time-points but the differences were not significant. Similarly, only a small decrease in radial diffusion was noticed in the genu of the corpus callosum of mice from group 1 (Fig. 6 A). In this region, the axial diffusion regained normal values as early as at the w12 + 3 time point (Fig. 6 B). The low FA values observed at w12 in cerebella of cuprizone-treated mice remained unchanged during the w12 to w12 + 12 time interval (Fig. 3 A). Radial diffusion remained elevated in this structure even after 12 weeks of normal diet (Fig. 6 E).
Histopathological examination was performed at different time points following DT-MRI measurements. Slight and insignificant recovery in the MBP signal could be detected in the w12 to w12 + 12 time interval in mouse brains from group 1 (Fig. 4 C,D,E). Few oligodendrocytes positive for CA II repopulated the corpus callosum at w12 + 6 and w12 + 12 (Fig. 4 D,E). In the cerebellum, weak recovery of D⊥ at w12 + 12, coincided with a small increase in signal coming from MBP+ fibers and CA II+ cells. The number of PCNA (see Fig. 8 A)- and BrdU-stained cells remained very low during the w12 to w12 + 12 interval and no colocalization with oligodendroglial markers (CA II or NG2) was observed in the corpus callosum of mice from group 1 (Figs. 7 C, 8 A). The number of myelinated axons remained low; no statistically significant modifications could be observed during the w12 to w12 + 12 time period (Fig. 5 I). A small recovery in the axonal diameter was detected (from 0.44 at w12 to 0.56 at w12 + 12) but the increase was not statistically significant due to large SDs (Fig. 5 J).
Figure 8.

A–C, Double immunostaining (CA II and PCNA) of the corpus callosum of mice from group 1 (A), group 2 (B), and group 3 (C). Numerous cells double stained with PCNA and CA II markers were present in the corpus callosum of T3-treated mice (B). The double-labeled cells were absent from the corpus callosum of chronically demyelinated mice (A) or control mice (C). Magnification ×600.
Figure 7.
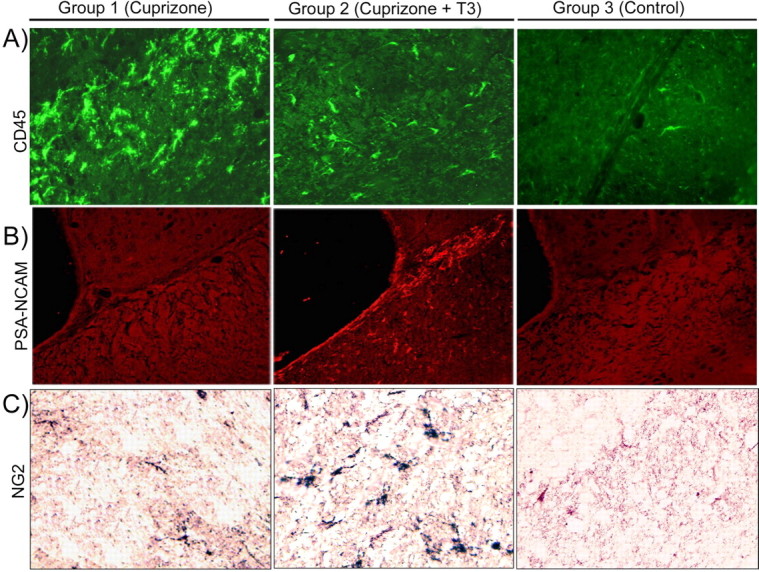
A–C, Brain section immunolabeling with anti-CD45 (A), anti-PSA-NCAM (B), and anti-NG2 (C) obtained at w12 + 3 in the corpus callosum of animals from groups 1, 2, and 3. We chose to present the data at w12 + 3 because they are the most representative for the comparison between mice receiving T3 treatment and remyelinating (group 2) and chronic demyelinated mice (group 1). Magnification ×400.
