

Abstract
Although transcriptional dysregulation is a critical pathogenic mechanism in Huntington's disease (HD), it is still not known how mutant huntingtin causes it. Here we show that alteration of histone monoubiquitylation is a key mechanism. Disrupted interaction of huntingtin with Bmi-1, a component of the hPRC1L E3 ubiquitin ligase complex, increases monoubiquityl histone H2A (uH2A) levels in a cell culture model of HD. Genes with expression that is repressed in transgenic R6/2 mouse brain have increased uH2A and decreased uH2B at their promoters, whereas actively transcribed genes show the opposite pattern. Reduction in uH2A reverses transcriptional repression and inhibits methylation of histone H3 at lysine 9 in cell culture. In contrast, reduction in uH2B induces transcriptional repression and inhibits methylation of histone H3 at lysine 4. This is the first report to demonstrate hPRC1L as a huntingtin-interacting histone modifying complex and a crucial role for histone monoubiquitylation in mammalian brain gene expression, which broadens our understanding of histone code. These findings also provide a rationale for targeting histone monoubiquitylation for therapy in HD.
Keywords: histone monoubiquitylation, huntingtin, Bmi-1, histone methylation, transcription, histone code
Introduction
Huntington's disease (HD) is caused by an expansion of a CAG trinucleotide repeat in the HD gene encoding an abnormally long polyglutamine within huntingtin protein (The Huntington's Disease Collaborative Research Group, 1993). Human HD and transgenic mouse models of HD show downregulation of particular genes including neurotransmitter receptors, second messengers, and calcium homeostasis molecules (Augood et al., 1997; Cha et al., 1998; Luthi-Carter et al., 2000; Hodges et al., 2006). It is probable that mutant huntingtin has its toxic effects by affecting the expression of genes that are important to neural functioning. However, the molecular mechanisms responsible for transcriptional dysregulation have not been defined.
Posttranslational modifications of histones including acetylation, methylation, phosphorylation, ubiquitylation, and SUMOylation appear to be key factors in turning specific genes on or off, which is defined as the “histone code” (Strahl and Allis, 2000; Jason et al., 2002; Shiio and Eisenman, 2003). Among these, histone monoubiquitylation is the least understood modification, occurring in the mostly monoubiquitylated form, mainly on histones H2A and H2B (Goldknopf et al., 1975; West and Bonner, 1980). In higher eukaryotes, H2A is monoubiquitylated at lysine 119 (Nickel and Davie, 1989). It is linked to gene silencing such as repression of Ultrabithorax gene in Drosophila and X-chromosome inactivation in female mammals (de Napoles et al., 2004; Wang et al., 2004). In contrast, H2B is monoubiquitylated at lysine 120 (Thorne et al., 1987). Several studies show that H2B monoubiquitylation is associated with actively transcribed chromatin (Nickel et al., 1989; Ng et al., 2003). However, the role for histone monoubiquitylation in the regulation of mammalian gene expression is primarily unknown.
Histone acetylation and methylation are involved in transcriptional dysregulation in HD (Steffan et al., 2001; Hockly et al., 2003; Ryu et al., 2006; Sadri-Vakili et al., 2007; Stack et al., 2007). However, histone acetylation is transient and does not adequately explain long-lasting transcriptional changes in HD. Interestingly, histone acetylation affects monoubiquityl histone H2A (uH2A) (Sadri-Vakili et al., 2007), a more durable modification, whereas uH2B controls histone methylation (Ng et al., 2002; Sun and Allis, 2002). Thus, histone monoubiquitylation is a potential bridge between histone acetylation and methylation, leading to changes in gene expression in HD. In addition, an increased recruitment of a transcriptional coactivator complex, STAGA/TFTC, is involved in transcriptional dysregulation of spinocerebellar ataxia type 7 (SCA7), another polyglutamine disease (Helmlinger et al., 2004, 2006a,b). Because SAGA, the yeast homolog for mammalian STAGA, contains Ubp8, an enzyme for histone H2B deubiquitylation (Henry et al., 2003), H2B deubiquitylation by Ubp8 might induce transcriptional alterations in SCA7.
In this study, we have investigated whether histone monoubiquitylation plays a role in transcriptional dysregulation. We have identified molecular mechanisms by which mutant huntingtin alters histone monoubiquitylation and show that uH2A and uH2B have opposite effects on transcription in mouse and cell culture models of HD. This is the first report to show that histone monoubiquitylation regulates mammalian brain gene expression.
Materials and Methods
Transgenic R6/2 mice.
Brains from R6/2 transgenic mice and wild-type littermate controls were used in experiments. R6/2 transgenic mice contain exon 1 of the HD gene with an expanded CAG repeat (150–200) under control of the human HD promoter (Mangiarini et al., 1996). We purchased R6/2 transgenic mice from The Jackson Laboratory [code B6CBA-TgN (HDexon1); Bar Harbor, ME] and have maintained the colony by backcrossing R6/2 males to C57BL/6XCBA F1 females. Four-, eight-, and twelve-week-old R6/2 transgenic and wild-type littermate controls were killed, and brains were rapidly removed and frozen in liquid isopentane. Brains were stored at −80°C until use. The guidelines for animal care and use were approved by the Massachusetts General Hospital Subcommittee on Research Animal Care.
Cell culture.
Striatal cell lines were established from wild-type and STHdh (Q111) knock-in embryonic mice (Trettel et al., 2000). STHdh cell lines express full-length versions of either wild-type (containing 7 glutamines, STHdh7/7) or mutant (111 glutamines, STHdh111/111) huntingtin. Cells were kept in DMEM (Invitrogen, Gaithersburg, MD) plus 10% fetal bovine serum at 33°C for propagation and were placed at 39°C for 48 h to stop their division. ST14a cells, previously derived from rat embryonic striatum, were used to generate inducible cell lines expressing the N-terminal fragment of huntingtin (Sipione et al., 2002). ST14A cells and all subclones proliferate at 33°C but become postmitotic at 39°C. Cells were grown in DMEM, supplemented as described previously (Cattaneo and Conti, 1998). Tet-free fetal calf serum (Clontech, Mountain View, CA) was used to supplement the medium for the inducible subclones (Tet-free medium).
Treatment of STHdh cells. STHdh7/7 cells kept at 39°C for 48 h were treated with 0.1–1 μm MG-132 (Calbiochem, La Jolla, CA) or 0.1–5 μm lactacystin (Calbiochem) for another 24 h.
Western blotting.
For Western blot analysis of uH2A, 10–40 μg of histone extracts, which were prepared as described by Ferrante and colleagues (Ferrante et al., 2004; Ryu and Ferrante, 2005) from wild-type and transgenic R6/2 mouse brains or STHdh cell lines, were resuspended in 2× tricine SDS sample buffer (Invitrogen, Carlsbad, CA). Samples were boiled at 100°C for 4 min and fractionated on a 10–20% tricine gel (Invitrogen) for 100 min at 120 V. Proteins were transferred to polyvinylidene difluoride (PVDF) membranes in transfer buffer (3% Tris base, 14.4% glycine, and 20% methanol) at 400 mA for 30 min, and the PVDF was then blocked with 3% milk in distilled water for 1 h before immunodetection with anti-uH2A at lysine 119 antibody (Upstate Biotechnology, Lake Placid, NY) at a dilution of 1:500 overnight at 4°C. Primary antibody incubation was followed by washes in distilled water before incubation with the secondary antibody (HRP-conjugated goat anti-rabbit IgG; Jackson ImmunoResearch, West Grove, PA) and visualization using the ECL detection system (NEN, Boston, MA). Protein membranes were stripped in stripping buffer (Pierce, Rockford, IL) for 30 min and reprobed with anti-H2A antibody (Upstate Biotechnology) at 1:1000 dilution overnight at 4°C.
For uH2B Western blot analysis, 100 μg of histone extracts from mouse brains or STHdh cell lines were immunoprecipitated with 2 μg of anti-histone H2B antibody (Cell Signaling Technology, Danvers, MA) overnight at 4°C and protein A-agarose beads (Upstate Biotechnology) for 1 h at 4°C. Immunoprecipitated complexes were then subjected to Western blot analysis with anti-ubiquitin antibody (Zymed, South San Francisco, CA) at 1:250 dilution overnight at 4°C. For an internal control, anti-H2B antibody (Chemicon, Temecula, CA) was used at 1:1000 dilution overnight at 4°C. Other antibodies used for Western blot analyses were as follows: Ring2 (provided by Dr. M. Vidal, Universidad Autonoma de Madrid, Madrid, Spain); Bmi-1 [mouse monoclonal antibody (Upstate Biotechnology) and rabbit polyclonal antibody (Proteintech Group, Chicago, IL)]; histone H1 (Upstate Biotechnology); glutathione S-transferase (GST; Chemicon); huntingtin (MAB 2166; Chemicon); β-actin (Sigma, St. Louis, MO); hBre1 (provided by Dr. R. Roeder, Rockefeller University, New York, NY); and acetylated histone H3 at lysines 9 and 14, methylated histone H3 at lysine 4 or at lysine 9, heterochromatin protein 1α (HP1α), and H3 (all from Upstate Biotechnology). Optical density of individual protein bands was analyzed by using an image analysis program (Alpha Innotech, San Leandro, CA). Arbitrary units for optical density were displayed on the y-axis of each densitometry histogram. Statistical analysis on the optical density of the protein bands was performed on the mean ± SEM, and levels of protein expression in these tissues were compared by t test.
