

Abstract
Phospholamban is an integral membrane protein that controls the calcium balance in cardiac muscle cells. As the function and regulation of this protein require the active involvement of low populated states in equilibrium with the native state, it is of great interest to acquire structural information about them. In this work we determine the conformations and populations of the ground state and the three main excited states of phospholamban by incorporating nuclear magnetic resonance (NMR) residual dipolar couplings (RDCs) as replica-averaged structural restraints in molecular dynamics simulations. We then provide a description of the manner in which phosphorylation at Ser16 modulates the activity of the protein by increasing the populations of its excited states. These results demonstrate that approach that we describe provides a detailed characterisation of the different states of phospholamban that determine the function and regulation of this membrane protein. We anticipate that the availability of conformational ensembles will provide opportunities for the development of therapeutic strategies to control the activity of phospholamban by modulating the relative populations of its conformational substates.
Introduction
As proteins in solution undergo conformational fluctuations, in addition to the native conformation they can populate other states with lower populations and higher free energies1–13. These excited states are often important in enzymatic reactions and molecular recognition events, as they include the conformations that are selected by ligands or binding partners5–8. NMR spectroscopy can provide detailed information about excited states at atomic level4–7. For instance, structural properties of these states can be obtained by the analysis of resonance line widths or by nuclear spin relaxation measurements4, 6, 9, 10. Another powerful approach exploits the introduction of partially aligned samples, which enables one to reintroduce anisotropy in the NMR observables and determine simultaneously the structure and dynamics of proteins14–17. The measurement of residual dipolar couplings (RDCs) makes it possible not only to characterize the ground states of the macromolecules but also to gain access to the structural features of excited states10, 18. This approach was recently illustrated in the case of membrane proteins by the characterization of the pH-triggered activated-state conformations of the influenza hemagglutinin fusion peptide11.
In this work, we apply this strategy to an integral membrane protein and determine the structures of the ground and excited states of phospholamban (PLN). PLN is a type-II membrane protein that in its unphosphorylated form binds and inhibits the sarcoplasmic reticulum Ca2+-ATPase (SERCA)19. PLN, which exists as a pentamer in the sarcoplasmic reticulum, disassembles into monomers to interact with SERCA19–21. When phosphorylated by protein kinase A (PKA) at Ser16, PLN inhibition is reversed, and the apparent Ca2+ affinity of SERCA increases without dissociating the PLN/SERCA complex22, 23. Dephosphorylation of PLN by protein phosphatase 1 ensures the cyclical regulation of this endogenous inhibitor19. PLN mutations are associated with the progression of heart failure24–26. Also, over-expression of a pseudo-phosphorylated form of PLN relieves the effect of heart failure in animal models by increasing SERCA function, making the SERCA/PLN complex a promising therapeutic target27, 28
Nuclear spin relaxation measurements revealed that PLN adopts in equilibrium an ensemble of conformations, and identified four domains within the protein29, 30: the amphipathic α-helix domain Ia (residues 1–16), the loop region (residues 17–22), the juxtamembrane domain Ib (residues 23–31), and the transmembrane domain II (residues 32–52). Domains Ib and II form a continuous α-helix that crosses the bilayer toward the lumen at a 24° angle with respect to the bilayer normal31. This angle is reduced to 11° in the pentameric form of PLN, which is believed to act as a storage state32, 33. Domain Ia and the loop region face the cytoplasm. Studies carried out in membrane-mimicking systems indicated that there are four different conformational states for the cytoplasmic region of PLN34–37: the T state, in which domain Ia is associated to the membrane, the T′ state, which is membrane associated with a partial unfolding of the α-helix in domain Ia, the R′ state, which is also membrane associated but unfolded, and the R state, which is completely unfolded and dissociated from the membrane34–37.
The NMR structure of the T state of PLN in its monomeric form in dodecylphosphocholine (DPC) micelles was recently determined by using interatomic distance restraints from 15N-edited NOESY spectra, dihedral angle restraints from chemical shifts, and hydrogen bond information from H/D exchange factors38. These structures were further refined with RDC data, which were augmented with paramagnetic relaxation enhancement (PRE) measurements to reduce the degeneracy from the RDC solutions and remove the ambiguity in the translational degree of freedom between the solution and membrane domains39. The structure of the T state in DPC micelle is similar to that of the same state in lipid bilayers, which was also recently determined by using a hybrid method in which solution NMR restraints from DPC micelles were combined with solid-state NMR restraints40, 41.
Increasing evidence indicate that the R state plays a central role in determining the function of PLN, as this is the state selected by PKA for phosphorylation, and its population is increased upon interaction with SERCA36, 42, 43. More importantly, phosphorylation-induced relief of SERCA inhibition has been found to be directly correlated to an increase in the population of excited (T′, R′ and R) states34, 44, 45. Therefore, PLN mutants with phosphomimetic mutations promoting the population of excited states are promising candidates for gene therapy28, 46, 47. As understanding the nature of the conformational equilibrium between the R and T states and the mechanism of the transition between them will help develop new therapeutic approaches to defective contractility cardiac disorders26, 46, it is important to determine the structures of these excited states and to characterise their conformational fluctuations. To achieve this goal, in this work we have defined the free energy landscape of PLN using an approach in which RDCs are used as replica-averaged structural restraints in molecular dynamics simulations48, 49. In this approach, the RDCs are calculated during the simulations using a structure-based method49 as averages over multiple replicas of the protein48, 49 in order to extract effectively the information about conformational fluctuations carried by the RDC data. Our results show that the cytoplasmic region of PLN, comprising domain Ia and the loop, explores a broad conformational space with four well-defined free energy minima, which correspond to the four states proposed in previous studies34–36. The analysis that we carried out further indicates that the statistical weights of these minima are shifted upon phosphorylation at Ser16 with a promotion of PLN to the excited states, in which domain Ia becomes more dynamic and unfolded.
Materials and Methods
Sample preparation
In this work we used AFA-PLN, a fully functional monomeric variant of PLN. AFA-PLN, which has three mutations in the transmembrane helix (C36A, C41F and C46A) with respect to the wild-type was recombinantly expressed and purified from E. coli as described previously50. A fourth mutation (S16E) was introduced by site-directed mutagenesis as described previously44. Phosphorylation at Ser16 was obtained by recombinantly expressed catalytic subunit of PKA to a 1:2000 PKA:PLN ratio as previously described34, 51. Acrylamide gels for RDC measurements were polymerized in glass tubes (5.7 mm inner diameter) from a mixture of 5.11% (w/v) acrylamide, 0.13% (w/v) bis-acrylamide, 0.1% (w/v) ammonium persulfate, 0.0031% (w/v) TEMED and 100 mM Tris-HCl at pH 8. For polymerization of negatively charged gels 25% of the acrylamide was substituted with 2-(acrylamido)-2-methyl-1-propanesulfonic acid (AMPS). The gels were washed twice in 50 mM NaH2PO4/Na2HPO4 buffer pH 6.5 and twice in ddH2O and subsequently cut into 10 mm pieces which were dehydrated at 42°C for 16h. NMR samples were prepared by reconstituting pS16-AFA-PLN into 100 mM dodecylphosphocholine (DPC) in buffer (6 M Guanidinium Hydrochloride (Gdn HCl), 20 mM NaHPO4, 120 mM NaCl, 0.01% NaN3, pH 6.0) to a protein concentration of 0.5 mM. The Gdn HCl was subsequently removed by dialysis against the same buffer (without Gdn HCl). For anisotropic samples, a dried acrylamide gel was rehydrated in protein sample for 16 h at 37 °C. A gel stretching apparatus (New Era Enterprises Inc)52 was used to transfer the gel into an open-ended NMR tube rendering the gel to stretch to about 1.8 times its original length. The RDC data obtained for PLN weakly aligned in charged and uncharged gels are reported in Tables S1 and S2. The two sets of RDCs are fairly independent, as indicated by their Q factor of 1.13 (RMSD 5.39 Hz, Fig. S1).