T3 treatment effects assessed by DT-MRI and histology
Injection of T3 hormone into mice of group 2 induced temporary hyperthyroidism, as attested by significantly high T3 level in the blood at w12 + 3 (data not shown). This hyperthyroidism was transient, since at w12 + 6 all mice had normal values of circulating T3 in the bloodstream. Administration of T3 however, influences the temporal evolution of DT-MRI derived parameter's values, calculated in the corpus callosum and cerebellum of cuprizone-treated mice. The abnormally low values displayed in the FA maps at w12 (Fig. 3 B) were progressively replaced by higher coefficients, reaching normal values in all investigated ROIs at w12 + 12 (Fig. 3 B). The most obvious period of recovery was the w12 to w12 + 6 interval in the genu and splenium of the corpus callosum while the cerebella of T3-treated mice displayed high FA in the maps by w12 + 3 (Fig. 3 B).
The radial diffusion measured from the splenium of the corpus callosum in the recovery phase showed a gradual decrease from w12 to reach normal values at w12 + 6 (Fig. 6 A), while the recovery was delayed in the genu of corpus callosum and reached normal values 6 weeks later at w12 + 12 (Fig. 6 B). The cerebellum showed a quick recovery in D⊥ values by w12 + 3 (Fig. 6 E). An estimate of the axial diffusion in the corpus callosum and cerebellum of T3-injected mice revealed normal values at each time point of the recovery phase (Fig. 6 B,D,F).
Histological examination at w12 + 3 of brain tissue of animals subjected to T3 treatment revealed a great number of CA II+ cells repopulating the corpus callosum and the cerebellum (Figs. 4, 9 K). The number of these cells increased further, at w12 + 6 (Fig. 4 G) and w12 + 12 (Fig. 4 H). At w12 + 3, MBP+ staining was already detected in both splenium and genu of the corpus callosum, and the staining intensity gradually increased with age during the recovery phase (Fig. 4 F,G,H). Reduced MBP labeling was seen in small areas along the corpus callosum reflecting irregular recovery of the oligodendrocyte population and myelin sheath formation.
The effect of T3 was not restricted to later stages of the oligodendrocyte lineage. Cuprizone intoxication resulted in significant reduction in the number of Olig2+ cells in the splenium of the corpus callosum that was reversed by T3 (Fig. 9 A–C,J). An increased number of PCNA+ and BrdU+ proliferating cells were detected in mouse brains of T3-treated mice at w12 + 3 (Fig. 8 B). Approximately 60% of the proliferating cells were also double stained for the CA II marker (Fig. 8 B). NG2-positive cells were also observed at the w12 + 3 and w12 + 6 (Fig. 7 C), as were a population of PSA-NCAM+ cells located all along the corpus callosum and in the subventricular zone of T3-treated mice (Fig. 7 B).
These increases in the number of early oligodendrocyte progenitor cells in T3-treated animals suggest this treatment modulates the expression of cues that influence induction or survival of early oligodendrocytes progenitors. A major regulator of early OPC development is the morphogen Shh (Orentas and Miller 1996; Davies and Miller, 2001) and in T3-treated animals increased expression of Shh was detectable in the subventricualr zone (data not shown) and particularly visible in the severely demyelinated areas as shown in splenium of corpus callosum (Fig. 9 D,E,F). The increase in Shh expression was correlated with a gradual increase in the number of Olig2+ cells (Fig. 9 A–C) after T3 treatment, predominantly observed in the corpus callosum (Fig. 9 J) but also in the subventricular zone (at w12 + 3: 39.4 Olig2+ cells/field of view in control group, 12.1 Olig2+ cells per field of view in cuprizone-treated group and 28.9 Olig2+ cells per field of view in cuprizone +T3-treated group) and cortex (at w12 + 3: 9.15 Olig2+ cells/field of view in control group, 4.14 Olig2+ cells/field of view in cuprizone-treated group and 11.1 Olig2+ cells/field of view in cuprizone + T3-treated group). The striatum region is much less affected. Importantly, the reappearance of Shh+ and Olig2+cells under the effect of T3 leads to the normalization of MBP expression (Fig. 9 G–I). These data suggest that T3 treatment provides an environment that is conducive for the subsequent development of OPCs as well as regulating their differentiation. In the corpus callosum of cuprizone-treated mice, T3 induces NG2+ cells (Fig. 10 B) and the majority of Olig2+ cells are CA II+ (Fig. 10 C,D) while the rest are astrocytes (data not shown).