Immunofluorescence.
Brain sections from wild-type and transgenic R6/2 were permeabilized with 0.5% Triton X-100 in 1× PBS for 20 min at room temperature (RT) and incubated with 50 μg/ml Affinipure Fab fragment goat anti-mouse IgM (for uH2A; Jackson ImmunoResearch) in 0.05% Tween 20 containing 1× PBS or 10% normal goat serum (NGS) in 1× PBS (for Ring2 and hBre1) for 1 h at RT. Then, sections were washed with 1× PBS and incubated in primary antibodies overnight at 4°C: anti-uH2A antibody (1:50 dilution with 0.05% Tween 20 containing PBS), anti-Ring2 antibody (1:500 dilution with 10% NGS in 1× PBS), and anti-hBre1 serum (1:250 dilution with 10% NGS in 1× PBS). Sections were washed with 1× PBS and incubated with secondary antibodies for 1 h at RT; for uH2A, fluorescein (FITC) Affinipure Fab fragment goat anti-mouse IgM (Jackson ImmunoResearch) was used at 1:200 dilution in 0.05% Tween 20 containing PBS. For Ring2 and hBre1, Cy3 goat anti-rabbit IgG (Jackson ImmunoResearch) was used at 1:1500 or 1:750 dilution in 1× PBS, respectively. Sections were washed again with 1× PBS and mounted onto glass slides. Stained sections were examined under a confocal microscope.
GST pull-down assay.
GST-Htt clones (HD20Q and HD53Q) were a generous gift from Dr. E. Wanker (Max-Planck Institute, Berlin, Germany). GST fusion proteins were prepared as described previously (Yohrling et al., 2003). Verification of protein synthesis was performed with SDS-PAGE and Coomassie Blue staining. One hundred micrograms of nuclear extracts from STHdh7/7 cells were incubated with 400 μg of each GST-agarose, GST-exon 1 Htt 20Q fusion protein-agarose, and GST-exon 1 Htt 53Q fusion protein-agarose in buffer containing 10 mm Tris-HCl, pH 7.5, 150 mm NaCl, 0.1 mm Na3VO4, 30 mm Na4P2O7, 50 mm NaF, 1% Triton X-100, and protease inhibitor cocktail tablets (Roche Applied Science, Indianapolis, IN) at 4°C overnight. Agarose beads were washed four times with the buffer above, and proteins were extracted by heating at 100°C for 4 min in 30 μl of 2× Tris-glycine sample buffer. The amount of Bmi-1 present in GST pull down was determined by Western blot analysis.
Coimmunoprecipitation.
Two hundred micrograms of nuclear extracts from STHdh7/7 cells were incubated with 1 μg of anti-Htt (MAB2166; Chemicon) or anti-Bmi-1 (rabbit polyclonal antibody; Proteintech Group) antibody and 30 μl of protein A-agarose beads (Upstate Biotechnology) at 4°C for 4–5 h. The immune complexes were washed four times with the buffer used for GST pull-down assay and extracted by heating at 100°C for 4 min in 30 μl of 2× Tris-glycine sample buffer. Then, the immune complexes were subjected to Western blot analysis for Bmi-1 or huntingtin protein.
Chromatin immunoprecipitation assay.
We have adapted the chromatin immunoprecipitation (ChIP) technique to the analysis of brain tissue and have recently published a detailed methodology for performing ChIP experiments (Chen-Plotkin et al., 2006). Briefly, brain tissue or cells were subjected to cross-linking with formaldehyde. After cross-linking, the samples were homogenized and subjected to sonication, cleaving the chromatin into segments between 200 and 1000 bp in length. The cross-linked, sonicated samples were subjected to immunoprecipitation with anti-Bmi-1 antibody (Proteintech Group) or anti-uH2A antibody (Upstate Biotechnology). For uH2B ChIP, we immunoprecipitated ChIP homogenates with 1 μg of anti-H2B antibody (Cell Signaling Technology, Beverly, MA). Then, the H2B-immunoprecipitated DNA samples were mixed in ChIP dilution buffer containing protease inhibitors (Chen-Plotkin et al., 2006) to give a 2 ml volume, followed by a second immunoprecipitation with 20 μg of anti-ubiquitin antibody (MAB1510; Chemicon). Specifically, antibody–histone–DNA complexes were pelleted with centrifugation and washed. Cross-links were reversed by a series of low- and high-salt washes. Proteins were degraded by the addition of proteinase K. DNA was extracted by phenol/chloroform and ethanol precipitation, followed by DNA quantitation. To verify the specificity of this uH2B ChIP technique, histone–DNA complexes after sequential ChIPs with anti-H2B and anti-ubiquitin antibodies were subjected to Western blot analyses for uH2A and H2B.
DNA quantification.
Immunoprecipitated DNA (from ChIP) was quantified using the DNA Quant System, which detects 10–500 pg/μl DNA (Promega, Madison, WI), according to the manufacturer's instructions. Briefly, a premix was made with a buffer solution containing sodium pyrophosphate, NDPK enzyme solution, and T4 DNA polymerase, aliquoted and mixed with either known DNA quantities (for a standard curve) or experimental unknown DNA quantities. The reaction was incubated for 10 min at 37°C, chilled on ice for 30 min before mixing with ENLITEN luciferase reagent. The light output was immediately measured in a luminometer.
Real-time PCR.
One microliter of ChIP-derived DNA was used as a template in 20 μl reactions containing 10 μl of 2× SYBR Green Master Mix (Applied Biosystems, Foster City, CA) and 0.5 μm of each primer. Real-time thermal cycling was performed using an iCycler thermal cycler (Bio-Rad, Hercules, CA), with continuous SYBR Green monitoring according to the manufacturer's recommendations, using iCycler software. Cycling parameters for all amplifications were as follows: 60 cycles of 95°C for 30 s, 57°C for 30 s, and 72°C for 45 s, followed by melt-curve analysis (55°C+ for 10 s for 80 cycles). All PCRs were performed in triplicate and included negative controls (no DNA) as well as positive controls (serial dilutions of known amounts of genomic DNA). Primer sequences for real-time PCR are provided in supplemental Table 1 (available at www.jneurosci.org as supplemental material).
Real-time PCR data analysis.
Target DNA sequence quantities were estimated from threshold amplification cycle numbers (Tc) using iCycler software. The Tc value is determined as the cycle at which fluorescence (measured in relative fluorescence units) rises above a baseline threshold. Melt-curve analysis was also performed on all PCR products. Products were differentiated from primer–dimers based on melting temperatures, and PCR samples that had <40% of the product melting at the appropriate temperature or product/primer–dimer ratios of <2:1 were deemed to have insufficient DNA for analysis. For each gene sequence studied, a ΔTc value was calculated for each sample by subtracting the value for the immunoprecipitated sample from the value for the corresponding input DNA to normalize for differences in ChIP sample aliquots before immunoprecipitation. DNA quantities were then expressed as percentages of corresponding input using the following equation: (antibody ChIP as a percentage of input) = 2(ΔTc) × 100. DNA quantities (normalized to input) were compared for immunoprecipitated versus mock-immunoprecipitated samples. Data were analyzed with a one-way ANOVA, followed by Fisher's least significant difference method.
Small interfering RNA transfection.
STHdh111/111 cells were transfected with small interfering RNA (siRNA) to mouse Ring2 (RNF2; Dharmacon, Lafayette, CO) or nontargeting control siRNA (Dharmacon) using Lipofectamine 2000 (Invitrogen) in OPTI-MEM I (Invitrogen) for 24–72 h. An siRNA to mouse hBre1 (RNF20; Dharmacon) was transfected into STHdh7/7 cells using Lipofectamine 2000 for 24–72 h. The final concentration of the siRNAs in cell culture medium was 100 nm.
RNA extraction and reverse transcription.
RNA was extracted from R6/2 dissected brain regions, STHdh cells, and ST14a cells using the RNeasy kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. Reverse transcription reactions were performed using the Supescript First-Strand Synthesis System for reverse transcription-PCRs using specific primers to quantify the amount of gene expression.
Results
uH2A is increased in transgenic R6/2 mouse brain
The transgenic R6/2 mouse model of HD expresses exon 1 of the human HD gene containing ∼150 CAG repeats (Mangiarini et al., 1996). We compared global levels of uH2A at lysine 119 (uH2A) in 4, 8, and 12 week transgenic R6/2 brains with those in wild-type brains by Western blot analyses of histone extracts with an antibody specific to uH2A. Levels of uH2A were increased in transgenic whole brains at ages of 8 and 12 weeks (Fig. 1A). Furthermore, uH2A levels were increased in the transgenic striatum, hippocampus, and cortex at all ages tested: 4, 8, and 12 weeks (Fig. 1B) (12 week data not shown). Brain sections of wild-type and transgenic mice (4, 8, and 12 weeks) were stained with the anti-uH2A antibody, and striatum and cortex were examined with confocal microscopy. uH2A immunoreactivity localized in nuclei and was increased in transgenic striatum and cortex of all ages examined (Fig. 1C) (4 and 8 week data not shown).
Figure 1.
