

Abstract
Although morphine induces both analgesia and dependence through μ-opioid receptors (MORs), the respective contributions of the intracellular effectors engaged by MORs remain unknown. To examine the contribution of G-protein-gated inwardly rectifying K+ (GIRK, Kir3) channels to morphine dependence and analgesia, we quantified naloxone-precipitated withdrawal behavior and morphine analgesia using GIRK knock-out (−/−) mice. The morphine withdrawal syndrome was strongly attenuated, whereas morphine analgesia was mostly preserved in mice lacking both GIRK2 and GIRK3 (GIRK2/3−/− mice). In acute slices containing the locus ceruleus (LC) from GIRK2/3−/− mice, the increase in spontaneous firing typically associated with morphine withdrawal was absent. Moreover, although morphine elicited normal presynaptic inhibition in the LC, postsynaptic GIRK currents were completely abolished in GIRK2/3−/− mice. Altogether, these data suggested that morphine-evoked postsynaptic inhibition of the LC was required for the induction of dependence. Consistent with this hypothesis, morphine withdrawal behavior was rescued in GIRK2/3−/− mice by ablation of adrenergic fibers using the neurotoxin N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine. Our data suggest that inhibition of adrenergic tone is required for the induction of dependence, and that channels containing GIRK2 and GIRK3 serve as an inhibitory gate.
Keywords: GIRK, G-protein-gated inwardly rectifying K+ channel, locus ceruleus, μ opioid receptor, noradrenaline, wild type, DSP4
Introduction
μ-Opioid receptors (MORs) couple to G-proteins of the Gi/o subfamily, leading to modulation of multiple intracellular effectors, including adenylyl cyclase, voltage-gated Ca2+ channels, and G-protein-gated inwardly rectifying K+ (GIRK) channels (Williams et al., 2001). GIRK channels mediate in large part the postsynaptic inhibitory effect linked to Gi/o-coupled receptors but do not seem to contribute significantly to presynaptic inhibition (Lüscher et al., 1997). Four mammalian GIRK subunits (GIRK1–4) have been cloned, and they combine to form functional heteromeric and homomeric channels (Wickman et al., 2002). Although GIRK2 can form homomeric GIRK channels (Inanobe et al., 1999), heteromeric channels containing both GIRK1 and GIRK2 are found in many neuron populations (Liao et al., 1996; Lüscher et al., 1997; Koyrakh et al., 2005; Marker et al., 2006). In addition, GIRK3 is widely expressed in the CNS and may contribute to GIRK currents in certain neuron populations, including the locus ceruleus (LC) (Torrecilla et al., 2002) and dopamine neurons of the ventral tegmental area (Cruz et al., 2004; Labouebe et al., 2007).
Several studies involving mice with mutant Girk genes have implicated GIRK channels in the acute analgesic effect of morphine (Ikeda et al., 2000; Mitrovic et al., 2003; Marker et al., 2004). In contrast, the role of GIRK channels in the effects of chronic opioid administration has not been explored.
Chronic exposure to morphine triggers adaptive phenomena such as tolerance and dependence. With tolerance, larger morphine doses have to be applied to obtain the initial effect, whereas dependence is apparent through the withdrawal syndrome triggered by the abrupt discontinuation of morphine exposure. On a cellular level, adaptive processes initiated by MOR activation, such as the recruitment of the adaptor protein β-arrestin2 and an uncoupling of the receptors from the G-proteins, can be observed within minutes (Borgland, 2001). The latter process seems to be involved in tolerance but not dependence, because the withdrawal syndrome is normal in the β-arrestin2−/− mouse (Bohn et al., 2000).
