

Abstract
Increasing evidence indicates that caspase activation and apoptosis are associated with a variety of neurodegenerative disorders, including Alzheimer's disease. We reported that anesthetic isoflurane can induce apoptosis, alter processing of the amyloid precursor protein (APP), and increase amyloid-β protein (Aβ) generation. However, the mechanism by which isoflurane induces apoptosis is primarily unknown. We therefore set out to assess effects of extracellular calcium concentration on isoflurane-induced caspase-3 activation in H4 human neuroglioma cells stably transfected to express human full-length APP (H4-APP cells). In addition, we tested effects of RNA interference (RNAi) silencing of IP3 receptor, NMDA receptor, and endoplasmic reticulum (ER) calcium pump, sacro-/ER calcium ATPase (SERCA1). Finally, we examined the effects of the NMDA receptor partial antagonist, memantine, in H4-APP cells and brain tissue of naive mice. EDTA (10 mm), BAPTA (10 μm), and RNAi silencing of IP3 receptor, NMDA receptor, or SERCA1 attenuated capase-3 activation. Memantine (4 μm) inhibited isoflurane-induced elevations in cytosolic calcium levels and attenuated isoflurane-induced caspase-3 activation, apoptosis, and cell viability. Memantine (20 mg/kg, i.p.) reduced isoflurane-induced caspase-3 activation in brain tissue of naive mice. These results suggest that disruption of calcium homeostasis underlies isoflurane-induced caspase activation and apoptosis. We also show for the first time that the NMDA receptor partial antagonist, memantine, can prevent isoflurane-induced caspase-3 activation and apoptosis in vivo and in vitro. These findings, indicating that isoflurane-induced caspase activation and apoptosis are dependent on cytosolic calcium levels, should facilitate the provision of safer anesthesia care, especially for Alzheimer's disease and elderly patients.
Keywords: Alzheimer's disease, anesthesia, isoflurane, apoptosis, calcium, memantine
Introduction
Cerebral deposition of amyloid β-protein (Aβ), derived from the amyloid precursor protein (APP), is a major pathological hallmark of Alzheimer's disease (AD) (for review, see Selkoe, 2001; Tanzi and Bertram, 2005). Increasing evidence suggests a role for caspase activation and apoptosis in AD neuropathogenesis (Gervais et al., 1999; LeBlanc et al., 1999; Lu et al., 2000; Eckert et al., 2003; Gastard et al., 2003; Zhao et al., 2003; Hitomi et al., 2004; Takuma et al., 2004).
Several studies showed the potential association of previous general anesthesia/surgery and risk of AD (Bohnen et al., 1994a,b; Muravchick and Smith, 1995). A previous study suggested that age of onset of AD was inversely related to anesthesia exposure before 50 years of age (Bohnen et al., 1994b). A recent study also reported that patients undergoing coronary artery bypass graft surgery under general anesthesia were at increased risk for AD compared with those having percutaneous transluminal coronary angioplasty under local anesthesia (Lee et al., 2005). Isoflurane, one of the most commonly used inhalation anesthetics, has been reported to enhance aggregation and cytotoxicity of Aβ (Eckenhoff et al., 2004) and induce caspase activation and apoptosis (Matsuoka et al., 2001; Jevtovic-Todorovic et al., 2003; Kvolik et al., 2005; Loop et al., 2005; Wei et al., 2005; Yon et al., 2005). Our recent studies have shown that a clinically relevant isoflurane treatment causes caspase-3 activation and apoptosis, decreases cell viability, affects APP processing, and increases Aβ generation in human neuroglioma cells stably transfected to express human APP (H4-APP cells) (Xie et al., 2006a,b, 2007). Moreover, isoflurane has been suggested to induce a vicious cycle of apoptosis and Aβ accumulation (Xie et al., 2007).
The mechanism by which isoflurane induces caspase activation and apoptosis remains unclear. Wei et al. (2005, 2008) reported that dantrolene, an endoplasmic reticulum (ER) ryanodine receptor antagonist, and inositol 1,4,5-trisphosphate (IP3) receptor knock-out can inhibit isoflurane-induced apoptosis, suggesting that abnormal calcium release from ER after isoflurane treatment may cause apoptosis (Wei et al., 2005, 2008). Here, we used a variety of treatments in vitro and in vivo to alter calcium homeostasis and assessed the effects of these treatments on isoflurane-induced caspase-3 activation in H4-APP cells. The IP3 receptor, located in the ER membrane, regulates release of calcium from the ER to the cytoplasm (Berridge, 1993). Activation of IP3 receptors causes elevated cytosolic calcium levels, leading to cell death (Hanson et al., 2004; Lindholm et al., 2006). A recent study by Zhang et al. (2006) showed that the endoplasmic reticulum calcium pump, sacro-/ER calcium ATPase (SERCA1), is required for calcium release-activated channel activity, and RNA interference (RNAi) knockdown of SERCA1 inhibits calcium release-activated channel activity. We therefore assessed effects of RNAi silencing of IP3 receptor, NMDA receptor, and SERCA1 on isoflurane-induced caspase-3 activation in H4-APP cells. Memantine is a relatively new FDA-approved drug for the treatment of AD, which acts as an uncompetitive (partial) antagonist of the NMDA receptor (for review, see Lipton, 2006). We assessed the effects of memantine on isoflurane-induced caspase-3 activation both in H4-APP cells and in naive mice.
Materials and Methods
Cell lines.
We used naive H4 human neuroglioma cells stably transfected to express full-length APP (FL-APP; H4-APP cells) in the studies. The cells were cultured in DMEM (high glucose) containing 9% heat-inactivated fetal calf serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mm l-glutamine, and 200 μg/ml G418.
Cell treatment.
Twenty-one percent O2, 5% CO2, and 2% isoflurane were delivered from an anesthesia machine to a sealed plastic box in a 37° Celsius incubator containing six-well plates seeded with one million cells in 1.5 ml of cell culture media as described in our previous studies (Xie et al., 2006a,b, 2007). Datex infrared gas analyzer (Puritan-Bennett, Tewksbury, MA) was used to continuously monitor the delivered CO2, O2, and isoflurane concentrations. We treated the cells with 2% isoflurane for 6 h, during which time the cells were incubated in serum-free media. In the interaction studies, the cells were treated with memantine (4 μm) 1 h before the treatment of 2% isoflurane. Low-calcium condition was created by using a calcium-free cell culture media (Invitrogen, Carlsbad, CA) and by adding EDTA (10 mm; Sigma-Aldrich, St. Louis, MO), a chelating compound (Rekasi et al., 2005), or BAPTA (10 μm; Sigma-Aldrich), an intracellular calcium chelator, to the media. To avoid off-target effects of RNAi, we used two sets of small interference RNAs (siRNAs) aimed at knockdown of NMDA receptor (NR)1 (first set, 3′GCCGGGAUCUUCCUGAUUUUU, 5′-PAAAUCAGGAAGAUCCCGGCUU; second set, 3′GGAGCACGCUGGACUCGUUUU, 5′PAACGAGUCCAGCGUGCUCCUU), IP3 receptor (first set, 3′GCAAUCACAUGUGGAAAUUUU, 5′-AAUUUCCACAUGUGAUUGCUU; second set, 3′UGGAAAGUCUGACCGAAUAUU, 5′PUAUUCGGUCAGACUUUCCAUU), and SERCA (first set, 3′ UCGCACAAGUCCAAGAUUGUU, 5′-CAAUCUUGGACUUGUGCGAUU; second set, 3′GGCCAAAGGUGUCUAUGAGUU, 5′-PCUCAUAGACACCUUUGGCCUU). These siRNAs and control siRNAs (3′UAGCGACUAAACACAUCAAUU) were obtained from Dharmacon (Lafayette, CO). siRNAs were transfected into cells by using electroporation (Amaxa Biosystems, Gaithersburg, MD) as described by Xie et al. (2005b). Briefly, we mixed 1 million cells, 100 μl of AMAXA electroporation transfection solution, and 10 μl of 20 μm siRNA together and then used the C-9 program in an AMAXA electroporation device for cell transfection. The transfected cells were then placed in one of the six-well plates containing 1.5 ml of cell culture media. The pretreated cells were then exposed to the isoflurane treatment.
Cell lysis and protein amount quantification.
Cell pellets were detergent-extracted on ice using immunoprecipitation buffer (10 mm Tris-HCl, pH 7.4, 150 mm NaCl, 2 mm EDTA, 0.5% Nonidet P-40) plus protease inhibitors (1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin A). The lysates were collected, centrifuged at 12,000 rpm for 10 min, and quantified for total proteins by a BCA protein assay kit (Pierce Technology Corporation, Iselin, NJ).
Cytosolic calcium measurement.
Cytosolic calcium levels were determined as described by Wei et al. (2008). Specifically, H4-APP cells were loaded with Fura-2 (Invitrogen), perfused with Tyrode buffer, and [Ca2+]i transients were recorded as changes in Fura-2 ratio (340/380 nm) using a spectrofluoroscope system (Ionoptix, Milton, MA). The cells were exposed to isoflurane (1 mm or 3.5%) with or without pretreatment with memantine (4 μm).
Mice treatment.