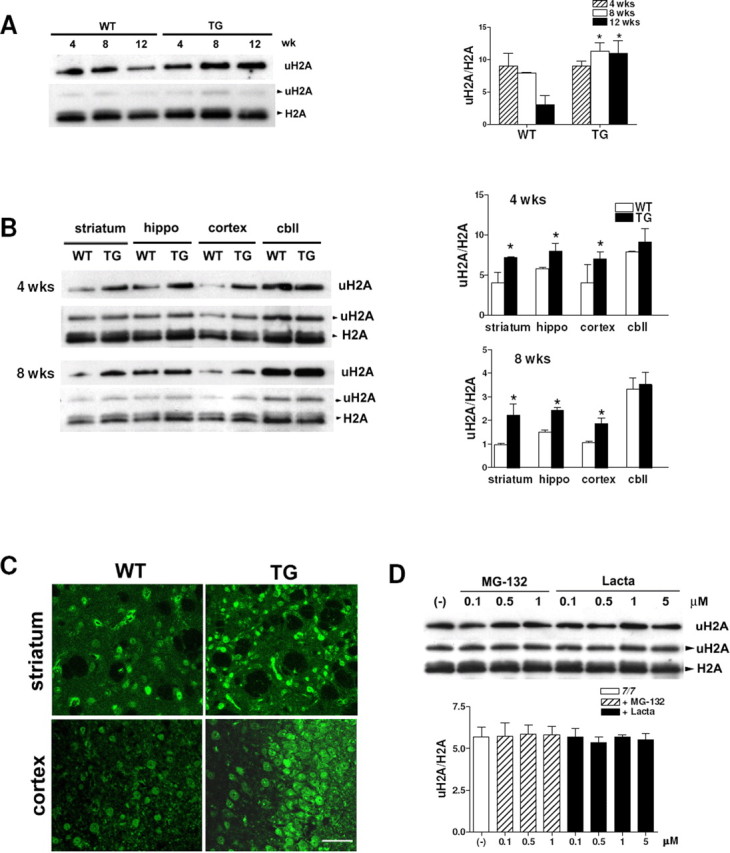
Increased uH2A in transgenic R6/2 brain. A, Top, Western blot analyses probed with anti-uH2A antibody showing increased global uH2A levels in transgenic R6/2 whole brains at the age of 8 and 12 weeks compared with wild type. Bottom, This blot was stripped and reprobed with anti-H2A antibody, which recognizes both monoubiquitylated and unmodified H2A, marked as uH2A and H2A, respectively. To quantify uH2A levels, intensities of uH2A bands in the top panel were normalized to those of H2A bands in the bottom panel (uH2A/H2A). Densitometry of uH2A/H2A is shown on the right (n = 4 animals per group; *p < 0.05). B, Western blots showing increased global uH2A levels in 4 and 8 week transgenic R6/2 striatum, hippocampus, and cortex, but not cerebellum. Densitometry of uH2A/H2A is shown on the right (n = 3 animals per group; *p < 0.05). C, Confocal images of 12 week R6/2 brain sections stained with anti-uH2A antibody (scale bar, 50 μm). D, Western blots showing no change in uH2A in STHdh7/7 cells treated with MG-132 (0.1–1 μm) or lactacystin (Lacta; 0.1–5 μm) for 24 h. Densitometry of uH2A/H2A is shown below (n = 4 independent experiments). Error bars indicate SEM. TG, Transgenic; WT, wild type; wk(s), week(s); hippo, hippocampus; cbll, cerebellum.
Proteasome inhibition does not affect uH2A levels
To examine whether increased uH2A levels in transgenic R6/2 brains are the result of impairment of the ubiquitin proteasome machinery that has been observed in HD (Valera et al., 2005), we treated full-length wild-type Htt-knock-in STHdh7/7 cells with proteasome inhibitors such as MG-132 (0.1–1 μm) and lactacystin (0.1–5 μm) for 24 h. Levels of uH2A were not affected, indicating that increased uH2A levels are not attributable to proteasome inhibition (Fig. 1D).
Bmi-1 binds to DNA more in the presence of mutant Htt
Recently, human Polycomb-repressive complex 1-like (hPRC1L; which is composed of several Polycomb group proteins including Ring1, Ring2, Bmi-1, and HPH2) was identified as an E3 ubiquitin ligase complex responsible for H2A monoubiquitylation (Wang et al., 2004). To examine whether a change in hPRC1L accounts for increased global levels of uH2A, we first examined Ring2, the catalytic domain of hPRC1L, by immunofluorescence and Western blot. Ring2 immunofluorescence was observed in nuclei, but there was no difference in subcellular localization or immunopositivity of Ring2 between wild-type and transgenic R6/2 striatum and cortex at 4, 8, or 12 weeks of age (data not shown). We also confirmed that there was no difference in nuclear Ring2 protein levels between 8 week wild-type and transgenic R6/2 by Western blot (Fig. 2A). Next, we looked at Bmi-1, the contribution of which to H2A monoubiquitylation is likely achieved through modulating the catalytic activity of Ring2 (Cao et al., 2005). Interestingly, we found that Bmi-1 was lower in nuclear extracts from 8 week transgenic striatum and cortex than those of wild type. But, it was detected more abundantly in histone extracts from 8 week transgenic striatum and cortex than those of wild type (Fig. 2A). Because histone extracts represent proteins present exclusively in nucleosomes such as histones, one possibility is that Bmi-1 protein binds more to nucleosomes in the presence of mutant Htt protein.
Figure 2.

Increased Bmi-1 binding to DNA in the presence of mutant huntingtin. A, Top, Western blot showing no difference in the Ring2 level in nuclear extracts (NE) between 8 week wild-type and transgenic R6/2 striatum and cortex. The blot was stripped and reprobed with anti-Bmi-1 antibody, which showed lower Bmi-1 in nuclear extracts from transgenic striatum and cortex. Densitometry of Ring2 and Bmi-1 is shown on the right (n = 3 animals per group; *p < 0.05). Bottom, Western blot showing higher Bmi-1 levels in histone extracts (HE) from 8 week transgenic R6/2 striatum and cortex. The blot was stripped and reprobed with anti-histone H1 antibody for an internal control. Bmi-1 bands were normalized to H1 bands (Bmi-1/H1), and densitometry of Bmi-1/H1 is shown on the right (n = 3 animals per group; *p < 0.05). B, GST pull-down assay showing Bmi-1 in nuclear extracts from STHdh7/7 cells binds to GST–exon 1 wild-type Htt (20Q) but less binds to GST–exon 1 mutant Htt (53Q). One-tenth of nuclear extracts used for the pull-down assay were loaded as the input. Dark bands in the GST lane are nonspecific ones. Immunoblotting of GST alone, 20Q, and 53Q proteins was performed with an anti-GST antibody. Then, Bmi-1 bands were normalized to GST bands (Bmi-1/GST.) Densitometry of Bmi-1/GST is shown on the right (n = 3 independent experiments; *p < 0.05, 20Q vs 53Q). C, Coimmunoprecipitation (IP) with anti-Htt antibody showing wild-type Htt in STHdh7/7 cells binds to Bmi-1, but mutant Htt in STHdh 111/111 does less. Another IP with anti-Bmi-1 antibody shows less Bmi-1 binding to mutant Htt than wild-type Htt. One-fifth of nuclear extracts used for the IPs were loaded as the input. Mouse (mIgG) and rabbit (rIgG) IgG are isotype-matched controls for the anti-Htt and anti-Bmi-1, respectively. Densitometry of Bmi-1 and Htt bands is shown below (n = 3 independent experiments; *p < 0.05). IB, Immunoblot. Error bars indicate SEM.
To address this possibility, we first examined whether there was an interaction between Htt and Bmi-1. In vitro GST pull-down assays using nuclear extracts from STHdh7/7 cells showed that exon 1 wild-type Htt (20Q) fusion protein bounds to Bmi-1 more than exon 1 mutant Htt (53Q) fusion protein (Fig. 2B). To see an endogenous interaction of Htt with Bmi-1, we immunoprecipitated nuclear extracts from wild-type Htt-expressing STHdh7/7 and mutant Htt-expressing (full-length mutant Htt-knock-in) STHdh111/111 cells with anti-Htt antibody and compared Bmi-1 in the immunoprecipitates by Western blot analyses. We found that wild-type Htt protein (from STHdh7/7 cells) bound to Bmi-1 more than mutant Htt (from STHdh111/111 cells) did. We also confirmed these Htt–Bmi-1 interactions by immunoprecipitating Bmi-1 and immunoblotting for Htt, which showed Bmi-1 bound more to wild-type Htt than to mutant Htt (Fig. 2C). Together, these findings suggest that wild-type Htt binds to Bmi-1 more than mutant Htt and, in the presence of mutant Htt, free Bmi-1 might be more accessible to bind to DNA. To address this idea, we performed ChIP with an anti-Bmi-1 antibody on STHdh7/7 and STHdh111/111 cells and quantified DNA amounts by normalizing the amounts of DNA bound by Bmi-1 to those of input DNA. We found that Bmi-1 binds to DNA more in STHdh111/111 cells than in STHdh7/7 cells (1.64 ± 0.10% of input DNA was bound by Bmi-1 in STHdh111/111 cells, whereas 1.05 ± 0.20% of input DNA was bound by Bmi-1 in STHdh7/7 cells). Thus, we suggest that Bmi-1 protein released from mutant Htt binds to DNA, which might contribute to increased uH2A levels in transgenic R6/2 brains.