NMR experiments
NMR experiments were conducted at 310 K on a Varian Inova spectrometer operating at a proton frequency of 599.548 MHz. 15N-1H and 13C′-15N RDCs were measured as the difference in splitting between isotropic (no gel, J) and anisotropic (stretched gel, J + RDC) samples from 2D TROSY based experiments53. 1704 complex points were acquired in the direct 1H dimension and 80 increments in the indirect 15N dimension with spectral widths of 10000 Hz and 1200 Hz for 1H and 15N respectively. Zero-filling was performed to a final matrix size of 16384×8192 points. 13C′-13Cα couplings were measured from uncoupled 3D HNCO experiments as the difference in J-splitting between isotropic and anisotropic samples. Experiments were acquired with 1664 points in the direct 1H dimension, and 40 and 32 increments in the 13C and 15N dimensions. The spectral widths were 10000 Hz, 1000 Hz and 1200 Hz for 1H, 13C and 15N, respectively. A recycle delay of 1.2 s was used for all experiments. All data was processed in NMRPipe and analyzed by Sparky.
Modeling of the PLN/DPC-micelle system
Molecular dynamics simulations with replica-averaged RDC restraints48, 49 (see below) were performed starting from one of the conformers of the NMR structural ensemble of PLN reconstituted in DPC micelles, which was previously obtained using NOEs, RDCs and PREs restraints incorporated into a simulated annealing protocol using XPLOR-NIH39. The NOE and PRE data, however, were not used as restraints in the molecular dynamics simulations described below. One structure from this ensemble was inserted to an equilibrated DPC micelle with 60 lipid molecules; the overall results of the sampling, however, did not depend on the choice of the initial structure, as it should be expected at convergence. The initial coordinates of the DPC micelle were obtained from T. C. Wong54. The size of the DPC micelle (19.8 ± 1.9 Å) was chosen to match the size estimated by SAXS measurements. The solvated DPC micelle was equilibrated for 0.5 ns before the insertion of PLN. A cylindrical space within the DPC micelle was created to accommodate for the transmembrane domain of PLN. PLN and the DPC micelle were then brought together and subjected to minimization and equilibration.
Molecular dynamics simulations were carried out using a modified version of the GROMACS package55 that implements RDC as structural restraints48, 49, 56 by using the Amber99SB force field57 with improved parameters for backbone58 and side-chains59. DPC parameters were derived by Tieleman et al.60. The system composed by PLN in a DPC-micelle was inserted in a cubic box of starting dimensions 70×70×70 Å3, and solvated with 9298 explicit TIP3P water molecules61. Bonds were constrained by the LINCS algorithm62. The particle-mesh Ewald (PME) method63 was used to account for the electrostatic contributions to non-bonded interactions with a grid spacing of 0.12 nm. The protonation states of pH-sensitive residues were set as follows: Arg and Lys as positively charged, Asp and Glu as negatively charged, and His as neutral. The net charge of the system was neutralized by the addition of Cl− and Na+ ions. The system was equilibrated with external temperature and pressure baths (NPT ensemble) by using the v-rescale64 and Berendsen65 algorithms, respectively, and coupling time steps of 0.1 ps and 1.0 ps, respectively.
Molecular dynamics simulations with replica-averaged RDC restraints
The use of NMR parameters as replica-averaged structural restraints in molecular dynamics simulations offers the possibility of interpreting the experimental measurements in terms of the maximum entropy principle66–68, and therefore, in the case presented in this work, to translate the RDC measurements into structural ensembles that represent the Boltzmann distributions of PLN and pS16-PLN. In this approach, the force field used in the molecular dynamics simulations is modified through the incorporation of the experimental information to modify the force field in the minimal manner that enables one to eliminate almost completely the deviations from the experimental data66–68.
Molecular dynamics simulations with replica-averaged RDC restraints were performed to determine the structural ensembles of PLN and pS16-PLN by adopting a structure-based calculation of the alignment tensor that accounted for the interaction between the PLN/micelle system and the alignment media48, 49.
In the approach that we used, the RDC restraints are imposed by adding a pseudo-energy term (ERDC) to a standard molecular mechanics force field (EFF)
| (1) |
The resulting force field (ETotal) is employed in molecular dynamics simulations, where the pseudo-energy term is given by
| (2) |
The restraints are imposed as averages over 16 replicas of the system48, 49. Although the maximum entropy principle justification for the use of replica-averaged restraints in principle holds for an infinitely large number of replicas66, 68, in practice excellent results are already obtained for relatively small numbers such as those used here48, 49, 66, 68. From previous studies we estimate that the errors in the determination of the populations should be below 4%48, 49. We also note that the number of replicas should be chosen according to arguments such as the convergence of the Shannon entropy (see e.g. Fig. 2 in Ref. 66), which are independent from the number of free energy minima of the system under investigation. For each replica the alignment tensors are independently computed48, 49. To save computational time, the tensors are computed individually on each replica every 250 steps, as they do not vary significantly over shorter intervals48, 49. An initial equilibration simulation at 310 K (the temperature at which PLN is active in its physiological environment, which as also the temperature at which the RDCs were recorded39) was run during which the agreement between calculated and experimental RDCs was allowed to converge to by progressively raising the weight α of the restraints. Subsequently, a series of 50 cycles of simulated annealing between 310 K and 500 K were carried out to sample the region of conformational space compatible with the RDC restraints. Each annealing cycle was carried out for a total of 8 ns (500 ps per replica) by using an integration step of 1 fs. After equilibration at 310 K, the final 100 ps of each annealing cycle were used to compute the equilibrated ensemble of 9600 structures by sampling the conformers from each replica at every 5 ps. The initial 20 cycles were discarded from the analyses. During each cycle, typically each replica explores (with different frequencies) the four minima on the free energy landscape.
The molecular dynamics trajectories for both PLN and pS16-PLN were generated by employing as structural restraints the RDC data measured in neutral gels (Fig. 1), while those measured in charged gels for were used for validation. The conformations of PLN in charged and uncharged gels are virtually identical, as illustrated by the overlay of the protein 1H-15N HSQC spectra spectra in the two alignment media (Fig. S2).
Figure 1.