The animal protocol was approved by the Massachusetts General Hospital Standing Committee on Animals. Sixteen female C57BL/6 mice (5–6 months of age) (The Jackson Laboratory, Bar Harbor, ME) were randomly assigned to an anesthesia or control group. Mice randomized to the anesthesia group (n = 8) received three sessions of 1.4% isoflurane in 100% oxygen for 2 h in an anesthetizing chamber. The mice were placed in 100% oxygen without isoflurane in the chamber for 10 min between each session. This isoflurane treatment allows the mice to have sufficient exposure to isoflurane while avoiding excessive accumulation of CO2. The control mice (n = 8) received 100% oxygen at an identical flow rate for 6 h and 20 min in an identical chamber. The mice breathed spontaneously, and anesthetic and oxygen concentrations were measured continuously (Datex, Puritan-Bennett; Ohmeda PPD, Madison, WI). Rectal temperature was measured intermittently, and the temperature of the anesthetizing chamber was controlled to maintain rectal temperature of the animals at 37 ± 0.5°C. Mean arterial blood pressure was measured noninvasively using a tail cuff (CODA2 system; Kent Scientific Corporation, Torrington, CT) in the anesthetized mice. This treatment of isoflurane did not significantly affect the blood pressure or blood gas of the mice (data not shown). Mice were killed by decapitation at the end of the experiments, the brain was removed rapidly, and the prefrontal cortex was dissected out and frozen in liquid nitrogen for subsequent processing for the determination of caspase-3 activation. For interaction studies, half of the anesthetized mice and half of the control mice received memantine (20 mg/kg) by intraperitoneal injection 1 h before the isoflurane treatment (Chen et al., 1998).
Brain tissue lysis and protein amount quantification.
The harvested brain tissues were homogenized on ice using immunoprecipitation buffer (10 mm Tris-HCl, pH 7.4, 150 mm NaCl, 2 mm EDTA, 0.5% Nonidet P-40) plus protease inhibitors (1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin A). The lysates were collected, centrifuged at 12,000 rpm for 10 min, and quantified for total proteins by a BCA protein assay kit (Pierce Technology Corporation).
Western blot analysis.
The harvested cells and brain tissues were subjected to Western blots as described by Xie et al. (2005a). A caspase-3 antibody (1:1000 dilution; Cell Signaling Technology, Beverly, MA) was used to recognize caspase-3 fragment (17–20 kDa) resulting from cleavage at asparate position 175 and FL-caspase-3 (35–40 kDa). Caspase-3 cleavage (activation) is based on the ratio of caspase-3 fragment to FL-caspase-3. Antibody anti-β-actin was used to visualize β-actin (42 kDa). Antibodies for IP3 receptor (1:1000; Millipore Bioscience Research Reagents, Temecula, CA), NR1 (1:1000; PhosphoSolutions, Aurora, CO), and SERCA1 (1:1000; Abcam, Cambridge, MA) were used to recognize IP3 receptor (250 kDa), NR1 (120 kDa), and SERCA1 (110 kDa). The quantitation of Western blots was performed as described by Xie et al. (2005a). Briefly, the intensity of signals was analyzed by using a Bio-Rad (Hercules, CA) image program (Quantity One) and an NIH Image version 1.37v. We quantified the Western blots using two steps. First, we used levels of β-actin to normalize (e.g., determining the ratio of FL-caspase-3 amount to β-actin amount) levels of proteins to control for loading differences in total protein amounts. Second, we presented changes in levels of proteins in the mice or cells treated with isoflurane as the percentage of those in the mice or cells treated with controls.
Cell viability assay.
3-(4, 5-Dimethylthiazol-2-yl)2,5diphenyltetrazolium bromide (MTT, Thiazolyl blue) is a water soluble tetrazolium salt. Dissolved MTT is converted to the insoluble purple formazan by cleavage of the tetrazolium ring by dehydrogenase enzymes. Active mitochondrion dehydrogenase (only in viable cells) will cause this conversion. We used a MTT assay kit (Sigma-Aldrich) to measure cell viability. The absorbency was measured with a spectrophotometer at a wavelength of 570 nm with background subtraction at 630 nm. Cell viability reduction suggests cell death.
Cell apoptosis assay.
Cell apoptosis was assessed by a cell death detection ELISA kit (Roche, Palo Alto, CA), which assays cytoplasmic histone-associated DNA fragmentation associated with cellular apoptosis.
Statistics.
Changes in caspase-3 activation were presented as a percentage of those of the control group. One hundred percent caspase-3 activation refers to control levels for purposes of comparison with experimental conditions. Data were expressed as mean ± SD. The number of samples varied from 3 to 10, and the samples were normally distributed. We used two-tailed t test to compare the difference between the experimental groups and control groups. p values <0.05 (* or #) and 0.01 (** or ##) were considered statistically significant.
Results
Memantine attenuates isoflurane-induced caspase-3 activation, cell viability reduction, and apoptosis in H4-APP cells
We reported previously that isoflurane can induce caspase-3 cleavage (activation) in H4-APP cells (Xie et al., 2006a,b, 2007). The underlying molecular mechanisms, however, remain primarily to be determined. Recent studies have shown that elevated levels of cytosolic calcium released from the ER may be at least partially responsible for isoflurane-induced caspase-3 activation and apoptosis (Wei et al., 2005, 2008). Memantine, a newly FDA approved drug for AD (Lipton, 2006), is a partial antagonist for the NMDA receptor, which can reduce influx of extracellular calcium. We therefore set out to assess whether memantine can attenuate isoflurane-induced caspase activation in H4-APP cells. Because caspase activation alone cannot report cell death (McLaughlin et al., 2003), we also monitored effects of isoflurane and memantine on cell viability and apoptosis in H4-APP cells.
Immunoblotting for caspase-3 revealed increases in caspase-3 fragment and decreases in FL-caspase-3 in H4-APP cells treated with 2% isoflurane compared with those cells in control conditions (Fig. 1A). Treatment with memantine plus isoflurane led to a visible decrease in caspase-3 activation compared with isoflurane treatment alone (Fig. 1A). Memantine treatment alone did not induce caspase-3 activation compared with control conditions (Fig. 1A).
Figure 1.

Memantine attenuates isoflurane-induced caspase-3 activation and apoptosis in H4-APP cells. A, Treatment with 2% isoflurane for 6 h (lane 4) induces caspase-3 cleavage (activation) compared with control conditions (lane 1) in H4-APP cells. Treatment with 4 μm memantine alone does not cause caspase-3 activation (lanes 2 and 3) compared with control condition (lane 1). However, treatment with isoflurane plus memantine (lanes 5 and 6) causes a lesser degree of caspase-3 activation compared with isoflurane treatment alone (lane 4). There is no significant difference in amounts of β-actin in all of the above treatments. B, The 2% isoflurane treatment (black) increases caspase-3 activation compared with control conditions (white), normalized to β-actin levels. Memantine treatment alone (gray) does not induce caspase-3 activation compared with control condition (white); however, memantine treatment (lined) attenuates the isoflurane-induced (black) caspase-3 activation, normalized to β-actin levels. C, Isoflurane treatment (2%) (black) decreases cell viability compared with control conditions (white). Memantine treatment alone (gray) does not affect cell viability compared with control conditions (white); however, memantine treatment (lined) attenuates the isoflurane-induced reduction in cell viability (black). D, Isoflurane treatment (2%) (black) induces apoptosis compared with control conditions (white). Memantine treatment alone (gray) does not induce apoptosis compared with control conditions (white); however, memantine treatment (lined) attenuates isoflurane-induced apoptosis (black). E, H4-APP cells contain NR1. Western blot analysis illustrates that anti-NR1 antibody (lanes 1–3) detects NR1 in H4-APP cells, whereas NR1 peptide (lanes 4–6) prevents anti-NR1 antibody from detecting NR1 in H4-APP cells. These results suggest that there is NR1 in H4-APP cells. Data are means ± SD (n = 3–9 for each experimental group). A t test is used to compare the difference between control condition and the isoflurane or memantine treatment. The asterisk indicates the difference between isoflurane and control condition on caspase-3 activation (*p = 0.015), cell viability (*p = 0.001), and apoptosis (*p = 0.001); the difference between saline and memantine on isoflurane-induced caspase-3 activation (#p = 0.013), cell viability (#p = 0.037), and apoptosis (#p = 0.001) is indicated.
Quantification of the results showed that isoflurane treatment led to a 184% increase in caspase-3 activation compared with control conditions (Fig. 1B) (p = 0.015). Memantine plus isoflurane yielded a significant reduction in isoflurane-induced caspase-3 activation (Fig. 1B): 101 versus 184% (p = 0.013). These results indicate that a clinically relevant concentration (2%) of isoflurane can induce apoptosis in H4-APP cells, and a clinically relevant concentration of memantine can attenuate isoflurane-induced caspase-3 activation (apoptosis). Memantine alone did not increase caspase-3 activation compared with control conditions (Fig. 1B).
MTT cell viability and cell apoptosis studies showed that isoflurane treatment led to a 26% reduction in cell viability (Fig. 1C) (p = 0.001) and a 137% increase in cell apoptosis (Fig. 1D) (p = 0.001) in H4-APP cells, compared with control conditions, respectively. Memantine treatment significantly attenuated the isoflurane-induced reduction in cell viability (Fig. 1C), 74 versus 95% (p = 0.037), and isoflurane-induced apoptosis (Fig. 1D), 137 versus 99% (p = 0.001). Meanwhile, memantine treatment alone had no effects on either cell viability or apoptosis compared with control conditions in H4-APP cells.