Dependence is a neuroadaptive state resulting from chronic morphine exposure that is characterized by changes in gene expression, opioid signaling, and synaptic transmission (Williams et al., 2001). At the behavioral level, dependence is typically studied indirectly by measuring the withdrawal syndrome precipitated by the abrupt termination of morphine exposure or administration of a MOR antagonist. In other words, dependence can be separated into an induction phase, during which morphine triggers adaptations, and an expression phase, which consists of the withdrawal syndrome. Attenuated withdrawal is predicted, therefore, if dependence is not induced or if a subject is incapable of expressing the withdrawal syndrome (e.g., resulting from a locomotor deficiency). At present, little is known about the structures and signaling relevant to the induction of morphine dependence. Our study suggests that MOR effectors other than GIRK channels primarily mediate systemic morphine analgesia, whereas GIRK channels, perhaps via the postsynaptic inhibition of the adrenergic neurons of the LC, constitute an inhibitory gate for the induction of dependence.
Materials and Methods
Reagents.
Morphine-HCl was obtained from Amino (Neuenhof, Switzerland), naloxone and 8-Br-cAMP were obtained from Tocris Cookson (Bristol, UK), and N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine (DSP4) and kynurenic acid were obtained from Sigma-Aldrich (St. Louis, MO).
Behavioral subjects and tests.
Procedures were performed with the permission of the Cantonal Veterinary Office of Geneva. Animals were kept on a 12 h light/dark cycle. Constitutive GIRK2−/− and GIRK3−/− mouse lines were generated as described previously (Signorini et al., 1997). For the studies described below, GIRK2−/− and GIRK3−/− mice were derived from crosses involving appropriate heterozygous mice. Wild-type (WT) siblings obtained from these crosses were used as controls for all studies. GIRK2/3−/− mice were generated using a three-step breeding strategy. GIRK3−/− and GIRK2+/− mice were crossed in round 1. GIRK2+/−/GIRK3+/− offspring from this cross were then bred in round 2 with GIRK3−/− mice. Male and female GIRK2+/−/GIRK3−/− offspring from the round 2 cross were then bred in round 3, yielding litters consisting of GIRK2+/+/GIRK3−/−, GIRK2+/−/GIRK3−/−, and GIRK2/3−/− mice. Two distinct cohorts of GIRK2/3−/− mice were generated over the course of this 3 year study. For cohort 1, the three-step breeding strategy began after backcrossing the GIRK2 and GIRK3 null alleles for 16 and 5 generations, respectively, against the C57BL/6 inbred mouse strain. For cohort 2, the three-step breeding strategy began after backcrossing the GIRK2 and GIRK3 null mutations for 16 and 11 generations, respectively, against the C57BL/6 mouse strain. Importantly, no differences in baseline or drug-induced behaviors were noted for GIRK2/3−/− mice in cohorts 1 and 2. Similarly, both the electrophysiological and behavioral data gleaned from GIRK3−/− mice obtained from heterozygous crosses, or as part of the three-step double-knock-out breeding strategy (cohort 1 or 2), were indistinguishable. Accordingly, data obtained from distinct mouse cohorts and breeding cages over the course of this study were simply pooled according to genotype. Genotypes were identified by PCR using genomic DNA isolated from tail biopsies. For all experiments, adolescent mice [postnatal day 20 (P20) to P35] were used. The animals were manipulated and habituated to the experimental environment for 1 week before the experiments to minimize interference from the stress response.
Tail-flick test.
Mice were immobilized in a small conic plastic cloth, the tail was subjected to radiant heat, and latencies to tail withdrawal were measured using a tail-flick analgesia meter (Columbus Instruments, Columbus, OH). The intensity of the heat source was adjusted to 7, so that the baseline responses were close to 2 s in WT mice. A cutoff time of 10 s was set to minimize tissue damage. Morphine was injected intraperitoneally 30–60 min before testing, and three latency measurements separated by at least 5 min were averaged. Morphine analgesia is represented as the percentage maximal possible effect (% MPE) = 100 × [(test latency − baseline latency)/(cutoff − baseline latency)]. An individual animal was given only a single injection to avoid possible interference caused by tolerance.
Motor activity.
Open-field environments [ENV-515; 17 (width) × 17 (length) × 12 (height) inches; MED Associates, St. Albans, VT], equipped with ventilated covers and three 16-beam infrared arrays were used to measure the distance traveled during a 90 min session under conditions of moderate light and sound attenuation, as described previously (Pravetoni and Wickman, 2008).