Figure 10.

A, B, Immunofluorescence of NG2 revealed with Alexa Fluor 546-conjugated anti-rabbit IgG in cuprizone-treated mice (A) and cuprizone+T3-treated mice (B) at w12 + 3. C, D, Double labeling of Olig2 immunoperoxidase detected with VIP in cell nuclei (dark color) and CA II immunofluorescence in corpus callosum of cuprizone+T3-treated mouse (C, D). CA II+ cells and Olig2+ cell nuclei (arrows) are predominantly present in demyelinated area. Magnification: A–C, ×400; D, ×900.
The number of CD45+ microglial cells decreased gradually in T3-treated mice at w12 + 3 (Fig. 7 A) and throughout the treatment period, demonstrating an effect of T3 on the levels of reactive microglia.
Ultrastructural analyses revealed a rapid and sustained effect of the T3, with an increase in the number of myelinated axons during the recovery phase (Fig. 5 F,G,H) as well as by the progressive increase in the axonal caliber that approximated control values by w12 + 12 in the corpus callosum while the cerebellum showed earlier recovery.
Discussion
Remyelination ultimately fails in the brains of the majority of MS patients and many studies have tried to elucidate the causes and design therapies to stimulate and induce recovery in MS plaques (Franklin, 2002). Particularly, chronic lesions are unresponsive to various applied treatments in human (Rovaris et al., 2006) and animal models (Franklin and Blakemore, 1997). The present study provides in vivo evidence that thyroid hormone therapy (T3) is effective in promoting remyelination in a chronically demyelinated mouse model of MS. The study successfully exploited the sensitivity of the in vivo noninvasive DT-MRI technique for longitudinal following the efficacy of T3-based therapeutic effects in cuprizone-induced demyelination.
Assessment of cuprizone effects in brain
The cuprizone is a well known gliotoxic agent acting as a selective and sensitive copper chelator. Several papers reported that animals treated with various doses of cuprizone develop different forms of neuropathology, involving in most cases demyelination (Matsushima and Morell, 2001). The exact mechanisms of toxin action are not well understood. It is known, however, that cuprizone administration alters the functioning of the mitochondria in the brain oligodendrocytes (Venturini, 1973). Long-term 0.2% cuprizone feeding produces an interesting model of MS that encompasses the progression of acute demyelinated lesions to a chronic state (Mason et al., 2004) that fail to recover after toxin withdrawal. Both oligodendrocytes and progenitors become progressively depleted in chronically demyelinated lesions (Mason et al., 2004). It is interesting to note that long term cuprizone treatment in mice does not affect thyroid hormones (T3 and T4) and the TSH blood circulating levels which exclude the possibility that demyelination in adult mice is the result of TH deprivation. Consequently, cuprizone demyelinated mice serve as excellent models for the development of in vivo noninvasive imaging methods, capable of diagnosis and following the course of demyelinating disease over time (Yu et al., 2004; Merkler et al., 2005; Sun et al., 2006). DT-MRI is one of the most powerful and sensitive techniques that proved its sensitivity in the case of myelin disorders in human (Ciccarelli et al., 2001; Agosta et al., 2005) and animal models (Song et al., 2005; Harsan et al., 2006, 2007; Deboy et al., 2007). The method has particular importance because it offers special insight in white matter pathology, providing specific information about the condition of myelin or axons. Analyses of radial diffusion calculated from the diffusion tensor shows correlations between increased values of this parameter and myelin abnormalities. In cuprizone-treated mice, the extent of demyelination was quantified by DT-MRI in several brain regions, and correlated with histological examination. Twelve weeks of intoxication with cuprizone resulted in greatly increased values of water diffusion radial to the callosal axons and slightly decreased values of the axial diffusion. Degeneration of hydrophobic layers of myelin, as confirmed by a lack of MBP labeling of brain sections as well as the loss of oligodendrocytes provides a free environment for water movement perpendicular to the tracts. Slight decreases of the axial diffusivity (p = 0.059) in both splenium and genu of the corpus callosum at w12 might express axonal damage that is often observed in MS demyelinated lesions and believed to be one of the reasons for recovery failure (Trapp et al., 1999). We previously demonstrated in a transgenic mouse model (Harsan et al. 2006) that the axonal morphology influences the axial diffusion values. Cuprizone-treated mice show also axonal modifications that could be related to the observed small modifications in the axial diffusivity. Injury at the axonal level in cuprizone model has been proposed previously (Stidworthy et al., 2003; Sun et al., 2006). The reported axonal damage by Sun and coworkers (Sun et al., 2006) in the corpus callosum of male mice, treated with cuprizone for 12 weeks, was not accompanied by modifications of the axial diffusion at the end of cuprizone treatment. The minor differences observed in our study may arise from the fact that we used female mice. Consistent with this hypothesis recent studies suggested that sex hormones have protective effects in male brains, whereas female white matter is more susceptible to cell death (Cerghet et al., 2006).