These results suggest that isoflurane can induce caspase-3 activation, cell viability reduction, and apoptosis in H4-APP cells, and memamtine can attenuate isoflurane-induced apoptotic cell damage, conceivably via antagonism of NMDA receptors. Furthermore, immunoblotting for NR1 specifically revealed that there are NMDA receptors in H4-APP cells (Fig. 1E), the cell lines in which memantine was able to attenuate effects of isoflurane on caspase-3 activation, cell viability reduction, and apoptosis. These findings further suggest that memantine may act on NMDA receptors to inhibit isoflurane-induced apoptotic cell damage.
Memantine inhibits isoflurane-induced elevation in cytosolic calcium levels in H4-APP cells
Given that excessive cytosolic calcium levels can trigger caspase activation and apoptosis (for review, see Mattson, 2007), and isoflurane may induce apoptosis via elevation of cytosolic calcium levels (Wei et al., 2005, 2008), we next asked whether memantine can inhibit the isoflurane-induced elevation in cytosolic calcium levels in H4-APP cells. We found that the isoflurane (1 mm or 3.5%) treatment can elevate cytosolic calcium levels in H4-APP cells (Fig. 2A). In contrast, in the H4-APP cells pretreated with memantine (4 μm), isoflurane failed to increase cytosolic calcium levels (Fig. 2B). These results suggest that memantine may inhibit the isoflurane-induced elevation in cytosolic calcium to attenuate isoflurane-induced caspase activation and apoptosis.
Figure 2.
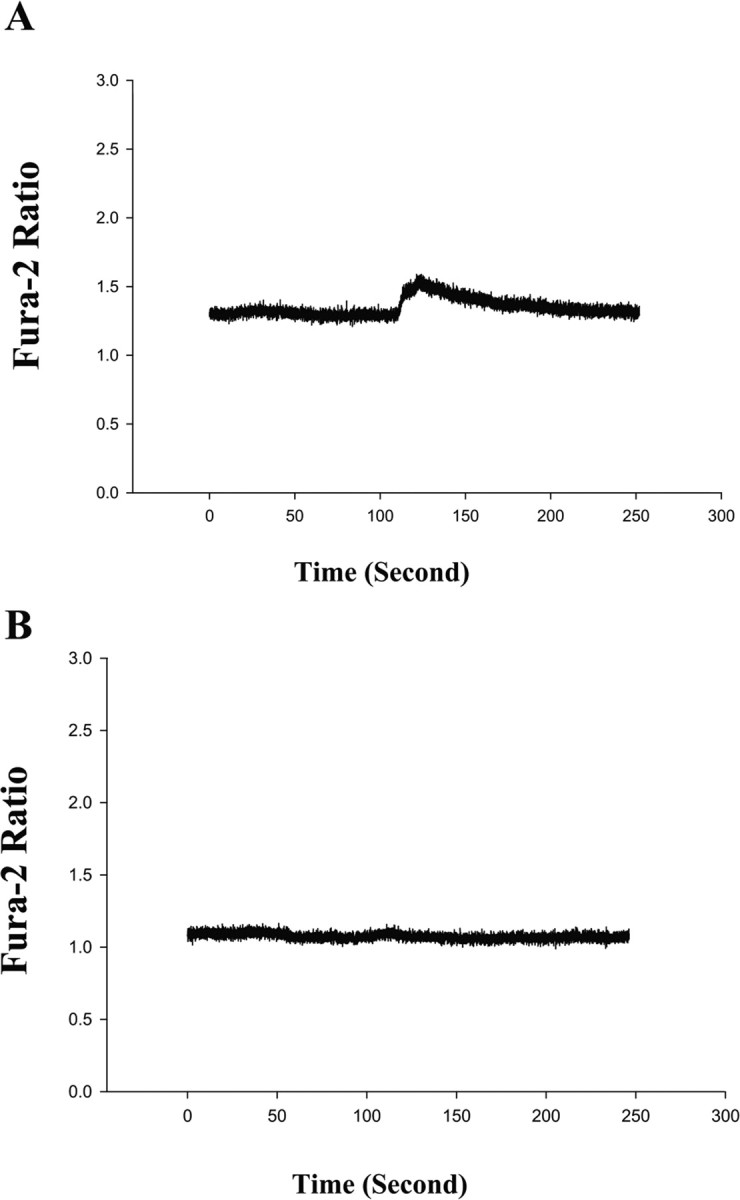
Isoflurane-induced elevation of cytosolic calcium levels can be inhibited by memantine in H4-APP cells. A, Isoflurane elevates cytosolic calcium levels in H4-APP cells. Isoflurane (1 mm; or 3.5%) was given to H4-APP cells at time 0. Cytosolic calcium levels were elevated after isoflurane treatment in H4-APP cells. B, Memantine inhibits isoflurane-induced elevation of cytosolic calcium levels in H4-APP cells. H4-APP cells were pretreated with 4 μm memantine for 5 min and then exposed to 1 mm (or 3.5%) isoflurane at time 0. Note that cytosolic calcium levels were not elevated by isoflurane in the memantine-pretreated H4-APP cells. These are representative tracings of two independent experiments.
Memantine attenuates isoflurane-induced caspase-3 activation in mouse brain
We next set out to assess the effects of memantine on isoflurane-induced caspase-3 activation in vivo in naive mice. After establishing that treatment with 1.4% isoflurane for 2 h did not significantly affect blood pressure and venous blood gas (data not shown), we exposed the mice to 1.4% isoflurane treatment for three 2 h sessions with 10 min between each session. This treatment enabled the mice to have enough isoflurane exposure yet avoid severe CO2 accumulation, because the mice reach a fully awake status between each session.
The three sessions of 1.4% isoflurane for 2 h induced caspase-3 activation in the mouse brain compared with the control conditions (Fig. 3A). Treatment with memantine (20 mg/kg, intraperitoneal route) plus isoflurane (three sessions of the treatment with 1.4% isoflurane for 2 h) led to a visible decrease in caspase-3 activation compared with isoflurane treatment alone (Fig. 3A). Treatment with memantine (20 mg/kg, intraperitoneal route) alone did not induce caspase-3 activation compared with the control condition (Fig. 3A).
Figure 3.

Memantine attenuates isoflurane-induced caspase-3 activation in naive mice. A, Isoflurane treatment (lane 3) induces caspase-3 cleavage (activation) compared with control conditions (lane 1) in naive mice. Treatment with memantine alone does not cause caspase-3 activation (lane 2) compared with control condition (lane 1). However, treatment with isoflurane plus memantine (lanes 4) causes a reduction in caspase-3 activation compared with isoflurane treatment alone (lane 3). There is no significant difference in the amounts of β-actin in all of the treatments. The Western blot, which shows the caspase-3 fragment (17 kDa) only, is the same Western blot with longer exposure time during the film development. B, Isoflurane treatment (black) increases caspase-3 activation compared with control conditions (white), normalized to β-actin levels, in naive mice. Memantine treatment alone (gray) does not induce caspase-3 activation compared with control condition (white); however, memantine treatment (lined) attenuates the isoflurane-induced (black) caspase-3 activation, normalized to β-actin levels. Data are means ± SD (n = 4 for each experimental group). A t test is used to compare the difference between control condition and the isoflurane or memantine treatment. *p = 0.034, difference between isoflurane and control condition on caspase-3 activation; #p = 0.020, difference between saline and memantine on isoflurane-induced caspase-3 activation.
Quantification of these results showed that the isoflurane treatment led to 177% increase in caspase-3 activation compared with control condition (Fig. 3B) (p = 0.034). Treatment with memantine plus isoflurane significantly reduced caspase-3 activation compared with isoflurane treatment alone (Fig. 3B) (p = 0.020; 177 vs 97%). Memantine treatment alone did not induce caspase-3 activation compared with the control condition (Fig. 3B). Collectively, all of these findings showed that the NMDA receptor partial antagonist, memantine, can attenuate isoflurane-induced apoptosis both in vivo and in vitro.
RNAi knockdown of NR1 reduces isoflurane-induced caspase-3 activation
Given that memantine can attenuate isoflurane-induced apoptosis, we next asked whether reductions in protein levels of NMDA receptors could also decrease isoflurane-induced apoptosis. We therefore assessed the effects of RNAi-mediated knockdown of NR1 on isoflurane-induced caspase-3 activation in H4-APP cells. RNAi silencing of NR1 significantly reduced NR1 levels (Fig. 4A,B), 100 versus 62% (p = 0.0439). Treatment with 2% isoflurane for 6 h induced caspase-3 activation (Fig. 4C, lanes 5 and 6, D, black) (p = 0.041). RNAi knockdown of NR1 (Fig. 4C, lanes 7 and 8, D, lined) attenuated isoflurane-induced caspase-3 activation in H4-APP cells, 168 versus 93% (p = 0.037). We repeated these experiments with different NR1 siRNAs and observed similar effects: RNAi knockdown of NR1 attenuated isoflurane-induced caspase-3 activation in H4-APP cells (data not shown). These findings suggest that the effects of RNAi knockdown of NR1 on isoflurane-induced caspase-3 activation are not likely to be caused by off-target effects of RNAi silencing of NR1. Collectively, these findings suggest that NR1 plays a role in isoflurane-induced caspase activation and apoptosis.
