

Abstract
Extinction of conditioned fear is an active learning process involving inhibition of fear expression. It has been proposed that fear extinction potentiates neurons in the infralimbic (IL) prefrontal cortex, but the cellular mechanisms underlying this potentiation remain unknown. It is also not known whether this potentiation occurs locally in IL neurons as opposed to IL afferents. To determine whether extinction enhances the intrinsic excitability of IL pyramidal neurons in layers II/III and V, we performed whole-cell patch-clamp recordings in slices from naive, conditioned, or conditioned-extinguished rats. We observed that conditioning depressed IL excitability compared with slices from naive animals, as evidenced by a decreased number of spikes evoked by injected current and an increase in the slow afterhyperpolarizing potential (sAHP). Extinction reversed these conditioning-induced effects. Furthermore, IL neurons from extinguished rats showed increased burst spiking compared with naive rats, which was correlated with extinction recall. These changes were specific to IL prefrontal cortex and were not observed in prelimbic prefrontal cortex. Together, these findings suggest that IL intrinsic excitability is reduced to allow for expression of conditioning memory and enhanced for expression of extinction memory through the modulation of Ca2+-gated K+ channels underlying the sAHP. Inappropriate modulation of these intrinsic mechanisms may underlie anxiety disorders, characterized by exaggerated fear and deficient extinction.
Keywords: amygdala, prelimbic, fear expression, medial prefrontal cortex, sAHP, bursting
Introduction
Inappropriate regulation of fear expression is a hallmark of anxiety disorders. One model to study fear regulation is extinction of auditory fear conditioning. Whereas acquisition of fear conditioning increases fear responses to the conditioned stimulus (CS), extinction decreases the fear responses. Extinction is thought to form a new inhibitory memory that suppresses the expression of conditioned fear. Several lines of evidence indicate that the infralimbic (IL) subregion of the medial prefrontal cortex (mPFC) is important for the inhibition of conditioned fear after extinction (Quirk et al., 2006). Electrical stimulation of IL prefrontal cortex during extinction training inhibits expression of conditioned fear (Milad et al., 2004; Vidal-Gonzalez et al., 2006). Moreover, extinction potentiates IL activity relative to conditioning, and this potentiation is correlated with expression of extinction memory (Herry and Garcia, 2002; Milad and Quirk, 2002; Barrett et al., 2003). In humans, similar observations have been made for a homolog of the rat IL, the ventral mPFC (Phelps et al., 2004; Kalisch et al., 2006; Milad et al., 2007), suggesting that rodent findings in extinction are applicable to humans.
Despite the abundant evidence linking IL activity with the inhibition of conditioned fear (Quirk et al., 2006), the cellular mechanisms that mediate the extinction-induced enhancement of IL activity have not been studied. It is not known, for example, whether extinction training alters IL neurons themselves or instead triggers neural plasticity in areas upstream from IL. Therefore, we tested the hypothesis that the extinction-induced cellular changes occur locally in IL neurons by examining the effects of training on intrinsic excitability. Growing evidence indicates that learning can alter the intrinsic excitability of neurons by affecting voltage-gated and calcium-gated channels present in spines, dendrites, and cell bodies (Giese et al., 2001; Daoudal and Debanne, 2003; Zhang and Linden, 2003; Disterhoft and Oh, 2006; Schulz, 2006). By enhancing or reducing dendritic filtering, learning-induced changes in excitability can increase or decrease the probability that a cell will fire in response to its inputs (Magee et al., 1998; Reyes, 2001; Disterhoft and Oh, 2006).
To determine whether fear conditioning or extinction modifies the intrinsic excitability of IL neurons, we combined behavioral analysis and patch-clamp recording. We compared four groups: rats that received fear conditioning only, rats that received conditioning and extinction, rats that received neither conditioning nor extinction and rats that received unpaired tones and shocks. Comparing these four groups allowed us to assess the effects of extinction independently from the effects of conditioning on cellular excitability.
Materials and Methods
Subjects.
The procedures were approved by the Institutional Animal Care and Use Committee of the Ponce School of Medicine in compliance with National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. Male Sprague Dawley rats [postnatal day 25 (P25) to P30] were transported from the Ponce School of Medicine colony to a satellite facility nearby, where they were group housed in transparent polyethylene cages inside a negative-pressure Biobubble (Colorado Clean Room, Ft. Collins, CO). Rats were maintained on a 12 h light/dark schedule with free access to food (standard laboratory rat chow) and water.
Behavioral apparatus.
Rats were fear conditioned, extinguished, and tested in a chamber of 25 × 29 × 28 cm with aluminum and Plexiglas walls (Coulbourn, Allentown, PA). The floor consisted of stainless-steel bars that could be electrified to deliver a mild shock. A speaker was mounted on the outside wall, and illumination was provided by a single overhead light. The chamber was situated inside a sound-attenuating box (Med Associates, Burlington, VT) with a ventilating fan, which produced an ambient noise level of 60 dB. The CS was a 4 kHz tone with duration of 30 s and an intensity of 80 dB. The unconditioned stimulus (US) was a 0.4 mA scrambled footshock, 0.5 s in duration, which coterminated with the tone during the conditioning phase. Between sessions, floor trays and shock bars were cleaned with soapy water, and the chamber walls were wiped with a damp cloth. Behavior was recorded with digital video cameras (Micro Video Products, Ontario, Canada).
Behavioral procedure.
On day 1, rats received three tone-shock pairings (conditioning phase). After matching for equivalent levels of freezing, conditioned rats were divided into the conditioned group (COND) and the extinguished group (EXT). On day 2, rats in the COND group remained in their home cages while rats in the EXT group were returned to the conditioning chamber and given 10 tone-alone trials (extinction phase). On day 3, both groups of rats received two tone-alone trials in the same chamber to test for recall of conditioning or extinction (test phase). A control group of rats, which was not conditioned (NAIVE), was tested with two tone-alone trials on day 3. An additional pseudoconditioned group (PSEUDO-COND) received three tones (interstimulus interval, 2 min) followed by three consecutive shocks (interstimulus interval, 0.5 s) at the end of the third tone interval. Rats were immediately removed from the chamber, and 48 h later, two test tones were given in a novel chamber to avoid expression of fear to the context that could alter the membrane properties. In all phases of the experiment, the interval between successive tones was variable, with an average of 2 min. All groups were tested on the same day to determine the long-term changes occurring in the mPFC that gate subsequent memory retrieval.
Slice preparation.
Immediately after the test tones on day 3, rats were deeply anesthetized with pentobarbital (150 mg/kg) and were perfused through the heart with ice-cold high-sucrose solution (in mm): 252 sucrose, 2 KCl, 1.25 NaH2PO4, 3 MgSO4, 26 NaHCO3, 20 glucose, and 1 CaCl2. Brains were quickly removed and placed in ice-cold artificial CSF (ACSF) containing the following (in mm): 126 NaCl, 3 KCl, 1.25 NaH2PO4, 1 MgSO4, 26 NaHCO3, 20 glucose, and 2 CaCl2 and bubbled with 95% O2 and 5% CO2. Coronal slices of the medial prefrontal cortex were cut at a thickness of 300 μm with a Vibratome 1000 Plus (Vibratome, St. Louis, MO). Slices were incubated at room temperature (21–23°C) in ACSF for at least 1 h before experiments. The NMDA receptor blocker MK-801 [(+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d]cyclohepten-5,10-imine maleate] (10 μm) was added during the incubation of slices to increase neuronal survival (Schurr et al., 1995).