The fractional anisotropy values are reduced in both splenium and genu of the corpus callosum, as well as in the cerebellum, mostly as a consequence of the great increase of the minor eigenvalues of the diffusion tensor (thus radial diffusivity), and correlate with the myelin loss as demonstrated by a lack of MBP reactivity.
The effects of 12 weeks of cuprizone insult are irreversible when no therapy is applied, and this is reflected in constantly high values of D⊥ in the corpus callosum and cerebella of demyelinated mice, long after cuprizone insult withdrawal. This is consistent with a permanent myelin and oligodendrocyte deficit as shown by a lack or weak expression of myelin and oligodendrocytic antigens.
Remyelination induced by T3-based therapy
Effective therapies in demyelination are likely to incorporate factors that play a role in normal myelination development. Thyroid hormones regulate brain development as attested by severe morphological, functional and cognitive impairments monitored in congenitally hypothyroid infants (Oppenheimer and Schwartz 1997). Particularly, they ensure normal oligodendrocyte maturation and myelination (McIntosh et al., 1981; Baas et al., 1997; Rodríguez-Peña 1999; Bury et al., 2002). There are two circulating thyroid hormones: thyroxin (T4) and T3, and their action is transduced by thyroid hormone receptors (TRs), expressed in different cell types, including precursors and mature oligodendrocytes. The effects of T4 hormone on remyelination were recently reported in an EAE rat model of immuno-inflammatory demyelination in which a severe oligodendrocyte population depletion is not clear (Calza et al., 2002; Fernandez et al., 2004; Calzà et al., 2005). Because the T4 will be further converted in T3 in the organism, and because the TRs affinity is much higher for T3, we chose to use directly T3 hormone injections as stimulating therapy leading to remyelination in chronically demyelinated mouse brain lesions. Despite that the T3 level increases several fold over physiological value immediately after T3 administration into mice, it becomes normal after 2–3 h.
Moreover, we quantified in vivo by DT-MRI the magnitude of remyelination at several time points in the recovery phase. The time course of radial diffusion in the splenium and genu of the corpus callosum correlates exactly with remyelination as assessed by MBP expression. The radial diffusion as well as the fractional anisotropy values normalized earlier in T3 treatment in the splenium (w12 + 6) than in the genu (w12 + 12) of the corpus callosum suggesting local regulation of the remyelination process. This pattern of myelination recapitulates the pattern of normal myelination in the mouse brain, implying that the regulatory mechanisms that control the timing of myelination are retained in the adult CNS.
Whereas the diffusion perpendicular to the tracts takes longer time to regain control values, the axial diffusion matches the normal values in both areas of the corpus callosum during early phases of recovery, suggesting a rapid recovery from acute axonal damage. Generally, acute axonal damage in MS is associated with areas of inflammation, and the hyperintensities observed in T2-weighted images at w12 in cuprizone-treated mice may represent important inflammatory processes or vasogenic edema as a result of breakdown of the blood brain barrier (BBB). Consistent with an anti-inflammatory effect of T3 is the observation that edema is quickly diminished during T3 treatment, while it persists in the brains of mice that were not subject to therapy. In cerebellum, the consequences of the T3 treatment occurred very rapidly, during the w12 to w12 + 3 period, thus paralleling the T3 injections. Such that, all DT-MRI parameters values were fully recovered at the w12 + 3 time point. This may reflect in part that the cerebellum is less affected during cuprizone treatment than the corpus callosum.