A) Comparison of the residual dipolar couplings of PLN (black) and pS16-PLN (red) measured in stressed polyacrylamide gels. B) Correlation between the average RDCs of domain II and the PLN-induced inhibition of SERCA, as assessed by the ΔpKCa values34 (see Materials and Methods) for PLN, pS16-PLN and S16E-PLN. C) Correlation between the standard deviations of the RDCs of domain II and the PLN-induced inhibition of SERCA, assessed as in panel (B).
As the approach that we used in this work was previously used only for globular proteins48, 49, in order to test its robustness in the case of membrane proteins solubilized in detergent micelles, we applied it to DsbB, a multispan membrane proteins reconstituted in DPC micelles and aligned in positively charged gels69. By using the available RDCs69 as replica-averaged structural restraints as described above, we found that the native state of DsbB that we determined is characterized by conformational fluctuations of smaller amplitude than PLN (Fig. S3A) and an overall remarkable agreement between experimental and calculated RDC values (Fig. S3).
Results and Discussion
Substate structure determination
In the free energy landscape view of protein behaviour, native states of proteins may often involve an equilibrium between different substates in rapid conformational exchange1–13. In these cases, the NMR measurement of an observable D reports on the average value of the observable across N substates
| (3) |
where k runs over the N substates, which have populations pk and values Dk of the observable D. In the case of a single substate (N=1), this problem corresponds to the standard structure determination problem, in which one determines the structure of a protein given a set of experimental data. More generally, however, the number N of substates is not known in advance and should also be determined from the data.
A question of great interest is whether, in the presence of multiple substates, given only the average value D one can determine the number of substates, their structures and the values of their populations. Although in principle a solution of this problem must exist, which corresponds to the true N, pk and Dk values, it remains to be established under which conditions, if at all, one can overcome the degeneracy of the solution, as multiple sets of N, pk and Dk values may correspond to a given D in Eq. (3).
Initial evidence that, at least in the case of two substates (N=2), there are approaches that can identify non-degenerate solution, was provided in a study in which two conformational substates of ribonuclease A were determined simultaneously from RDC data56 or from chemical shift data70. These results were obtained by using the NMR data as replica-averaged structural restraints in molecular dynamics simulations, as this procedure represents an optimal way to generate structural ensembles according to the maximum entropy principle, which in practice means that given a force field and a set of experimental data, this procedure results in an ensemble of conformations most compatible with both the force field and the experimental data, without making any further assumption.
In order to validate the results, in favourable cases, one can measure directly the NMR parameters of the substates, i.e. the Dk values, and thus verify directly whether the substate structure determination procedure has been carried out correctly. A most powerful approach to achieve this result is to use relaxation dispersion methods, which have been used to determine directly the structures of excited states of proteins with low populations that exchange on the millisecond timescale with the most populated state4, 9, 10. It should also be noted that a very important validation is represented by the value of N, since this value may be known experimentally through a variety of means. If the procedure results in a value of N consistent with independent observations, one has at least an indication of the validity of the calculations.
In this work we apply the substate structure determination approach to find the substates of phospholamban using residual dipolar couplings as replica-averaged structural restraints in molecular dynamics simulations, as the initial evidence that we have acquired indicates that this procedure may represent an effective implementation to solve the substate structure determination problem.
Residual dipolar couplings of PLN and pS16-PLN
We studied the conformational properties of PLN in the monomeric state, which was stabilised by mutating three transmembrane Cys residues (C36A, C41F, C46A)71; the resulting mutational variant has an activity identical to that of wild-type PLN71. Relaxation measurements of PLN identified four domains with different backbone dynamics29. These four domains also reflect specific patterns in the 15N-1H RDC profiles with the region spanning domain Ia exhibiting positive RDC values with an average of 2.14 Hz and a standard deviation of 3.00 Hz, whereas the flexible region covering the loop and the domain Ib presents slightly negative RDC values with an average of −1.16 Hz and a standard deviation of 4.31 Hz39 (Fig. 1). A different RDC pattern is associated with the membrane-spanning domain II, which shows stronger positive RDC values with an average of 6.52 Hz and a standard deviation of 7.90 Hz39 (Fig. 1). The errors on the 15N-1H RDCs were determined as the standard error of three measurements using three separate sample preparations. For the 13C′-15N and 13C′-13Cα RDCs the errors were estimated as
| (4) |
where Δυani and Δυiso are the anisotropic (in stretched gels) and isotropic (with no gels) linewidths, respectively, and σani and σiso are the anisotropic and isotropic signal-to-noise ratios, respectively.
RDC measurements of a phosphorylated form of PLN (pS16-PLN) show significant variations within domain II with a substantial reduction of the average and standard deviation (2.46 Hz and 5.49 Hz, respectively, Fig. 1). These findings indicate that phosphorylation of Ser16 has strong effects on the structure and dynamics of PLN, which is consistent with previous studies51. Since Ser16 phosphorylation does not affect the dynamics of domain II, the reduced intensities of 15N-1H RDCs likely arise from an increased dispersion of the mutual orientations of the dynamic domains of PLN.
It has also been previously established that the pseudo-phosphorylation of PLN at Ser16, as realized by the S16E mutation, leads to partial loss of SERCA inhibition34. Consistently, the RDCs for the S16E mutant are intermediate between those of PLN and pS16-PLN (Fig. 1B), with average and standard deviation in domain II of 5.18 and 5.38 Hz, respectively (Fig. 1B,C). The approximately linear correlation between RDCs and inhibitory function (Fig. 1B), which was measured by the ΔpKCa values34 (determined as pKCaSERCA-pKCaSERCA+PLN), provides further support to the view that the RDCs are sensitive to structural fluctuations that are integral to PLN function.
Determination of the structures of the four substates of PLN
To account for the conformational fluctuations of PLN, RDCs corresponding to 15N-1H, 13C′-15N and 13C′-13Cα bond vectors were used as replica-averaged structural restraints in molecular dynamics simulations48, 49 to determine a structural ensemble of this protein (see Materials and Methods). The calculations were started from a structure previously determined by NOEs, RDCs and PREs39 (Fig. 2a), although the NOEs and PREs were not used as restraints in the calculations themselves.
Figure 2.
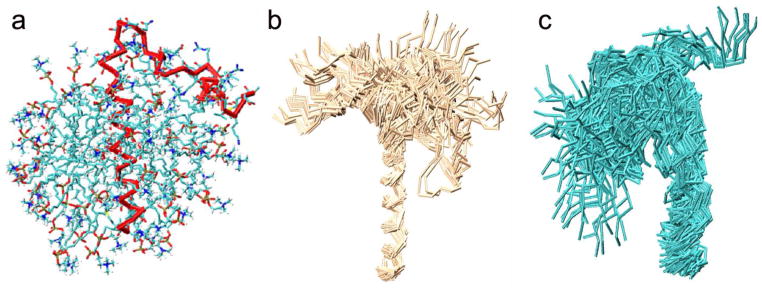
Structural ensembles of PLN and pS16-PLN. (a) Starting model of PLN in a DPC micelle. (b) Ensembles of conformations of PLN. (c) Ensembles of conformations of pS16-PLN.