Recordings were performed blind with respect to group assignment. Slices were transferred to a submersion recording chamber mounted and perfused at 2–3 ml/min with room temperature ACSF. Neurons were visualized with infrared video microscopy using a 40× water immersion objective on an upright E600FN microscope (Nikon Instruments, Melville, NY). Whole-cell recordings were done with glass pipettes with a resistance of 3–5 MΩ when filled with an internal solution containing the following (in mm): 150 KMeSO4, 10 KCl, 0.1 EGTA, 10 HEPES, 0.3 GTP, and 0.2 ATP, pH 7.3 (291 mOsm).
Current-clamp recordings.
Whole-cell current-clamp recordings were obtained from the soma of mPFC pyramidal neurons located in layers II/III and V of the IL and prelimbic (PL) subregions. Cells were held in current-clamp mode at −70 mV, and action potential discharges in response to the injection of depolarizing current pulses were recorded with a patch-clamp amplifier (MultiClamp 700A; Molecular Devices, Sunnyvale, CA). Recordings were filtered at 4 kHz, digitized at 10 kHz, and saved to computer using pClamp9 (Molecular Devices). Membrane potentials (Vm) were not corrected for the junction potential of 9 mV. The series resistance was equal across groups (NAIVE, 26 ± 1.2; COND, 24 ± 2.0; EXT, 25 ± 1.4). The input resistance (Rin) was measured from a 5 mV, 50 ms depolarizing pulse in voltage-clamp mode at a holding potential of −60 mV.
To examine the effects of training on membrane excitability, mPFC pyramidal neurons were injected with an 800 ms depolarizing current pulses ranging from 40 to 470 pA at 10 pA increments with an intertrial interval of 5 s. The number of action potentials evoked by each current intensity was counted from individual responses. We measured the amplitude of the fast afterhyperpolarizing potentials (fAHPs) in the second and third current evoked spikes within the 800 ms pulse. The fAHP was measured by subtracting the voltage at the peak of the fAHP from the threshold potential for spike initiation. The medium afterhyperpolarizing potentials (mAHPs) and slow afterhyperpolarizing potentials (sAHPs) were measured after the end of the 800 ms pulse. The mAHP was measured as the peak of the AHP, and the sAHP was measured as the average potential during a 50 ms period beginning 280 ms after the end of the 800 ms depolarizing pulse (Sah and Faber, 2002). To block the sAHP, 100 μm norepinephrine was bath applied for 7 min with 100 μm ascorbic acid to prevent oxidation of norepinephrine. For these experiments, the internal solution contained the following (in mm): 115 K-gluconate, 20 KCl, 10 HEPES, 0.3 GTP, 0.2 ATP, and 10 phosphocreatine, pH 7.3 (291 mOsm).
To measure the voltage sag that has been correlated with the activation of the hyperpolarization-activated cation currents (Ih), we injected a hyperpolarizing current pulse of −20 pA in current-clamp mode. The voltage sag was calculated by subtracting the average steady-state voltage during a 100 ms period beginning at 645 ms after the beginning of the hyperpolarizing step minus the peak of the hyperpolarization.
Morphological analysis.
In all experiments, 5 mm biocytin was included in the recording solution to label the neurons for post hoc morphological identification of IL or PL pyramidal neurons (see Fig. 2A). At the end of the electrophysiological recordings, the slices were fixed overnight in 4% paraformaldehyde. Neurons were subsequently visualized with a standard advidin-biotin peroxidase procedure (Vectastain ABC kit; Vector Laboratories, Burlingame, CA) as described previously (Porter et al., 2001) and visualized with bright-field microscopy. Neurons that were not located in the IL or PL cortices or that were not pyramidal-shaped with obvious apical dendrites were excluded from the analysis.
Figure 2.
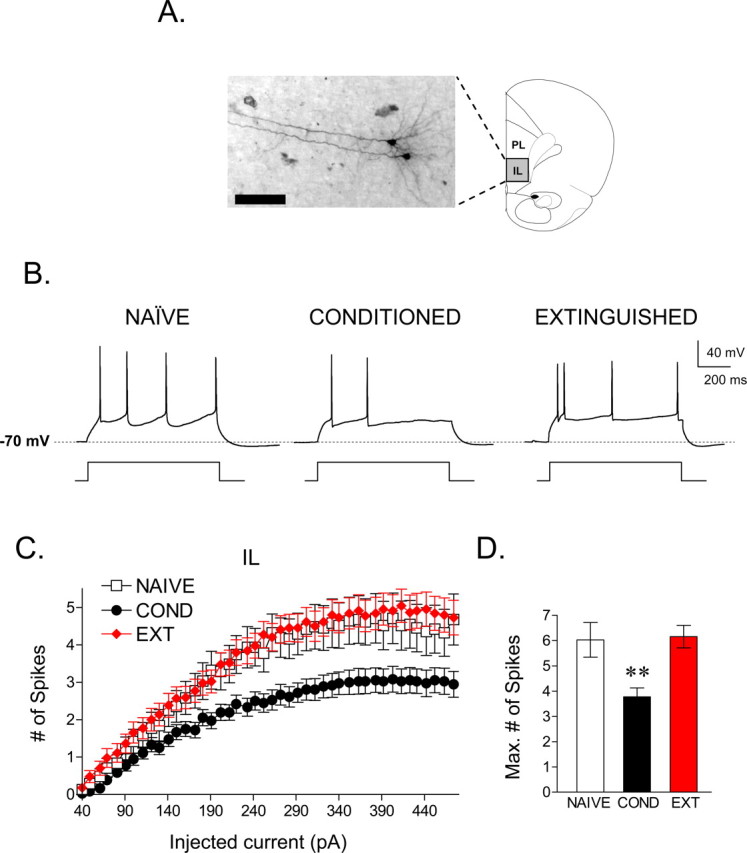
Fear conditioning depressed the intrinsic excitability of IL pyramidal neurons, and extinction reversed this depression. A, Photomicrograph showing two pyramidal cells located in the infralimbic cortex that were recorded sequentially. B, Single traces demonstrating that cells from the COND group (n = 35) fired fewer spikes compared with the cells from the NAIVE (n = 31) and EXT (n = 43) groups in response to a 220 pA current pulse. C, Depolarizing steps at different current intensities evoked significantly fewer spikes in the COND group than the NAIVE and EXT groups. D, Bar graph showing that the maximum number of evoked spikes at any current step. The COND group was significantly reduced compared with the NAIVE and EXT groups (**p < 0.01). Scale bar, 100 μm.
Statistical analysis.
The percentage of time spent freezing (Blanchard and Blanchard, 1972) was used as a measure of conditioned fear. Freezing is the cessation of all movements except respiration. The total time spent freezing during the 30 s tone was scored from videotape with a digital stopwatch by observers blinded with respect to experimental group. The electrophysiological data were analyzed using Clampfit (Molecular Devices). Student's t test or one-way ANOVA (STATISTICA; Statsoft, Tulsa, OK) were used to analyze the behavioral and electrophysiological data. After a significant main effect, post hoc tests were performed with Tukey's honestly significant difference tests. Values are reported as the mean ± SEM.