Figure 4.

RNAi knockdown of NR1 reduces isoflurane-induced caspase-3 activation in H4-APP cells. A, NR1 siRNA (lanes 3 and 4) reduces protein levels of NR1 compared with control siRNA treatment (lanes 1 and 2). There is no significant difference in amounts of β-actin in control or NR1 siRNA-treated cells. B, Quantitation of the Western blots shows that NR1 siRNA treatment (black) reduces protein levels of NR1 compared with control siRNA treatment (white). A t test is used to compare difference between control siRNA and NR1 siRNA treatment in reducing NR1 protein levels (*p = 0.0439). C, Treatment with control siRNA plus isoflurane (lanes 5 and 6) induces caspase-3 activation compared with control siRNA plus control condition (lanes 1 and 2). NR1 siRNA alone (lanes 3 and 4) does not induce caspase-3 activation. However, treatment with NR1 siRNA plus isoflurane (lanes 7 and 8) induces a lesser degree of caspase-3 activation compared with a treatment with control siRNA plus isoflurane (lanes 5 and 6). The Western blot, which shows the caspase-3 fragment (17 kDa) only, is the same Western blot with longer exposure time during the film development. D, Quantification of the Western blot shows that control condition plus NR1 siRNA treatment (gray) dose not induce caspase-3 activation compared with control condition plus control siRNA treatment (white). The treatment with isoflurane plus control siRNA (black) induces caspase-3 activation compared with control condition plus control siRNA treatment (white). NR1 siRNA treatment (lined) attenuates the isoflurane-induced (black) caspase-3 activation. Data are means ± SD (n = 3–6 for each experimental group). A t test is used to compare the difference. *p = 0.041, difference between isoflurane and control condition on caspase-3 activation; #p = 0.037, difference between control siRNA and NR1 siRNA on isoflurane-induced caspase-3 activation.
Reduced extracellular and intracellular calcium levels attenuate isoflurane-induced caspase-3 activation
Excitotoxic neuronal cell damage can be exacerbated by hyperactivation of NMDA receptors, which results in excessive calcium influx and subsequent free radical formation (for review, see Lipton, 2006). Isoflurane has been reported to induce apoptosis by elevating cytosolic calcium levels (Wei et al., 2005). Given that memantine and RNAi knockdown of NR1 attenuated isoflurane-induced apoptosis, we next addressed whether this involves partial inhibition of calcium influx by exposing H4-APP cells to low-calcium levels and/or treating with the chelator EDTA. Treatment with isoflurane plus low calcium led to a visible reduction in caspase-3 activation compared with treatment with isoflurane under normal calcium conditions (Fig. 5A). Treatment with isoflurane plus low calcium plus EDTA led to even less caspase-3 activation compared with either isoflurane alone or isoflurane plus low calcium (Fig. 5A). Quantitation of these results revealed that treatment with isoflurane plus low calcium (Fig. 5B, gray) led to a 19% reduction (p = 0.046) in caspase-3 activation compared with isoflurane plus normal calcium condition (Fig. 5B, white). Treatment with isoflurane plus low calcium and EDTA (Fig. 5B, black) led to an even greater (68%; p = 0.015) reduction in caspase-3 activation compared with the treatment with isoflurane plus normal calcium (Fig. 5B, white).
Figure 5.

EDTA, low calcium, and BAPTA attenuate isoflurane-induced caspase-3 activation in H4-APP cells. A, Treatment with isoflurane plus low-calcium condition (lanes 3 and 4) yields a reduction in caspase-3 cleavage (activation) compared with isoflurane treatment alone (lanes 1 and 2) in H4-APP cells. Treatment with isoflurane plus low-calcium condition and EDTA (lanes 5 and 6) causes an even lesser degree of caspase-3 activation compared with either isoflurane treatment alone (lanes 1 and 2) or the treatment of isoflurane plus low-calcium condition (lanes 3 and 4). There is no significant difference in the amounts of β-actin in all of the above treatments in H4-APP cells. The Western blot, which shows the caspase-3 fragment (17 kDa) only, is the same Western blot with longer exposure time during the film development. B, Caspase-3 activation assessed by quantifying the ratio of caspase-3 fragment to FL-caspase-3 in the Western blots. The treatment with isoflurane and low-calcium condition (gray) induces a reduction in caspase-3 activation compared with isoflurane treatment alone (white). The treatment with isoflurane plus low-calcium condition and EDTA (black) triggers an even lesser degree of caspase-3 activation compared with either isoflurane treatment alone (white) or the treatment with isoflurane plus low-calcium condition (gray). C, Treatment with DMSO plus isoflurane (lanes 5 and 6) induces caspase-3 activation compared with DMSO plus control condition (lanes 1 and 2). BAPTA alone (lanes 3 and 4) does not induce caspase-3 activation. However, treatment with BAPTA plus isoflurane (lanes 7 and 8) induces a lesser degree of caspase-3 activation compared with a treatment with DMSO plus isoflurane (lanes 5 and 6). D, Quantification of the Western blot shows that control condition plus BAPTA treatment (gray) dose not induce caspase-3 activation compared with control condition plus DMSO treatment (white). The treatment with isoflurane plus DMSO (black) induces caspase-3 activation compared with control condition plus DMSO treatment (white). BAPTA treatment (lined) attenuates the isoflurane-induced (black) caspase-3 activation. Data are means ± SD (n = 3–6 for each experimental group). A t test is used to compare the difference. *p = 0.046, difference between isoflurane plus low-calcium condition and isoflurane plus normal calcium condition; *p = 0.015, difference between isoflurane plus normal calcium condition and isoflurane plus low-calcium condition and EDTA; #p = 0.029, difference between isoflurane plus low-calcium condition and isoflurane plus low-calcium condition and EDTA; *p = 0.0103, difference between isoflurane and control condition on caspase-3 activation; #p = 0.045, difference between BAPTA and control treatment on isoflurane-induced caspase-3 activation.
To further confirm that isoflurane-induced caspase-3 activation is associated with cytosolic calcium levels, we assessed effects of BAPTA, an intracellular calcium chelator, on isoflurane-induced caspase-3 activation in H4-APP cells. Treatment with 2% isoflurane for 6 h induced caspase-3 activation (Fig. 5C, lanes 5 and 6, D, black) (p = 0.0102). BAPTA (Fig. 5C, lanes 7 and 8, D, lined) attenuated isoflurane-induced caspase-3 activation in H4-APP cells, 185 versus 154% (p = 0.045). Collectively, these results suggest that the isoflurane-induced apoptosis is dependent on both external and cytosolic calcium concentration.
RNAi silencing of IP3 receptor and SERCA1 attenuate isoflurane-induced caspase-3 activation
We next asked whether isoflurane-induced apoptosis is dependent on internal calcium released from ER via the IP3 receptor and SERCA1. We first established RNAi knockdown of IP3 receptor in H4-APP cells using siRNA treatment (Fig. 6A). Treatment with isoflurane plus control siRNA led to a visible increase in caspase-3 activation compared with the treatment with control condition plus control siRNA (Fig. 6A). RNAi silencing of IP3 receptor alone did not induce caspase-3 activation compared with the treatment with control condition plus control siRNA (Fig. 6A). However, treatment with isoflurane plus IP3 receptor siRNA attenuated caspase-3 activation compared with the treatment with isoflurane plus control siRNA (Fig. 6A). Quantification of these data showed that RNAi knockdown of IP3 receptor led to a 33% reduction in protein levels of IP3 receptor compared with control siRNA treatment (Fig. 6B) (p = 0.026). Treatment with IP3 receptor siRNA alone had no effect on caspase-3 activation (Fig. 6C). Although treatment with isoflurane plus control siRNA led to a 292% increase (p = 0.001) in caspase-3 activation compared with the control, treatment with isoflurane plus IP3 receptor siRNA (Fig. 6C) led to less caspase-3 activation than did treatment with isoflurane plus control siRNA, 222 versus 292% (p = 0.035). These results suggest that knockdown of IP3 receptor via RNAi can attenuate isoflurane-induced caspase-3 activation (apoptosis). Next, we assessed the effects of RNAi-mediated knockdown of SERCA1 on isoflurane-induced caspase-3 activation in H4-APP cells. RNAi-mediated knockdown of SERCA1 reduced SERCA1 levels by 33% (p = 0.021) (Fig. 7A,B). RNAi knockdown of SERCA1 (Fig. 7C,D) inhibited isoflurane-induced caspase-3 activation in H4-APP cells, 194% (control siRNA) versus 117% (SERCA1 siRNA) (p = 0.012). To avoid off-target effects of RNAi knockdown of IP3 receptor and SERCA, we repeated these experiment with different IP3 receptor or SERCA siRNAs and observed similar effects: RNAi knockdown of IP3 receptor and SERCA attenuated isoflurane-induced caspase-3 activation in H4-APP cells (data not shown). Collectively, these findings suggest that isoflurane induction of caspase activation and apoptosis is dependent on calcium release from the ER.
Figure 6.