Association of uH2A with gene promoters inversely correlates with transcriptional activities of the genes in R6/2
To study whether uH2A is relevant to transcriptional dysregulation in HD, we performed ChIP with an anti-uH2A antibody and quantitative real-time PCR with primers for the promoter regions of mouse D2 dopamine receptor (Drd2), preproenkephalin (Penk1), presomatostatin (Sst), the NR1 subunit of the NMDA receptor (Grin1), and β-actin (Actb) genes; expression of Drd2, Penk1, and Sst genes are known to be downregulated in HD, whereas Grin1 and Actb expression are not changed (Luthi-Carter et al., 2000). Whole brains from 8 week transgenic R6/2 mice showed a significant increase in uH2A association with Drd2, Penk1, and Sst genes. However, there was no difference in uH2A association with Grin1 gene between wild-type and transgenic brains (supplemental Fig. S1A, available at www.jneurosci.org as supplemental material). When we examined each brain region including striatum, hippocampus, cortex, and cerebellum, we found decreased uH2A association with Sst gene in 4 week transgenic cortex (Fig. 3A). Interestingly, in 8 and 12 week transgenic brain regions, we observed increased uH2A association with the Drd2, Penk1, and Sst gene promoters. In particular, striatum, which is the most affected area in HD, consistently showed increased uH2A association with all downregulated genes tested. However, uH2A association with Grin1 and Actb gene promoters was not different between wild-type and transgenic brains (Fig. 3B,C) (Penk1 and Actb not shown). These findings suggest that increased uH2A binding to gene promoters is a marker for transcriptional repression, which becomes more prominent over the disease course. To investigate how uH2A is related to other gene changes, we analyzed uH2A enrichment at promoters of (1) genes that are expressed in wild-type brains but not transgenic brains (“expressed-WT-only”) [inhibin β-C (Inhbc) and DnaJ (Hsp40) homolog, subfamily B, member 3 (Dnajb3)], (2) genes that are expressed in transgenic brains but not wild-type brains (“expressed-Tg-only”) [GABAA subunit theta (Gabt) and DNA methyltransferase 1 associated protein 1 (Dmap1)], or (3) genes that are expressed more highly in transgenic brains (“upregulated-Tg”) [cholecystokinin (CCK)]. Very interestingly, we observed increased uH2A association with Inhbc and Dnajb3 gene promoters but decreased uH2A association with Gabt, Dmap1, and CCK gene promoters in transgenic brain regions compared with wild type (Fig. 3D) (Dmap1 data not shown). Together, uH2A association with gene promoters appears to be specifically related to gene transcriptional activities (i.e., increased uH2A with “downregulated-Tg” and expressed-WT-only genes but decreased uH2A with upregulated-Tg and expressed-Tg-only genes in R6/2 brains).
Figure 3.

In vivo ChIP demonstrates that uH2A association with gene promoters inversely correlates with transcriptional activity in R6/2. A, No difference in uH2A association with Drd2, Sst, and Grin1 gene promoters between 4 week wild-type and transgenic R6/2 brain regions, except for decreased uH2A association with Sst gene promoter in transgenic cortex (n = 3 animals per group; *p < 0.05). uH2A enrichment in Grin1 gene promoter in hippocampus (hippo) was below a detection limit in both wild-type and transgenic mice. B, Increased uH2A association with Drd2 gene promoter in 8 week transgenic striatum, hippocampus, and cortex but not cerebellum (cbll) compared with wild-type. Increased uH2A association with Sst gene promoter in 8 week transgenic striatum and cortex, but not in hippocampus or cerebellum (n = 3 animals per group; *p < 0.05). C, Increased uH2A association with Drd2 gene promoters in all brain regions of 12 week transgenic R6/2 compared with wild type but increased uH2A association with Sst gene promoter in 12 week R6/2 striatum and hippocampus (n = 3 animals per group; *p < 0.05). D, Increased uH2A association with expressed-WT-only Inhbc and Dnaj3b gene promoters but decreased uH2A association with expressed-Tg-only Gabt and upregulated-Tg CCK gene promoters in all brain regions of 12 week transgenic mice, except for CCK in transgenic cerebellum, compared with wild type (n = 3 animals per group; *p < 0.05). Error bars indicate SEM. TG, Transgenic; WT, wild type; hippo, hippocampus; cbll, cerebellum; IP, immunoprecipitation.
Association of uH2A with promoters of downregulated genes is increased in mutant Htt-inducible cells and full-length mutant Htt knock-in cells
To study whether increased uH2A association with downregulated genes is a universal phenomenon in HD, we performed uH2A ChIP on parental ST14a (12.4), wild-type Htt-inducible (HD19), or mutant Htt-inducible (HD43) cells in the absence or presence of the inducer (0.5 μg/ml doxycycline). HD43 cells (105 glutamines) and HD19 cells (26 glutamines) overexpressed the N-terminal 548 amino acid fragment of mutant or wild-type Htt, respectively (supplemental Fig. S1B, available at www.jneurosci.org as supplemental material). ChIP and real-time PCR showed that there was increased uH2A association with Drd2 and Penk1 gene promoters in HD43 cells compared with that in 12.4 and HD19 cells when expression of Htt was induced by doxycycline. However, there was no difference in association of uH2A with Grin1 and Actb genes between doxycycline-added HD43 and HD19 cells (supplemental Fig. S1C, available at www.jneurosci.org as supplemental material).
We also analyzed uH2A association with gene promoters in another HD cell model, STHdh cells. Full-length mutant Htt-knock-in STHdh111/111 cells have different sets of downregulated genes from transgenic R6/2 brains, which include vitamin D receptor (Vdr), transcription factor 7 (Tcf7), inhibin β-b (Inhbb), and NADPH-dehydrogenase (Dhrs4) (Sadri-Vakili et al., 2007). We found increased uH2A association with those genes that are downregulated in STHdh111/111 cells. However, uH2A association with Actb (unchanged gene) promoter was equivalent in STHdh7/7 and STHdh111/111 cells (Fig. 4A).
Figure 4.

Increased uH2A association with genes downregulated in a cell culture model of HD and Ring2 knockdown-mediated reversal of transcriptional repression. A, Increased uH2A association with Vdr, Tcf7, Inhbb, and Dhrs4 gene promoters in STHdh111/111 cells compared with STHdh7/7 cells (n = 3 independent experiments; *p < 0.05). B, Western blot showing a reduced Ring2 protein level in STHdh111/111 cells transfected with 100 nm Ring2 siRNA (Ring2) for 24–72 h. Mock or 100 nm nontargeting control siRNA (NTC)-transfected cells do not show a significant change in Ring2 protein level. For an internal control, the blot was stripped and reprobed for β-actin. Ring2 bands were normalized to β-actin bands (Ring2/β-actin), and densitometry of Ring2/β-actin is shown below (n = 3 independent experiments; *p < 0.05, Ring2 vs NTC). C, Western blot showing reduction in the uH2A level in STHdh111/111 cells transfected with 100 nm Ring2 siRNA for 24–48 h. Densitometry of uH2A/H2A is shown below (n = 4 independent experiments; *p < 0.05, Ring2 vs NTC). D, Quantitative real-time PCR showing increased mRNA levels of VDR, TCF7, and Inhibin b in STHdh111/111 cells transfected with 100 nm Ring2 siRNA for 24 h, which is equivalent to those in STHdh7/7 cells. mRNA levels of each gene normalized to that of β-actin gene are indicated as VDR/β-actin, TCF7/β-actin, and Inhibin b/β-actin. Mock or NTC-transfected cells do not show any changes in mRNA levels of those genes (n = 4 independent experiments; *p < 0.05, Ring2 vs NTC). Error bars indicate SEM. IP, Immunoprecipitation.
Ring2 knockdown rescues transcriptional repression in mutant Htt-knock-in cells
To examine whether gene downregulation seen in HD can be corrected by reducing uH2A levels, we used an siRNA targeting Ring2 protein in STHdh111/111 cells. Ring2 siRNA knocked down Ring2 protein expression compared with mock and nontargeting control siRNA-transfected cells for all the time points examined (Fig. 4B). We confirmed that there was a dramatic decrease in uH2A levels in cells transfected with Ring2 siRNA for 24–48 h and that there was no change in total H2A levels in transfected cells (Fig. 4C). To examine mRNA levels in Ring2 siRNA-transfected cells, cDNAs from these cells were subjected to quantitative real-time PCR. Transcript levels were normalized to β-actin. Transcriptional repression of vitamin D receptor (VDR), transcription factor 7 (TCF7), and inhibin β-b (Inhibin b) was reversed in Ring2 siRNA-transfected cells (Fig. 4D). Together, these findings indicate that increased uH2A induces transcriptional repression and that uH2A is likely to play a causal role in transcriptional changes seen in HD.
uH2B is decreased in transgenic R6/2 brain
In contrast to uH2A, histone H2B monoubiquitylated at lysine 120 (uH2B) corresponds to actively transcribed chromatin (Nickel et al., 1989). We have analyzed global levels of uH2B by immunoprecipitating histone extracts from wild-type and transgenic R6/2 brains with an anti-H2B antibody and immunoblotting with an anti-ubiquitin (Ub) antibody. uH2B levels were very low, undetectable in 8 week transgenic whole brain (Fig. 5A). In 4 and 8 week transgenic mice, uH2B levels were undetectable in striatum and hippocampus but decreased in cerebellum compared with wild type (Fig. 5B) (8 week data not shown). To examine whether there is an alteration in hBre1, which is the E3 ubiquitin ligase specific to H2B (Kim et al., 2005; Zhu et al., 2005) to account for the decreased uH2B levels in transgenic mice, we stained brain sections from wild-type and transgenic mice at 4, 8, and 12 weeks of age with anti-hBre1 serum. hBre1 immunofluorescence was observed mainly in the peripheries of nuclei, but there was no difference in subcellular localization or protein level of hBre1, between wild-type and transgenic striatum and cortex areas of all the ages examined (Fig. 5C) (4 and 8 week data not shown).
