

Abstract
Mitochondrial dysfunction and oxidative stress contribute to motor neuron degeneration in amyotrophic lateral sclerosis (ALS). Recent reports indicate that astrocytes expressing the mutations of superoxide dismutase-1 (SOD1) may contribute to motor neuron injury in ALS. Here, we provide evidence that mitochondrial dysfunction in SOD1G93A rat astrocytes causes astrocytes to induce apoptosis of motor neurons. Mitochondria from SOD1G93A rat astrocytes displayed a defective respiratory function, including decreased oxygen consumption, lack of ADP-dependent respiratory control, and decreased membrane potential. Protein 3-nitrotyrosine was detected immunochemically in mitochondrial proteins from SOD1G93A astrocytes, suggesting that mitochondrial defects were associated with nitroxidative damage. Furthermore, superoxide radical formation in mitochondria was increased in SOD1G93A astrocytes. Similar defects were found in mitochondria isolated from the spinal cord of SOD1G93A rats, and pretreatment of animals with the spin trap 5,5-dimethyl-1-pyrroline N-oxide restored mitochondrial function, forming adducts with mitochondrial proteins in vivo. As shown previously, SOD1G93A astrocytes induced death of motor neurons in cocultures, compared with nontransgenic ones. This behavior was recapitulated when nontransgenic astrocytes were treated with mitochondrial inhibitors. Remarkably, motor neuron loss was prevented by preincubation of SOD1G93A astrocytes with antioxidants and nitric oxide synthase inhibitors. In particular, low concentrations (∼10 nm) of two mitochondrial-targeted antioxidants, ubiquinone and carboxy-proxyl nitroxide, each covalently coupled to a triphenylphosphonium cation (Mito-Q and Mito-CP, respectively), prevented mitochondrial dysfunction, reduced superoxide production in SOD1G93A astrocytes, and restored motor neuron survival. Together, our results indicate that mitochondrial dysfunction in astrocytes critically influences motor neuron survival and support the potential pharmacological utility of mitochondrial-targeted antioxidants in ALS treatment.
Keywords: mitochondria, ALS, astrocytes, SOD1, free radicals, antioxidants
Introduction
Mitochondrial dysfunction and oxidative stress have been involved in amyotrophic lateral sclerosis (ALS) pathogenesis [for review, see Dupuis et al. (2004), Bruijn et al. (2004), and Manfredi and Xu (2005)]. Previous reports described decreased mitochondrial respiratory activity in spinal cords from transgenic (Tg) superoxide dismutase-1 (SOD1)G93A mice (Jung et al., 2002; Mattiazzi et al., 2002) and in cellular models of the disease (Menzies et al., 2002). However, it is unknown whether mitochondrial dysfunction in ALS affects non-neuronal cells in addition to motor neurons. In mutant SOD1 chimeric mice, toxicity to motor neurons requires damage from mutant SOD1 acting within non-neuronal cells (Clement et al., 2003). Most importantly, non-neuronal cells that do not express mutant SOD1 extend survival of mutant-expressing motor neurons, supporting a role for glia in ALS pathology and raising the concept of the so-called nonautonomous nature of motor neuron death in ALS (Clement et al., 2003).
Astrocytic activation occurs in both human and animal models of ALS (Barbeito et al., 2004; Bruijn et al., 2004). We have shown that astrocytes may contribute to motor neuron loss. Reactive astrocytes upregulate nitric oxide (·NO) production and NGF (nerve growth factor) expression, which induces motor neuron death through p75 receptor signaling (Cassina et al., 2002, 2005; Pehar et al., 2004). These effects can be prevented by increasing antioxidant defenses in astrocytes either by activation of Nrf2 transcription factor or antioxidant treatment (Vargas et al., 2005). Remarkably, astrocytes expressing SOD1 mutations become neurotoxic for motor neurons (Vargas et al., 2006; Nagai et al., 2007), suggesting a common link between SOD1 mutations, oxidative stress, and astrocyte-induced neuronal death.
Herein, we provide experimental evidence that mitochondria in SOD1G93A-expressing astrocytes display severe impairment of oxygen consumption and ADP-dependent respiratory control, which is associated with enhanced mitochondrial superoxide formation and local oxidative and nitrative damage [referred to herein as nitroxidative stress; for review, see Peluffo and Radi (2007)]. In addition, mitochondrial dysfunction results in decreased ability to sustain motor neuron survival and can be prevented by mitochondrial-targeted antioxidants. Together, our results suggest that mitochondrial activity in astrocytes greatly influences their subsequent ability to regulate motor neuron survival.
Materials and Methods
Materials.
JC-1, 3,8-phenanthridine diamine-hexyl triphenylphosphonium iodide (MitoSOX Red), and MitoTracker Deep Red were provided by Invitrogen (Carlsbad, CA). Culture media and serum were from Invitrogen. 5,5-Dimethyl-1-pyrroline N-oxide (DMPO) was from Alexis Biochemicals (San Diego, CA) and further purified by double distillation. Antibodies to DMPO–protein adducts and protein 3-nitrotyrosine were raised as previously described (Cassina et al., 2000; Mason, 2004). The manganese porphyrin manganese(III) meso-tetrakis[(N-ethyl)pyridinium-2-yl]porphyrin (MnTE-2-PyP), mito-ubiquinone (Mito-Q; a mixture of mitoquinol [10-(3,6-dihydroxy-4,5-dimethoxy-2-methylphenyl) decyl] triphenylphosphonium bromide and mitoquinone [10-(4,5-dimethoxy-2-methyl-3,6-dioxo-1,4-cyclohexadien-1-yl)decyl] triphenylphosphonium bromide) and mito-carboxy-proxyl nitroxide (Mito-CP) were kindly provided by Dr. Ines Batinic-Haberle (Duke University Medical Center, Durham, NC) (Batinic-Haberle et al., 1998), Dr. Michael Murphy (Medical Research Council, Dunn Human Nutrition Unit, Cambridge, UK) (Kelso et al., 2001), and Dr. Balaranan Kalyanaraman (Medical College of Wisconsin, Milwaukee, WI) (Jauslin et al., 2003), respectively. All other reagents were from Sigma (St. Louis, MO).
Animals.
Procedures using laboratory animals were in accordance with the international guidelines for the use of live animals and were approved by the Institutional Animal Committee. In this study, Sprague Dawley SOD1WT transgenic rats, which overexpressed the wild-type form of SOD1, were obtained from Dr. Pak Chan (Stanford University, Palo Alto, CA) and remain healthy for at least 2 years (Chan et al., 1998). Sprague Dawley SOD1G93A L26H rats, which develop motor neuron disease between 104 and 121 d of age, were kindly provided by Dr. David S. Howland (Wyeth Research, Princeton, NJ) (Howland et al., 2002). Onset typically appeared as hindlimb abnormal gait affecting one limb first (considered as early symptomatic) and progress very quickly (1–2 d) to overt hindlimb paralysis. Within 1–2 d, the second hindlimb is involved, although animals could still ambulate through the use of forepaws. Affected rats typically reached end-stage disease very quickly, in an average of 11 d after onset of symptoms.
Cell culture.
Primary transgenic SOD1G93A, SOD1WT, and nontransgenic astrocytes cultures were prepared either from spinal cords or brain of 1- to 2-d-old rat pups according to the procedures of Saneto and De Vellis (1987) with minor modifications (Cassina et al., 2002). The neonates were previously genotyped by PCR, and neonates bearing the SOD1 mutation were used for preparing SOD1G93A-bearing astrocyte cultures and the nontransgenic littermates as control cultures. Astrocytes were plated at a density of 2 × 104 cells/cm2 and maintained in DMEM supplemented with 10% fetal bovine serum, HEPES (3.6 g/L), penicillin (100 IU/ml), and streptomycin (100 μg/ml). Astrocyte monolayers were >98% pure as determined by GFAP immunoreactivity and devoid of OX42-positive microglial cells. Motor neurons were obtained from embryonic day 15 (E15) rat spinal cord by a combination of metrizamide-gradient centrifugation and immunopanning with the monoclonal antibody IgG192 against p75NTR (Henderson et al., 1993). For coculture experiments, only wild-type motor neurons were plated on rat astrocyte monolayers at a density of 300 cells/cm2 and maintained for 48 h in L15 medium supplemented with 0.63 mg/ml bicarbonate, 5 μg/ml insulin, 0.1 mg/ml conalbumin, 0.1 mm putrescine, 30 nm sodium selenite, 20 nm progesterone, 20 mm glucose, 100 IU/ml penicillin, 100 μg/ml streptomycin, and 2% horse serum, fixed with paraformaldehyde at 4% plus 0.1% glutaraldehyde in PBS at 4°C for 15 min, and then processed for immunocytochemistry as described previously (Cassina et al., 2002).