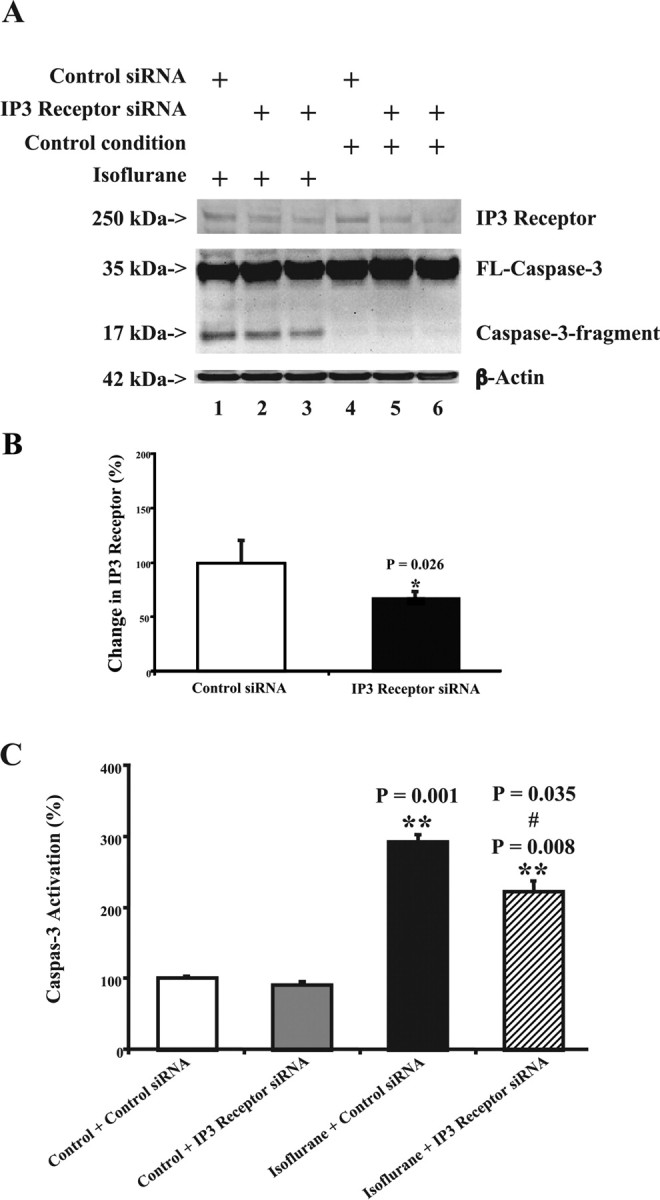
RNAi knockdown of IP3 receptor reduces isoflurane-induced caspase-3 activation in H4-APP cells. A, IP3 receptor siRNA (lanes 2, 3, 5, and 6) reduces the protein levels of IP3 receptor compared with control siRNA treatment (lanes 1 and 4). The treatment with control siRNA plus isoflurane (lane 1) induces caspase-3 activation compared with control siRNA plus control condition (lane 4). The treatment of IP3 receptor siRNA plus control condition (lanes 5 and 6) does not induce caspase-3 activation compared with control siRNA plus control condition (lane 4). However, the treatment with IP3 receptor siRNA plus isoflurane (lanes 2 and 3) yields a reduction in caspase-3 activation compared with the treatment with control siRNA plus isoflurane (lane 1). B, Quantitation of the Western blots shows that IP3 receptor siRNA treatment (black) reduces the protein levels of IP3 receptor compared with control siRNA treatment (white). A t test is used to compare the difference between control siRNA and IP3 receptor siRNA treatment in reducing IP3 receptor protein levels (*p = 0.026). C, Control condition plus IP3 siRNA treatment (gray) dose not induce caspase-3 activation compared with control condition plus control siRNA treatment (white). The treatment with either isoflurane plus control siRNA (black) or isoflurane plus IP3 receptor siRNA (lined) causes caspase-3 activation compared with control condition plus control siRNA treatment (white). However, IP3 receptor siRNA treatment (lined) attenuates the isoflurane-induced (black) caspase-3 activation. Data are means ± SD (n = 3–6 for each experimental group). A t test is used to compare the difference. **p = 0.001, **p = 0.008, the difference between isoflurane and control condition on caspase-3 activation; #p = 0.035, the difference between control siRNA and IP3 receptor siRNA on the isoflurane-induced caspase-3 activation.
Figure 7.

RNAi knockdown of SERCA1 reduces isoflurane-induced caspase-3 activation in H4-APP cells. A, SERCA1 siRNA (lanes 3 and 4) reduces protein levels of SERCA1 compared with control siRNA treatment (lanes 1 and 2). There is no significant difference in amounts of β-actin in control or SERCA1 siRNA-treated cells. B, Quantitation of the Western blots shows that SERCA1 siRNA treatment (black) reduces protein levels of SERCA1 compared with control siRNA treatment (white). A t test is used to compare the difference between control siRNA and SERCA1 siRNA treatment in reducing SERCA1 protein levels (*p = 0.021). C, Treatment with control siRNA plus isoflurane (lanes 4 and 5) induces caspase-3 activation compared with control siRNA plus control condition (lane 1). SERCA1 siRNA alone (lanes 2 and 3) does not induce caspase-3 activation. However, treatment with SERCA1 siRNA plus isoflurane (lanes 6 and 7) induces a reduction in caspase-3 activation compared with the treatment with control siRNA plus isoflurane (lanes 4 and 5). The Western blot, which shows the caspase-3 fragment (17 kDa) only, is the same Western blot with longer exposure time during the film development. D, Quantification of the Western blot shows that control condition plus SERCA1 siRNA treatment (gray) dose not induce caspase-3 activation compared with control condition plus control siRNA treatment (white). Treatment with isoflurane plus control siRNA (black) induces caspase-3 activation compared with control condition plus control siRNA treatment (white). SERCA1 siRNA treatment (lined) attenuates the isoflurane-induced caspase-3 activation (black). Data are means ± SD (n = 4 for each experimental group). A t test is used to compare the difference. **p = 0.001, difference between isoflurane and control condition on caspase-3 activation; #p = 0.012, difference between control siRNA and SERCA1 siRNA on isoflurane-induced caspase-3 activation.
Discussion
The commonly used inhalation anesthetic isoflurane has been shown previously to induce caspase activation, apoptosis, and enhanced Aβ generation in cultured cells and brain slices (Matsuoka et al., 2001; Jevtovic-Todorovic et al., 2003; Eckenhoff et al., 2004; Loop et al., 2005; Wei et al., 2005; Wise-Faberowski et al., 2005; Xie et al., 2006a,b, 2007). However, the mechanism by which isoflurane induces caspase activation and apoptosis is not clear. It has been suggested that isoflurane may trigger abnormal calcium release from ER to induce caspase activation and apoptosis (Wei et al., 2005, 2008). We show that reductions in extracellular calcium concentration can attenuate isoflurane-induced caspase-3 activation in H4-APP cells, as does RNAi silencing of the IP3 receptor, NR1, and SERCA1. Normal NMDA receptor activity is necessary for induction of long-term potentiation (LTP), a form of synaptic plasticity associated with learning and memory. Hyperactivity of the NMDA receptor, after certain pathological conditions, can engender excessive neuronal calcium influx leading to cellular damage [e.g., apoptosis (for review, see Lipton, 2006)]. Here, we have shown that the partial NMDA receptor antagonist memantine can attenuate isoflurane-induced caspase-3 activation in H4-APP cells. Given that caspase-3 activation alone may not suggest cell damage but rather may be essential for neuroprotection in preconditioning (McLaughlin et al., 2003), we also measured effects of isoflurane and memantine on cell viability and apoptosis. We found that memantine can attenuate isoflurane-induced apoptosis and cell viability reduction, which suggests that memantine can inhibit isoflurane-induced apoptotic cell damage. Memantine also attenuated isoflurane-induced elevated cytosolic calcium levels in H4-APP cells and isoflurane-induced caspase-3 activation in the mouse brain in vivo. Collectively, these findings indicate that isoflurane-induced apoptotic cell damage is driven by calcium dyshomeostasis and can be pharmacologically modulated by memantine.
Isoflurane has been reported to affect synapse function by acting on the ion channels, including sodium, potassium, and calcium channels, associated with neurotransmitter receptors such as nicotinic, serotonin type 3, GABAA, glycine, and glutamate receptors (Franks and Lieb, 1998; Mennerick et al., 1998; Narahashi et al., 1998) (for review, see Campagna et al., 2003). Specifically, Westphalen and Hemmings (2006) illustrated that isoflurane can enhance basal release of GABA and inhibit basal release of glutamate from isolated rat cortical nerve terminals, and intracellular calcium buffering can limit the isoflurane-induced inhibition of basal glutamate release. Hollmann et al. (2001) reported that clinically relevant concentrations of isoflurane, sevoflurane, or desflurane can reduce current associated with NR1/NR2A and NR1/NR2B expressed recombinantly in Xenopus oocytes with a reversible, concentration-dependent, and voltage-insensitive manner. Isoflurane has also been reported to block NMDA-stimulated currents in cultured hippocampus neurons (Yang and Zorumski, 1991) and to attenuate glutamate-dependent intraneuronal translocation of Ca2+ (Puil et al., 1990). Recent studies showed that isoflurane can block NMDA receptor-mediated current (Solt et al., 2006), and NMDA receptor antagonist (+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate (MK-801) can decrease the minimum alveolar concentration of isoflurane (Eger et al., 2006). In addition, isoflurane has been reported to attenuate LTP in hippocampus slices from mice, and GABA antagonist picrotoxin can inhibit the effects of isoflurane (Simon et al., 2001). Collectively, these findings suggest that isoflurane may inhibit NMDA neurotransmission. However, other studies suggested that isoflurane does not inhibit NMDA transmission (Pearce et al., 1989); in fact, a recent study showed that isoflurane may actually increase NMDA receptor activity by enhancing phosphorylation at S896 of the NR1 subunit of NMDA receptor (Zhan et al., 2006). We therefore postulate that isoflurane may affect NMDA neurotransmission with a concentration- and duration-dependent manner. With low concentration and short duration of treatment, isoflurane may inhibit NMDA neurotransmission-associated calcium influx to produce neuroprotection effects (e.g., preconditioning). With high concentration and long duration of treatment, however, isoflurane may potentiate NMDA neurotransmission-associated calcium influx to trigger cell damage (e.g., apoptosis). Future studies will be necessary to assess the effects of different concentrations and durations of isoflurane treatments on NMDA neurotransmission and NMDA neurotransmission-associated calcium influx to further test this hypothesis.