Figure 5.
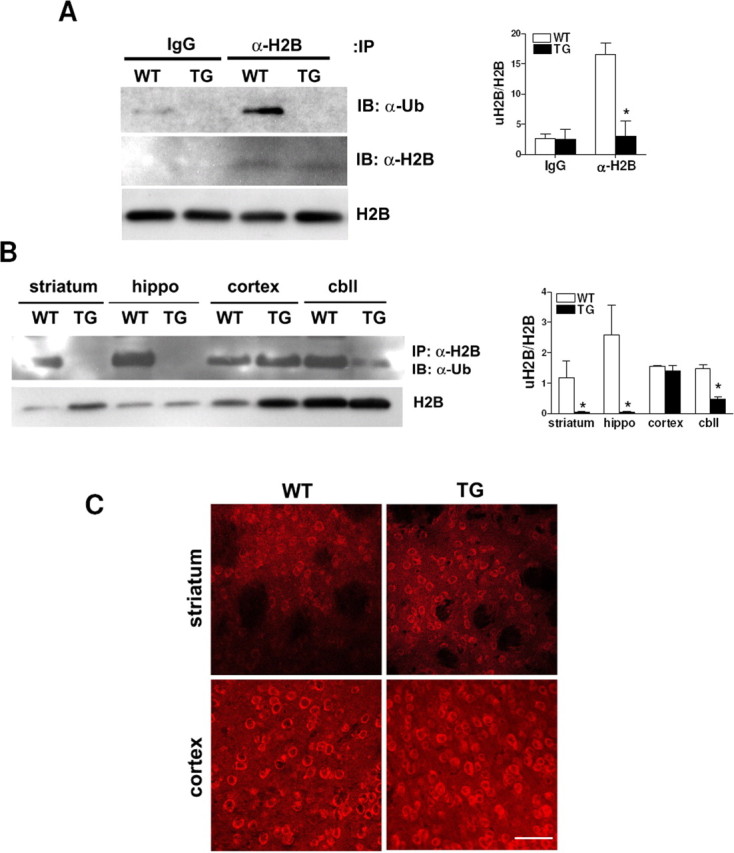
uH2B is decreased in transgenic R6/2 brain. A, Western blot showing that the global uH2B level in 8 week transgenic R6/2 whole brains was below a detection limit of immunoprecipitation (IP) with IgG or anti-H2B antibody followed by immunoblotting with anti-ubiquitin antibody (IB: α-Ub). This blot was stripped and reprobed with anti-H2B antibody, which showed that the same amounts of H2B were pulled down from wild-type and transgenic mice (IB: α-H2B). Western blot analysis of histone extracts also demonstrated H2B expression levels were equal in wild-type and transgenic mice (H2B). Densitometry of uH2B/H2B is shown on the right (n = 4 animals per group; *p < 0.05). B, Western blot showing global uH2B levels are undetectable in 4 week transgenic striatum and hippocampus and low in 4 week transgenic cerebellum. Western blot analysis of histone extracts from each brain region of 4 week mice demonstrated equal expression levels of H2B in wild-type and transgenic mice (H2B). Densitometry of uH2B/H2B is shown on the right (n = 4 animals per group; *p < 0.05). C, Confocal images of 12 week wild-type and transgenic brains stained with anti-hBre1 serum (scale bar, 50 μm). Error bars indicate SEM. TG, Transgenic; WT, wild type; hippo, hippocampus; cbll, cerebellum.
Association of uH2B with gene promoters positively correlates with transcriptional activities of the genes in R6/2
To study whether the decreased global uH2B level is related to transcriptional dysregulation in HD, we examined uH2B association with gene promoters by performing sequential ChIPs with anti-H2B and then anti-Ub antibodies. The specificity of this uH2B ChIP technique was verified by subjecting the immunoprecipitated histone–DNA complexes to a Western analysis for uH2A; no uH2A was detected in uH2B ChIP samples (supplemental Fig. S1D, available at www.jneurosci.org as supplemental material). uH2B had decreased association with the Drd2 gene promoter in 8 week transgenic whole brain, but there was no difference in its association with the Grin1 and Actb promoters between wild-type and transgenic brains (supplemental Fig. S1E, available at www.jneurosci.org as supplemental material). Although decreased uH2B levels were detected as early as 4 weeks of age (Fig. 5B), uH2B association with the Drd2 and Sst gene promoters was increased in striatum but decreased in hippocampus of 4 week transgenic mice compared with wild type (Fig. 6A) (Sst data not shown). However, decreased uH2B association with Drd2, Penk1, and Sst was observed in 8 and 12 week transgenic striatum, hippocampus, and cortex, but not cerebellum. There was no difference in uH2B association with Grin1 and Actb genes in each brain region between wild-type and transgenic mice (Fig. 6B,C) (8 week Sst and Grin1 and 12 week Penk1 and Actb data not shown). These results demonstrate that uH2B association with the downregulated genes decreases over the disease course. We also analyzed uH2B enrichment at promoters of expressed-WT-only (Inhbc and Dnajb3), expressed-Tg-only (Gabt and Dmap1), or upregulated-Tg (CCK) genes in 8 week R6/2 brains and found decreased uH2B association with Inhbc and Dnajb3 genes but increased association with Gabt, Dmap1, and CCK genes in 8 week transgenic brain regions (Fig. 6D) (Dmap1 data not shown). Together, uH2B association with gene promoters appears to positively correlate with transcriptional activities of the genes tested. These findings demonstrate that H2B monoubiquitylation is involved in transcriptional dysregulation in HD in the same way as H2A monoubiquitylation, which affects not only gene expression levels (upregulation or downregulation) but also genes being turned on and off (expressed-WT-only or expressed-Tg-only).
Figure 6.

In vivo ChIP demonstrates that uH2B association with gene promoters positively correlates with transcriptional activity in R6/2. A, uH2B association with Drd2 gene promoter was increased in striatum but decreased in hippocampus of 4 week transgenic R6/2. No difference in uH2B association with Actb gene promoter between wild-type and transgenic mice (n = 3 animals per group; *p < 0.05). B, Decreased uH2B association with Drd2 and Penk1 gene promoters in 8 week transgenic striatum, hippocampus, and cortex, but not cerebellum, compared with wild type (n = 3 animals per group; *p < 0.05). C, Decreased uH2B association with Drd2 and Sst gene promoters in 12 week transgenic striatum, hippocampus, and cortex, but not cerebellum, compared with wild type (n = 3 animals per group; *p < 0.05). D, Decreased uH2B association with expressed-WT-only Inhbc and Dnaj3b gene promoters but increased uH2B association with expressed-Tg-only Gabt and upregulated-Tg CCK gene promoters in 8 week transgenic striatum, hippocampus, and cortex, but not cerebellum, compared with wild type (n = 4 animals per group; *p < 0.05). Error bars indicate SEM. TG, Transgenic; WT, wild type; hippo, hippocampus; cbll, cerebellum.
Association of uH2B with promoters of downregulated genes is decreased in mutant Htt-inducible cells and full-length mutant Htt knock-in cells
To study whether decreased uH2B association with downregulated genes is a universal phenomenon in HD, we performed uH2B ChIP on parental ST14a (12.4), wild-type Htt-inducible (HD19), or mutant Htt-inducible (HD43) cells in the absence or presence of 0.5 μg/ml doxycycline. Immunoprecipitated DNAs were subjected to real-time PCR with primers for the promoter regions of rat Drd2, Penk1, Grin1, and Actb genes. In the presence of doxycycline, there was decreased uH2B association with Drd2 and Penk1 in HD43 cells compared with 12.4 and HD19 cells. However, there was no difference in association of uH2B with Grin1 and Actb genes between HD43 and HD19 cells (supplemental Fig. S1F, available at www.jneurosci.org as supplemental material). Association of uH2B with genes downregulated (Vdr, Tcf7, Inhbb, and Dhrs4) in STHdh111/111 cells was also decreased, but uH2B association with Actb (unchanged gene) promoter was equivalent in STHdh7/7 and STHdh111/111 cells (Fig. 7A).
Figure 7.