Cell treatment.
Confluent astrocyte monolayers were transferred to supplemented L15 media prior to treatment. Astrocyte monolayers were incubated with rotenone (2 μm), antimycin A (5 μm), sodium azide (5 μm), fluorocitrate (250–100 μm), DMPO (75 mm), MnTE-2-PyP (10 μm), Mito-Q (10–100 nm), or Mito-CP (10–100 nm) for 24 h before mitochondrial purification or motor neuron plating after washing twice with PBS supplemented with (in mm) 0.8 MgCl2, 1 CaCl2, and 5 glucose.
Mitochondrial preparation.
Intact spinal cord or astrocyte culture mitochondria were prepared by differential centrifugation as described previously (Cassina and Radi, 1996). Briefly, rats were anesthetized, and the spinal cord or cerebral cortex was removed and washed extensively, minced, and homogenized with a small tissue grinder. Tissue fragments were disrupted using a Potter-Elvehjem homogenizer in homogenization buffer containing 0.3 m sucrose, 5 mm morpholinepropanesulfonic acid, 5 mm potassium phosphate, 1 mm EGTA, and 0.1% bovine serum albumin (BSA). Confluent astrocyte monolayers were cell scraped in respiration buffer, homogenized, and centrifuged at 1500 × g, and mitochondria were isolated from the supernatant by centrifugation at 13,000 × g. Mitochondrial pellets were resuspended in minimal volume of homogenization buffer.
Oxygen consumption studies.
Mitochondrial respiration was measured polarographically using a Cole-Parmer (Vernon Hills, IL) oximeter fitted with a water-jacketed Clark-type electrode (model 5300; YSI, Yellow Springs, OH) in an incubation chamber of 1.6 ml capacity as described previously (Cassina and Radi, 1996). Briefly, oxygen consumption studies were performed in homogenization buffer at 37°C, pH 7.4, at 0.2–0.5 mg/ml mitochondria. Glutamate–malate (2.5 mm each) and succinate (5 mm) were used to quantify complex I- and II-dependent respiration, respectively. Rotenone (2 μm) and antimycin A (AA; 2 μm) were used as complex I and III inhibitors, respectively. For each complex respiration, we also tested the addition of the ATPase inhibitor oligomycin (5 μm) and the mitochondrial uncoupler carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). ADP was used to evaluate mitochondrial coupling. The respiratory control ratio (RCR) was calculated as the ratio of oxygen consumption rates after (state 3) and before (state 4) ADP addition. The purity of the mitochondrial fraction was confirmed by measuring cytochrome c oxidase and lactate dehydrogenase activities as markers of mitochondria and cytosol, respectively (Radi et al., 1991).
Motor neuron counting.
Motor neurons on the astrocyte monolayer were identified by p75 immunocytochemistry as previously described (Cassina et al., 2002). Motor neuron survival was assessed by counting all p75-positive cells displaying neurites longer than four cell diameters in an area of 0.90 cm2 along a diagonal in 24-well plates. The mean density of motor neurons in control cocultures was 88 ± 5 cells/cm2.
Determination of superoxide generation by microscopy.
Astrocyte monolayers were switched from 10% FBS to low serum (2% horse serum) for 48 h before analysis. Cultured cells were transferred to the heated stage (37°C) of a Zeiss (Thornwood, NY) LSM510 confocal microscope with constant 5% CO2 and imaged with a 63× oil-immersion objective using 405 nm excitation (Robinson et al., 2006). Untreated, live astrocytes were imaged to set instrument parameters and minimize autofluorescence from 400 nm excitation. Astrocytes were then incubated with 0.3 μm MitoSOX Red for 15 min and washed, and the media were replaced. After MitoSOX Red images were obtained, astrocytes were incubated with 4′,6′-diamidino-2-phenylindole (DAPI) and 3.5 nm MitoTracker Deep Red.
Flow cytometric analysis of superoxide production and mitochondrial membrane potential.
The probe JC-1 was used to measure mitochondrial membrane potential (Almeida el al, 2001) and MitoSOX Red to measure mitochondrial superoxide (Mukhopadhyay et al., 2007). Cells were mildly trypsinized and resuspended at a density of 4 × 105 cells/ml in prewarmed (37°C) PBS solution supplemented with 20 mm glucose and 2% FBS, pH 7.4, containing JC1 (2 μm) or MitoSOX Red (2.5 μm). After a 15 min incubation period at 37°C in the dark, cell suspensions were centrifuged and resuspended in probe-free solution. Data acquisition was performed in a CyAn ADP cytometer (Dako, High Wycombe, UK). Astrocytes were gated to exclude subcellular debris as differentiated by forward and side scatter. Probes were excited with a 488 nm laser. JC-1 monomer fluorescence signal was detected on FL1 (green fluorescence, centered around 530 nm), and JC-1 aggregates on the FL2 detector (red fluorescence, centered around 575 nm). Mean fluorescence intensity values for FL1 and FL2 were obtained for all experiments. The relative aggregate/monomer (red/green) fluorescence intensity values were used for data presentation. These values were expressed in a percentage scale, with the red/green fluorescence ratio values of nontransgenic astrocytes (3.7 ± 0.30) considered as 100% and the red/green fluorescence ratio values of FCCP-treated transgenic astrocytes considered as 0%. MitoSOX Red fluorescence was detected in FL2 (red fluorescence, centered around 575 nm) and expressed as a percentage of nontransgenic cells.
Determination of mitochondrial membrane potential.
Astrocyte monolayers were rinsed twice with prewarmed Hank's solution and incubated with 30 μm JC-1 in Hank's solution for 30 min at 37°C. Cells were rinsed, mounted, and kept at 37°C. Epifluorescence was examined using a Nikon (Tokyo, Japan) Eclipse TE 200 microscope with the appropriate filters. Fluorescence was monitored in a multifunctional microplate reader Fluostar Galaxy from BMG Labtech (Winooski, VT) using 490 nm excitation/535 nm emission for the monomeric form and 570 nm excitation/595 nm emission for the aggregate of JC-1. Mitochondrial membrane potential is shown as a ratio of the fluorescence of aggregate (aqueous phase) and monomer (membrane-bound) forms of JC-1 and was pooled with data obtained by cytometry.
Immunohistochemistry.
Early symptomatic SOD1G93A rats (105–120 d) were injected 48 and 24 h before perfusion with 200 mg/kg DMPO intraperitoneally. Under deep anesthesia (35 mg/kg pentobarbital), rats were transcardially perfused with 0.9% saline followed by 4% paraformaldehyde fixative. The spinal cords were removed and embedded in Paraplast. Five micrometer tissue sections were preincubated with 0.3% hydrogen peroxide in methanol, washed, and permeabilized with 0.1% Triton X-100 in PBS for 15 min and blocked for 2 h with 10% goat serum, 2% BSA, and 0.1% Triton X-100 in PBS. The primary polyclonal anti-DMPO 1:400 was diluted in blocking solution and incubated overnight at 4°C. After washing with PBS, cultures were incubated with secondary anti-rabbit biotinylated antibody 1:250 in blocking solution for 1 h and then with streptavidin–horseradish peroxidase 1 μg/ml in Tris-HCl, pH 7.4 (both from Jackson ImmunoResearch, West Grove, PA), for 1 h at room temperature. Development was performed with 0.5 mg/ml DAB solution and 0.01% (v/v) hydrogen peroxide in 0.05 m Tris-HCl. The slides were counterstained with hematoxylin. Images were captured by a Nikon digital camera coupled to a Nikon Eclipse TE 200 epifluorescence microscope.