Several methods have been proposed to exploit the information provided by RDC measurements to characterise the dynamics of proteins49, 56, 72–78. Although the majority of these approaches have been used to assess conformational fluctuations of relatively small amplitude, we have recently shown that RDCs can be employed to describe large-scale structural dynamics as those associated with the functional fluctuations of enzymes48, 49. The method that we used is based on the treatment of RDCs as replica-averaged restraints in molecular dynamics simulations in which the RDCs are calculated from the shape and charge of each individual structure in the ensemble79. By using this method we have shown that it is possible to reproduce the conformational equilibria among different states populated by proteins48, 49.
The structural ensemble of PLN that we obtained was determined by enforcing an agreement between experimental and back-calculated RDC values (Fig. S4), resulting in Q factors of 0.12, 0.16 and 0.16 for 15N-1H, 13C′-15N and 13C′-13Cα bond vectors, respectively. In order to validate this ensemble, we used it to back-calculate the values of NMR parameters that were not used as restraints in the structure calculations. First, we used RDC data recorded in negatively charged acrylamide/bis acrylamide gels (Fig. S5A,B); these data were not employed as restraints in the calculations, thereby providing an independent assessment of the quality of the structures obtained. By using a structure-based alignment method to back-calculate the RDC values48, 49 (see Materials and Methods), the ensemble results in an excellent agreement with charged-gels RDC values. We found, however, a poorer agreement when we employed an alternative method based on the fitting of the experimental RDC values to the structures (the SVD method80, Fig. S6). These findings underscore the importance of using a structured-based method of alignment in the presence of large conformational fluctuations of proteins. In the structure-based approach that we followed48, 49 (see Materials and Methods), none of the structures of the ensemble is required to match individually the experimental data, because the values of the latter are averaged over a heterogeneous ensemble of structural states and cannot be attributed to single conformation48, 49. By contrast, in the SVD method, the alignment tensor of individual conformations is fitted by requiring an optimal match with the RDC data, a procedure that may not be very accurate if the RDC data themselves refer to an ensemble of structurally different conformations48. The calculations carried out by applying the RDC restraints to single conformations resulted in poorer agreement with the RDC measurements (Fig. S7). Further validation of the structural ensemble that we determined was obtained by comparing experimental and back-calculated chemical shifts, which resulted in a very good agreement (Fig. S5c).
One of the main characteristics of the structural ensemble that we determined is the presence of a well-defined C-terminal α-helical domain in the hydrophobic region of the micelle. This α-helix, which exhibits an average RMS fluctuation of 1 Å on the Cα atoms, does not resemble the curved shape that was reported previously for pentameric PLN81, but is in agreement with the conformation of the same pentameric PLN derived from solid-state NMR measurement in lipid bilayers32 and from NOEs, RDCs and PREs39 for the AFA-PLN monomer in micelles. This difference arises because of the averaging procedure that we used in this work, which accounts for the conformational fluctuations of PLN without assuming the presence of a well-defined average structure of the entire molecule (Fig. 1b). The structures that we obtained indicate that the N-terminal region of PLN is highly heterogeneous and assumes a large variety of orientations on the micelle surface compared to the C-terminal α-helical frame (Fig. 1b). The analysis of the structural ensemble shows that the C-terminal domain II adopts a stable α-helical conformation (Fig. 3a), while the N-terminal domain Ia adopts a less populated α-helical conformation that deviates from the canonical α-helical parameters (Fig. 3a).
Figure 3.

Comparison of the residue-specific α-helical populations of PLN and pS16-PLN. Black and red colors indicate PLN and pS16-PLN, respectively. The location of the four domains of PLN is indicated in the top of each panel. A) Local root mean square deviation (RMSD) from an ideal α-helix. The RMSD is calculated by using a running 4-residue window. The RMSD of the window is assigned to the average position of the residues. B) α-helix populations calculated by using δ2D82, by employing the measured chemical shifts of PLN and pS16-PLN. The errors in the per-residue populations estimated with the δ2D method has been reported to be below 8%82.
Validation of the structures of PLN using chemical shifts
The α-helical features described above are in good agreement with the conformational analysis carried out with δ2D program82, which employs chemical shifts for an accurate determination of secondary structure populations (Fig. 3b). The correspondence between the results obtained independently using either RDC information (Fig. 3a) or chemical shift information (Fig. 3b) about the α-helical features of PLN provides support for the validity of the conformations that we determined for PLN.
As the structures of the four substates of PLN that we have determined were obtained using RDC values averaged over all of them (see Eq. (3)), a stringent validation would be provided by comparing the structures with independent experimental data measured specifically for the individual substates. As these substates, however, are in fast exchange, it is very challenging to perform such measurements. We have therefore exploited the observation that the population of the R state can be progressively increased by altering the sequence of PLN, either by mutations or by truncations. In particular, we compare here the chemical shifts back-calculated for the T and R states, which were calculated as averages over all the conformations of the two states, respectively and those experimentally measured for a peptide corresponding to the cytoplasmic domain of PLN, which is unstructured42. The results indicate that the cytoplasmic domain in the R state has chemical shifts intermediate between those of the T state, which is ordered, and of an unstructured state (Fig. 4).
Figure 4.

Comparison of the chemical shifts back-calculated from the structures of the T and R states, respectively, with those experimentally measured42 for an unstructured peptide corresponding to the cytoplasmic domain of PLN. The analysis the differences between the chemical shifts of the T state and the peptide (green), and between the R state and the peptide (orange) indicates that the R state exhibits a degree of structural order intermediate between that of the T state and of the unstructured peptide.
Effect of phosphorylation on the conformational properties of PLN
In order to characterise the structural effects of phosphorylation at Ser16 on the backbone dynamics of PLN, we repeated the structure calculations using RDC measured on the pS16-PLN variant. The resulting ensemble of conformations had Q factors of 0.09, 0.10 and 0.11 for 15N-1H and 13C′-15N and 13C′-13Cα, respectively. Very good agreement is also found for the validation of the ensemble with independent data of RDC measured in charged gels and chemical shifts (Fig. S5). The calculations were performed by using the same protocol as employed for PLN, but with a phosphoserine at position 16.
Our findings indicate that pS16-PLN exhibits conformational fluctuations of larger amplitude than those of PLN. These fluctuations significantly increase in particular in the C-terminal α-helix (domain II) with an average RMS fluctuation of 2.24 Å (Fig. 3). The orientations of the N-terminal domain compared to the C-terminal frame are significantly more dispersed than in the PLN ensemble (Fig. 2c) with a decrease in the α-helical content of the N-terminal helix (domain Ia), which is now populated at 40% (Fig. 3). Hence, the structural ensemble of pS16-PLN suggests that phosphorylation at Ser16 dramatically alters the conformational fluctuations of PLN and destabilises the secondary structure elements.