It is also possible that isoflurane can induce apoptosis via non-NMDA neurotransmission-associated calcium influx. Indeed, El Beheiry et al. (2007) showed that reduction in exogenous calcium concentration and selective blockade of L-type calcium channel with nifedipine can reduce the isoflurane-induced suppression of evoked dendritic field EPSPs in hippocampus slides from old rats. These findings suggest that isoflurane can act on different types of calcium channels to affect cell functions. It will be interesting in future studies to test the effects of various calcium channel blockers, including nifedipine (L-type), ω-conotoxin GVIA (N-type), and ω-conotoxin MVIIC (P/Q-type) (El Beheiry et al., 2007), on isoflurane-induced apoptosis and Aβ generation.
Memantine, a newly FDA approved drug for AD treatment, is an uncompetitive (partial) antagonist of the NMDA receptor (for review, see Lipton, 2006). Memantine has a low affinity for the NMDA receptor channel pore and thus has a fast off-rate compared with other NMDA receptor antagonists (e.g., MK-801). This character of memantine enables memantine to only enter the NMDA receptor channel when the channel is opened by an agonist. Therefore, memantine will not accumulate in the NMDA receptor channel and will not interfere with normal synaptic transmission associated with NMDA receptor (Lipton, 1993; Lipton and Rosenberg, 1994; Chen and Lipton, 1997) (for review, see Lipton, 2006). Memantine, an NMDA receptor uncompetitive (partial) antagonist, is different from NMDA noncompetitive antagonists, including MK-801, ketamine, and phencyclidine, in clinical acceptance, because of the fact that memantine can preferentially block NMDA receptor-operated channels when they are excessively open while relatively sparing normal neurotransmission (for review, see Lipton, 2006). Our current findings showing that NMDA receptors exist in H4-APP cells (Figs. 1E, 4A), and that the NMDA partial antagonist memantine can attenuate isoflurane-induced apoptotic cell damage in H4-APP cells and in naive mice, suggest that memantine may act on NMDA receptors to inhibit isoflurane-induced apoptotic cell damage. Moreover, these findings suggest that memantine may be used to pharmacologically intervene with isoflurane-induced apoptosis and subsequent neurotoxicity in patients, especially elderly and AD patients.
Previous studies have suggested that isoflurane can induce apoptosis by activating the ryanodine receptor in the ER to facilitate calcium release from ER to the cytoplasm (Wei et al., 2005). IP3 receptors and SERCA1 can also regulate cytosolic calcium levels (Berridge, 1993; Zhang et al., 2006). Activation of IP3 receptors can cause elevated cytosolic calcium levels, leading to cell death (Hanson et al., 2004; Lindholm et al., 2006). RNAi knockdown of SERCA1 can inhibit calcium influx and calcium release-activated calcium channel activity. We therefore assessed the roles of the IP3 receptor and SERCA1 in isoflurane-induced caspase-3 activation. Knockdown of IP3 receptor and SERCA1 in H4-APP cells attenuated isoflurane-induced caspase-3 activation, suggesting that isoflurane can induce apoptosis by activating IP3 receptor and/or SERCA1, facilitating calcium release from the ER to the cytoplasm.
Although isoflurane has been reported to induce caspase activation and to cause apoptosis (Matsuoka et al., 2001; Eckenhoff et al., 2004; Kvolik et al., 2005; Loop et al., 2005; Wei et al., 2005; Xie et al., 2006a,b, 2007), other reports suggest that isoflurane can protect against apoptosis (Zaugg et al., 2000; Tyther et al., 2001; Wise-Faberowski et al., 2001; de Klaver et al., 2002; Kawaguchi et al., 2004; Wise-Faberowski et al., 2004; Gray et al., 2005). This difference could be attributable to the use of different cell lines (e.g., rat cardiac cells vs human neural cells) or differences in duration and concentration of isoflurane exposure (Xie et al., 2006a,b). A transient and moderate elevation of cytosolic calcium levels after a lower concentration of isoflurane treatment for a short duration could provide cytoprotection via upregulating host preconditioning response (Bickler et al., 2005; Bickler and Fahlman, 2006; Zhan et al., 2006). However, prolonged exposure to a high concentration of isoflurane may maintain IP3 receptor in an open status, thereby elevating cytosolic calcium levels and ultimately leading to cell damage (Orrenius et al., 2003; Paschen and Mengesdorf, 2005).
Our in vitro and limited in vivo studies as well as other findings suggest that isoflurane may affect AD neuropathogenesis; however, it is necessary to perform further determination of the in vivo relevance of these effects, including in vivo apoptosis (e.g., terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling) studies, given that caspase activation alone may not represent apoptotic cell damage (McLaughlin et al., 2003), before we can conclude that the inhalation anesthetic isoflurane promotes AD neuropathogenesis in humans.
In conclusion, we have shown that decreases in cytosolic calcium levels by reduction in extracellular and intracellular calcium levels, treatment with the NMDA antagonist memantine, or RNAi silencing of the IP3 receptor, NR1, or SERCA1 can attenuate isoflurane-induced apoptosis. These findings suggest a therapeutic strategy for preventing potential isoflurane-associated neurotoxicity (and Aβ generation) based on reducing cytosolic calcium levels. Moreover, we found that this can be achieved pharmacologically by using memantine. In the future studies aimed at further defining the molecular mechanism by which isoflurane induces caspase activation and apoptosis, we will assess effects of isoflurane on cytosolic calcium levels and whether the isoflurane-induced caspase activation and apoptosis are dependent on cytosolic calcium levels. Given the aging of the population and growing numbers of patients with AD who require surgery and general anesthesia, more studies in elucidating the molecular mechanism by which isoflurane induces apoptosis and potentiates Aβ generation are warranted. Our current study indicating that isoflurane-induced apoptosis is dependent on cytosolic calcium levels will hopefully lead to the provision of safer anesthesia care, especially for AD and elderly patients.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant NS048140, the American Geriatrics Society Jahnigen Award, the William F. Milton Fund from Harvard University, and the Investigator-Initiated Research Grant from the Alzheimer's Association (Z.X.); by NIH Grants AG014713 and MH60009 and the Cure Alzheimer's Fund (R.E.T.); by NIH Grant GM077057 and the American Geriatrics Society Jahnigen Award (D.J.C.); and by NIH Grant AG20253 (G.C.). The cost of anesthetic isoflurane and partial salary support of Y.D. and B.Z. were generously provided by the Department of Anesthesia and Critical Care in Massachusetts General Hospital and Harvard Medical School. We thank Dr. Stuart Forman (Associate Professor of Anesthesia, Department of Anesthesia and Critical Care, Massachusetts General Hospital and Harvard Medical School, Boston, MA) for the scientific discussion and technical help for the studies. These studies were performed in Massachusetts General Hospital and Harvard Medical School.