Western blot analysis.
Protein samples were separated on a 10% SDS-polyacrylamide gel and transferred to nitrocellulose membrane (GE Healthcare, Piscataway, NJ) overnight at 100 mA at 4°C. For DMPO Western blotting, membranes were blocked for 1 h in Tris-buffered saline (TBS), 2.5% BSA, and 2.5% casein and then 1 h incubation with primary anti-DMPO antibody at 1:5000 dilution in the same buffer plus 0.2% Tween 20 (washing buffer). After washing, the membrane was incubated with peroxidase-conjugated secondary antibody (Bio-Rad, Hercules, CA) diluted in 0.3% Tween 20 plus 0.1% bovine serum albumin for 1 h. For nitrotyrosine Western blotting, membranes were blocked for 1 h in TBS, 0.6% Tween 20, and 5% bovine serum albumin, followed by 1 h incubation with primary anti-nitrotyrosine antibody at 1:2000 dilution in the same buffer (Brito et al., 1999). After washing with 0.6% Tween 20 in TBS, the membrane was incubated with peroxidase-conjugated secondary antibody (Bio-Rad) diluted in 0.3% Tween 20 plus 0.1% bovine serum albumin for 1 h. Then both membranes were washed and developed using the ECL chemiluminescent detection system (GE Healthcare). Densitometric analysis was performed using the NIH ImageJ program.
Statistics.
Each experiment was repeated at least three times, and data are reported as the mean ± SEM. Comparison of the means was performed by one-way ANOVA. Pairwise tests between any two means used the Student–Newman–Keuls test, and differences were declared statistically significant if p < 0.05. When normality test failed, comparison of the means was performed by one-way ANOVA on ranks followed by the Kruskal–Wallis test. All statistics were performed using the SigmaStat Software (SPSS, Chicago, IL).
Results
Decreased mitochondrial respiration associated with oxidative stress in the SOD1G93A rat spinal cord
To assess whether the electron transport chain (ETC) function was altered in mitochondria from SOD1G93A rats, we measured oxygen consumption in mitochondria preparations from the spinal cord. We selected early symptomatic animals (120 d of age) to perform the study. We stimulated mitochondrial respiration by either glutamate–malate or succinate, which supply reducing equivalents through complex I and II, respectively (Fig. 1A,B; supplemental Fig. S1, available at www.jneurosci.org as supplemental material). In this assay, addition of ADP increases oxygen consumption only in well coupled mitochondria preparations. Here, glutamate–malate or succinate induced oxygen consumption in mitochondria from both transgenic and nontransgenic rats. However, the addition of ADP increased oxygen consumption only in mitochondria from nontransgenic spinal cords, indicating that coupling, and therefore the ability to synthesize ATP, was significantly impaired in spinal cord mitochondria from symptomatic SOD1G93A rats (Fig. 1A,B). Remarkably, such defect could be prevented by the systemic administration of the spin trap DMPO (50 mg/kg) to symptomatic animals 48 and 24 h before mitochondrial isolation (Fig. 1A,B). DMPO is a nitroxide that reacts with a variety of radicals, including protein radicals, to form DMPO–protein radical adducts. We then explored whether DMPO was able to trap mitochondrial protein radicals in vivo using a specific antibody that recognizes DMPO nitrone–protein adducts (Detweiler et al., 2002). Indeed, in biological systems the unstable protein–DMPO nitroxide radical adduct can be oxidized by one electron to a stable and electron paramagnetic resonance-silent protein–DMPO nitrone adduct, which can be recognized immunochemically (Detweiler et al., 2005). Western blot analysis showed an increase in DMPO–protein adducts in mitochondrial protein extracts from DMPO-treated SOD1G93A rat spinal cords compared with DMPO-treated nontransgenic animals (Fig. 1C). In agreement, immunohistochemistry of spinal cord sections revealed an increase in DMPO–protein adducts staining in both neuropil and motor neurons from transgenic animals, whereas a low background was observed in nontransgenic ones (Fig. 1D).
Figure 1.
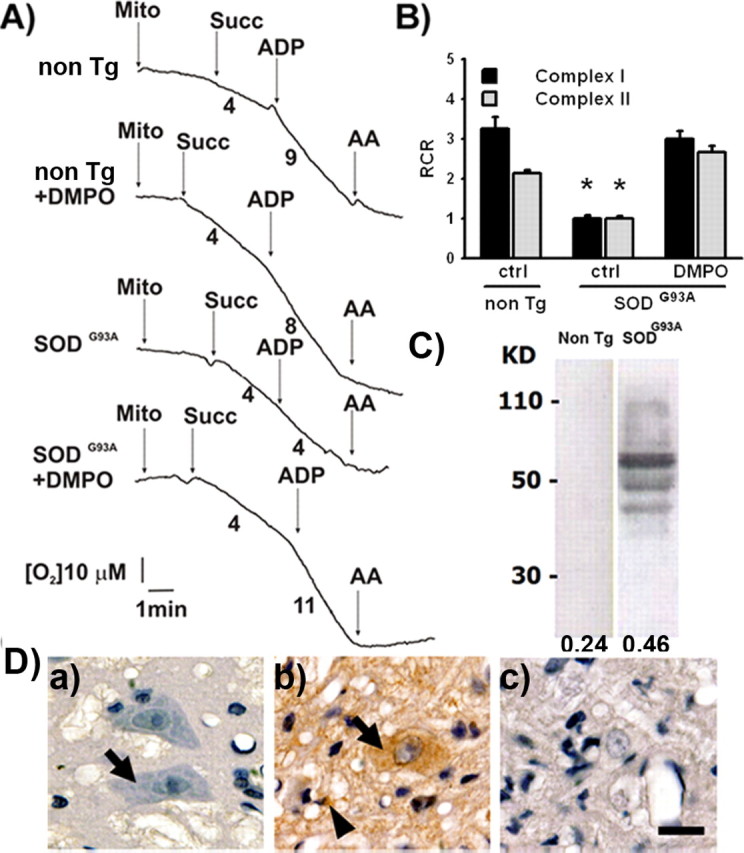
Respiratory complex II-dependent oxygen consumption in spinal cord mitochondria. A, Representative state-4 and state-3 oxygen consumption curves in the presence of succinate (Succ; 5 mm) in intact rat spinal cord mitochondria (0.5 mg/ml). Numbers under the traces represent micromolar O2 per minute. B, Calculated respiratory control ratio for spinal cord mitochondria from the indicated treatments. Data are mean ± SEM from three independent experiments. *p < 0.05, significantly different from non-Tg control. C, Immunoblotting of mitochondrial protein from the lumbar spinal cord of DMPO-injected rats. Ten micrograms of mitochondrial protein were seeded per lane in 13% acrylamide gel. Anti-DMPO antibody was used at 1:5000. Numbers on the left indicate molecular weights, and numbers on the bottom indicate the mean optical density for each lane. This is a representative blot from three independent experiments. D, Immunohistochemistry in sections of the anterior horn of DMPO-injected nontransgenic rats (a), DMPO-injected early symptomatic SOD1G93A rats (120 d of age; b), and vehicle-injected SOD1G93A rats (c). Note the intense punctuate immunolabeling in both motor neurons (arrows) and surrounding glial cells (arrowheads) in b compared with lack of labeling in a. DMPO was injected intraperitoneally (50 mg/kg) 48 and 24 h before mitochondrial isolation, and anti-DMPO was used at 1:400. Scale bar, 20 μm. Images are representative from three independent experiments.