Free energy landscapes of PLN and pS16-PLN
By projecting the structural ensembles onto specific order parameters it is possible to identify the main features of the free energy landscapes of PLN and pS16-PLN. We choose two order parameters that account for independent characteristics of the PLN ensembles. As the present study is aimed at the characterization of structural states connected to folding and unfolding events of PLN, as well as its adsorption and detachment from the surface of micelles, we chose as reaction coordinates the number of residues in α-helical conformations at the N-terminus (domains Ia and Ib) and the number of contacts between the N-terminal region of the protein (residues 1–30) and the micelle (Fig. 5). These coordinates were chosen because they report on the most relevant conformational features that distinguish the R and T states. This projection identifies four structural forms closely resembling the four PLN states previously proposed based on chemical shift analysis and EPR methods34, and provides molecular details of these low-populated conformational that are crucial for PLN function.
Figure 5.

Comparison of the free energy landscapes of PLN and pS16-PLN. The free energy is given as a function of the number of contacts among backbone atoms of the N-terminal region of the protein (residues 1–30) and DPC molecules and of the number of α-helical residues in the Ia and Ib domains. Upon phosphorylation at Ser16 the R and R′ states increase in population. A representative structure of the R state has been deposited in the PDB (2LPF).
The most populated free energy basin of the PLN ensemble presents an α-helical structure in the Ia and Ib domains and a large number of contacts with the micelle surface (Fig. 5); this state exhibits the structural features of the T state of PLN. A second basin, corresponding to the T′ state, presents a large number of contacts with the micelle but reduced α-helical content (Fig. 5). A minor basin, R′, still presents a large fraction of contacts with the micelle but lacks any secondary structure element within the Ia and Ib domains (Fig. 5). Such an unfolded state corresponds to the R′ state as previously hypothesized based on chemical shifts analysis34. Finally, a low-populated state in PLN does not show secondary structure formation at the N-terminal region of the protein and has a reduced number of contacts with the micelle. This conformation represents an excited state, the R state, in which the N-terminus of the protein is unfolded and exposed to the bulk solvent (Fig. 5).
We also repeated the molecular dynamics simulations described above without RDC restraints. This procedure resulted in very different free energy landscapes exhibiting only two substates (Fig. S8). Therefore this procedure would give N=2 in Eq. (3) instead of N=4, which is known independently to be the case34–37. These results demonstrate that the appearance of the T′, R′ and R states of PLN is not a consequence of the force field used in the molecular dynamics simulations, but of the replica-averaged RDC restraints added to the force field itself. We conclude that our method samples more accurately the different conformational states of proteins and that procedure we followed is capable of reproducing statistical weights of metastable states and conformational equilibria in proteins48, 49.
We then repeated the analysis for pS16-PLN. Our analysis indicates that phosphorylation at Ser16 induces a redistribution of the Boltzmann weights of the four states of PLN (Fig. 5). We found that the T state of pS16-PLN has a decreased population (37%) with respect to the unphosphorylated form (55%), and that the R′ and R states significantly increase in population (from 7% to 13% and from 4% to 12%, respectively). The result that the R state trebles its population upon phosphorylation is consistent with previous NMR34 and EPR36 results. Also, the increase in the population of the excited state is directly correlated to the loss of inhibitory function upon phosphorylation34, 36, 44. The reduction in the number of contacts for the R state is more pronounced for pS16-PLN, indicating a higher tendency of the N-terminal region to detach from the micelle surface (Fig. 6). This finding could arise from charge repulsion introduced upon phosphorylation.
Figure 6.

Comparison of the R states of PLN and pS16-PLN. The R state is nearly three-fold more populated in pS16-PLN (red curve) than in PLN (black curve), and is characterised by a smaller number of contacts between the cytoplasmic residues (domain Ia, loop and domain Ib), and the micelle surface.
In summary, we identified four structural states in conformational equilibrium within the heterogeneous ensemble of PLN in the presence of micelles, thus providing information on the structural features of the conformational states that are crucial for the biological activity of PLN, although in the presence of lipid bilayers some details, in particular the populations of the different states, may be expected to be different. In this context, the redistribution of the populations of these four states upon phosphorylation illustrate the type of conformational equilibria that dictate the biological properties of this protein.
Conclusions
The conformational equilibria between the different states of PLN are particularly relevant to determine the biological function of this protein. In the absence of a membrane, the cytoplasmic domains of PLN remain intrinsically disordered, while membranes and membrane-mimicking systems induce them to form more ordered states (the T and T′ states). The various conformational states of PLN have different activities, as indicated by biochemical assays, NMR and EPR studies, which suggest that the conformational fluctuations of PLN have a prominent role in the regulation of the activity of SERCA. Through these fluctuations PLN occupies more disordered states (R and R′), whose characterization at the atomic level has been very challenging because of their low populations. In this study, we have demonstrated that the incorporation of residual dipolar couplings as replica-averaged structural restraints in molecular dynamics simulations provides an accurate free energy landscape of PLN, and enables one to characterize the four basins corresponding to the T, T′, R and R′ states, which were previously identified by chemical shift trajectories in unfolding experiments34, and to determine the corresponding conformations at atomic-level resolution.
These results open the possibility to correlate the population levels of the excited states of PLN with the biological function of this protein. We have recently shown that populating the R state is a condition for PLN to be selected by protein kinase A for phosphorylation42. When the equilibrium is shifted toward the T state, the phosphorylation efficiency drops dramatically, whereas an increase in the population of the R state raises the rate of phosphorylation at Ser16. Since the cytoplasmic domain of PLN interacts with at least seven different partners19, we anticipate that these proteins may select and bind different members in the structural ensemble of PLN.
The results that we have presented suggest novel opportunities for the identification of novel loss-of-function (dominant-negative) PLN analogs, which have been proposed as a possible therapeutic strategy for defective contractility cardiac disorders26, 46. Support for this view has been provided recently by the demonstration that it is possible to tune the conformational fluctuations of PLN to regulate the function of SERCA and in turn muscle contractility34, 44. An increase in the population of the R state of PLN by site-specific mutations localized in the dynamic loop or using phosphomimetic mutations (S16E and S16D) was shown to lead to an enhancement of the apparent Ca2+ affinity of SERCA, with significant benefits for cardiac contractility. We anticipate that the availability of the structures of the excited states of PLN determined in this work will help devise even more effective dominant-negative mutants and identify small molecules capable of regulating the conformational equilibria of PLN.
Supplementary Material
Acknowledgments
Funding information
This work was supported by grants from the EPSRC (ADS), EMBO (ADS), Marie Curie (ADS), BBSRC (RWM and MV) and NIH (GM64742 to GV).