References
- Berridge MJ. Inositol trisphosphate and calcium signalling. Nature. 1993;361:315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- Bickler PE, Fahlman CS. The inhaled anesthetic, isoflurane, enhances Ca2+-dependent survival signaling in cortical neurons and modulates MAP kinases, apoptosis proteins and transcription factors during hypoxia. Anesth Analg. 2006;103:419–429. doi: 10.1213/01.ane.0000223671.49376.b2. table of contents. [DOI] [PubMed] [Google Scholar]
- Bickler PE, Zhan X, Fahlman CS. Isoflurane preconditions hippocampal neurons against oxygen-glucose deprivation: role of intracellular Ca2+ and mitogen-activated protein kinase signaling. Anesthesiology. 2005;103:532–539. doi: 10.1097/00000542-200509000-00016. [DOI] [PubMed] [Google Scholar]
- Bohnen N, Warner MA, Kokmen E, Kurland LT. Early and midlife exposure to anesthesia and age of onset of Alzheimer's disease. Int J Neurosci. 1994a;77:181–185. doi: 10.3109/00207459408986029. [DOI] [PubMed] [Google Scholar]
- Bohnen NI, Warner MA, Kokmen E, Beard CM, Kurland LT. Alzheimer's disease and cumulative exposure to anesthesia: a case-control study. J Am Geriatr Soc. 1994b;42:198–201. doi: 10.1111/j.1532-5415.1994.tb04952.x. [DOI] [PubMed] [Google Scholar]
- Campagna JA, Miller KW, Forman SA. Mechanisms of actions of inhaled anesthetics. N Engl J Med. 2003;348:2110–2124. doi: 10.1056/NEJMra021261. [DOI] [PubMed] [Google Scholar]
- Chen HS, Lipton SA. Mechanism of memantine block of NMDA-activated channels in rat retinal ganglion cells: uncompetitive antagonism. J Physiol (Lond) 1997;499:27–46. doi: 10.1113/jphysiol.1997.sp021909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HS, Wang YF, Rayudu PV, Edgecomb P, Neill JC, Segal MM, Lipton SA, Jensen FE. Neuroprotective concentrations of the N-methyl-d-aspartate open-channel blocker memantine are effective without cytoplasmic vacuolation following post-ischemic administration and do not block maze learning or long-term potentiation. Neuroscience. 1998;86:1121–1132. doi: 10.1016/s0306-4522(98)00163-8. [DOI] [PubMed] [Google Scholar]
- de Klaver MJ, Manning L, Palmer LA, Rich GF. Isoflurane pretreatment inhibits cytokine-induced cell death in cultured rat smooth muscle cells and human endothelial cells. Anesthesiology. 2002;97:24–32. doi: 10.1097/00000542-200207000-00005. [DOI] [PubMed] [Google Scholar]
- Eckenhoff RG, Johansson JS, Wei H, Carnini A, Kang B, Wei W, Pidikiti R, Keller JM, Eckenhoff MF. Inhaled anesthetic enhancement of amyloid-beta oligomerization and cytotoxicity. Anesthesiology. 2004;101:703–709. doi: 10.1097/00000542-200409000-00019. [DOI] [PubMed] [Google Scholar]
- Eckert A, Marques CA, Keil U, Schussel K, Muller WE. Increased apoptotic cell death in sporadic and genetic Alzheimer's disease. Ann NY Acad Sci. 2003;1010:604–609. doi: 10.1196/annals.1299.113. [DOI] [PubMed] [Google Scholar]
- Eger EI, Jr, Liao M, Laster MJ, Won A, Popovich J, Raines DE, Solt K, Dutton RC, Cobos FV, Jr, Sonner JM. Contrasting roles of the N-methyl-d-aspartate receptor in the production of immobilization by conventional and aromatic anesthetics. Anesth Analg. 2006;102:1397–1406. doi: 10.1213/01.ANE.0000219019.91281.51. [DOI] [PubMed] [Google Scholar]
- El Beheiry H, Ouanounou A, Carlen PL. L-type calcium channel blockade modifies anesthetic actions on aged hippocampal neurons. Neuroscience. 2007;147:117–126. doi: 10.1016/j.neuroscience.2007.03.031. [DOI] [PubMed] [Google Scholar]
- Franks NP, Lieb WR. Which molecular targets are most relevant to general anaesthesia? Toxicol Lett. 1998;100–101:1–8. doi: 10.1016/s0378-4274(98)00158-1. [DOI] [PubMed] [Google Scholar]
- Gastard MC, Troncoso JC, Koliatsos VE. Caspase activation in the limbic cortex of subjects with early Alzheimer's disease. Ann Neurol. 2003;54:393–398. doi: 10.1002/ana.10680. [DOI] [PubMed] [Google Scholar]
- Gervais FG, Xu D, Robertson GS, Vaillancourt JP, Zhu Y, Huang J, LeBlanc A, Smith D, Rigby M, Shearman MS, Clarke EE, Zheng H, Van Der Ploeg LH, Ruffolo SC, Thornberry NA, Xanthoudakis S, Zamboni RJ, Roy S, Nicholson DW. Involvement of caspases in proteolytic cleavage of Alzheimer's amyloid-beta precursor protein and amyloidogenic A beta peptide formation. Cell. 1999;97:395–406. doi: 10.1016/s0092-8674(00)80748-5. [DOI] [PubMed] [Google Scholar]
- Gray JJ, Bickler PE, Fahlman CS, Zhan X, Schuyler JA. Isoflurane neuroprotection in hypoxic hippocampal slice cultures involves increases in intracellular Ca2+ and mitogen-activated protein kinases. Anesthesiology. 2005;102:606–615. doi: 10.1097/00000542-200503000-00020. [DOI] [PubMed] [Google Scholar]
- Hanson CJ, Bootman MD, Roderick HL. Cell signalling: IP3 receptors channel calcium into cell death. Curr Biol. 2004;14:R933–R935. doi: 10.1016/j.cub.2004.10.019. [DOI] [PubMed] [Google Scholar]
- Hitomi J, Katayama T, Eguchi Y, Kudo T, Taniguchi M, Koyama Y, Manabe T, Yamagishi S, Bando Y, Imaizumi K, Tsujimoto Y, Tohyama M. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Abeta-induced cell death. J Cell Biol. 2004;165:347–356. doi: 10.1083/jcb.200310015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollmann MW, Liu HT, Hoenemann CW, Liu WH, Durieux ME. Modulation of NMDA receptor function by ketamine and magnesium. Part II: interactions with volatile anesthetics. Anesth Analg. 2001;92:1182–1191. doi: 10.1097/00000539-200105000-00020. [DOI] [PubMed] [Google Scholar]
- Jevtovic-Todorovic V, Hartman RE, Izumi Y, Benshoff ND, Dikranian K, Zorumski CF, Olney JW, Wozniak DF. Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. J Neurosci. 2003;23:876–882. doi: 10.1523/JNEUROSCI.23-03-00876.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi M, Drummond JC, Cole DJ, Kelly PJ, Spurlock MP, Patel PM. Effect of isoflurane on neuronal apoptosis in rats subjected to focal cerebral ischemia. Anesth Analg. 2004;98:798–805. doi: 10.1213/01.ane.0000105872.76747.f6. table of contents. [DOI] [PubMed] [Google Scholar]
- Kvolik S, Glavas-Obrovac L, Bares V, Karner I. Effects of inhalation anesthetics halothane, sevoflurane, and isoflurane on human cell lines. Life Sci. 2005;77:2369–2383. doi: 10.1016/j.lfs.2004.12.052. [DOI] [PubMed] [Google Scholar]
- LeBlanc A, Liu H, Goodyer C, Bergeron C, Hammond J. Caspase-6 role in apoptosis of human neurons, amyloidogenesis, and Alzheimer's disease. J Biol Chem. 1999;274:23426–23436. doi: 10.1074/jbc.274.33.23426. [DOI] [PubMed] [Google Scholar]
- Lee TA, Wolozin B, Weiss KB, Bednar MM. Assessment of the emergence of Alzheimer's disease following coronary artery bypass graft surgery or percutaneous transluminal coronary angioplasty. J Alzheimers Dis. 2005;7:319–324. doi: 10.3233/jad-2005-7408. [DOI] [PubMed] [Google Scholar]
- Lindholm D, Wootz H, Korhonen L. ER stress and neurodegenerative diseases. Cell Death Differ. 2006;13:385–392. doi: 10.1038/sj.cdd.4401778. [DOI] [PubMed] [Google Scholar]
- Lipton SA. Prospects for clinically tolerated NMDA antagonists: open-channel blockers and alternative redox states of nitric oxide. Trends Neurosci. 1993;16:527–532. doi: 10.1016/0166-2236(93)90198-u. [DOI] [PubMed] [Google Scholar]
- Lipton SA. Paradigm shift in neuroprotection by NMDA receptor blockade: memantine and beyond. Nat Rev Drug Discov. 2006;5:160–170. doi: 10.1038/nrd1958. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Rosenberg PA. Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med. 1994;330:613–622. doi: 10.1056/NEJM199403033300907. [DOI] [PubMed] [Google Scholar]
- Loop T, Dovi-Akue D, Frick M, Roesslein M, Egger L, Humar M, Hoetzel A, Schmidt R, Borner C, Pahl HL, Geiger KK, Pannen BH. Volatile anesthetics induce caspase-dependent, mitochondria-mediated apoptosis in human T lymphocytes in vitro. Anesthesiology. 2005;102:1147–1157. doi: 10.1097/00000542-200506000-00014. [DOI] [PubMed] [Google Scholar]
- Lu DC, Rabizadeh S, Chandra S, Shayya RF, Ellerby LM, Ye X, Salvesen GS, Koo EH, Bredesen DE. A second cytotoxic proteolytic peptide derived from amyloid beta-protein precursor. Nat Med. 2000;6:397–404. doi: 10.1038/74656. [DOI] [PubMed] [Google Scholar]