Results
To assess the effects of both conditioning and extinction on cell excitability, three groups of rats were examined (Fig. 1A): a conditioned group, COND (n = 14), that received auditory fear conditioning but no extinction; an extinguished group, EXT (n = 15), that received both conditioning and extinction; and an untrained group, NAIVE (n = 12), that received neither conditioning nor extinction. Figure 1B shows the average tone-induced freezing for all three groups. On day 1, rats in the COND and EXT groups acquired similar levels of conditioned freezing (COND, 71%; EXT, 72%). On day 2, rats in the EXT group showed gradual within-session extinction across 10 trials to a final freezing level of 24%. On day 3, rats in the COND group showed high levels of freezing to the test tones, whereas rats in the EXT group showed low levels of freezing, indicating good recall of extinction. NAIVE rats showed no freezing to the test tones. Average freezing levels to test tones on day 3 were 70, 22, and 1% for COND, EXT, and NAIVE groups, respectively. One-way ANOVA showed a significant main effect of group (F(2,38) = 55.94; p < 0.001), and post hoc comparisons indicated that the COND group showed more freezing compared with the EXT and NAIVE groups (p < 0.001). In addition, rats in the EXT group froze significantly more than NAIVE controls (p = 0.008), indicating that although extinction training reduced fear expression, rats in the EXT group still showed some conditioned fear.
Figure 1.

Experimental design. A, Four experimental groups were studied. In the conditioned group (COND; n = 14), rats received three tone-shock pairs on day 1 and were left in their home cages on day 2. The extinction group (EXT; n = 15) received three tone-shock pairings on day 1 and 10 tones alone on day 2 in the same chamber. A naive control group (NAIVE; n = 12) received neither conditioning nor extinction. On day 1, the pseudoconditioned group (PSEUDO-COND; n = 6) received three tones and three shocks in an unpaired manner. All groups received two test tones on day 3 and then were killed immediately. B, Mean percentage of freezing for all groups across the behavioral procedure. As expected, rats in the COND group showed higher levels of freezing to the test tones, compared with the NAIVE, PSEUDO-COND, and EXT groups (**p < 0.01).
Conditioning depresses IL excitability
Immediately after the test tones on day 3, coronal slices of the mPFC were prepared, and the intrinsic excitability of pyramidal neurons was assessed with whole-cell patch-clamp recordings. All recorded neurons were filled with biocytin and identified as pyramidal neurons based on their morphology. A total of 238 neurons were recorded, 109 of which were located in IL, and 129 were located in PL. Neurons were located in layers II/III (IL, 22; PL, 15) and layer V (IL, 87; PL, 114). We intentionally targeted cells in layer V because of their projections to subcortical structures, including the amygdala (Gabbott et al., 2005). IL cells located in layers II/III and V showed similar physiological properties (layers II/III: Vm, −55 ± 1; Rin, 355 ± 35; layer V: Vm, −54 ± 0.6; Rin, 321 ± 15) and similar responses to conditioning and extinction. For these reasons, the data from superficial and deep layers were combined. IL cells were distributed across the three experimental groups (NAIVE, 31; COND, 35; EXT, 43). Figure 2A shows a photomicrograph of two representative IL pyramidal neurons. Neither the Vm nor the Rin of recorded IL neurons significantly differed across groups (for the electrophysiological properties of IL neurons, see Table 1).
Table 1.
Electrophysiological properties of IL neurons
| NAIVE (n = 31) | COND (n = 35) | EXT (n = 43) | |
|---|---|---|---|
| Vm (mV) | −53 ± 0.76 | −56 ± 1.00 | −57 ± 0.84 |
| Rin (MΩ) | 330 ± 27 | 287 ± 18 | 305 ± 26 |
| Rheobase (pA) | 103 ± 10 | 105 ± 9 | 92 ± 6 |
| mAHP (mV) | −4.6 ± 0.38 | −5.4 ± 0.39 | −4.5 ± 0.27 |
| sAHP (mV) | −1.9 ± 0.26 | −3.5 ± 0.33b | −1.9 ± 0.24 |
| Voltage sag (mV) | 1.17 ± 0.12 | 1.2 ± 0.13 | 0.99 ± 0.07 |
| Peak amplitude (mV)a | 49 ± 0.82 | 47 ± 1.28 | 48 ± 0.66 |
| Half width (ms)a | 1.1 ± 0.03 | 1.0 ± 0.03 | 1.0 ± 0.02 |
| Threshold (mV)a | −38 ± 0.79 | −36 ± 0.99 | −38 ± 0.62 |
aFirst spike from the trace that showed the maximum number of evoked spikes.
bThe average data in the group is significantly different from NAIVE controls (p < 0.05).
To determine the relative excitability of the neurons in each group, we measured the number of action potentials elicited by a series of depolarizing current pulses (Fig. 2B,C). In all cells, the response to injected current was measured from a holding potential of −70 mV. Consistent with the lack of difference in Rin among the groups, no difference was observed in the amount of depolarizing current necessary to evoke an action potential (rheobase) or in the spike threshold (Table 1). However, the number of spikes that could be elicited in conditioned rats was significantly reduced compared with the NAIVE group. Extinction reversed this effect, because the number of spikes in the EXT group was similar to the NAIVE group. Repeated measures ANOVA across all three groups revealed a main effect of group (F(2,108) = 5.70; p = 0.004) and a significant current by group interaction (F(60,3240) = 2.19; p < 0.001). Post hoc comparisons indicated that cells in the COND group generated significantly fewer spikes than both NAIVE and EXT groups at all intensities between 190 and 470 pA (p ≤ 0.03). The maximum number of spikes elicited by any size current pulse for each cell (Fig. 2D) was significantly reduced in the COND group by ∼40% compared with the NAIVE group (NAIVE, 6.03 spikes; COND, 3.77 spikes; EXT, 6.15 spikes). One-way ANOVA showed a main effect of group (F(2,108) = 6.55; p = 0.002), and post hoc comparisons indicated that the COND group was significantly lower than both the NAIVE (p = 0.01) and EXT (p = 0.004) groups. The NAIVE and EXT groups were not different from each other (p = 0.99). Despite the difference in spike count, there was no difference in the average intensity needed to evoke the maximum number of spikes between the groups (NAIVE, 254 ± 14; COND, 229 ± 13; EXT, 245 ± 15; p = 0.46).
Extinction increases bursting in IL
Neurons in the COND group fired fewer spikes, whereas neurons in the NAIVE and EXT groups showed a similar number of evoked spikes, suggesting that extinction reversed conditioning-induced changes. However, as apparent in the examples of Figure 2B, extinction training also tended to reduce the first interspike interval (ISI) resulting in the generation of high-frequency spike doublets or bursts. Figure 3A shows additional examples demonstrating that the ISIs were shorter in the EXT group compared with the NAIVE group or the COND group. A comparison of the first ISI at the current eliciting the maximum number of spikes showed a significant reduction in the EXT group compared with NAIVE controls (NAIVE, 83 ms; EXT, 46 ms; t = 2.45; df, 67; p = 0.016) (Fig. 3B). No significant group differences were observed for the second or third ISIs (p > 0.05 for both). Because the number of spikes could influence the ISIs, we compared the results of the NAIVE and EXT groups, which fired the same number of spikes. Consistent with our initial analysis, the first ISI in traces showing three spikes was still significantly shorter in the EXT group compared with the NAIVE group (NAIVE, 156 ms; EXT, 116 ms; t = 2.21; df, 56; p < 0.05). In addition, 50% of the cells in the EXT group exhibited a first ISI <30 ms (Fig. 3C), corresponding to previously defined rates of mPFC burst firing in vivo (>33 Hz) (Laviolette et al., 2005; Burgos-Robles et al., 2007). In contrast, only 24% of NAIVE cells and 18% of COND cells showed first ISIs <30 ms (χ2, 11.4; df, 2; p < 0.01). Therefore, although extinction training restored the conditioning-induced depression in IL excitability, it also induced bursting not seen in the untrained group.
Figure 3.