Antioxidants and NOS inhibition restored the uncoupled mitochondrial activity in cultured SOD1G93A-expressing astrocytes
To determine whether mitochondrial respiration was affected in SOD1G93A astrocytes, we isolated mitochondria from pure astrocyte cultures obtained from nontransgenic, SOD1WT, or SOD1G93A transgenic neonate rats, which overexpressed either wild-type or G93A mutation of SOD1, respectively. We used the cerebral cortex instead of the spinal cord as a source to provide sufficient yield of mitochondria for respiratory experiments. Mitochondria from SOD1G93A-bearing astrocytes showed a decrease in both complex I- and II-dependent respiration rate and no increase in oxygen consumption in response to ADP compared with mitochondria from nontransgenic or SOD1WT (Fig. 2A,B; supplemental Fig. S1, available at www.jneurosci.org as supplemental material). The defect was comparable with that observed in whole spinal cord mitochondrial preparations (Fig. 1A,B). When SOD1G93A astrocytes were preincubated with DMPO (75 mm) for 48 h before mitochondria isolation, the respiratory control rate was partially reestablished. The detection of oxidative damage in mitochondria associated with organelle dysfunction described in Figure 1 prompted us to test the recently designed mitochondrial-targeted antioxidants Mito-Q and Mito-CP. These compounds have been developed by covalently coupling to triphenylphosphonium cation, thereby allowing these compounds be preferentially taken up by mitochondria (Kelso et al., 2001). They have been shown to be effective in preventing oxidative damage at substantially lower concentrations than other antioxidants (Jauslin et al., 2003; Dhanasekaran et al., 2005). Transgenic astrocytes preincubated with Mito-Q (20 nm) for 24 h recovered the ability to increase oxygen consumption after addition of ADP and restored the RCR (and therefore coupling) to control values more effectively than DMPO (Fig. 2A,B). The preincubation of non-Tg astrocytes with Mito-Q did not affect mitochondrial respiration (data not shown). We have previously described that SOD1G93A transgenic astrocytes produce more nitric oxide than nontransgenic ones (Vargas et al., 2006). Because nitric oxide may be contributing to oxidative damage, we then pretreated transgenic astrocytes with the nitric oxide synthase (NOS) inhibitor l-N-nitroarginine methyl ester (l-NAME; 5 mm) and also found that this restores the RCR (Fig. 2A). Western blot analysis of mitochondrial protein extracts from the same mitochondrial samples used for the oxygen consumption studies revealed changes in protein 3-nitrotyrosine immunodetection, a well established biomarker of nitric oxide-derived oxidants (Radi, 2004). The samples from SOD1G93A-bearing astrocytes exhibited an increase in 3-nitrotyrosine-immunoreactive bands compared with nontransgenic ones. Furthermore, Mito-Q-treated SOD1G93A astrocytes showed a decrease in 3-nitrotyrosine immunoreactivity (Fig. 2C), indicating that Mito-Q treatment diminished nitroxidative damage to mitochondrial proteins.
Figure 2.

Antioxidants and NOS inhibition reestablished mitochondrial respiration and decreased protein 3-nitrotyrosine levels in mitochondrial proteins from SOD1G93A astrocytes. A, Representative oxygen consumption curves by mitochondria isolated from cultured cerebral cortex astrocytes prepared from non-Tg or transgenic SOD1WT or SOD1G93A rat neonates treated as indicated. Mitochondrial respiration was evaluated as indicated in Figure 1. DMPO (75 mm, 48 h), Mito-Q (10 nm, 24 h), or l-NAME (5 mm, 24 h) were added to the culture medium of SOD1G93A before mitochondrial isolation. Numbers under the traces indicate the absolute values of state-3 and -4 respiration in micromolar O2 per minute. B, Calculated respiratory control ratio for complex I and II of astrocyte mitochondria from the indicated groups. Data are mean ± SEM from four independent experiments. *p < 0.05, significantly different from non-Tg control. C, Protein 3-nitrotyrosine immunoblotting of mitochondrial proteins from the same samples used to measure oxygen consumption in A. Thirty micrograms of mitochondrial protein were seeded per lane in 13% acrylamide gel. Numbers on the left indicate molecular weights, and numbers on the bottom indicate the mean optical density for each lane. This is a representative blot from three independent experiments.
Superoxide generation and membrane potential determination in SOD1G93A astrocyte mitochondria
To evaluate mitochondrial oxidant formation in SOD1G93A astrocytes, cultures were incubated with MitoSOX Red, a specific and mitochondrial-targeted detection probe for superoxide radical (O2·−) (Robinson et al., 2006). SOD1G93A transgenic astrocytes showed an approximate twofold increase in fluorescence emission from the superoxide product of MitoSOX Red as detected using the selective excitation wavelength at 405 nm (Fig. 3A). Mitochondrial superoxide generation was measured simultaneously in a large population of astrocytes by flow cytometry as has been recently published (Mukhopadhyay et al., 2007), using 5 μm antimycin A, a complex III inhibitor that increases mitochondrial superoxide production, as a positive control (Fig. 3B). The mean fluorescence intensity showed increase in SOD1G93A astrocytes compared with SOD1WT or non-Tg astrocytes. The increment was reduced to the nontransgenic levels when SOD1G93A astrocytes were treated with Mito-Q (Fig. 3B). However, basal levels of fluorescence are likely to reflect direct redox interactions of MitoSOX with ETC components in addition to the O2·−-dependent reactions. To further characterize the mitochondrial dysfunction, we used the JC1 fluorescent probe to measure mitochondrial membrane potential by flow cytometry as previously published (Almeida et al., 2001). The ratio of the fluorescence of aggregate and monomer forms of JC-1 also reflected a decrease in mitochondrial membrane potential in SOD1G93A transgenic astrocytes (Fig. 3C). The ability to concentrate the JC-1 probe in mitochondria was equally decreased in both cortical and spinal cord astrocytes (supplemental Fig. S2, available at www.jneurosci.org as supplemental material).
Figure 3.
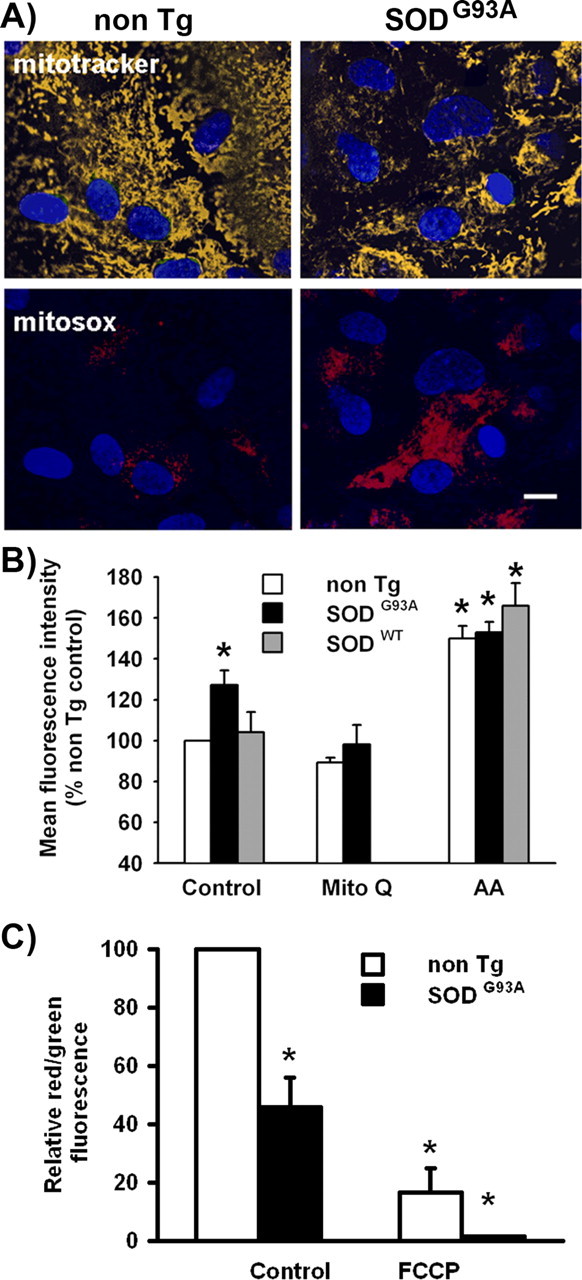
Increased superoxide generation and decreased mitochondrial membrane potential (ΔΨ) in SOD1G93A astrocytes. A, Fluorescence micrographs of cultured astrocytes with MitoTracker Deep Red (top row, yellow) and MitoSOX Red (bottom row, red). Astrocyte monolayers were treated as indicated in Materials and Methods. Nuclei are stained with DAPI (blue). Scale bar, 100 μm. B, Data are mean fluorescence intensity of MitoSOX Red emission measured by flow cytometry in nontransgenic (white bars), SOD1G93A (black bars), or SOD1WT (gray bars) astrocyte cultures treated for 24 h with Mito-Q or antimycin A as positive control. Data are mean ± SEM from three independent determinations. *p < 0.05, significantly different from non-Tg control. C, Mitochondrial membrane potential as a ratio of the fluorescence of aggregate and monomer forms of JC-1 in SOD1G93A (black bars) and nontransgenic (white bars) astrocytes in control and after addition of the uncoupler FCCP. These values were expressed in a percentage scale, with the red/green fluorescence ratio values of nontransgenic astrocytes (3.7 ± 0.30) considered as 100% and the red/green fluorescence ratio values of FCCP-treated transgenic astrocytes considered as 0%. Data are mean ± SEM from three independent experiments. *p < 0.05, significantly different from non-Tg control.