Withdrawal.
Animals were given twice-daily injections of morphine at increasing doses (20, 40, 60, 80, 100 mg/kg, i.p.) over a period of 5 d. On the morning of day 6, mice were injected with morphine (100 mg/kg, i.p.), and withdrawal was precipitated 2 h later with naloxone (1 mg/kg, i.p.). Where noted, mice were injected with DSP4 (50 mg/kg, i.p.) 3–5 d before the first morphine injection. For 10 min before and 30 min after naloxone administration, withdrawal signs (jumping, sniffing, tremor, paw tremor, teeth chattering, wet-dog shakes, diarrhea) were quantified by two independent experimenters blind to genotype. The impact of gender, genotype, and drug treatment on each individual sign was analyzed. Subsequently, a global withdrawal score was calculated for each animal after subtraction of the signs observed during baseline. The behavior of WT mice was used as reference and was given a global score of 100 points, with each sign receiving equal weight. Behaviors measured in GIRK−/− mice were counted and normalized to WT for each symptom. The sum of points was then averaged for all animals of the same genotype and condition.
Electrophysiology.
Horizontal slices (250 μm) of the brainstem were made from mice in cooled artificial CSF (ACSF) containing the following (in mm): 119 NaCl, 2.5 KCl, 1.3 MgCl2, 2.5 CaCl2, 1.0 NaH2PO4, 26.2 NaHCO3, and 11 glucose bubbled with 95% O2/5% CO2. Slices were warmed to 34°C and transferred after 1 h to the recording chamber superfused (2.5 ml/min) with ACSF. Visualized whole-cell voltage-clamp recording techniques were used to measure holding currents and synaptic responses of LC neurons at −63 mV (corrected for a calculated liquid junction potential of −13 mV). Inhibitory synaptic currents were obtained by extracellular stimulation (0.1 ms duration, 0.05 Hz) in the presence of kynurenic acid (2 mm). The internal solution contained the following (in mm): 130 K-Gluconate, 4 MgCl2, 1.1 EGTA, 5 HEPES, 3.4 Na2ATP, 10 Na2-Creatine-phosphate, and 0.1 Na3GTP. The calculated reversal potential for Cl− was −71.7 mV. Currents were amplified, filtered at 1 kHz, digitized at 5 kHz, and saved directly to a computer hard disk. Cell membrane and access resistance were monitored with each sweep, and the experiment was terminated if the access resistance changed >20%. Spontaneous firing of LC neurons was recorded using a pipette in cell-attached, current-clamp mode; traces were filtered at 10 kHz and digitized at 20 kHz.
Measurement of noradrenaline.
Mice were killed 4–10 d after DSP4 injection, and the frontal cortex was rapidly dissected. Tissue was homogenized by sonication in 1 ml of 0.1 m perchloric acid containing dihydroxybenzylamide (DHBA; Sigma-Aldrich) and centrifuged at 13,000 × g for 15 min. Supernatants were then filtered through 0.45 μm Ultrafree-MC centrifugal filter units (Millipore Corporation, Billerica, MA) and stored at −70°C. Catecholamine measurements were performed by HPLC with amperometric detection similar to that described previously (Grouzmann et al., 2003). Acidified tissues extracts (1 ml) were spiked with 40,000 ng of DHBA as internal standard to assess recovery. The separation was achieved on a reversed-phase column Nucleosil (5 mm C-18, 25 cm, 4.6 mm; Macherey-Nagel, Oensingen, Switzerland) using a 50 mm sodium acetate buffer mobile phase containing 20 mm citric acid, 0.135 mm EDTA, 1 mm dibutylamine, and 3.8 mm sodium octyl sulfonate, as an ion-pairing agent, and 7% methanol at a flow rate of 0.9 ml/min. The electrochemical detector (Decade; Antec, Leyden, The Netherlands) was set at +0.8 V. The following order of elution was observed: noradrenaline (NA), adrenaline, DHBA, and dopamine. The recovery was close to 100%, and the quantification limit was 5 pg per injection. The interassay coefficients of variation were 11–12%.