Footnotes
Figures S1–S8 and Tables S1–S2. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Frauenfelder H, Sligar SG, Wolynes PG. The energy landscapes and motions of proteins. Science. 1991;254:1598–1603. doi: 10.1126/science.1749933. [DOI] [PubMed] [Google Scholar]
- 2.Dobson CM. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 3.Karplus M, Kuriyan J. Molecular dynamics and protein function. Proc Natl Acad Sci USA. 2005;102:6679–6685. doi: 10.1073/pnas.0408930102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neudecker P, Robustelli P, Cavalli A, Walsh P, Lundstrom P, Zarrine-Afsar A, Sharpe S, Vendruscolo M, Kay LE. Structure of an intermediate state in protein folding and aggregation. Science. 2012;336:362–366. doi: 10.1126/science.1214203. [DOI] [PubMed] [Google Scholar]
- 5.Boehr DD, Nussinov R, Wright PE. The role of dynamic conformational ensembles in biomolecular recognition. Nat Chem Biol. 2009;5:789–796. doi: 10.1038/nchembio.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baldwin AJ, Kay LE. NMR spectroscopy brings invisible protein states into focus. Nat Chem Biol. 2009;5:808–814. doi: 10.1038/nchembio.238. [DOI] [PubMed] [Google Scholar]
- 7.Kalodimos CG. NMR reveals novel mechanisms of protein activity regulation. Protein Sci. 2011;20:773–782. doi: 10.1002/pro.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vendruscolo M. Excited-state control of protein activity. J Mol Biol. 2011;412:153–154. doi: 10.1016/j.jmb.2011.07.047. [DOI] [PubMed] [Google Scholar]
- 9.Bouvignies G, Vallurupalli P, Hansen DF, Correia BE, Lange O, Bah A, Vernon RM, Dahlquist FW, Baker D, Kay LE. Solution structure of a minor and transiently formed state of a T4 lysozyme mutant. Nature. 2011;477:111–114. doi: 10.1038/nature10349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Korzhnev DM, Religa TL, Banachewicz W, Fersht AR, Kay LE. A transient and low-populated protein-folding intermediate at atomic resolution. Science. 2010;329:1312–1316. doi: 10.1126/science.1191723. [DOI] [PubMed] [Google Scholar]
- 11.Lorieau JL, Louis JM, Schwieters CD, Bax A. pH-triggered, activated-state conformations of the influenza hemagglutinin fusion peptide revealed by NMR. Proc Natl Acad Sci USA. 2012;109:19994–19999. doi: 10.1073/pnas.1213801109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marsh JA, Teichmann SA, Forman-Kay JD. Probing the diverse landscape of protein flexibility and binding. Curr Op Struct Biol. 2012;22:643–650. doi: 10.1016/j.sbi.2012.08.008. [DOI] [PubMed] [Google Scholar]
- 13.Guerry P, Salmon L, Mollica L, Roldan JLO, Markwick P, van Nuland NAJ, McCammon JA, Blackledge M. Mapping the population of protein conformational energy sub-states from NMR dipolar couplings. Angew Chem Intl Ed. 2013;52:3181–3185. doi: 10.1002/anie.201209669. [DOI] [PubMed] [Google Scholar]
- 14.Tjandra N, Bax A. Direct measurement of distances and angles in biomolecules by NMR in a dilute liquid crystalline medium. Science. 1997;278:1111–1114. doi: 10.1126/science.278.5340.1111. [DOI] [PubMed] [Google Scholar]
- 15.Tolman JR, Flanagan JM, Kennedy MA, Prestegard JH. NMR evidence for slow collective motions in cyanometmyoglobin. Nat Struct Biol. 1997;4:292–297. doi: 10.1038/nsb0497-292. [DOI] [PubMed] [Google Scholar]
- 16.Bax A. Weak alignment offers new NMR opportunities to study protein structure and dynamics. Protein Sci. 2003;12:1–16. doi: 10.1110/ps.0233303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blackledge M. Recent progress in the study of biomolecular structure and dynamics in solution from residual dipolar couplings. Prog Nucl Mag Res Spec. 2005;46:23–61. [Google Scholar]
- 18.Vallurupalli P, Hansen DF, Kay LE. Structures of invisible, excited protein states by relaxation dispersion NMR spectroscopy. Proc Natl Acad Sci USA. 2008;105:11766–11771. doi: 10.1073/pnas.0804221105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.MacLennan DH, Kranias EG. Phospholamban: A crucial regulator of cardiac contractility. Nat Rev Mol Cell Biol. 2003;4:566–577. doi: 10.1038/nrm1151. [DOI] [PubMed] [Google Scholar]
- 20.Cornea RL, Autry JM, Chen ZH, Jones LR. Reexamination of the role of the leucine/isoleucine zipper residues of phospholamban in inhibition of the ca2+ pump of cardiac sarcoplasmic reticulum. J Biol Chem. 2000;275:41487–41494. doi: 10.1074/jbc.M008195200. [DOI] [PubMed] [Google Scholar]
- 21.Cornea RL, Jones LR, Autry JM, Thomas DD. Mutation and phosphorylation change the oligomeric structure of phospholamban in lipid bilayers. Biochemistry. 1997;36:2960–2967. doi: 10.1021/bi961955q. [DOI] [PubMed] [Google Scholar]
- 22.Wegener AD, Simmerman HKB, Lindemann JP, Jones LR. Phospholamban phosphorylation in intact ventricles - phosphorylation of serine-16 and threonine-17 in response to beta-adrenergic stimulation. J Biol Chem. 1989;264:11468–11474. [PubMed] [Google Scholar]
- 23.James ZM, McCaffrey JE, Torgersen KD, Karim CB, Thomas DD. Protein-protein interactions in calcium transport regulation probed by saturation transfer electron paramagnetic resonance. Bioph J. 2012;103:1370–1378. doi: 10.1016/j.bpj.2012.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmitt JP, Kamisago M, Asahi M, Li GH, Ahmad F, Mende U, Kranias EG, MacLennan DH, Seidman JG, Seidman CE. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science. 2003;299:1410–1413. doi: 10.1126/science.1081578. [DOI] [PubMed] [Google Scholar]
- 25.Haghighi K, Kolokathis F, Gramolini AO, Waggoner JR, Pater L, Lynch RA, Fan GC, Tsiapras D, Parekh RR, Dorn GW, MacLennan DH, Kremastinos DT, Kranias EG. A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proc Natl Acad Sci USA. 2006;103:1388–1393. doi: 10.1073/pnas.0510519103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haghighi K, Kolokathis F, Pater L, Lynch RA, Asahi M, Gramolini AO, Fan GC, Tsiapras D, Hahn HS, Adamopoulos S, Liggett SB, Dorn GW, MacLennan DH, Kremastinos DT, Kranias EG. Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J Clin Invest. 2003;111:869–876. doi: 10.1172/JCI17892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoshijima M, Ikeda Y, Iwanaga Y, Minamisawa S, Date MO, Gu YS, Iwatate M, Li MX, Wang LL, Wilson JM, Wang YB, Ross J, Chien KR. Chronic suppression of heart-failure progression by a pseudophosphorylated mutant of phospholamban via in vivo cardiac raav gene delivery. Nat Med. 2002;8:864–871. doi: 10.1038/nm739. [DOI] [PubMed] [Google Scholar]