Astrocytes fail to support motor neuron survival after mitochondrial dysfunction: protective action of mitochondrial-targeted antioxidants
We then analyzed whether SOD1G93A astrocytic mitochondrial dysfunction could affect motor neuron survival in a coculture model of ALS previously described by our group (Cassina et al., 2002). A feeder layer of SOD1G93A astrocytes decreased the survival of nontransgenic motor neurons by 50% compared with a feeder layer of nontransgenic astrocytes (Fig. 4) or with SOD1WT astrocytes (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). Pretreatment of SOD1G93A astrocytes with the NOS inhibitor l-NAME (5 mm), the spin trap DMPO (75 mm), or the metalloporphyrin catalytic antioxidant MnTE-2-PyP (10 μm) before motor neuron plating enhanced motor neuron survival to the level of that obtained on a feeder layer of nontransgenic astrocytes (Fig. 4). On the other hand, induction of mitochondrial dysfunction by pretreatment of nontransgenic astrocytes with different ETC inhibitors decreased motor neuron survival in coculture conditions (Fig. 4), which was reversed by l-NAME and MnTE treatment (data not shown). We used rotenone (2 μm), antimycin A (5 μm), and sodium azide (5 μm), complex I, complex III, and complex IV inhibitors, respectively. Furthermore, we used fluorocitrate (100–250 μm), which blocks the activity of Krebs cycle enzyme aconitase and has been recently shown to induce inhibition of astrocyte mitochondria and increase vulnerability of cocultured neurons to glutamate toxicity (Voloboueva et al., 2007). The ETC inhibitors did not affect astrocyte viability at the concentration used (data not shown). Preincubation of SOD1G93A-bearing astrocytes with Mito-CP or Mito-Q for 24 h, at much lower concentrations than used for compounds in Figure 4, resulted in increased motor neuron survival in cocultures to a degree similar to that achieved over a nontransgenic astrocyte monolayer (Fig. 5). Indeed, the pretreatment of nontransgenic astrocytes with both compounds improved the motor neuron survival obtained in the control condition (Fig. 5).
Figure 4.

Motor neuron survival in the presence of astrocytes. Motor neuron survival 48 h after plating either on nontransgenic or SOD1G93A astrocyte monolayers as indicated. The non-Tg astrocyte monolayers were previously treated with rotenone (Rot; 2 μm), antimycin A (AA; 5 μm), sodium azide (Az; 5 μm) or fluorocitrate (FC; 100 μm) and the SOD1G93A astrocyte monolayers with l-NAME (5 mm), DMPO (75 mm), or MnTE (10 μm) for 24 h as indicated and washed, and then a nontransgenic motor neuron suspension was added. Motor neuron counting was performed as described in Materials and Methods. Data are presented as a percentage of the non-Tg control group. Data are mean ± SEM from three independent experiments. *p < 0.05, significantly different from non-Tg control.
Figure 5.

Restoration of motor neuron survival by mitochondrial-targeted antioxidants. Motor neuron survival 48 h after plating either on nontransgenic or SOD1G93A astrocyte monolayers as indicated. The non-Tg or the SOD1G93A-bearing monolayers were previously treated with Mito-Q (10–100 nm) or Mito-CP (10–100 nm) for 24 h as indicated and washed, and then a nontransgenic motor neuron suspension was added. Dotted lines indicate the 100 and 55% motor neuron survival obtained in control conditions over a nontransgenic or SOD1G93A astrocyte feeder layer, respectively. Data are presented as described in Figure 4. Data are mean ± SEM from three independent experiments. *p < 0.05, significantly different from non-Tg control.
Discussion
Herein, we provide evidence that mitochondrial dysfunction linked to nitroxidative stress in SOD1G93A astrocytes promotes the death of neighboring motor neurons. Previous studies have shown decreased mitochondrial function in SOD1G93A ALS mice (Jung et al., 2002; Mattiazzi et al., 2002) and motor neuron cell lines transfected with ALS mutated forms of SOD1 (Menzies et al., 2002; Fukada et al., 2004). Our findings indicate for the first time that mitochondrial defects and enhanced generation of oxidants including O2·− are occurring in cultured SOD1G93A astrocytes but not in SOD1WT astrocytes, which cause SOD1G93A astrocytes to become neurotoxic for motor neurons. Furthermore, this can be prevented by mitochondrial-targeted antioxidants and NOS inhibitors. Together, these results support the idea that nitroxidative stress-induced mitochondrial defects in astrocytes may contribute to disease pathogenesis.
Defective mitochondrial respiration in SOD1G93A astrocytes seems to be associated with nitroxidative stress as shown by: (1) the extensive formation of DMPO–protein adducts and protein 3-nitrotyrosine in isolated mitochondria from both spinal cord and astrocyte cultures and (2) the fact the spin trap DMPO, a NOS inhibitor, and, in particular, low concentrations of mitochondrial-targeted antioxidants (Mito-Q and Mito-CP) completely reversed mitochondrial dysfunction in vitro and/or in vivo. Use of DMPO allowed us to combine the specificity of spin trapping and the sensitivity of antigen-antibody interactions to detect the in vivo formation of protein radicals. The immunodetection of protein–DMPO radical adducts has been used before for Western blotting of isolated proteins (Detweiler et al., 2005), but here we show for the first time that it is also suitable for Western blotting and immunohistochemistry in vivo, after systemic DMPO administration to animals. In fact, this approach provides a new tool to detect free radical damage in vivo, extending the utility of other markers, including 3-nitrotyrosine (Abe et al., 1997; Bruijn et al., 1997) and hydroxynonenal–protein adducts (Pedersen et al., 1998). It is not fully clear how trapping protein radicals with DMPO was beneficial to the cells; most likely, the formation of protein radical–DMPO adducts will block free radical-propagating reactions (Nauser et al., 2005). Accordingly, DMPO treatment has been shown to increase the lifespan in SOD1G93A transgenic mice (Liu et al., 2002). Overall, the results with DMPO further support the impact of oxidative stress in ALS pathology and prompted us to evaluate the efficacy of more specific redox-based therapies.