Extinction increased bursting in IL neurons. A, Representative traces from NAIVE, COND, and EXT groups. B, Group data showing that extinction training reduced the duration of the first ISI, compared with other groups. C, Cumulative percentages showing that more cells in the EXT group had ISIs of <30 ms compared with cells in the NAIVE and COND groups. D, For each rat in the extinction group, the first ISI for all cells was averaged. Across rats, this value was significantly correlated with freezing levels at test (r = 0.72; p < 0.001), indicating that IL bursting is correlated with extinction recall (NAIVE vs EXT, *p < 0.05).
To investigate whether bursting in IL may be a determinant of extinction memory, we calculated the correlation between the first ISIs and freezing levels at test, in rats given extinction. As shown in Figure 3D, ISI duration was correlated significantly with freezing at test (r = 0.72; p < 0.001), suggesting that IL bursting facilitates fear inhibition.
Conditioning and extinction modify the afterhyperpolarizing potentials in IL neurons
Intrinsic excitability is regulated by different classes of potassium channels (Magee and Carruth, 1999; Yuan and Chen, 2006; Yue and Yaari, 2006; Bean, 2007). For example, activation of Ca2+-dependent potassium channels hyperpolarizes the membrane, making it less likely that the cells will reach threshold and fire subsequent action potentials (Sah, 1996; Lancaster et al., 2001). The AHP generated by the activation of these Ca2+-dependent potassium channels consist of three distinct components: fast, medium, and slow (Sah and Faber, 2002). The fAHP that follows single action potentials lasts tens of milliseconds. In contrast, the mAHP and sAHP have much slower kinetics and are activated after prolonged depolarization. The fAHP after the first evoked spike was contaminated with the passive change in membrane potential from the current injection (Duvarci and Pare, 2007) and could not be analyzed. We therefore measured the fAHPs evoked by the second and third spikes (Fig. 4A1). ANOVA revealed a main effect of group in the second fAHP (F(2,108) = 8.18; p < 0.001) (Fig. 4A2) but not third fAHP (F(2,108) = 1.86; p = 0.16). Post hoc comparisons for the fAHP of the second spike indicated that the NAIVE and COND groups were not different from each other (p = 0.99). However, cells from the EXT group showed significantly reduced fAHP amplitudes compared with both NAIVE and COND groups (both p values <0.01) (Fig. 4A2).
Figure 4.
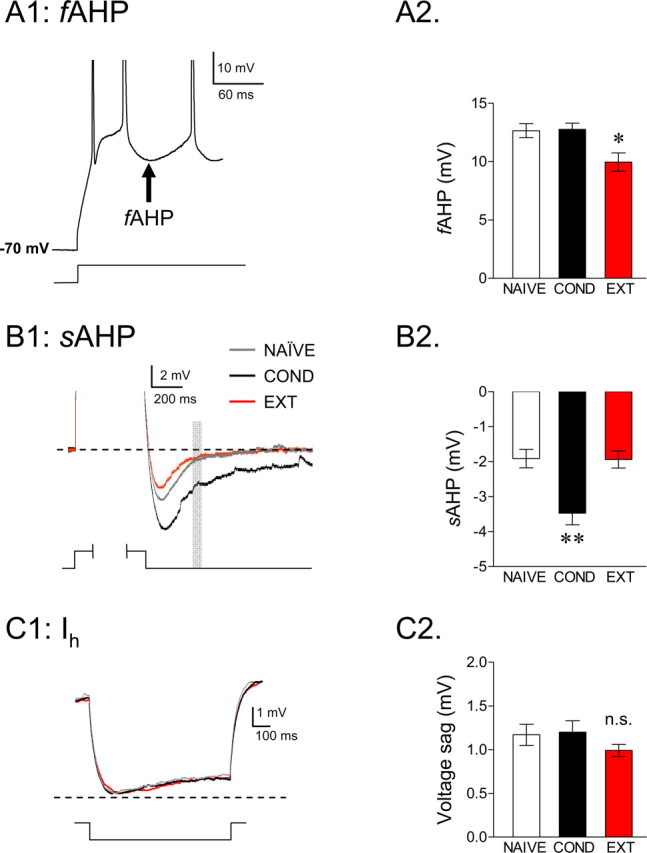
Training-induced alterations in IL afterhyperpolarizing potentials. A1, Example trace showing the fAHP measured after the second and third spikes of an 800 ms current step. A2, Group data showing that extinction reduced the fAHP after the second evoked spike. B1, Example traces showing the voltage change in response to 800 ms depolarizing pulse used to elicit an mAHP and an sAHP (spikes are truncated). The mAHP was measured at the peak hyperpolarization, and the gray box indicates where the sAHP was measured. B2, Group data for the sAHP showing that conditioning enhanced the sAHP relative to the other two groups. C1, Averaged traces for the three groups showing the voltage response to a hyperpolarizing current step of −20 pA. C2, No difference in the resulting voltage sag was observed between the groups, suggesting that training had no effect on the activation of H-channels (**p < 0.01; *p < 0.05).
The mAHP and sAHP have been shown to be correlated inversely with average firing rate (Lorenzon and Foehring, 1995). Because the amplitude of the sAHP depends on the number of spikes (Abel et al., 2004), we measured the mAHP and sAHP in IL neurons from traces in which cells fired the same number of spikes to determine whether they were affected by conditioning or extinction. Because the majority of cells from the COND group fired a maximum of two spikes, we limited our analysis to traces showing two spikes. The AHP was measured at two time points: at the negative peak (mAHP) and 280 ms after the end of the current pulse (sAHP) (Fig. 4B1). The mAHP showed no difference between groups (Table 1), but the sAHP was significantly enhanced in the COND group compared with the NAIVE controls (main effect, F(2,97) = 9.99; p < 0.001; COND vs NAIVE, p = 0.001) (Fig. 4B2). The enhancement of the sAHP in the COND group is consistent with conditioning-induced depression of IL excitability. In line with the extinction-mediated reversal of spike count, extinction training reversed the conditioning-induced increase in the sAHP. The sAHP in the EXT group was significantly lower than in the COND group (p < 0.001) and not significantly different from the NAIVE group (p = 0.99) (Fig. 4B2). These results show that training-induced changes in IL excitability are correlated with the changes in the sAHP
Conditioning-induced enhancement of the sAHP could explain the decrease in IL excitability observed after fear conditioning. To further test this possibility, in a separate experiment, we pharmacologically reduced the sAHP by bath applying 100 μm norepinephrine to IL cells from the three experimental groups (NAIVE, n = 9; COND, n = 9; EXT, n = 6). Consistent with our previous report (Mueller et al., 2008), norepinephrine significantly reduced the sAHP in all groups (paired t test; t = 3.55; p < 0.05 for all groups). This manipulation also abolished between-group differences in spike count (NAIVE, 6.5 spikes; COND, 6.5 spikes; EXT, 5.4 spikes; F(2,21) = 0.14; p = 0.86), consistent with the idea that conditioning-induced decreases in excitability are mediated by alterations in the sAHP.
In contrast to the sAHP and fAHP, no differences were found in the voltage sags in response to hyperpolarizing current pulses, suggesting that conditioning and extinction did not affect activation of the H-current (Fig. 4C1,C2; Table 1).