Decreased uH2B associations with genes downregulated in a cell culture model of HD and hBre1 knockdown-mediated transcriptional repression. A, Decreased uH2B association with Vdr, Tcf7, Inhbb, and Dhrs4 gene promoters in STHdh111/111 cells compared with that in STHdh7/7 cells (n = 3 independent experiments; *p < 0.05). IP, Immunoprecipitation. B, Western blot showing decreased levels of hBre1 and uH2B in STHdh7/7 cells transfected with 100 nm hBre1siRNA (hBre1) for 24–72 h. Mock or nontargeting control siRNA (NTC)-transfected cells do not affect them. β-Actin and H2B were used as internal controls for hBre1 and uH2B, respectively. Densitometry of hBre1/β-actin and uH2B/H2B is shown below (n = 3 independent experiments; *p < 0.05, hBre1 vs NTC). C, Quantitative real-time PCR showing that decreased mRNA levels of VDR, TCF7, and Inhibin b in STHdh7/7 cells transfected with 100 nm hBre1 siRNA for 48 h, which is equivalent to those in STHdh111/111cells. mRNA levels of each gene normalized to that of β-actin gene are indicated as VDR/β-actin, TCF7/β-actin, and Inhibin b/β-actin (n = 4 independent experiments; *p < 0.05, hBre1 vs NTC). Error bars indicate SEM.
hBre1 knockdown induces transcriptional repression in wild-type Htt knock-in cells
To determine whether uH2B alters gene expression, we manipulated uH2B levels by knocking down hBre1, the E3 ubiquitin ligase. The hBre1 siRNA reduced hBre1 and uH2B levels in STHdh7/7cells, whereas mock and nontargeting control siRNA did not. There was no change in β-actin and H2B levels in hBre1 siRNA-transfected cells (Fig. 7B). Levels of mRNA encoding VDR, TCF7, and Inhibin b were decreased in hBre1 siRNA-transfected cells. Mock and nontargeting control siRNA-transfected cells did not affect mRNA levels (Fig. 7C). These findings demonstrate that decreasing uH2B is sufficient to induce transcriptional repression and that uH2B is likely to play a causal role in transcriptional changes seen in HD.
uH2A affects methylation of histone H3 at lysine 9, whereas uH2B affects methylation of histone H3 at lysine 4
It would be important to understand how uH2A and uH2B are linked to each other and other histone modifications such as acetylation and methylation. Acetylation of histone H3 at lysines 9 and 14 (AcH3) and methylation of histone H3 at lysine 4 (methyl H3K4) are known to be involved in transcriptional activation (Roth et al., 2001; Santos-Rosa et al., 2002). Moreover, methyl H3K4 is known to be regulated by uH2B (Ng et al., 2002; Sun and Allis, 2002). In contrast, methylation of histone H3 at lysine 9 (methyl H3K9) plays a role in transcriptional repression by recruiting heterochromatin protein 1 (HP1) (Bannister et al., 2001; Lachner et al., 2001).
We analyzed uH2A, uH2B, AcH3, methyl H3K4, methyl H3K9, and HP1α levels in cells transfected with Ring2 or hBre1 siRNA by performing Western blot analyses. Reducing uH2A with Ring2 siRNA did not change uH2B, AcH3, and methyl H3K4 levels (supplemental Fig. S2A, available at www.jneurosci.org as supplemental material). However, transfection with Ring2 siRNA significantly decreased di- and tri-methyl H3K9 and HP1α (Fig. 8A). These results suggest that uH2A contributes to transcriptional repression by regulating proteins involved in heterochromatin formation such as methyl H3K9 and HP1α. However, reducing uH2B with hBre1 siRNA did not affect uH2A, AcH3, methyl H3K9, or HP1α levels but decreased di- and tri-methyl H3K4 (Fig. 8B and supplemental Fig. S2B, available at www.jneurosci.org as supplemental material) (HP1α not shown). Together, uH2A and uH2B are independent, and they do not affect acetylation of histone H3. However, histone monoubiquitylation regulates methylation of histone H3 at different lysine residues, which might play a role in determining transcriptional activities.
Figure 8.

uH2A regulates methylation of histone H3 at lysine 9 and recruitment of HP1α, whereas uH2B regulates methylation of histone H3 at lysine 4. A, Western blots showing decreased histone H3 di- and tri-methylation at lysine 9 (H3K9me2 and H3K9me3, respectively) and HP1α in STHdh111/111 cells transfected with Ring2 siRNA for 24–48 h. H3 was used as an internal control. Densitometry of H3K9me2/H3, H3K9me3/H3, and HP1α/H3 is on the right (n = 3 independent experiments; *p < 0.05, Ring2 vs NTC). B, Western blots showing decreased histone H3 di-and tri-methylation at lysine 4 (H3K4me2 and H3K4me3) but no change in monomethyl histone H3 at lysine 4 (H3K4me) in STHdh7/7cells transfected with hBre1 siRNA for 24–72 h. H2B was used as an internal control. Densitometry of H3K4me/H2B, H3K4me2/H2B, and H3K4me3/H2B is shown on the right (n = 3 independent experiments; *p < 0.05, hBre1 vs NTC). Error bars indicate SEM.
Discussion
Transcriptional changes in human HD and transgenic mouse models of HD suggest that mutant Htt exerts its toxic effects by disrupting gene expression important for neuronal survival and function. Although several pieces of evidence show that histone acetylation and methylation contribute to HD pathogenesis (McCampbell et al., 2001; Steffan et al., 2001; Hockly et al., 2003; Ryu et al., 2006; Sadri-Vakili et al., 2007; Stack et al., 2007), histone monoubiquitylation has not yet been studied. Given the known functions of uH2A (transcriptional repression) and H2B (transcriptional activation), we investigated whether histone monoubiquitylation plays a role in transcriptional dysregulation in HD. We addressed five major questions: (1) Do levels of uH2A and H2B differ between wild-type and transgenic R6/2 brains? (2) Does huntingtin affect monoubiquitylation of histone H2A and H2B? (3) Do levels of monoubiquityl histones at gene promoters correlate with transcriptional activity? (4) Does histone monoubiquitylation induce transcriptional changes? (5) Does histone monoubiquitylation affect other histone modifications?
We found increased uH2A levels in transgenic R6/2 mouse striatum, hippocampus, and cortex at all the ages examined. Proteasome impairment does not appear to be involved in the regulation of the uH2A level (Fig. 1). To understand the molecular mechanisms underlying increased uH2A in the presence of mutant Htt protein, we examined Ring2 and Bmi-1 of the mammalian Polycomb-repressive complex 1, termed hPRC1L, because it has an H2A-K119 ubiquitin E3 ligase activity. Ring2 is the catalytic subunit of hPRC1L, and Bmi-1 positively regulates H2A monoubiquitylation in a highly cooperative manner with Ring2 (Cao et al., 2005). Whereas there were no differences in subcellular localization, immunoreactivity, and nuclear protein levels of Ring2, there was a difference in Bmi-1 between wild-type and transgenic R6/2 (Fig. 2). On the basis of in vitro binding experiments with recombinant GST proteins and in vivo coimmunoprecipitation, wild-type Htt was found to bind to Bmi-1, whereas mutant Htt bound less, which might result in more Bmi-1 binding to DNA (Fig. 2). We thus identify hPRC1L as a histone modifying complex that interacts with huntingtin. Additionally, our study provides a clue to understanding normal physiological functions of wild-type Htt, which is that wild-type Htt might regulate transcriptional activity by limiting the availability of Bmi-1 binding to gene promoters.
We also found decreased uH2B levels in transgenic R6/2 brains (Fig. 5). As is the case with Ring2 for uH2A, subcellular localization or immunoreactivity of the E3 ubiquitin ligase for H2B, termed hBre1, was not different between wild-type and transgenic R6/2 brains. We did not observe an interaction between Htt and hBre1 with GST pull-down assays using the recombinant GST-Htt proteins and nuclear extracts from STHdh7/7 cells (data not shown). The ubiquitin protease Ubp8 mediates deubiquitylation of histone H2B in Saccharomyces cerevisiae (Henry et al., 2003). In Drosophila, the ubiquitin-specific protease 7–guanosine 5′-monophosphate synthetase complex catalyzes the selective deubiquitylation of histone H2B but not H2A (van der Knaap et al., 2005). However, the mammalian ubiquitinase specific to uH2B has not yet been identified. Thus, activity of hBre1 or changes in yet-unidentified uH2B-ubiquitinase would be interesting venues to investigate in HD.
Although global levels of uH2A were increased and global levels of uH2B were decreased in transgenic mouse brain, their levels in the promoters of specific single genes varied depending on transcriptional activities of genes. The promoters of the downregulated-Tg and expressed-WT-only genes in R6/2 mice have increased uH2A association and decreased uH2B association, which was observed mostly after 4 weeks of age (Figs. 3, 6). However, association of uH2A or uH2B with the unchanged genes in R6/2 was compatible between wild-type and transgenic R6/2. This relationship of monoubiquityl histones with the downregulated genes was recapitulated in cell culture models of HD, in both Htt-inducible cell lines (ST14a) and full-length Htt knock-in cells (STHdh) (Figs. 4, 7, and supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Furthermore, we found that the promoters of the upregulated-Tg and expressed-Tg-only genes in R6/2 have decreased uH2A and increased uH2B association. Therefore, our results provide strong evidence that uH2A and uH2B levels at promoters could be used as a marker for transcriptional activity of genes; transcriptionally inactive genes have high uH2A and low uH2B levels at their promoters, whereas transcriptionally active gene promoters have low uH2A and high uH2B levels.