Previously, it has been shown that SOD1G93A astrocytes are toxic for motor neurons in coculture via diffusible factors (Vargas et al., 2006; Nagai et al., 2007). A mechanism involving oxidative stress, increased ·NO production, and decreased glutathione levels has been proposed (Vargas et al., 2006). Here, we show evidence that defective mitochondrial respiration leads to the establishment of a neurotoxic astrocyte phenotype. Decrease of mitochondrial respiration in nontransgenic astrocyte cultures by ETC inhibitors, particularly azide-dependent inhibition of cytochrome c oxidase and fluorocitrate-dependent inhibition of aconitase, resulted in decreased motor neuron survival, suggesting that mitochondrial function in astrocytes is necessary to support motor neuron survival. Furthermore, restoration of astrocytic mitochondrial respiration with antioxidants Mito-Q and Mito-CP prevented the neurotoxic phenotype. In addition, we have recently shown that these mitochondrial-targeted antioxidants are effective in preventing cell death in pure motor neuron cultures challenged with proapoptotic stimuli that trigger ROS formation (Pehar et al., 2007). As a first step toward mitochondria-specific therapies, the Murphy and Kalyanaraman laboratories have developed bioactive molecules delivered to mitochondria by covalent attachment to the triphenylphosphonium cation through an alkyl chain (Kelso et al., 2001; Dhanasekaran et al., 2005). The delocalized positive charge of these lipophilic cations enables them to permeate lipid bilayers easily and to possibly accumulate several hundredfold within mitochondria. This selective uptake greatly increases the efficacy and specificity of molecules designed to interact with mitochondria while also decreasing harmful side reactions. In this work, we show that Mito-Q is effective in restoring mitochondrial respiration and preventing neurotoxicity in SOD1G93A-bearing astrocytes at much smaller doses than those used for other antioxidants. The effects of Mito-Q are likely to be combined effects, resulting in decreased steady-state levels of superoxide (decrease of MitoSOX oxidation) and peroxynitrite (decrease of mitochondrial protein nitration). Additional studies are required to further define the specific protective mechanism at the cellular level, but the mitochondrial-targeted antioxidants provide a new potential therapy to be tested in ALS models.
One potential functional consequence of mitochondrial dysfunction in ALS is the impairment of energy metabolism and increased generation of oxidants. Overexpression of SOD1G93A in a cell line reproduces mitochondrial abnormal morphology and decreased respiration rate (Menzies et al., 2002) and makes the cells highly dependent on glycolysis, suggesting that SOD1G93A expression may induce a switch to anaerobic metabolism to maintain ATP levels. Such observations in a cell line might be relevant to understand the impact of SOD1G93A in astrocytes that are known to switch from aerobic to glycolytic metabolism in response to ETC inhibition by ·NO (Almeida et al., 2001). Recently, we showed overproduction of ·NO in SOD1G93A astrocytes (Vargas et al., 2006). In addition, here we demonstrate that there is an increase in mitochondrial O2·− formation in SOD1G93A astrocytes using a novel and specific mitochondrial-targeted fluorescent probe. Because mutant SOD1 has been shown to mediate aberrant oxidative chemistry in neuronal cells (Estevez et al., 1999; Zimmerman et al., 2007), increased ·NO and O2·− would facilitate the formation of peroxynitrite, which can damage key mitochondrial enzymes (Radi et al., 2002; Quijano et al., 2005). The mutated forms of SOD1 can be found inside mitochondria (Jaarsma et al., 2001; Higgins et al., 2002; Liu et al., 2004; Vijayvergiya et al., 2005) and may shift the redox state of respiratory complexes (Ferri et al., 2006) or disrupt the association of cytochrome c with the inner membrane (Kirkinezos et al., 2005). Furthermore, aggregates of wild-type and mutant SOD1 in the mitochondria from transgenic mouse spinal cord correlate with exacerbation of the disease (Deng et al., 2006). All the previous mechanisms may contribute to increase nitroxidative damage inside mitochondria. Accordingly, DMPO, l-NAME, Mito-Q, and MnTE-2-PyP restored mitochondrial respiration and also prevented astrocyte-derived neurotoxicity. It has been recently shown that MnTE-2-PyP in the Mn3+ oxidation state is reduced by complex I and complex II of the mitochondrial ETC, and the reduced (Mn2+) complex reacts and decomposes peroxynitrite, suggesting that Mn-porphyrins may act as peroxynitrite reduction catalysts in vivo and protect mitochondria from oxidative damage (Ferrer-Sueta et al., 2006; Spasojević et al., 2007). These results emphasize the key role of nitroxidative stress in astrocytes in the pathogenesis of neurodegenerative diseases.
It needs to be established whether compounds that ameliorate mitochondrial dysfunction might be beneficial in ALS. Compounds that can improve the bioenergetics of mitochondria such as Mn-porphyrins have been shown to increase the lifespan in SOD1G93A transgenic mice (Wu et al., 2003). Our results support that astrocytes might be a target for redox-based therapies in ALS. In summary, we have shown that astrocytes from SOD1G93A transgenic animals exhibiting an altered mitochondrial activity are neurotoxic for motor neurons. The restoration of mitochondrial function after mitochondrial-targeted antioxidant treatment opens hope for more specific tools to treat a wide range of neurodegenerative disorders.
Footnotes
This work was supported by grants from the Howard Hughes Medical Institute and the International Centre for Genetic Engineering and Biotechnology (R.R.) and by the Amyotrophic Lateral Sclerosis Association and Comisión Sectorial de Investigación Científica (P.C.). We thank Pablo Díaz Amarillo for genotyping the rats, Mariela González for technical assistance, and Mark Levy for making the wild-type SOD astrocytes. We especially thank Dr. Mariela Bollati for assistance in flow cytometry. We thank the Imaging Core of the Environmental Health Science Center (ES00240), Linus Pauling Institute. Rafael Radi is a Howard Hughes International Research Scholar.
References
- Abe K, Pan LH, Watanabe M, Konno H, Kato T, Itoyama Y. Upregulation of protein-tyrosine nitration in the anterior horn cells of amyotrophic lateral sclerosis. Neurol Res. 1997;19:124–128. doi: 10.1080/01616412.1997.11740784. [DOI] [PubMed] [Google Scholar]
- Almeida A, Almeida J, Bolanos JP, Moncada S. Different responses of astrocytes and neurons to nitric oxide: the role of glycolytically generated ATP in astrocyte protection. Proc Natl Acad Sci USA. 2001;98:15294–15299. doi: 10.1073/pnas.261560998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbeito LH, Pehar M, Cassina P, Vargas MR, Peluffo H, Viera L, Estevez AG, Beckman JS. A role for astrocytes in motor neuron loss in amyotrophic lateral sclerosis. Brain Res Brain Res Rev. 2004;47:263–274. doi: 10.1016/j.brainresrev.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Batinic-Haberle I, Benov L, Spasojevic I, Fridovich I. The ortho effect makes manganese(III) meso-tetrakis(N-methylpyridinium-2-yl)porphyrin a powerful and potentially useful superoxide dismutase mimic. J Biol Chem. 1998;273:24521–24528. doi: 10.1074/jbc.273.38.24521. [DOI] [PubMed] [Google Scholar]