Conditioning-induced effects on IL excitability are caused by associative learning
Conditioned rats showed reduced excitability compared with NAIVE controls. However, because NAIVE rats received no shocks whatsoever, reduced excitability in COND rats might be caused by shock presentations rather than a tone-shock association. To examine this possibility, pseudoconditioned rats (PSEUDO-COND; n = 6 rats) were given three tones and three shocks in an unpaired manner. Two days later, they received two test tones and were prepared for slice recording. Rats in the PSEUDO-COND group showed no freezing to the test tones. In response to increasing current pulses, cells from the PSEUDO-COND (n = 24 cells) group responded similarly to the NAIVE group, firing more spikes than the COND group (Fig. 5A). Consistent with this, the maximum number of spikes in the PSEUDO-COND group (5.9 spikes) was no different from the NAIVE group (5.0 spikes) (p = 0.98) and was significantly higher than the COND group (3.2 spikes) (F(2,88) = 6.04; p ≤ 0.02) (Fig. 5B). Paralleling spike findings, the sAHP in the PSEUDO-COND group (−1.7 mV) was similar to the NAIVE group (−1.9 mV) and significantly smaller than the COND group (−3.5 mV) in traces showing two spikes (main effect, F(2,80) = 9.96; p < 0.001) (Fig. 5C). In addition, the first ISI in the PSEUDO-COND group (78.6 ± 12) was not different from the NAIVE (82.5 ± 16) and COND (118.4 ± 23) groups (main effect, F(2,82) = 1.46; p = 0.23). These findings indicate that the conditioning-induced effects on membrane excitability are caused by associative learning and not by shock presentations per se.
Figure 5.

Conditioning-induced effects on IL excitability are caused by associative learning and not by shock presentations. A, Depolarizing steps at different current intensities evoked significantly fewer spikes in the COND group than in the PSEUDO-COND (n = 24 cells) and NAIVE groups. B, Bar graph showing that the maximum number of evoked spikes in IL neurons at any current step. The maximum number of evoked spikes was significantly reduced in the COND group compared with the PSEUDO-COND and NAIVE groups. C, Average data showing that the sAHP in cells from the COND group was significantly enhanced compared with cells from the PSEUDO-COND and NAIVE groups (**p < 0.01).
Conditioning and extinction have no effect on PL membrane excitability
Finally, we examined whether conditioning or extinction alter the intrinsic excitability of PL cortex (for the electrophysiological properties of PL neurons, see Table 2) (NAIVE, n = 27; COND, n = 48; EXT, n = 54). In contrast to IL neurons, PL neurons did not show a significant group difference in the number of spikes evoked by increasing current steps (F(2,126) = 0.59; p = 0.55) (Fig. 6A). The maximum number of spikes evoked in the three groups also did not significantly differ (NAIVE, 6.1; COND, 5.5; EXT, 6.4; F(2,126) = 1.32; p = 0.27) (Fig. 6B). Furthermore, neither the first interspike interval (p = 0.32) nor the sAHP (p = 0.53) in traces showing two spikes differed across groups (Fig. 6C,D). These findings indicate that learning did not alter the excitability of PL neurons. Therefore, the training-induced changes in intrinsic excitability were regionally specific.
Table 2.
Electrophysiological properties of PL neurons
| NAIVE (n = 27) | COND (n = 48) | EXT (n = 54) | |
|---|---|---|---|
| Vm (mV) | −55 ± 0.98 | −57 ± 0.77 | −58 ± 0.67 |
| Rin (MΩ) | 295 ± 26 | 252 ± 15 | 243 ± 18 |
| Rheobase (pA) | 102 ± 8.5 | 114 ± 10.5 | 126 ± 7.8 |
| mAHP (mV) | −4.2 ± 0.23 | −3.8 ± 0.29 | −4.3 ± 0.23 |
| sAHP (mV) | −1.9 ± 0.23 | −2.0 ± 0.24 | −1.8 ± 0.19 |
| Voltage sag (mV) | 0.94 ± 0.06 | 0.78 ± 0.06 | 0.89 ± 0.07 |
| Peak amplitude (mV)a | 36 ± 1.4 | 36 ± 1.3 | 35 ± 1.9 |
| Half width (ms)a | 1.2 ± 0.04 | 1.1 ± 0.03 | 1.1 ± 0.03 |
| Threshold (mV)a | −39 ± 0.81 | −37 ± 0.66 | −36 ± 0.59 |
aFirst spike from the trace that showed the maximum number of evoked spikes.
Figure 6.

PL neurons did not show significant changes in intrinsic excitability with conditioning or extinction. A, Depolarizing steps at different current intensities evoked a similar number of PL spikes in the three groups. B, Bar graph showing no difference in the maximum number of evoked spikes in PL subregion between the groups. C, Average data showing that the duration of ISIs in the NAIVE and EXT groups did not differ for the PL subregion. D, Average data for the sAHP showing no difference between groups for the PL subregion.
Discussion
We combined classical fear conditioning with patch-clamp electrophysiology to explore the cellular mechanisms of prefrontal control over the expression of conditioned fear. We showed for the first time that: (1) fear conditioning depressed IL intrinsic excitability and increased the sAHP; (2) extinction returned IL excitability and sAHP to preconditioning levels; and (3) extinction also decreased the fAHP and introduced a bursting component not seen in the untrained group. These findings indicate that conditioning and extinction alter the intrinsic excitability of IL projection neurons in opposite directions to modulate differentially the expression of conditioned fear responses.
Previous studies have suggested that IL activity is necessary for the recall of extinction memory but not for conditioning. Electrolytic lesions or pharmacological inactivation of IL had no effect on conditioning or extinction training but impaired subsequent recall of extinction memory (Morgan et al., 1993; Quirk et al., 2000; Lebron et al., 2004; Sierra-Mercado et al., 2006). Furthermore, IL neurons showed enhanced responses to tone presentations only during recall of extinction (Milad and Quirk, 2002). In contrast, we observed that fear conditioning depressed IL intrinsic excitability, suggesting that IL activity inhibits the expression of conditioned fear, even before extinction training. Consistent with our data, previous experiments performed in rodents (Garcia et al., 1999; Herry and Garcia, 2002) and humans (Phelps et al., 2004; Kalisch et al., 2006; Milad et al., 2007) have shown that acquisition of fear conditioning depresses mPFC activity. Together, these findings suggest that IL output may serve as a tonic brake on fear expression that is temporarily released by conditioning via intrinsic cellular mechanisms. What cellular mechanisms underlie the conditioning-induced depression in IL excitability? Previous studies of other cortical areas have reported a reduced sAHP after acquisition of various types of learning, such as water maze (Oh et al., 2003), trace eye blink conditioning (Moyer et al., 1996; Thompson et al., 1996), and operant conditioning (Saar et al., 1998). The sAHP limits repetitive firing of neurons; therefore, reductions in the sAHP would be expected to increase spiking (Faber and Sah, 2003) that could facilitate learning (Stackman et al., 2002; Tzounopoulos and Stackman, 2003). In contrast, we observed that fear conditioning enhanced the sAHP and depressed IL excitability. The depression of IL intrinsic excitability after fear conditioning would be expected to reduce the feedforward inhibition of amygdala central nucleus output neurons (Royer and Pare, 2002; Quirk et al., 2003; Pare et al., 2004), thereby increasing the expression of conditioned fear.
We observed that extinction training reversed the conditioning-induced effects on IL intrinsic excitability. Similarly, other studies have shown that extinction reverses conditioning-induced cellular changes (Lin et al., 2003; Kim et al., 2007). Furthermore, in our experiments, extinction also enhanced bursting, indicating that extinction did not simply reverse the effects of conditioning on intrinsic excitability but also introduced unique cellular changes. There are several possible mechanisms for the extinction-induced increase in bursting. Neuronal excitability can be enhanced by closing large-conductance Ca2-activated K potassium channels (Shao et al., 1999; Traub et al., 2003; Brenner et al., 2005) or A-type potassium channels (Perez et al., 2006; Takeda et al., 2006; Wang and Schreurs, 2006), both of which are expressed in cortical neurons (Jin et al., 2000; Dong and White, 2003) and have been linked to increased bursting (Magee and Carruth, 1999; Jin et al., 2000; Traub et al., 2003; Gu et al., 2007). Interestingly, a decrease in A-type currents has been correlated with invertebrate learning (Yamoah et al., 2005) and hippocampal long-term potentiation (Frick et al., 2004). Additional experiments are underway to determine whether A-type or other potassium currents are reduced after extinction training.