- Matsuoka H, Kurosawa S, Horinouchi T, Kato M, Hashimoto Y. Inhalation anesthetics induce apoptosis in normal peripheral lymphocytes in vitro. Anesthesiology. 2001;95:1467–1472. doi: 10.1097/00000542-200112000-00028. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Calcium and neurodegeneration. Aging Cell. 2007;6:337–350. doi: 10.1111/j.1474-9726.2007.00275.x. [DOI] [PubMed] [Google Scholar]
- McLaughlin B, Hartnett KA, Erhardt JA, Legos JJ, White RF, Barone FC, Aizenman E. Caspase 3 activation is essential for neuroprotection in preconditioning. Proc Natl Acad Sci USA. 2003;100:715–720. doi: 10.1073/pnas.0232966100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mennerick S, Jevtovic-Todorovic V, Todorovic SM, Shen W, Olney JW, Zorumski CF. Effect of nitrous oxide on excitatory and inhibitory synaptic transmission in hippocampal cultures. J Neurosci. 1998;18:9716–9726. doi: 10.1523/JNEUROSCI.18-23-09716.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muravchick S, Smith DS. Parkinsonian symptoms during emergence from general anesthesia. Anesthesiology. 1995;82:305–307. doi: 10.1097/00000542-199501000-00039. [DOI] [PubMed] [Google Scholar]
- Narahashi T, Aistrup GL, Lindstrom JM, Marszalec W, Nagata K, Wang F, Yeh JZ. Ion channel modulation as the basis for general anesthesia. Toxicol Lett. 1998;100–101:185–191. doi: 10.1016/s0378-4274(98)00184-2. [DOI] [PubMed] [Google Scholar]
- Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–565. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- Paschen W, Mengesdorf T. Endoplasmic reticulum stress response and neurodegeneration. Cell Calcium. 2005;38:409–415. doi: 10.1016/j.ceca.2005.06.019. [DOI] [PubMed] [Google Scholar]
- Pearce RA, Stringer JL, Lothman EW. Effect of volatile anesthetics on synaptic transmission in the rat hippocampus. Anesthesiology. 1989;71:591–598. doi: 10.1097/00000542-198910000-00019. [DOI] [PubMed] [Google Scholar]
- Puil E, el-Beheiry H, Baimbridge KG. Anesthetic effects on glutamate-stimulated increase in intraneuronal calcium. J Pharmacol Exp Ther. 1990;255:955–961. [PubMed] [Google Scholar]
- Rekasi Z, Czompoly T, Schally AV, Boldizsar F, Varga JL, Zarandi M, Berki T, Horvath RA, Nemeth P. Antagonist of growth hormone-releasing hormone induces apoptosis in LNCaP human prostate cancer cells through a Ca2+-dependent pathway. Proc Natl Acad Sci USA. 2005;102:3435–3440. doi: 10.1073/pnas.0410006102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Simon W, Hapfelmeier G, Kochs E, Zieglgansberger W, Rammes G. Isoflurane blocks synaptic plasticity in the mouse hippocampus. Anesthesiology. 2001;94:1058–1065. doi: 10.1097/00000542-200106000-00021. [DOI] [PubMed] [Google Scholar]
- Solt K, Eger EI, Jr, Raines DE. Differential modulation of human N-methyl-d-aspartate receptors by structurally diverse general anesthetics. Anesth Analg. 2006;102:1407–1411. doi: 10.1213/01.ane.0000204252.07406.9f. [DOI] [PubMed] [Google Scholar]
- Takuma H, Tomiyama T, Kuida K, Mori H. Amyloid beta peptide-induced cerebral neuronal loss is mediated by caspase-3 in vivo. J Neuropathol Exp Neurol. 2004;63:255–261. doi: 10.1093/jnen/63.3.255. [DOI] [PubMed] [Google Scholar]
- Tanzi RE, Bertram L. Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective. Cell. 2005;120:545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Tyther R, Fanning N, Halligan M, Wang J, Redmond HP, Shorten G. The effect of the anaesthetic agent isoflurane on the rate of neutrophil apoptosis in vitro. Ir J Med Sci. 2001;170:41–44. doi: 10.1007/BF03167720. [DOI] [PubMed] [Google Scholar]
- Wei H, Kang B, Wei W, Liang G, Meng QC, Li Y, Eckenhoff RG. Isoflurane and sevoflurane affect cell survival and BCL-2/BAX ratio differently. Brain Res. 2005;1037:139–147. doi: 10.1016/j.brainres.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Wei H, Liang G, Yang H, Wang Q, Hawkins B, Madesh M, Wang S, Eckenhoff RG. The common inhalational anesthetic isoflurane induces apoptosis via activation of inositol 1,4,5-trisphosphate receptors. Anesthesiology. 2008;108:251–260. doi: 10.1097/01.anes.0000299435.59242.0e. [DOI] [PubMed] [Google Scholar]
- Westphalen RI, Hemmings HC., Jr Volatile anesthetic effects on glutamate versus GABA release from isolated rat cortical nerve terminals: basal release. J Pharmacol Exp Ther. 2006;316:208–215. doi: 10.1124/jpet.105.090647. [DOI] [PubMed] [Google Scholar]
- Wise-Faberowski L, Raizada MK, Sumners C. Oxygen and glucose deprivation-induced neuronal apoptosis is attenuated by halothane and isoflurane. Anesth Analg. 2001;93:1281–1287. doi: 10.1097/00000539-200111000-00051. [DOI] [PubMed] [Google Scholar]
- Wise-Faberowski L, Aono M, Pearlstein RD, Warner DS. Apoptosis is not enhanced in primary mixed neuronal/glial cultures protected by isoflurane against N-methyl-d-aspartate excitotoxicity. Anesth Analg. 2004;99:1708–1714. doi: 10.1213/01.ANE.0000136474.35627.FF. table of contents. [DOI] [PubMed] [Google Scholar]
- Wise-Faberowski L, Zhang H, Ing R, Pearlstein RD, Warner DS. Isoflurane-induced neuronal degeneration: an evaluation in organotypic hippocampal slice cultures. Anesth Analg. 2005;101:651–657. doi: 10.1213/01.ane.0000167382.79889.7c. table of contents. [DOI] [PubMed] [Google Scholar]
- Xie Z, Romano DM, Tanzi RE. Effects of RNAi-mediated silencing of PEN-2, APH-1a, and nicastrin on wild-type vs FAD mutant forms of presenilin 1. J Mol Neurosci. 2005a;25:67–77. doi: 10.1385/JMN:25:1:067. [DOI] [PubMed] [Google Scholar]
- Xie Z, Romano DM, Tanzi RE. RNA interference-mediated silencing of X11alpha and X11beta attenuates amyloid beta-protein levels via differential effects on beta-amyloid precursor protein processing. J Biol Chem. 2005b;280:15413–15421. doi: 10.1074/jbc.M414353200. [DOI] [PubMed] [Google Scholar]
- Xie Z, Dong Y, Maeda U, Alfille P, Culley DJ, Crosby G, Tanzi RE. The common inhalation anesthetic isoflurane induces apoptosis and increases amyloid beta protein levels. Anesthesiology. 2006a;104:988–994. doi: 10.1097/00000542-200605000-00015. [DOI] [PubMed] [Google Scholar]
- Xie Z, Dong Y, Maeda U, Moir R, Inouye SK, Culley DJ, Crosby G, Tanzi RE. Isoflurane-induced apoptosis: a potential pathogenic link between delirium and dementia. J Gerontol A Biol Sci Med Sci. 2006b;61:1300–1306. doi: 10.1093/gerona/61.12.1300. [DOI] [PubMed] [Google Scholar]
- Xie Z, Dong Y, Maeda U, Moir RD, Xia W, Culley DJ, Crosby G, Tanzi RE. The inhalation anesthetic isoflurane induces a vicious cycle of apoptosis and amyloid beta-protein accumulation. J Neurosci. 2007;27:1247–1254. doi: 10.1523/JNEUROSCI.5320-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Zorumski CF. Effects of isoflurane on N-methyl-d-aspartate gated ion channels in cultured rat hippocampal neurons. Ann NY Acad Sci. 1991;625:287–289. doi: 10.1111/j.1749-6632.1991.tb33851.x. [DOI] [PubMed] [Google Scholar]
- Yon JH, Daniel-Johnson J, Carter LB, Jevtovic-Todorovic V. Anesthesia induces neuronal cell death in the developing rat brain via the intrinsic and extrinsic apoptotic pathways. Neuroscience. 2005;135:815–827. doi: 10.1016/j.neuroscience.2005.03.064. [DOI] [PubMed] [Google Scholar]
- Zaugg M, Jamali NZ, Lucchinetti E, Shafiq SA, Siddiqui MA. Norepinephrine-induced apoptosis is inhibited in adult rat ventricular myocytes exposed to volatile anesthetics. Anesthesiology. 2000;93:209–218. doi: 10.1097/00000542-200007000-00032. [DOI] [PubMed] [Google Scholar]
- Zhan X, Fahlman CS, Bickler PE. Isoflurane neuroprotection in rat hippocampal slices decreases with aging: changes in intracellular Ca2+ regulation and N-methyl-d-aspartate receptor-mediated Ca2+ influx. Anesthesiology. 2006;104:995–1003. doi: 10.1097/00000542-200605000-00016. [DOI] [PubMed] [Google Scholar]
- Zhang SL, Yeromin AV, Zhang XH, Yu Y, Safrina O, Penna A, Roos J, Stauderman KA, Cahalan MD. Genome-wide RNAi screen of Ca(2+) influx identifies genes that regulate Ca(2+) release-activated Ca(2+) channel activity. Proc Natl Acad Sci USA. 2006;103:9357–9362. doi: 10.1073/pnas.0603161103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M, Su J, Head E, Cotman CW. Accumulation of caspase cleaved amyloid precursor protein represents an early neurodegenerative event in aging and in Alzheimer's disease. Neurobiol Dis. 2003;14:391–403. doi: 10.1016/j.nbd.2003.07.006. [DOI] [PubMed] [Google Scholar]