- Brito C, Naviliat M, Tiscornia AC, Vuillier F, Gualco G, Dighiero G, Radi R, Cayota AM. Peroxynitrite inhibits T lymphocyte activation and proliferation by promoting impairment of tyrosine phosphorylation and peroxynitrite-driven apoptotic death. J Immunol. 1999;162:3356–3366. [PubMed] [Google Scholar]
- Bruijn LI, Beal MF, Becher MW, Schulz JB, Wong PC, Price DL, Cleveland DW. Elevated free nitrotyrosine levels, but not protein-bound nitrotyrosine or hydroxyl radicals, throughout amyotrophic lateral sclerosis (ALS)-like disease implicate tyrosine nitration as an aberrant in vivo property of one familial ALS-linked superoxide dismutase 1 mutant. Proc Natl Acad Sci USA. 1997;94:7606–7611. doi: 10.1073/pnas.94.14.7606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruijn LI, Miller TM, Cleveland DW. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci. 2004;27:723–749. doi: 10.1146/annurev.neuro.27.070203.144244. [DOI] [PubMed] [Google Scholar]
- Cassina A, Radi R. Differential inhibitory action of nitric oxide and peroxynitrite on mitochondrial electron transport. Arch Biochem Biophys. 1996;328:309–316. doi: 10.1006/abbi.1996.0178. [DOI] [PubMed] [Google Scholar]
- Cassina AM, Hodara R, Souza JM, Thomson L, Castro L, Ischiropoulos H, Freeman BA, Radi R. Cytochrome c nitration by peroxynitrite. J Biol Chem. 2000;275:21409–21415. doi: 10.1074/jbc.M909978199. [DOI] [PubMed] [Google Scholar]
- Cassina P, Peluffo H, Pehar M, Martinez-Palma L, Ressia A, Beckman JS, Estevez AG, Barbeito L. Peroxynitrite triggers a phenotypic transformation in spinal cord astrocytes that induces motor neuron apoptosis. J Neurosci Res. 2002;67:21–29. doi: 10.1002/jnr.10107. [DOI] [PubMed] [Google Scholar]
- Cassina P, Pehar M, Vargas MR, Castellanos R, Barbeito AG, Estevez AG, Thompson JA, Beckman JS, Barbeito L. Astrocyte activation by fibroblast growth factor-1 and motor neuron apoptosis: implications for amyotrophic lateral sclerosis. J Neurochem. 2005;93:38–46. doi: 10.1111/j.1471-4159.2004.02984.x. [DOI] [PubMed] [Google Scholar]
- Chan PH, Kawase M, Murakami K, Chen SF, Li Y, Calagui B, Reola L, Carlson E, Epstein CJ. Overexpression of SOD1 in transgenic rats protects vulnerable neurons against ischemic damage after global cerebral ischemia and reperfusion. J Neurosci. 1998;18:8292–8299. doi: 10.1523/JNEUROSCI.18-20-08292.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement AM, Nguyen MD, Roberts EA, Garcia M, Boillee S, Rule M, McMahon AP, Doucette W, Siwek D, Ferrante RJ, Brown RH, Julien JP, Goldstein LS, Cleveland DW. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science. 2003;302:113–117. doi: 10.1126/science.1086071. [DOI] [PubMed] [Google Scholar]
- Deng HX, Shi Y, Furukawa Y, Zhai H, Fu R, Liu E, Gorrie GH, Khan MS, Hung WY, Bigio EH, Lukas T, Dal Canto MC, O'Halloran TV, Siddique T. Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc Natl Acad Sci USA. 2006;103:7142–7147. doi: 10.1073/pnas.0602046103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detweiler CD, Deterding LJ, Tomer KB, Chignell CF, Germolec D, Mason RP. Immunological identification of the heart myoglobin radical formed by hydrogen peroxide. Free Radic Biol Med. 2002;33:364–369. doi: 10.1016/s0891-5849(02)00895-x. [DOI] [PubMed] [Google Scholar]
- Detweiler CD, Lardinois OM, Deterding LJ, de Montellano PR, Tomer KB, Mason RP. Identification of the myoglobin tyrosyl radical by immuno-spin trapping and its dimerization. Free Radic Biol Med. 2005;38:969–976. doi: 10.1016/j.freeradbiomed.2004.12.031. [DOI] [PubMed] [Google Scholar]
- Dhanasekaran A, Kotamraju S, Karunakaran C, Kalivendi SV, Thomas S, Joseph J, Kalyanaraman B. Mitochondria superoxide dismutase mimetic inhibits peroxide-induced oxidative damage and apoptosis: role of mitochondrial superoxide. Free Radic Biol Med. 2005;39:567–583. doi: 10.1016/j.freeradbiomed.2005.04.016. [DOI] [PubMed] [Google Scholar]
- Dupuis L, Gonzalez de Aguilar JL, Oudart H, de Tapia M, Barbeito L, Loeffler JP. Mitochondria in amyotrophic lateral sclerosis: a trigger and a target. Neurodegener Dis. 2004;1:245–254. doi: 10.1159/000085063. [DOI] [PubMed] [Google Scholar]
- Estevez AG, Crow JP, Sampson JB, Reiter C, Zhuang Y, Richardson GJ, Tarpey MM, Barbeito L, Beckman JS. Induction of nitric oxide-dependent apoptosis in motor neurons by zinc-deficient superoxide dismutase. Science. 1999;286:2498–2500. doi: 10.1126/science.286.5449.2498. [DOI] [PubMed] [Google Scholar]
- Ferrer-Sueta G, Hannibal L, Batinic-Haberle I, Radi R. Reduction of manganese porphyrins by flavoenzymes and submitochondrial particles: a catalytic cycle for the reduction of peroxynitrite. Free Rad Biol Med. 2006;41:503–512. doi: 10.1016/j.freeradbiomed.2006.04.028. [DOI] [PubMed] [Google Scholar]
- Ferri A, Cozzolino M, Crosio C, Nencini M, Casciati A, Butler Gralla E, Rotilio G, Selverstone VJ, Carri MT. Familial ALS-superoxide dismutases associate with mitochondria and shift their redox potentials. Proc Natl Acad Sci USA. 2006;103:13860–13865. doi: 10.1073/pnas.0605814103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukada K, Zhang F, Vien A, Cashman NR, Zhu H. Mitochondrial proteomic analysis of a cell line model of familial amyotrophic lateral sclerosis. Mol Cell Proteomics. 2004;12:1211–1223. doi: 10.1074/mcp.M400094-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson CE, Camu W, Mettling C, Gouin A, Poulsen K, Karihaloo M, Rullamas J, Evans T, McMahon SB, Armanini MP, Berkemeier L, Phillips HS, Rosenthal A. Neurotrophins promote motor neuron survival and are present in embryonic limb bud. Nature. 1993;363:266–270. doi: 10.1038/363266a0. [DOI] [PubMed] [Google Scholar]
- Higgins CM, Jung C, Ding H, Xu Z. Mutant Cu, Zn superoxide dismutase that causes motoneuron degeneration is present in mitochondria in the CNS. J Neurosci. 2002;22(RC215):1–6. doi: 10.1523/JNEUROSCI.22-06-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howland DS, Liu J, She Y, Goad B, Maragakis NJ, Kim B, Erickson J, Kulik J, DeVito L, Psaltis G, DeGennaro LJ, Cleveland DW, Rothstein JD. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS) Proc Natl Acad Sci USA. 2002;99:1604–1609. doi: 10.1073/pnas.032539299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaarsma D, Rognoni F, van Duijn W, Verspaget HW, Haasdijk ED, Holstege JC. CuZn superoxide dismutase (SOD1) accumulates in vacuolated mitochondria in transgenic mice expressing amyotrophic lateral sclerosis-linked SOD1 mutations. Acta Neuropathol (Berl) 2001;102:293–305. doi: 10.1007/s004010100399. [DOI] [PubMed] [Google Scholar]
- Jauslin ML, Meier T, Smith RA, Murphy MP. Mitochondria-targeted antioxidants protect Friedreich Ataxia fibroblasts from endogenous oxidative stress more effectively than untargeted antioxidants. FASEB J. 2003;17:1972–1974. doi: 10.1096/fj.03-0240fje. [DOI] [PubMed] [Google Scholar]
- Jung C, Higgins CM, Xu Z. Mitochondrial electron transport chain complex dysfunction in a transgenic mouse model for amyotrophic lateral sclerosis. J Neurochem. 2002;83:535–545. doi: 10.1046/j.1471-4159.2002.01112.x. [DOI] [PubMed] [Google Scholar]