We observed that bursting in IL neurons was positively correlated with expression of extinction memory, suggesting that IL bursting is necessary to inhibit fear. IL bursting would be expected to enhance the impact of IL projections onto target neurons (Lisman, 1997). Therefore, after extinction, bursting would increase the likelihood that IL neurons would activate inhibitory intercalated cells in the amygdala to inhibit fear expression (Quirk et al., 2003; Pare et al., 2004). We recently observed that IL bursting shortly after extinction in vivo predicts extinction recall the following day and is NMDA receptor dependent (Burgos-Robles et al., 2007). In the current study, we observed an increase in intrinsic (synapse-independent) bursting in IL neurons 24 h after extinction training. Because blockade of NMDA receptors (Burgos-Robles et al., 2007) or protein synthesis (Santini et al., 2004; Mueller et al., 2008) in IL prefrontal cortex impairs extinction memory, one possibility is that the NMDA receptor-mediated increase in bursting immediately after extinction training is maintained until the following day to allow recall of the extinction memory. Consistent with this possibility, NMDA receptor activation in slices enhances intrinsic excitability (Armano et al., 2000; Daoudal and Debanne, 2003; Xu et al., 2005) in a protein synthesis-dependent manner (Xu et al., 2005). Additional experiments are needed to determine whether NMDA blockers or protein synthesis inhibitors given during extinction training prevent extinction-induced enhancement of intrinsic bursting in IL prefrontal cortex.
Our results support the hypothesis that modulation of prefrontal excitability controls the expression of conditioned fear (Milad et al., 2006). Therefore, it is possible that excessive depression of IL excitability underlies posttraumatic stress disorder (PTSD), in which patients exhibit exaggerated fear expression and poor extinction (Rauch et al., 2006). In line with this possibility, previous work has shown that mPFC is hyporesponsive in normal subjects who fail to recall extinction memory (Milad et al., 2007) and in PTSD patients exposed to traumatic reminders (Bremner et al., 1999; Shin et al., 2004). Pharmacological enhancers of mPFC neuronal excitability (Wrubel et al., 2007; Zushida et al., 2007) might eventually be used to facilitate exposure-based treatments for PTSD.
Footnotes
This work was supported by National Institutes of Health Grants R01-MH058883 (G.J.Q.), S06-GM08236 (G.J.Q.), and S06-GM08239 (J.T.P.). We thank Marian Sepulveda for assistance with the behavioral studies and Drs. Denis Pare and Hugh T. Blair for comments on this manuscript.
References
- Abel HJ, Lee JC, Callaway JC, Foehring RC. Relationships between intracellular calcium and afterhyperpolarizations in neocortical pyramidal neurons. J Neurophysiol. 2004;91:324–335. doi: 10.1152/jn.00583.2003. [DOI] [PubMed] [Google Scholar]
- Armano S, Rossi P, Taglietti V, D'Angelo E. Long-term potentiation of intrinsic excitability at the mossy fiber-granule cell synapse of rat cerebellum. J Neurosci. 2000;20:5208–5216. doi: 10.1523/JNEUROSCI.20-14-05208.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett D, Shumake J, Jones D, Gonzalez-Lima F. Metabolic mapping of mouse brain activity after extinction of a conditioned emotional response. J Neurosci. 2003;23:5740–5749. doi: 10.1523/JNEUROSCI.23-13-05740.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean BP. The action potential in mammalian central neurons. Nat Rev Neurosci. 2007;8:451–465. doi: 10.1038/nrn2148. [DOI] [PubMed] [Google Scholar]
- Blanchard DC, Blanchard RJ. Innate and conditioned reactions to threat in rats with amygdaloid lesions. J Comp Physiol Psychol. 1972;81:281–290. doi: 10.1037/h0033521. [DOI] [PubMed] [Google Scholar]
- Bremner JD, Staib LH, Kaloupek D, Southwick SM, Soufer R, Charney DS. Neural correlates of exposure to traumatic pictures and sound in Vietnam combat veterans with and without posttraumatic stress disorder: a positron emission tomography study. Biol Psychiatry. 1999;45:806–816. doi: 10.1016/s0006-3223(98)00297-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner R, Chen QH, Vilaythong A, Toney GM, Noebels JL, Aldrich RW. BK channel beta4 subunit reduces dentate gyrus excitability and protects against temporal lobe seizures. Nat Neurosci. 2005;8:1752–1759. doi: 10.1038/nn1573. [DOI] [PubMed] [Google Scholar]
- Burgos-Robles A, Vidal-Gonzalez I, Santini E, Quirk GJ. Consolidation of fear extinction requires NMDA receptor-dependent bursting in the ventromedial prefrontal cortex. Neuron. 2007;53:871–880. doi: 10.1016/j.neuron.2007.02.021. [DOI] [PubMed] [Google Scholar]
- Daoudal G, Debanne D. Long-term plasticity of intrinsic excitability: learning rules and mechanisms. Learn Mem. 2003;10:456–465. doi: 10.1101/lm.64103. [DOI] [PubMed] [Google Scholar]
- Disterhoft JF, Oh MM. Learning, aging and intrinsic neuronal plasticity. Trends Neurosci. 2006;29:587–599. doi: 10.1016/j.tins.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Dong Y, White FJ. Dopamine D1-class receptors selectively modulate a slowly inactivating potassium current in rat medial prefrontal cortex pyramidal neurons. J Neurosci. 2003;23:2686–2695. doi: 10.1523/JNEUROSCI.23-07-02686.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvarci S, Pare D. Glucocorticoids enhance the excitability of principal basolateral amygdala neurons. J Neurosci. 2007;27:4482–4491. doi: 10.1523/JNEUROSCI.0680-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber ES, Sah P. Calcium-activated potassium channels: multiple contributions to neuronal function. Neuroscientist. 2003;9:181–194. doi: 10.1177/1073858403009003011. [DOI] [PubMed] [Google Scholar]
- Frick A, Magee J, Johnston D. LTP is accompanied by an enhanced local excitability of pyramidal neuron dendrites. Nat Neurosci. 2004;7:126–135. doi: 10.1038/nn1178. [DOI] [PubMed] [Google Scholar]
- Gabbott PL, Warner TA, Jays PR, Salway P, Busby SJ. Prefrontal cortex in the rat: projections to subcortical autonomic, motor, and limbic centers. J Comp Neurol. 2005;492:145–177. doi: 10.1002/cne.20738. [DOI] [PubMed] [Google Scholar]
- Garcia R, Vouimba RM, Baudry M, Thompson RF. The amygdala modulates prefrontal cortex activity relative to conditioned fear. Nature. 1999;402:294–296. doi: 10.1038/46286. [DOI] [PubMed] [Google Scholar]
- Giese KP, Peters M, Vernon J. Modulation of excitability as a learning and memory mechanism: a molecular genetic perspective. Physiol Behav. 2001;73:803–810. doi: 10.1016/s0031-9384(01)00517-0. [DOI] [PubMed] [Google Scholar]