In cuprizone induced chronic demyelination, treatment with T3 appears to act to promote the appearance of OPCs as well as regulate their differentiation. This hypothesis is supported by several observations. First, there is a local increase in the expression of Shh (Orentas and Miller, 1996; Davies and Miller, 2001), a known inducer of OPCs. Second, three weeks after initiation of T3 treatment a substantial number of CA II+ oligodendrocytes repopulated the corpus callosum and cerebellum and the number of Olig2+-CA II+ cells was increased over nontreated controls. Third, there was an increase in cell proliferation in T3-treated animals and 60% of PCNA+ proliferating cells were double stained for CA II. Together these studies suggest that T3 promotes early stages of oligodendrocyte development. This hypothesis is consistent with the observation that thyroid hormone may act on neural stem cells, favoring the generation of more oligodendrocytes (Rogister et al., 1999).
Oligodendrocyte precursors are highly motile and the stream of PSA-NCAM+ migrating cells observed in the brains of recovering animals suggests also an impact of T3 on cell migration from the germinative subventricular zone, while the rapid remyelination induced by T3 in the current model and the upregulation of the expression of MBP (Calza et al., 2002), suggest that T3 promotes OPC differentiation or formation of the myelin sheath by oligodendrocytes. Finally, the reduction in microglial activation suggests that T3 treatment reduces inflammatory responses either directly or indirectly and that all these factors combine to promote repair in conditions of chronic demyelination. The current studies clearly suggest that T3 treatment induces formation of new myelin sheaths, which constitute new barriers for water diffusion in the brain. This is sensitively monitored by DT-MRI that provides means to estimate in vivo the extent of the recovery. These results raise the possibility of future applications of therapy for MS based not only on T3, which may have undesirable off target effects, but with future brain specific thyromimetics or other related molecules inducers of the endogenous potential of adult brain remyelination.
Footnotes
This work was supported by grants from the Foundation of European Leukodystrophies Association to M.S.G. and National Institutes of Health Grant NS 30800 to R.H.M. We thank M. Bury and D. Lam for their valuable technical assistance. Guinea pig anti-mouse TSH antibody and TSH reference preparation used in the TSH assay were obtained from Dr. A. F. Parlow, National Hormone and Peptide Program, Harbor–University of California, Los Angeles Medical Center, Los Angeles, CA.
References
- Agosta F, Benedetti B, Rocca MA, Valsasina P, Rovaris M, Comi G, Filippi M. Quantification of cervical cord pathology in primary progressive MS using diffusion tensor MRI. Neurology. 2005;64:631–635. doi: 10.1212/01.WNL.0000151852.15294.CB. [DOI] [PubMed] [Google Scholar]
- Baas D, Bourbeau D, Sarliève LL, Ittel ME, Dussault JH, Puymirat J. Oligodendrocyte maturation and progenitor cell proliferation are independently regulated by thyroid hormone. Glia. 1997;19:324–332. doi: 10.1002/(sici)1098-1136(199704)19:4<324::aid-glia5>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Basser PJ, Mattiello J, LeBihan D. Estimation of the effective self-diffusion tensor from the NMR spin echo. J Magn Reson B. 1994;103:247–254. doi: 10.1006/jmrb.1994.1037. [DOI] [PubMed] [Google Scholar]
- Billon N, Tokumoto Y, Forrest D, Raff M. Role of thyroid hormone receptors in timing oligodendrocyte differentiation. Dev Biol. 2001;235:110–120. doi: 10.1006/dbio.2001.0293. [DOI] [PubMed] [Google Scholar]