- Kelso GF, Porteous CM, Coulter CV, Hughes G, Porteous WK, Ledgerwood EC, Smith RA, Murphy MP. Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties. J Biol Chem. 2001;276:4588–4596. doi: 10.1074/jbc.M009093200. [DOI] [PubMed] [Google Scholar]
- Kirkinezos IG, Bacman SR, Hernandez D, Oca-Cossio J, Arias LJ, Perez-Pinzon MA, Bradley WG, Moraes CT. Cytochrome c association with the inner mitochondrial membrane is impaired in the CNS of G93A-SOD1 mice. J Neurosci. 2005;25:164–172. doi: 10.1523/JNEUROSCI.3829-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Lillo C, Jonsson PA, Vande Velde C, Ward CM, Miller TM, Subramaniam JR, Rothstein JD, Marklund S, Andersen PM, Brannstrom T, Gredal O, Wong PC, Williams DS, Cleveland DW. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron. 2004;43:5–17. doi: 10.1016/j.neuron.2004.06.016. [DOI] [PubMed] [Google Scholar]
- Liu R, Li B, Flanagan SW, Oberley LW, Gozal D, Qiu M. Increased mitochondrial antioxidative activity or decreased oxygen free radical propagation prevent mutant SOD1-mediated motor neuron cell death and increase amyotrophic lateral sclerosis-like transgenic mouse survival. J Neurochem. 2002;80:488–500. doi: 10.1046/j.0022-3042.2001.00720.x. [DOI] [PubMed] [Google Scholar]
- Manfredi G, Xu Z. Mitochondrial dysfunction and its role in motor neuron degeneration in ALS. Mitochondrion. 2005;2:77–87. doi: 10.1016/j.mito.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Mason RP. Using anti-5,5-dimethyl-1-pyrroline N-oxide (anti-DMPO) to detect protein radicals in time and space with immuno-spin trapping. Free Radic Biol Med. 2004;36:1214–1223. doi: 10.1016/j.freeradbiomed.2004.02.077. [DOI] [PubMed] [Google Scholar]
- Mattiazzi M, D'Aurelio M, Gajewski CD, Martushova K, Kiaei M, Beal MF, Manfredi G. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J Biol Chem. 2002;277:29626–29633. doi: 10.1074/jbc.M203065200. [DOI] [PubMed] [Google Scholar]
- Menzies FM, Cookson MR, Taylor RW, Turnbull DM, Chrzanowska-Lightowlers ZM, Dong L, Figlewicz DA, Shaw PJ. Mitochondrial dysfunction in a cell culture model of familial amyotrophic lateral sclerosis. Brain. 2002;125:1522–1533. doi: 10.1093/brain/awf167. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay P, Rajesh M, Yoshihiro K, Hasko G, Pacher P. Simple quantitative detection of mitochondrial superoxide production in live cells. Biochem Biophys Res Commun. 2007;358:203–208. doi: 10.1016/j.bbrc.2007.04.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, Przedborski S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. 2007;5:615–622. doi: 10.1038/nn1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nauser T, Koppenol WH, Gebicki JM. The kinetics of oxidation of GSH by protein radicals. Biochem J. 2005;392:693–701. doi: 10.1042/BJ20050539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen WA, Fu W, Keller JN, Markesbery WR, Appel S, Smith RG, Kasarskis E, Mattson MP. Protein modification by the lipid peroxidation product 4 hydroxynonenal in the spinal cords of amyotrophic lateral sclerosis patients. Ann Neurol. 1998;44:819–824. doi: 10.1002/ana.410440518. [DOI] [PubMed] [Google Scholar]
- Pehar M, Cassina P, Vargas MR, Castellanos R, Viera L, Beckman JS, Estevez AG, Barbeito L. Astrocytic production of nerve growth factor in motor neuron apoptosis: implications for amyotrophic lateral sclerosis. J Neurochem. 2004;89:464–473. doi: 10.1111/j.1471-4159.2004.02357.x. [DOI] [PubMed] [Google Scholar]
- Pehar M, Vargas MR, Robinson KM, Cassina P, Díaz-Amarilla PJ, Hagen TM, Radi R, Barbeito L, Beckman JS. Mitochondrial superoxide production and nuclear factor erythroid 2-related factor 2 activation in p75 neurotrophin receptor-induced motor neuron apoptosis. J Neurosci. 2007;27:7777–7785. doi: 10.1523/JNEUROSCI.0823-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peluffo G, Radi R. Biochemistry of protein tyrosine nitration in cardiovascular pathology. Cardiovasc Res. 2007;75:291–302. doi: 10.1016/j.cardiores.2007.04.024. [DOI] [PubMed] [Google Scholar]
- Quijano C, Cassina A, Castro L, Rodríguez M, Radi R. Peroxynitrite: a mediator of nitric-oxide dependent mitochondrial dysfunction in pathology. In: Cadenas E, Lamas S, editors. Nitric oxide, cell signaling, and gene expression. New York: Marcel Dekker; 2005. pp. 99–143. [Google Scholar]
- Radi R. Nitric oxide, oxidants, and protein tyrosine nitration. Proc Natl Acad Sci USA. 2004;101:4003–4008. doi: 10.1073/pnas.0307446101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radi R, Turrens JF, Chang LY, Bush KM, Crapo JD, Freeman BA. Detection of catalase in rat heart mitochondria. J Biol Chem. 1991;266:22028–22034. [PubMed] [Google Scholar]
- Radi R, Cassina A, Hodara R, Quijano C, Castro L. Peroxynitrite reactions and formation in mitochondria. Free Radic Biol Med. 2002;33:1451–1464. doi: 10.1016/s0891-5849(02)01111-5. [DOI] [PubMed] [Google Scholar]
- Robinson KM, Janes MS, Pehar M, Monette JS, Ross MF, Hagen TM, Murphy MP, Beckman JS. Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc Natl Acad Sci USA. 2006;103:15038–15043. doi: 10.1073/pnas.0601945103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saneto RP, De Vellis J. Neuronal and glial cells: cell culture of the central nervous system. In: Turner AJ, Brachelard HS, editors. Neurochemistry: a practical approach. Washington, DC: IRL; 1987. pp. 27–63. [Google Scholar]
- Spasojević I, Chen Y, Noel TJ, Yu Y, Cole MP, Zhang L, Zhao Y, St Clair DK, Batinić-Haberle I. Mn porphyrin-based superoxide dismutase (SOD) mimic, MnIIITE-2-PyP5+, targets mouse heart mitochondria. Free Radic Biol Med. 2007;42:1193–1200. doi: 10.1016/j.freeradbiomed.2007.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas MR, Pehar M, Cassina P, Martinez-Palma L, Thompson JA, Beckman JS, Barbeito L. Fibroblast growth factor-1 induces heme oxygenase-1 via nuclear factor erythroid 2-related factor 2 (Nrf2) in spinal cord astrocytes: consequences for motor neuron survival. J Biol Chem. 2005;280:25571–25579. doi: 10.1074/jbc.M501920200. [DOI] [PubMed] [Google Scholar]
- Vargas MR, Pehar M, Cassina P, Beckman JS, Barbeito L. Increased glutathione biosynthesis by Nrf2 activation in astrocytes prevents p75NTR-dependent motor neuron apoptosis. J Neurochem. 2006;97:687–696. doi: 10.1111/j.1471-4159.2006.03742.x. [DOI] [PubMed] [Google Scholar]
- Vijayvergiya C, Beal MF, Buck J, Manfredi G. Mutant superoxide dismutase 1 forms aggregates in the brain mitochondrial matrix of amyotrophic lateral sclerosis mice. J Neurosci. 2005;25:2463–2470. doi: 10.1523/JNEUROSCI.4385-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voloboueva LA, Suh SW, Swanson RA, Giffard RG. Inhibition of mitochondrial function in astrocytes: implications for neuroprotection. J Neurochem. 2007;102:1383–1394. doi: 10.1111/j.1471-4159.2007.4634.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu AS, Kiaei M, Aguirre N, Crow JP, Calingasan NY, Browne SE, Beal MF. Iron porphyrin treatment extends survival in a transgenic animal model of amyotrophic lateral sclerosis. J Neurochem. 2003;85:142–150. doi: 10.1046/j.1471-4159.2003.01639.x. [DOI] [PubMed] [Google Scholar]
- Zimmerman MC, Oberley LW, Flanagan SW. Mutant SOD1-induced neuronal toxicity is mediated by increased mitochondrial superoxide levels. J Neurochem. 2007;102:609–618. doi: 10.1111/j.1471-4159.2007.04502.x. [DOI] [PubMed] [Google Scholar]