- Gu N, Vervaeke K, Storm JF. BK potassium channels facilitate high-frequency firing and cause early spike frequency adaptation in rat CA1 hippocampal pyramidal cells. J Physiol (Lond) 2007;580:859–882. doi: 10.1113/jphysiol.2006.126367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herry C, Garcia R. Prefrontal cortex long-term potentiation, but not long-term depression, is associated with the maintenance of extinction of learned fear in mice. J Neurosci. 2002;22:577–583. doi: 10.1523/JNEUROSCI.22-02-00577.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W, Sugaya A, Tsuda T, Ohguchi H, Sugaya E. Relationship between large conductance calcium-activated potassium channel and bursting activity. Brain Res. 2000;860:21–28. doi: 10.1016/s0006-8993(00)01943-0. [DOI] [PubMed] [Google Scholar]
- Kalisch R, Korenfeld E, Stephan KE, Weiskopf N, Seymour B, Dolan RJ. Context-dependent human extinction memory is mediated by a ventromedial prefrontal and hippocampal network. J Neurosci. 2006;26:9503–9511. doi: 10.1523/JNEUROSCI.2021-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Lee S, Park K, Hong I, Song B, Son G, Park H, Kim WR, Park E, Choe HK, Kim H, Lee C, Sun W, Kim K, Shin KS, Choi S. Amygdala depotentiation and fear extinction. Proc Natl Acad Sci USA. 2007;104:20955–20960. doi: 10.1073/pnas.0710548105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster B, Hu H, Ramakers GM, Storm JF. Interaction between synaptic excitation and slow afterhyperpolarization current in rat hippocampal pyramidal cells. J Physiol (Lond) 2001;536:809–823. doi: 10.1111/j.1469-7793.2001.00809.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laviolette SR, Lipski WJ, Grace AA. A subpopulation of neurons in the medial prefrontal cortex encodes emotional learning with burst and frequency codes through a dopamine D4 receptor-dependent basolateral amygdala input. J Neurosci. 2005;25:6066–6075. doi: 10.1523/JNEUROSCI.1168-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebron K, Milad MR, Quirk GJ. Delayed recall of fear extinction in rats with lesions of ventral medial prefrontal cortex. 2004;11:544–548. doi: 10.1101/lm.78604. [DOI] [PubMed] [Google Scholar]
- Lin C-H, Yeh S-H, Leu T-H, Chang W-C, Wang S-T, Gean P-W. Identification of calcineurin as a key signal in the extinction of fear memory. J Neurosci. 2003;23:1574–1579. doi: 10.1523/JNEUROSCI.23-05-01574.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE. Bursts as a unit of neural information: making unreliable synapses reliable. Trends Neurosci. 1997;20:38–43. doi: 10.1016/S0166-2236(96)10070-9. [DOI] [PubMed] [Google Scholar]
- Lorenzon NM, Foehring RC. Alterations in intracellular calcium chelation reproduce developmental differences in repetitive firing and afterhyperpolarizations in rat neocortical neurons. Brain Res Dev Brain Res. 1995;84:192–203. doi: 10.1016/0165-3806(94)00171-u. [DOI] [PubMed] [Google Scholar]
- Magee J, Hoffman D, Colbert C, Johnston D. Electrical and calcium signaling in dendrites of hippocampal pyramidal neurons. Annu Rev Physiol. 1998;60:327–346. doi: 10.1146/annurev.physiol.60.1.327. [DOI] [PubMed] [Google Scholar]
- Magee JC, Carruth M. Dendritic voltage-gated ion channels regulate the action potential firing mode of hippocampal CA1 pyramidal neurons. J Neurophysiol. 1999;82:1895–1901. doi: 10.1152/jn.1999.82.4.1895. [DOI] [PubMed] [Google Scholar]
- Milad MR, Quirk GJ. Neurons in medial prefrontal cortex signal memory for fear extinction. Nature. 2002;420:70–74. doi: 10.1038/nature01138. [DOI] [PubMed] [Google Scholar]
- Milad MR, Vidal-Gonzalez I, Quirk GJ. Electrical stimulation of medial prefrontal cortex reduces conditioned fear in a temporally specific manner. Behav Neurosci. 2004;118:389–394. doi: 10.1037/0735-7044.118.2.389. [DOI] [PubMed] [Google Scholar]
- Milad MR, Rauch SL, Pitman RK, Quirk GJ. Fear extinction in rats: implications for human brain imaging and anxiety disorders. Biol Psychol. 2006;73:61–71. doi: 10.1016/j.biopsycho.2006.01.008. [DOI] [PubMed] [Google Scholar]
- Milad MR, Wright CI, Orr SP, Pitman RK, Quirk GJ, Rauch SL. Recall of fear extinction in humans activates the ventromedial prefrontal cortex and hippocampus in concert. Biol Psychiatry. 2007;62:446–454. doi: 10.1016/j.biopsych.2006.10.011. [DOI] [PubMed] [Google Scholar]
- Morgan MA, Romanski LM, LeDoux JE. Extinction of emotional learning: contribution of medial prefrontal cortex. Neurosci Lett. 1993;163:109–113. doi: 10.1016/0304-3940(93)90241-c. [DOI] [PubMed] [Google Scholar]
- Moyer JR, Jr, Thompson LT, Disterhoft JF. Trace eyeblink conditioning increases CA1 excitability in a transient and learning-specific manner. J Neurosci. 1996;16:5536–5546. doi: 10.1523/JNEUROSCI.16-17-05536.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller D, Porter JT, Quirk GJ. Noradrenergic signaling in infralimbic cortex increases cell excitability and strengthens memory for fear extinction. J Neurosci. 2008;28:369–375. doi: 10.1523/JNEUROSCI.3248-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh MM, Kuo AG, Wu WW, Sametsky EA, Disterhoft JF. Watermaze learning enhances excitability of CA1 pyramidal neurons. J Neurophysiol. 2003;90:2171–2179. doi: 10.1152/jn.01177.2002. [DOI] [PubMed] [Google Scholar]
- Pare D, Quirk GJ, Ledoux JE. New vistas on amygdala networks in conditioned fear. J Neurophysiol. 2004;92:1–9. doi: 10.1152/jn.00153.2004. [DOI] [PubMed] [Google Scholar]
- Perez MF, White FJ, Hu XT. Dopamine D(2) receptor modulation of K(+) channel activity regulates excitability of nucleus accumbens neurons at different membrane potentials. J Neurophysiol. 2006;96:2217–2228. doi: 10.1152/jn.00254.2006. [DOI] [PubMed] [Google Scholar]
- Phelps EA, Delgado MR, Nearing KI, LeDoux JE. Extinction learning in humans: role of the amygdala and vmPFC. Neuron. 2004;43:897–905. doi: 10.1016/j.neuron.2004.08.042. [DOI] [PubMed] [Google Scholar]
- Porter JT, Johnson CK, Agmon A. Diverse types of interneurons generate thalamus-evoked feedforward inhibition in the mouse barrel cortex. J Neurosci. 2001;21:2699–2710. doi: 10.1523/JNEUROSCI.21-08-02699.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ, Russo GK, Barron JL, Lebron K. The role of ventromedial prefrontal cortex in the recovery of extinguished fear. J Neurosci. 2000;20:6225–6231. doi: 10.1523/JNEUROSCI.20-16-06225.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ, Likhtik E, Pelletier JG, Pare D. Stimulation of medial prefrontal cortex decreases the responsiveness of central amygdala output neurons. J Neurosci. 2003;23:8800–8807. doi: 10.1523/JNEUROSCI.23-25-08800.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ, Garcia R, Gonzalez-Lima F. Prefrontal mechanisms in extinction of conditioned fear. Biol Psychiatry. 2006;60:337–343. doi: 10.1016/j.biopsych.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Rauch SL, Shin LM, Phelps EA. Neurocircuitry models of posttraumatic stress disorder and extinction: human neuroimaging research–past, present, and future. Biol Psychiatry. 2006;60:376–382. doi: 10.1016/j.biopsych.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Reyes A. Influence of dendritic conductances on the input-output properties of neurons. Annu Rev Neurosci. 2001;24:653–675. doi: 10.1146/annurev.neuro.24.1.653. [DOI] [PubMed] [Google Scholar]