Reduction in uH2A by siRNA-mediated knockdown of Ring2 corrected mRNA levels of certain genes known to be downregulated in STHdh111/111 cells (Fig. 4). In contrast, reduction in uH2B by hBre1 siRNA induced transcriptional repression of those genes in STHdh7/7cells (Fig. 7). Therefore, we successfully demonstrated that increased uH2A and decreased uH2B indeed induce transcriptional changes in our cell culture model of HD. These findings strongly suggest that uH2A and uH2B play a causal role in transcriptional dysregulation in human HD as well as R6/2 transgenic mice. In addition, the histone monoubiquitylation pathway could be a potential therapeutic target for HD. For instance, correcting uH2A level by targeting Ring2 protein might rescue transcriptional abnormalities and thereby the phenotype of HD.
The molecular mechanisms underlying uH2A-mediated-transcriptional repression, particularly in mammalian systems remain primarily unknown. Recently, Cul4 (E3 ubiquitin ligase) was shown to play a role in the assembly of closed heterochromatin by recruiting Clr4 (histone H3 methyltransferase) and HP1 into nucleosomes in yeast (Jia et al., 2005). It was also demonstrated that H2A is a substrate for Cul4 in the context of DNA damage repair (Kapetanaki et al., 2006). Interestingly, we found that uH2A functions upstream of di- and tri-methyl histone H3 at lysine 9 (H3K9) and HP1α (Fig. 8). Both di- and tri-methylation of H3K9 are predominant marks of heterochromatin, and HP1α is associated mainly with centromeric heterochromatin in mammalian cells (Schotta et al., 2002; Peters et al., 2003). Because heterochromatin is linked to transcriptional repression, we hypothesize that increased uH2A in certain DNA regions favors heterochromatin and results in transcriptional repression.
In contrast, the molecular mechanisms by which uH2B regulates transcriptional activation are relatively well known. uH2B functions upstream of methylation of histone H3 at lysine 4 (H3K4) and then at lysine 79 (H3K79); specifically, it is required for the transition from monomethylation to subsequent methylation of H3K4 (Shahbazian et al., 2005). In concordance, our results showed that hBre1-mediated uH2B affects di- and tri-methylation of H3K4 (Fig. 8). But uH2A and uH2B do not affect each other or acetylation of histone H3 at lysines 9 and 14 (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Together, we identified the new relationships between histone modifications; monoubiquityl H2A and H2B are independent from each other but function upstream of histone H3 methylation, albeit at different lysine residues.
We provide a comprehensive evaluation of the possibility that histone monoubiquitylation plays a crucial role in mutant Htt-mediated transcriptional dysregulation. Our data indicate that monoubiquitylation of histone H2A and deubiquitylation of H2B are two key alterations responsible for transcriptional changes in HD. This mechanism appears to be achieved through a huntingtin-interacting histone modifying complex, hPRC1L, and methylation of histone H3, which broadens our understanding of the histone code. This is the first report to demonstrate involvement of uH2A and uH2B in mammalian brain gene expression. Our findings also imply that histone monoubiquitylation is a potential target for HD therapy.
Footnotes
This work was supported by grants from the National Institute of Neurological Disorders and Stroke (NS38106, NS45242), the Huntington's Disease Society of America (HDSA) Coalition for the Cure (J.-H.J.C.), the Glendorn Foundation (J.-H.J.C.), the Massachusetts Biomedical Research Corporation (Tosteson Postdoctoral Fellowship to M.-O.K.), and HDSA (research fellowship to M.-O.K.). We thank Dr. Miguel Vidal (Universidad Autonoma de Madrid, Madrid, Spain) and Jaehoon Kim and Dr. Robert Roeder (Rockefeller University, New York, NY) for the generous gift of anti-Ring2 antibody and anti-hBre1 serum. We thank Dr. Elena Cattaneo (University of Milan, Milan, Italy) and Dr. Marcy MacDonald for the generous gift of ST14a and STHdh cell lines, respectively.
References
- Augood SJ, Faull RL, Emson PC. Dopamine D1 and D2 receptor gene expression in the striatum in Huntington's disease. Ann Neurol. 1997;42:215–221. doi: 10.1002/ana.410420213. [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410:120–124. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- Cao R, Tsukada Y, Zhang Y. Role of Bmi-1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Mol Cell. 2005;20:845–854. doi: 10.1016/j.molcel.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Cattaneo E, Conti L. Generation and characterization of embryonic striatal conditionally immortalized ST14A cells. J Neurosci Res. 1998;53:223–234. doi: 10.1002/(SICI)1097-4547(19980715)53:2<223::AID-JNR11>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Cha JH, Kosinski CM, Kerner JA, Alsdorf SA, Mangiarini L, Davies SW, Penney JB, Bates GP, Young AB. Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human Huntington disease gene. Proc Natl Acad Sci USA. 1998;95:6480–6485. doi: 10.1073/pnas.95.11.6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen-Plotkin AS, Sadri-Vakili G, Yohrling GJ, Braveman MW, Benn CL, Glajch KE, Dirocco DP, Farrell LA, Krainc D, Gines S, Macdonald ME, Cha JH. Decreased association of the transcription factor Sp1 with genes downregulated in Huntington's disease. Neurobiol Dis. 2006;22:233–241. doi: 10.1016/j.nbd.2005.11.001. [DOI] [PubMed] [Google Scholar]
- de Napoles M, Mermoud JE, Wakao R, Tang YA, Endoh M, Appanah R, Nesterova TB, Silva J, Otte AP, Vidal M, Koseki H, Brockdorff N. Polycomb group proteins Ring1A/B link ubiquitylation of histone H2A to heritable gene silencing and X inactivation. Dev Cell. 2004;7:663–676. doi: 10.1016/j.devcel.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Ryu H, Kubilus JK, D'Mello S, Sugars KL, Lee J, Lu P, Smith K, Browne S, Beal MF, Kristal BS, Stavrovskaya IG, Hewett S, Rubinsztein DC, Langley B, Ratan RR. Chemotherapy for the brain: the antitumor antibiotic mithramycin prolongs survival in a mouse model of Huntington's disease. J Neurosci. 2004;24:10335–10342. doi: 10.1523/JNEUROSCI.2599-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldknopf IL, Taylor CW, Baum RM, Yeoman LC, Olson MO, Prestayko AW, Busch H. Isolation and characterization of protein A24, a “histone-like” non-histone chromosomal protein. J Biol Chem. 1975;250:7182–7187. [PubMed] [Google Scholar]
- Helmlinger D, Hardy S, Sasorith S, Klein F, Robert F, Weber C, Miguet L, Potier N, Van-Dorsselaer A, Wurtz JM, Mandel JL, Tora L, Devys D. Ataxin-7 is a subunit of GCN5 histone acetyltransferase-containing complexes. Hum Mol Genet. 2004;13:1257–1265. doi: 10.1093/hmg/ddh139. [DOI] [PubMed] [Google Scholar]
- Helmlinger D, Hardy S, Eberlin A, Devys D, Tora L. Both normal and polyglutamine-expanded ataxin-7 are components of TFTC-type GCN5 histone acetyltransferase-containing complexes. Biochem Soc Symp. 2006a:155–163. doi: 10.1042/bss0730155. [DOI] [PubMed] [Google Scholar]
- Helmlinger D, Hardy S, Abou-Sleymane G, Eberlin A, Bowman AB, Gansmuller A, Picaud S, Zoghbi HY, Trottier Y, Tora L, Devys D. Glutamine-expanded ataxin-7 alters TFTC/STAGA recruitment and chromatin structure leading to photoreceptor dysfunction. PLoS Biol. 2006b;4:e67. doi: 10.1371/journal.pbio.0040067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry KW, Wyce A, Lo WS, Duggan LJ, Emre NC, Kao CF, Pillus L, Shilatifard A, Osley MA, Berger SL. Transcriptional activation via sequential histone H2B ubiquitylation and deubiquitylation, mediated by SAGA-associated Ubp8. Genes Dev. 2003;17:2648–2663. doi: 10.1101/gad.1144003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockly E, Richon VM, Woodman B, Smith DL, Zhou X, Rosa E, Sathasivam K, Ghazi-Noori S, Mahal A, Lowden PA, Steffan JS, Marsh JL, Thompson LM, Lewis CM, Marks PA, Bates GP. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington's disease. Proc Natl Acad Sci USA. 2003;100:2041–2046. doi: 10.1073/pnas.0437870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges A, Strand AD, Aragaki AK, Kuhn A, Sengstag T, Hughes G, Elliston LA, Hartog C, Goldstein DR, Thu D, Hollingsworth ZR, Collin F, Synek B, Holmans PA, Young AB, Wexler NS, Delorenzi M, Kooperberg C, Augood SJ, Faull RL, et al. Regional and cellular gene expression changes in human Huntington's disease brain. Hum Mol Genet. 2006;15:965–977. doi: 10.1093/hmg/ddl013. [DOI] [PubMed] [Google Scholar]