- Blakemore WF. The response of oligodendrocytes to chemical injury. Acta Neurol Scand Suppl. 1984;100:33–38. [PubMed] [Google Scholar]
- Bury F, Carré JL, Vega S, Ghandour MS, Rodriguez-Peña A, Langley K, Sarliève LL. Coexpression of thyroid hormone receptor isoforms in mouse oligodendrocytes. J Neurosci Res. 2002;67:106–113. doi: 10.1002/jnr.10111. [DOI] [PubMed] [Google Scholar]
- Calza L, Fernandez M, Giuliani A, Aloe L, Giardino L. Thyroid hormone activates oligodendrocyte precursors and increases a myelin-forming protein and NGF content in the spinal cord during experimental allergic encephalomyelitis. Proc Natl Acad Sci U S A. 2002;99:3258–3263. doi: 10.1073/pnas.052704499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calzà L, Fernandez M, Giuliani A, D'Intino G, Pirondi S, Sivilia S, Paradisi M, Desordi N, Giardino L. Thyroid hormone and remyelination in adult central nervous system: a lesson from an inflammatory-demyelinating disease. Brain Res Brain Res Rev. 2005;48:339–346. doi: 10.1016/j.brainresrev.2004.12.022. [DOI] [PubMed] [Google Scholar]
- Cerghet M, Skoff RP, Bessert D, Zhang Z, Mullins C, Ghandour MS. Proliferation and death of oligodendrocytes and myelin proteins are differentially regulated in male and female rodents. J Neurosci. 2006;26:1439–1447. doi: 10.1523/JNEUROSCI.2219-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccarelli O, Werring DJ, Wheeler-Kingshott CA, Barker GJ, Parker GJ, Thompson AJ, Miller DH. Investigation of MS normal-appearing brain using diffusion tensor MRI with clinical correlations. Neurology. 2001;56:926–933. doi: 10.1212/wnl.56.7.926. [DOI] [PubMed] [Google Scholar]
- Davies JE, Miller RH. Local sonic hedgehog signaling regulates oligodendrocyte precursor appearance in multiple ventricular domains in the chick metencephalon. Dev Biol. 2001;233:513–525. doi: 10.1006/dbio.2001.0224. [DOI] [PubMed] [Google Scholar]
- DeBoy CA, Zhang J, Dike S, Shats I, Jones M, Reich DS, Mori S, Nguyen T, Rothstein B, Miller RH, Griffin JT, Kerr DA, Calabresi PA. High resolution diffusion tensor imaging of axonal damage in focal inflammatory and demyelinating lesions in rat spinal cord. Brain. 2007;130:2199–2210. doi: 10.1093/brain/awm122. [DOI] [PubMed] [Google Scholar]
- Fernandez M, Giuliani A, Pirondi S, D'Intino G, Giardino L, Aloe L, Levi-Montalcini R, Calzà L. Thyroid hormone administration enhances remyelination in chronic demyelinating inflammatory disease. Proc Natl Acad Sci U S A. 2004;101:16363–16368. doi: 10.1073/pnas.0407262101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin RJ. Why does remyelination fail in multiple sclerosis? Nat Rev Neurosci. 2002;3:705–714. doi: 10.1038/nrn917. [DOI] [PubMed] [Google Scholar]
- Franklin RJ, Blakemore WF. To what extent is oligodendrocyte progenitor migration a limiting factor in the remyelination of multiple sclerosis lesions? Mult Scler. 1997;3:84–87. doi: 10.1177/135245859700300205. [DOI] [PubMed] [Google Scholar]
- Harsan L, Jalabi W, Grucker D, Ghandour MS. New insights on neuronal alterations in jimpy mutant brain. Neurochem Res. 2004;29:943–952. doi: 10.1023/b:nere.0000021238.00299.93. [DOI] [PubMed] [Google Scholar]
- Harsan LA, Poulet P, Guignard B, Steibel J, Parizel N, de Sousa PL, Boehm N, Grucker D, Ghandour MS. Brain dysmyelination and recovery assessment by noninvasive in vivo diffusion tensor magnetic resonance imaging. J Neurosci Res. 2006;83:392–402. doi: 10.1002/jnr.20742. [DOI] [PubMed] [Google Scholar]