- Royer S, Pare D. Bidirectional synaptic plasticity in intercalated amygdala neurons and the extinction of conditioned fear responses. Neuroscience. 2002;115:455–462. doi: 10.1016/s0306-4522(02)00455-4. [DOI] [PubMed] [Google Scholar]
- Saar D, Grossman Y, Barkai E. Reduced after-hyperpolarization in rat piriform cortex pyramidal neurons is associated with increased learning capability during operant conditioning. Eur J Neurosci. 1998;10:1518–1523. doi: 10.1046/j.1460-9568.1998.00149.x. [DOI] [PubMed] [Google Scholar]
- Sah P. Ca(2+)-activated K+ currents in neurones: types, physiological roles and modulation. Trends Neurosci. 1996;19:150–154. doi: 10.1016/s0166-2236(96)80026-9. [DOI] [PubMed] [Google Scholar]
- Sah P, Faber ES. Channels underlying neuronal calcium-activated potassium currents. Prog Neurobiol. 2002;66:345–353. doi: 10.1016/s0301-0082(02)00004-7. [DOI] [PubMed] [Google Scholar]
- Santini E, Ge H, Ren K, Pena de Ortiz S, Quirk GJ. Consolidation of fear extinction requires protein synthesis in the medial prefrontal cortex. J Neurosci. 2004;24:5704–5710. doi: 10.1523/JNEUROSCI.0786-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz DJ. Plasticity and stability in neuronal output via changes in intrinsic excitability: it's what's inside that counts. J Exp Biol. 2006;209:4821–4827. doi: 10.1242/jeb.02567. [DOI] [PubMed] [Google Scholar]
- Schurr A, Payne RS, Rigor BM. Protection by MK-801 against hypoxia-, excitotoxin-, and depolarization-induced neuronal damage in vitro. Neurochem Int. 1995;26:519–525. doi: 10.1016/0197-0186(94)00148-n. [DOI] [PubMed] [Google Scholar]
- Shao LR, Halvorsrud R, Borg-Graham L, Storm JF. The role of BK-type Ca2+-dependent K+ channels in spike broadening during repetitive firing in rat hippocampal pyramidal cells. J Physiol (Lond) 1999;521:135–146. doi: 10.1111/j.1469-7793.1999.00135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin LM, Orr SP, Carson MA, Rauch SL, Macklin ML, Lasko NB, Peters PM, Metzger LJ, Dougherty DD, Cannistraro PA, Alpert NM, Fischman AJ, Pitman RK. Regional cerebral blood flow in the amygdala and medial prefrontal cortex during traumatic imagery in male and female Vietnam veterans with PTSD. Arch Gen Psychiatry. 2004;61:168–176. doi: 10.1001/archpsyc.61.2.168. [DOI] [PubMed] [Google Scholar]
- Sierra-Mercado D, Jr, Corcoran KA, Lebron-Milad K, Quirk GJ. Inactivation of the ventromedial prefrontal cortex reduces expression of conditioned fear and impairs subsequent recall of extinction. Eur J Neurosci. 2006;24:1751–1758. doi: 10.1111/j.1460-9568.2006.05014.x. [DOI] [PubMed] [Google Scholar]
- Stackman RW, Hammond RS, Linardatos E, Gerlach A, Maylie J, Adelman JP, Tzounopoulos T. Small conductance Ca2+-activated K+ channels modulate synaptic plasticity and memory encoding. J Neurosci. 2002;22:10163–10171. doi: 10.1523/JNEUROSCI.22-23-10163.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda M, Tanimoto T, Ikeda M, Nasu M, Kadoi J, Yoshida S, Matsumoto S. Enhanced excitability of rat trigeminal root ganglion neurons via decrease in A-type potassium currents following temporomandibular joint inflammation. Neuroscience. 2006;138:621–630. doi: 10.1016/j.neuroscience.2005.11.024. [DOI] [PubMed] [Google Scholar]
- Thompson LT, Moyer JR, Jr, Disterhoft JF. Transient changes in excitability of rabbit CA3 neurons with a time course appropriate to support memory consolidation. J Neurophysiol. 1996;76:1836–1849. doi: 10.1152/jn.1996.76.3.1836. [DOI] [PubMed] [Google Scholar]
- Traub RD, Buhl EH, Gloveli T, Whittington MA. Fast rhythmic bursting can be induced in layer 2/3 cortical neurons by enhancing persistent Na+ conductance or by blocking BK channels. J Neurophysiol. 2003;89:909–921. doi: 10.1152/jn.00573.2002. [DOI] [PubMed] [Google Scholar]
- Tzounopoulos T, Stackman R. Enhancing synaptic plasticity and memory: a role for small-conductance Ca(2+)-activated K+ channels. Neuroscientist. 2003;9:434–439. doi: 10.1177/1073858403259282. [DOI] [PubMed] [Google Scholar]
- Vidal-Gonzalez I, Vidal-Gonzalez B, Rauch SL, Quirk GJ. Microstimulation reveals opposing influences of prelimbic and infralimbic cortex on the expression of conditioned fear. Learn Mem. 2006;13:728–733. doi: 10.1101/lm.306106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Schreurs BG. Characteristics of IA currents in adult rabbit cerebellar Purkinje cells. Brain Res. 2006;1096:85–96. doi: 10.1016/j.brainres.2006.04.048. [DOI] [PubMed] [Google Scholar]
- Wrubel KM, Barrett D, Shumake J, Johnson SE, Gonzalez-Lima F. Methylene blue facilitates the extinction of fear in an animal model of susceptibility to learned helplessness. Neurobiol Learn Mem. 2007;87:209–217. doi: 10.1016/j.nlm.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Xu J, Kang N, Jiang L, Nedergaard M, Kang J. Activity-dependent long-term potentiation of intrinsic excitability in hippocampal CA1 pyramidal neurons. J Neurosci. 2005;25:1750–1760. doi: 10.1523/JNEUROSCI.4217-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamoah EN, Levic S, Redell JB, Crow T. Inhibition of conditioned stimulus pathway phosphoprotein 24 expression blocks the reduction in A-type transient K+ current produced by one-trial in vitro conditioning of Hermissenda. J Neurosci. 2005;25:4793–4800. doi: 10.1523/JNEUROSCI.5256-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan LL, Chen X. Diversity of potassium channels in neuronal dendrites. Prog Neurobiol. 2006;78:374–389. doi: 10.1016/j.pneurobio.2006.03.003. [DOI] [PubMed] [Google Scholar]
- Yue C, Yaari Y. Axo-somatic and apical dendritic Kv7/M channels differentially regulate the intrinsic excitability of adult rat CA1 pyramidal cells. J Neurophysiol. 2006;95:3480–3495. doi: 10.1152/jn.01333.2005. [DOI] [PubMed] [Google Scholar]
- Zhang W, Linden DJ. The other side of the engram: experience-driven changes in neuronal intrinsic excitability. Nat Rev Neurosci. 2003;4:885–900. doi: 10.1038/nrn1248. [DOI] [PubMed] [Google Scholar]
- Zushida K, Sakurai M, Wada K, Sekiguchi M. Facilitation of extinction learning for contextual fear memory by PEPA: a potentiator of AMPA receptors. J Neurosci. 2007;27:158–166. doi: 10.1523/JNEUROSCI.3842-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]