- Jason LJ, Moore SC, Lewis JD, Lindsey G, Ausio J. Histone ubiquitination: a tagging tail unfolds? BioEssays. 2002;24:166–174. doi: 10.1002/bies.10038. [DOI] [PubMed] [Google Scholar]
- Jia S, Kobayashi R, Grewal SI. Ubiquitin ligase component Cul4 associates with Clr4 histone methyltransferase to assemble heterochromatin. Nat Cell Biol. 2005;7:1007–1013. doi: 10.1038/ncb1300. [DOI] [PubMed] [Google Scholar]
- Kapetanaki MG, Guerrero-Santoro J, Bisi DC, Hsieh CL, Rapic-Otrin V, Levine AS. The DDB1-CUL4ADDB2 ubiquitin ligase is deficient in xeroderma pigmentosum group E and targets histone H2A at UV-damaged DNA sites. Proc Natl Acad Sci USA. 2006;103:2588–2593. doi: 10.1073/pnas.0511160103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Hake SB, Roeder RG. The human homolog of yeast BRE1 functions as a transcriptional coactivator through direct activator interactions. Mol Cell. 2005;20:759–770. doi: 10.1016/j.molcel.2005.11.012. [DOI] [PubMed] [Google Scholar]
- Lachner M, O'Carroll D, Rea S, Mechtler K, Jenuwein T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 2001;410:116–120. doi: 10.1038/35065132. [DOI] [PubMed] [Google Scholar]
- Luthi-Carter R, Strand A, Peters NL, Solano SM, Hollingsworth ZR, Menon AS, Frey AS, Spektor BS, Penney EB, Schilling G, Ross CA, Borchelt DR, Tapscott SJ, Young AB, Cha JH, Olson JM. Decreased expression of striatal signaling genes in a mouse model of Huntington's disease. Hum Mol Genet. 2000;9:1259–1271. doi: 10.1093/hmg/9.9.1259. [DOI] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- McCampbell A, Taye AA, Whitty L, Penney E, Steffan JS, Fischbeck KH. Histone deacetylase inhibitors reduce polyglutamine toxicity. Proc Natl Acad Sci USA. 2001;98:15179–15184. doi: 10.1073/pnas.261400698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng HH, Xu RM, Zhang Y, Struhl K. Ubiquitination of histone H2B by Rad6 is required for efficient Dot1-mediated methylation of histone H3 lysine 79. J Biol Chem. 2002;277:34655–34657. doi: 10.1074/jbc.C200433200. [DOI] [PubMed] [Google Scholar]
- Ng HH, Robert F, Young RA, Struhl K. Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol Cell. 2003;11:709–719. doi: 10.1016/s1097-2765(03)00092-3. [DOI] [PubMed] [Google Scholar]
- Nickel BE, Davie JR. Structure of polyubiquitinated histone H2A. Biochemistry. 1989;28:964–968. doi: 10.1021/bi00429a007. [DOI] [PubMed] [Google Scholar]
- Nickel BE, Allis CD, Davie JR. Ubiquitinated histone H2B is preferentially located in transcriptionally active chromatin. Biochemistry. 1989;28:958–963. doi: 10.1021/bi00429a006. [DOI] [PubMed] [Google Scholar]
- Peters AH, Kubicek S, Mechtler K, O'Sullivan RJ, Derijck AA, Perez-Burgos L, Kohlmaier A, Opravil S, Tachibana M, Shinkai Y, Martens JH, Jenuwein T. Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol Cell. 2003;12:1577–1589. doi: 10.1016/s1097-2765(03)00477-5. [DOI] [PubMed] [Google Scholar]
- Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Annu Rev Biochem. 2001;70:81–120. doi: 10.1146/annurev.biochem.70.1.81. [DOI] [PubMed] [Google Scholar]
- Ryu H, Ferrante RJ. Emerging chemotherapeutic strategies for Huntington's disease. Expert Opin Emerg Drugs. 2005;10:345–363. doi: 10.1517/14728214.10.2.345. [DOI] [PubMed] [Google Scholar]
- Ryu H, Lee J, Hagerty SW, Soh BY, McAlpin SE, Cormier KA, Smith KM, Ferrante RJ. ESET/SETDB1 gene expression and histone H3 (K9) trimethylation in Huntington's disease. Proc Natl Acad Sci USA. 2006;103:19176–19181. doi: 10.1073/pnas.0606373103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadri-Vakili G, Bouzou B, Benn CL, Kim MO, Chawla P, Overland RP, Glajch KE, Xia E, Qiu Z, Hersch SM, Clark TW, Yohrling GJ, Cha JH. Histones associated with downregulated genes are hypo-acetylated in Huntington's disease models. Hum Mol Genet. 2007;16:1293–1306. doi: 10.1093/hmg/ddm078. [DOI] [PubMed] [Google Scholar]
- Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NC, Schreiber SL, Mellor J, Kouzarides T. Active genes are tri-methylated at K4 of histone H3. Nature. 2002;419:407–411. doi: 10.1038/nature01080. [DOI] [PubMed] [Google Scholar]
- Schotta G, Ebert A, Krauss V, Fischer A, Hoffmann J, Rea S, Jenuwein T, Dorn R, Reuter G. Central role of Drosophila SU(VAR)3–9 in histone H3–K9 methylation and heterochromatic gene silencing. EMBO J. 2002;21:1121–1131. doi: 10.1093/emboj/21.5.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahbazian MD, Zhang K, Grunstein M. Histone H2B ubiquitylation controls processive methylation but not monomethylation by Dot1 and Set1. Mol Cell. 2005;19:271–277. doi: 10.1016/j.molcel.2005.06.010. [DOI] [PubMed] [Google Scholar]
- Shiio Y, Eisenman RN. Histone sumoylation is associated with transcriptional repression. Proc Natl Acad Sci USA. 2003;100:13225–13230. doi: 10.1073/pnas.1735528100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipione S, Rigamonti D, Valenza M, Zuccato C, Conti L, Pritchard J, Kooperberg C, Olson JM, Cattaneo E. Early transcriptional profiles in huntingtin-inducible striatal cells by microarray analyses. Hum Mol Genet. 2002;11:1953–1965. doi: 10.1093/hmg/11.17.1953. [DOI] [PubMed] [Google Scholar]
- Stack EC, Del Signore SJ, Luthi-Carter R, Soh BY, Goldstein DR, Matson S, Goodrich S, Markey AL, Cormier K, Hagerty SW, Smith K, Ryu H, Ferrante RJ. Modulation of nucleosome dynamics in Huntington's disease. Hum Mol Genet. 2007;16:1164–1175. doi: 10.1093/hmg/ddm064. [DOI] [PubMed] [Google Scholar]
- Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, Kurokawa R, Housman DE, Jackson GR, Marsh JL, Thompson LM. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413:739–743. doi: 10.1038/35099568. [DOI] [PubMed] [Google Scholar]
- Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- Sun ZW, Allis CD. Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeast. Nature. 2002;418:104–108. doi: 10.1038/nature00883. [DOI] [PubMed] [Google Scholar]
- The Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Thorne AW, Sautiere P, Briand G, Crane-Robinson C. The structure of ubiquitinated histone H2B. EMBO J. 1987;6:1005–1010. doi: 10.1002/j.1460-2075.1987.tb04852.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trettel F, Rigamonti D, Hilditch-Maguire P, Wheeler VC, Sharp AH, Persichetti F, Cattaneo E, MacDonald ME. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum Mol Genet. 2000;9:2799–2809. doi: 10.1093/hmg/9.19.2799. [DOI] [PubMed] [Google Scholar]
- Valera AG, Diaz-Hernandez M, Hernandez F, Ortega Z, Lucas JJ. The ubiquitin-proteasome system in Huntington's disease. Neuroscientist. 2005;11:583–594. doi: 10.1177/1073858405280639. [DOI] [PubMed] [Google Scholar]
- van der Knaap JA, Kumar BR, Moshkin YM, Langenberg K, Krijgsveld J, Heck AJ, Karch F, Verrijzer CP. GMP synthetase stimulates histone H2B deubiquitylation by the epigenetic silencer USP7. Mol Cell. 2005;17:695–707. doi: 10.1016/j.molcel.2005.02.013. [DOI] [PubMed] [Google Scholar]
- Wang H, Wang L, Erdjument-Bromage H, Vidal M, Tempst P, Jones RS, Zhang Y. Role of histone H2A ubiquitination in Polycomb silencing. Nature. 2004;431:873–878. doi: 10.1038/nature02985. [DOI] [PubMed] [Google Scholar]
- West MH, Bonner WM. Histone 2B can be modified by the attachment of ubiquitin. Nucleic Acids Res. 1980;8:4671–4680. doi: 10.1093/nar/8.20.4671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yohrling GJ, Farrell LA, Hollenberg AN, Cha JH. Mutant huntingtin increases nuclear corepressor function and enhances ligand-dependent nuclear hormone receptor activation. Mol Cell Neurosci. 2003;23:28–38. doi: 10.1016/s1044-7431(03)00032-0. [DOI] [PubMed] [Google Scholar]
- Zhu B, Zheng Y, Pham AD, Mandal SS, Erdjument-Bromage H, Tempst P, Reinberg D. Monoubiquitination of human histone H2B: the factors involved and their roles in HOX gene regulation. Mol Cell. 2005;20:601–611. doi: 10.1016/j.molcel.2005.09.025. [DOI] [PubMed] [Google Scholar]