- Harsan LA, Poulet P, Guignard B, Parizel N, Skoff RP, Ghandour MS. Astrocytic hypertrophy in dysmyelination influences the diffusion anisotropy of white matter. J Neurosci Res. 2007;85:935–944. doi: 10.1002/jnr.21201. [DOI] [PubMed] [Google Scholar]
- Jalabi W, Boehm N, Grucker D, Ghandour MS. Recovery of myelin after induction of oligodendrocyte cell death in postnatal brain. J Neurosci. 2005;25:2885–2894. doi: 10.1523/JNEUROSCI.2748-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SA, Jolson DM, Cuta KK, Mariash CN, Anderson GW. Triiodothyronine is a survival factor for developing oligodendrocytes. Mol Cell Endocrinol. 2003;199:49–60. doi: 10.1016/s0303-7207(02)00296-4. [DOI] [PubMed] [Google Scholar]
- Kang SM, Cho MS, Seo H, Yoon CJ, Oh SK, Choi YM, Kim DW. Efficient induction of oligodendrocytes from human embryonic stem cells. Stem Cells. 2007;25:419–424. doi: 10.1634/stemcells.2005-0482. [DOI] [PubMed] [Google Scholar]
- Komoly S. Experimental demyelination caused by primary oligodendrocyte dystrophy. Regional distribution of the lesions in the nervous system of mice. Ideggyogy Sz. 2005;58:40–43. [PubMed] [Google Scholar]
- Kornek B, Lassmann H. Neuropathology of multiple sclerosis-new concepts. Brain Res Bull. 2003;61:321–326. doi: 10.1016/s0361-9230(03)00095-9. [DOI] [PubMed] [Google Scholar]
- Le Bihan D. Looking into the functional architecture of the brain with diffusion MRI. Nat Rev Neurosci. 2003;4:469–480. doi: 10.1038/nrn1119. [DOI] [PubMed] [Google Scholar]
- Le Bihan D, Breton E, Lallemand D, Grenier P, Cabanis E, Laval-Jeantet M. MR imaging of intravoxel incoherent motions: application to diffusion and perfusion in neurologic disorders. Radiology. 1986;161:401–407. doi: 10.1148/radiology.161.2.3763909. [DOI] [PubMed] [Google Scholar]
- Ludwin SK. Central nervous system demyelination and remyelination in the mouse: an ultrastructural study of cuprizone toxicity. Lab Invest. 1978;39:597–612. [PubMed] [Google Scholar]
- Mason JL, Toews A, Hostettler JD, Morell P, Suzuki K, Goldman JE, Matsushima GK. Oligodendrocytes and progenitors become progressively depleted within chronically demyelinated lesions. Am J Pathol. 2004;164:1673–1682. doi: 10.1016/S0002-9440(10)63726-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushima GK, Morell P. The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain Pathol. 2001;11:107–116. doi: 10.1111/j.1750-3639.2001.tb00385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh GH, Howard DA, Mano MT, Wellby ML, Hetzel BS. Iodine deficiency and brain development in the rat. Aust J Biol Sci. 1981;34:427–433. doi: 10.1071/bi9810427. [DOI] [PubMed] [Google Scholar]
- Merkler D, Boretius S, Stadelmann C, Ernsting T, Michaelis T, Frahm J, Brück W. Multicontrast MRI of remyelination in the central nervous system. NMR Biomed. 2005;18:395–403. doi: 10.1002/nbm.972. [DOI] [PubMed] [Google Scholar]
- Mori S, Zhang J. Principles of diffusion tensor imaging and its applications to basic neuroscience research. Neuron. 2006;51:527–539. doi: 10.1016/j.neuron.2006.08.012. [DOI] [PubMed] [Google Scholar]
- Oppenheimer JH, Schwartz HL. Molecular basis of thyroid hormone-dependent brain development. Endocr Rev. 1997;18:462–475. doi: 10.1210/edrv.18.4.0309. [DOI] [PubMed] [Google Scholar]
- Orentas DM, Miller RH. The origin of spinal cord oligodendrocytes is dependent on local influences from the notochord. Dev Biol. 1996;177:43–53. doi: 10.1006/dbio.1996.0143. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. Ed 2. New York: Academic; 2001. [Google Scholar]