- 28.Kho C, Lee A, Hajjar RJ. Altered sarcoplasmic reticulum calcium cycling-targets for heart failure therapy. Nat Rev Card. 2012;9:717–733. doi: 10.1038/nrcardio.2012.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Metcalfe EE, Zamoon J, Thomas DD, Veglia G. H-1/N-15 heteronuclear NMR spectroscopy shows four dynamic domains for phospholamban reconstituted in dodecylphosphocholine micelles. Bioph J. 2004;87:1205–1214. doi: 10.1529/biophysj.103.038844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Traaseth NJ, Veglia G. Probing excited states and activation energy for the integral membrane protein phospholamban by NMR cpmg relaxation dispersion experiments. Bioch Bioph Acta Biomem. 2010;1798:77–81. doi: 10.1016/j.bbamem.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Traaseth NJ, Buffy JJ, Zamoon J, Veglia G. Structural dynamics and topology of phospholamban in oriented lipid bilayers using multidimensional solid-state NMR. Biochemistry. 2006;45:13827–13834. doi: 10.1021/bi0607610. [DOI] [PubMed] [Google Scholar]
- 32.Verardi R, Shi L, Traaseth NJ, Walsh N, Veglia G. Structural topology of phospholamban pentamer in lipid bilayers by a hybrid solution and solid-state NMR method. Proc Natl Acad Sci USA. 2011;108:9101–9106. doi: 10.1073/pnas.1016535108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Becucci L, Cembran A, Karim CB, Thomas DD, Guidelli R, Gao JL, Veglia G. On the function of pentameric phospholamban: Ion channel or storage form? Bioph J. 2009;96:L60–L62. doi: 10.1016/j.bpj.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gustavsson M, Traaseth NJ, Karim CB, Lockamy EL, Thomas DD, Veglia G. Lipid-mediated folding/unfolding of phospholamban as a regulatory mechanism for the sarcoplasmic reticulum ca(2+)-ATPase. J Mol Biol. 2011;408:755–765. doi: 10.1016/j.jmb.2011.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karim CB, Kirby TL, Zhang ZW, Nesmelov Y, Thomas DD. Phospholamban structural dynamics in lipid bilayers probed by a spin label rigidly coupled to the peptide backbone. Proc Natl Acad Sci USA. 2004;101:14437–14442. doi: 10.1073/pnas.0402801101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karim CB, Zhang ZW, Howard EC, Torgersen KD, Thomas DD. Phosphorylation-dependent conformational switch in spin-labeled phospholamban bound to serca. J Mol Biol. 2006;358:1032–1040. doi: 10.1016/j.jmb.2006.02.051. [DOI] [PubMed] [Google Scholar]
- 37.Gustavsson M, Traaseth NJ, Veglia G. Probing ground and excited states of phospholamban in model and native lipid membranes by magic angle spinning NMR spectroscopy. Bioch Bioph Acta Biomem. 2012;1818:146–153. doi: 10.1016/j.bbamem.2011.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zamoon J, Mascioni A, Thomas DD, Veglia G. NMR solution structure and topological orientation of monomeric phospholamban in dodecylphosphocholine micelles. Bioph J. 2003;85:2589–2598. doi: 10.1016/s0006-3495(03)74681-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shi L, Traaseth NJ, Verardi R, Gustavsson M, Gao JL, Veglia G. Paramagnetic-based NMR restraints lift residual dipolar coupling degeneracy in multidomain detergent-solubilized membrane proteins. J Am Chem Soc. 2011;133:2232–2241. doi: 10.1021/ja109080t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Traaseth NJ, Shi L, Verardi R, Mullen DG, Barany G, Veglia G. Structure and topology of monomeric phospholamban in lipid membranes determined by a hybrid solution and solid-state NMR approach. Proc Natl Acad Sci USA. 2009;106:10165–10170. doi: 10.1073/pnas.0904290106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shi L, Traaseth NJ, Verardi R, Cembran A, Gao JL, Veglia G. A refinement protocol to determine structure, topology, and depth of insertion of membrane proteins using hybrid solution and solid-state NMR restraints. J Biomol NMR. 2009;44:195–205. doi: 10.1007/s10858-009-9328-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Masterson LR, Yu T, Shi L, Wang Y, Gustavsson M, Mueller MM, Veglia G. Camp-dependent protein kinase a selects the excited state of the membrane substrate phospholamban. J Mol Biol. 2011;412:155–164. doi: 10.1016/j.jmb.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zamoon J, Nitu F, Karim C, Thomas DD, Veglia G. Mapping the interaction surface of a membrane protein: Unveiling the conformational switch of phospholamban in calcium pump regulation. Proc Natl Acad Sci USA. 2005;102:4747–4752. doi: 10.1073/pnas.0406039102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ha KN, Traaseth NJ, Verardi R, Zamoon J, Cembran A, Karim CB, Thomas DD, Veglia G. Controlling the inhibition of the sarcoplasmic ca2+-ATPase by tuning phospholamban structural dynamics. J Biol Chem. 2007;282:37205–37214. doi: 10.1074/jbc.M704056200. [DOI] [PubMed] [Google Scholar]
- 45.Traaseth NJ, Thomas DD, Veglia G. Effects of ser16 phosphorylation on the allosteric transitions of phospholamban/ca2+-ATPase complex. J Mol Biol. 2006;358:1041–1050. doi: 10.1016/j.jmb.2006.02.047. [DOI] [PubMed] [Google Scholar]
- 46.Raake PWJ, Tscheschner H, Reinkober J, Ritterhoff J, Katus HA, Koch WJ, Most P. Gene therapy targets in heart failure: The path to translation. Clin Pharm Therap. 2011;90:542–553. doi: 10.1038/clpt.2011.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ha KN, Gustavsson M, Veglia G. Tuning the structural coupling between the transmembrane and cytoplasmic domains of phospholamban to control sarcoplasmic reticulum ca2+-ATPase (serca) function. J Muscle Res Cell Mot. 2012;33:485–492. doi: 10.1007/s10974-012-9319-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.De Simone A, Montalvao RW, Vendruscolo M. Determination of conformational equilibria in proteins using residual dipolar couplings. J Chem Theor Comp. 2011;11:4189–4195. doi: 10.1021/ct200361b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Montalvao RW, De Simone A, Vendruscolo M. Determination of structural fluctuations of proteins from structure-based calculations of residual dipolar couplings. J Biomol NMR. 2012;53:281–292. doi: 10.1007/s10858-012-9644-3. [DOI] [PubMed] [Google Scholar]
- 50.Buck B, Zamoon J, Kirby TL, DeSilva TM, Karim C, Thomas D, Veglia G. Overexpression, purification, and characterization of recombinant ca-ATPase regulators for high-resolution solution and solid-state NMR studies. Prot Exp Pur. 2003;30:253–261. doi: 10.1016/s1046-5928(03)00127-x. [DOI] [PubMed] [Google Scholar]
- 51.Metcalfe EE, Traaseth NJ, Veglia G. Serine 16 phosphorylation induces an order-to-disorder transition in monomeric phospholamban. Biochemistry. 2005;44:4386–4396. doi: 10.1021/bi047571e. [DOI] [PubMed] [Google Scholar]
- 52.Chou JJ, Gaemers S, Howder B, Louis JM, Bax A. A simple apparatus for generating stretched polyacrylamide gels, yielding uniform alignment of proteins and detergent micelles. J Biomol NMR. 2001;21:377–382. doi: 10.1023/a:1013336502594. [DOI] [PubMed] [Google Scholar]