- Pluchino S, Furlan R, Martino G. Cell-based remyelinating therapies in multiple sclerosis: evidence from experimental studies. Curr Opin Neurol. 2004;17:247–255. doi: 10.1097/00019052-200406000-00003. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Peña A. Oligodendrocyte development and thyroid hormone. J Neurobiol. 1999;40:497–512. doi: 10.1002/(sici)1097-4695(19990915)40:4<497::aid-neu7>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Rogister B, Ben-Hur T, Dubois-Dalcq M. From neural stem cells to myelinating oligodendrocytes. Mol Cell Neurosci. 1999;14:287–300. doi: 10.1006/mcne.1999.0790. [DOI] [PubMed] [Google Scholar]
- Rovaris M, Confavreux C, Furlan R, Kappos L, Comi G, Filippi M. Secondary progressive multiple sclerosis: current knowledge and future challenges. Lancet Neurol. 2006;5:343–354. doi: 10.1016/S1474-4422(06)70410-0. [DOI] [PubMed] [Google Scholar]
- Sarliève LL, Rodríguez-Peña A, Langley K. Expression of thyroid hormone receptor isoforms in the oligodendrocyte lineage. Neurochem Res. 2004;29:903–922. doi: 10.1023/b:nere.0000021235.83952.9a. [DOI] [PubMed] [Google Scholar]
- Schoonover CM, Seibel MM, Jolson DM, Stack MJ, Rahman RJ, Jones SA, Mariash CN, Anderson GW. Thyroid hormone regulates oligodendrocyte accumulation in developing rat brain white matter tracts. Endocrinology. 2004;145:5013–5020. doi: 10.1210/en.2004-0065. [DOI] [PubMed] [Google Scholar]
- Song SK, Sun SW, Ju WK, Lin SJ, Cross AH, Neufeld AH. Diffusion tensor imaging detects and differentiates axon and myelin degeneration in mouse optic nerve after retinal ischemia. Neuroimage. 2003;20:1714–1722. doi: 10.1016/j.neuroimage.2003.07.005. [DOI] [PubMed] [Google Scholar]
- Song SK, Yoshino J, Le TQ, Lin SJ, Sun SW, Cross AH, Armstrong RC. Demyelination increases radial diffusivity in corpus callosum of mouse brain. Neuroimage. 2005;26:132–140. doi: 10.1016/j.neuroimage.2005.01.028. [DOI] [PubMed] [Google Scholar]
- Stidworthy MF, Genoud S, Suter U, Mantei N, Franklin RJ. Quantifying the early stages of remyelination following cuprizone-induced demyelination. Brain Pathol. 2003;13:329–339. doi: 10.1111/j.1750-3639.2003.tb00032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streckfuss F, Hamann I, Schomburg L, Michaelis M, Sapin R, Klein MO, Köhrle J, Schweizer U. Hepatic deiodinase activity is dispensable for the maintenance of normal circulating thyroid hormone levels in mice. Biochem Biophys Res Commun. 2005;337:739–745. doi: 10.1016/j.bbrc.2005.09.102. [DOI] [PubMed] [Google Scholar]
- Sun SW, Liang HF, Trinkaus K, Cross AH, Armstrong RC, Song SK. Noninvasive detection of cuprizone induced axonal damage and demyelination in the mouse corpus callosum. Magn Reson Med. 2006;55:302–308. doi: 10.1002/mrm.20774. [DOI] [PubMed] [Google Scholar]
- Tansey FA, Zhang H, Cammer W. Expression of carbonic anhydrase II mRNA and protein in oligodendrocytes during toxic demyelination in the young adult mouse. Neurochem Res. 1996;21:411–416. doi: 10.1007/BF02527704. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Ransohoff R, Rudick R. Axonal pathology in multiple sclerosis: relationship to neurologic disability. Curr Opin Neurol. 1999;12:295–302. doi: 10.1097/00019052-199906000-00008. [DOI] [PubMed] [Google Scholar]
- Venturini G. Enzymic activities and sodium, potassium and copper concentrations in mouse brain and liver after cuprizone treatment in vivo. J Neurochem. 1973;21:1147–1151. doi: 10.1111/j.1471-4159.1973.tb07569.x. [DOI] [PubMed] [Google Scholar]
- Yu O, Steibel J, Mauss Y, Guignard B, Eclancher B, Chambron J, Grucker D. Remyelination assessment by MRI texture analysis in a cuprizone mouse model. Magn Reson Imaging. 2004;22:1139–1144. doi: 10.1016/j.mri.2004.08.017. [DOI] [PubMed] [Google Scholar]