- 53.Permi P, Rosevear PR, Annila A. A set of hnco-based experiments for measurement of residual dipolar couplings in N-15, C-13, (h-2)-labeled proteins. J Biomol NMR. 2000;17:43–54. doi: 10.1023/a:1008372624615. [DOI] [PubMed] [Google Scholar]
- 54.Wymore T, Gao XF, Wong TC. Molecular dynamics simulation of the structure and dynamics of a dodecylphosphocholine micelle in aqueous solution. J Mol Struct. 1999;485:195–210. [Google Scholar]
- 55.Hess B, Kutzner C, van der Spoel D, Lindahl E. Gromacs 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theor Comp. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 56.De Simone A, Richter B, Salvatella X, Vendruscolo M. Toward an accurate determination of free energy landscapes in solution states of proteins. J Am Chem Soc. 2009;131:3810–3811. doi: 10.1021/ja8087295. [DOI] [PubMed] [Google Scholar]
- 57.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Best RB, Hummer G. Optimized molecular dynamics force fields applied to the helix-coil transition of polypeptides. J Phys Chem B. 2009;113:9004–9015. doi: 10.1021/jp901540t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lindorff-Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO, Shaw DE. Improved side-chain torsion potentials for the Amber ff99sb protein force field. Proteins. 2010;78:1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tieleman DP, van der Spoel D, Berendsen HJC. Molecular dynamics simulations of dodecylphosphocholine micelles at three different aggregate sizes: Micellar structure and chain relaxation. J Phys Chem B. 2000;104:6380–6388. [Google Scholar]
- 61.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 62.Hess B, Bekker H, Berendsen HJC, Fraaije J. Lincs: A linear constraint solver for molecular simulations. J Comp Chem. 1997;18:1463–1472. [Google Scholar]
- 63.Darden T, York D, Pedersen L. Particle mesh ewald - an n. Log(n) method for ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 64.Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J Chem Phys. 2007;126:014101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- 65.Berendsen HJC, Postma JPM, Vangunsteren WF, Di Nola A, Haak JR. Molecular-dynamics with coupling to an external bath. J Chem Phys. 1984;81:3684–3690. [Google Scholar]
- 66.Cavalli A, Camilloni C, Vendruscolo M. Molecular dynamics simulations with replica-averaged structural restraints generate structural ensembles according to the maximum entropy principle. J Chem Phys. 2013;138:094112. doi: 10.1063/1.4793625. [DOI] [PubMed] [Google Scholar]
- 67.Pitera JW, Chodera JD. On the use of experimental observations to bias simulated ensembles. J Chem Theor Comp. 2012;8:3445–3451. doi: 10.1021/ct300112v. [DOI] [PubMed] [Google Scholar]
- 68.Roux B, Weare J. On the statistical equivalence of restrained-ensemble simulations with the maximum entropy method. J Chem Phys. 2013;138:084107. doi: 10.1063/1.4792208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhou YP, Cierpicki T, Jimenez RHF, Lukasik SM, Ellena JF, Cafiso DS, Kadokura H, Beckwith J, Bushweller JH. NMR solution structure of the integral membrane enzyme dsbb: Functional insights into dsbb-catalyzed disulfide bond formation. Mol Cell. 2008;31:896–908. doi: 10.1016/j.molcel.2008.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Camilloni C, Robustelli P, De Simone A, Cavalli A, Vendruscolo M. Characterization of the conformational equilibrium between the two major substates of rnase a using NMR chemical shifts. J Am Chem Soc. 2012;134:3968–3971. doi: 10.1021/ja210951z. [DOI] [PubMed] [Google Scholar]
- 71.Karim CB, Marquardt CG, Stamm JD, Barany G, Thomas DD. Synthetic null-cysteine phospholamban analogue and the corresponding transmembrane domain inhibit the ca-ATPase. Biochemistry. 2000;39:10892–10897. doi: 10.1021/bi0003543. [DOI] [PubMed] [Google Scholar]
- 72.Clore GM, Schwieters CD. Amplitudes of protein backbone dynamics and correlated motions in a small alpha/beta protein: Correspondence of dipolar coupling and heteronuclear relaxation measurements. Biochemistry. 2004;43:10678–10691. doi: 10.1021/bi049357w. [DOI] [PubMed] [Google Scholar]
- 73.Clore GM, Schwieters CD. How much backbone motion in ubiquitin is required to account for dipolar coupling data measured in multiple alignment media as assessed by independent cross-validation? J Am Chem Soc. 2004;126:2923–2938. doi: 10.1021/ja0386804. [DOI] [PubMed] [Google Scholar]
- 74.Bouvignies G, Markwick P, Bruscheweiler R, Blackledge M. Simultaneous determination of protein backbone structure and dynamics from residual dipolar couplings. J Am Chem Soc. 2006;128:15100–15101. doi: 10.1021/ja066704b. [DOI] [PubMed] [Google Scholar]
- 75.Iwahara J, Zweckstetter M, Clore GM. NMR structural and kinetic characterization of a homeodomain diffusing and hopping on nonspecific DNA. Proc Natl Acad Sci USA. 2006;103:15062–15067. doi: 10.1073/pnas.0605868103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lange OF, Lakomek NA, Fares C, Schroder GF, Walter KFA, Becker S, Meiler J, Grubmuller H, Griesinger C, de Groot BL. Recognition dynamics up to microseconds revealed from an RDC-derived ubiquitin ensemble in solution. Science. 2008;320:1471–1475. doi: 10.1126/science.1157092. [DOI] [PubMed] [Google Scholar]
- 77.Fenwick RB, Esteban-Martin S, Richter B, Lee D, Walter KFA, Milovanovic D, Becker S, Lakomek NA, Griesinger C, Salvatella X. Weak long-range correlated motions in a surface patch of ubiquitin involved in molecular recognition. J Am Chem Soc. 2011;133:10336–10339. doi: 10.1021/ja200461n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Huang JR, Grzesiek S. Ensemble calculations of unstructured proteins constrained by RDC and PRE data: A case study of urea-denatured ubiquitin. J Am Chem Soc. 2010;132:694–705. doi: 10.1021/ja907974m. [DOI] [PubMed] [Google Scholar]
- 79.Zweckstetter M, Bax A. Prediction of sterically induced alignment in a dilute liquid crystalline phase: Aid to protein structure determination by NMR. J Am Chem Soc. 2000;122:3791–3792. [Google Scholar]
- 80.Losonczi JA, Andrec M, Fischer MWF, Prestegard JH. Order matrix analysis of residual dipolar couplings using singular value decomposition. J Mag Res. 1999;138:334–342. doi: 10.1006/jmre.1999.1754. [DOI] [PubMed] [Google Scholar]
- 81.Oxenoid K, Chou JJ. The structure of phospholamban pentamer reveals a channel-like architecture in membranes. Proc Natl Acad Sci USA. 2005;102:10870–10875. doi: 10.1073/pnas.0504920102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Camilloni C, De Simone A, Vranken WF, Vendruscolo M. Determination of secondary structure populations in disordered states of proteins using nuclear magnetic resonance chemical shifts. Biochemistry. 2012;51:2224–2231. doi: 10.1021/bi3001825. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.