Statistics.
Compiled data are expressed as mean ± SEM. For statistical comparisons of two conditions, the nonparametric Mann–Whitney or Wilcoxon matched tests were used. For multiple comparisons, a two-way ANOVA followed by Bonferroni's posttest or the nonparametric Kruskal–Wallis test followed by Dunn's posttests were applied (Instat, version 3.0; GraphPad Software, San Diego, CA). All data represent observations from at least three different animals.
Results
Morphine analgesia and withdrawal in GIRK−/− mice
We first measured thermal nociception and morphine analgesia using a radiant heat tail-flick test. We found that the baseline tail-flick latency was significantly shorter for GIRK2−/− and GIRK2/3−/− mice compared with WT mice, but there was no difference between the two GIRK−/− lines (WT, 2.0 ± 0.1 s; GIRK2−/−, 1.5 ± 0.1 s; GIRK2/3−/−, 1.5 ± 0.1 s; n ≥ 14 for each genotype; p < 0.001 vs WT) (Fig. 1a). The dose–response curve describing morphine analgesia was shifted slightly to the right for GIRK2−/− mice (WT, EC50, 6.9 ± 0.3 mg/kg, n ≥ 12; GIRK2−/−, EC50, 14.3 ± 1.2 mg/kg, n ≥ 10; p < 0.001) (Fig. 1b); no additional shift of the dose–response curve was observed for GIRK2/3−/− mice (EC50, 16.2 ± 0.6, n ≥ 10; p > 0.05 vs GIRK2−/−) (Fig. 1b, triangles). In an independent series of experiments, we adjusted the radiant heat of the tail-flick test to yield similar baseline latencies for each animal tested. This occluded differences in baseline nociception (Mitrovic et al., 2003; Marker et al., 2004) and permitted measurement of morphine analgesia at equi-effective stimuli. In this study, a slight reduction in morphine potency was detected in GIRK2−/− and GIRK2/3−/− mice, but analgesic efficacy was preserved (supplemental Fig. S1a, available at www.jneurosci.org as supplemental material). Similar outcomes were obtained in a separate study of morphine analgesia involving the hot-plate assay (supplemental Fig. S1b,c, available at www.jneurosci.org as supplemental material). These observations are consistent with those of previous studies implicating GIRK2-containing channels in systemic opioid analgesia (Ikeda et al., 2000; Marker et al., 2002, 2004; Mitrovic et al., 2003), with the exception that at this age of the mice, no significant influence of gender on baseline nociception was observed. Moreover, morphine analgesia was only different in WT male and female mice for the hot plate assay but not in the tail-flick test (supplemental Fig. S2a–d, available at www.jneurosci.org as supplemental material). Because gender differences were small and no longer present in GIRK−/− mice, data from male and female mice were pooled within genotypes.
Figure 1.

Normal analgesic efficacy of morphine in GIRK2/3−/− mice. a, Baseline latency measurements for naive animals in the tail-flick test. In GIRK2−/− and GIRK2/3−/− mice, baseline latencies are shorter than in WT mice. b, Dose–response curves for morphine-mediated prolongation of tail-flick latencies. In GIRK2−/− and GIRK2/3−/− mice, morphine dose–response curves were shifted to the right, indicating a decreased potency but preserved efficacy. ***p < 0.001.
We next investigated the effect of GIRK subunit ablation on the naloxone-precipitated withdrawal syndrome after 5 d of morphine exposure. The withdrawal syndrome was assessed by quantifying seven distinct withdrawal signs (Fig. 2 a–g) across genotypes and gender. We did not observe a significant impact of gender on the appearance of any single withdrawal sign, and as such, data from male and female animals were pooled within genotypes (supplemental Table 1, available at www.jneurosci.org as supplemental material). We did observe, however, that GIRK2−/− animals exhibited a significantly increased number of escape jumps and wet-dog shakes than WT counterparts (Fig. 2a,b). Hyperactivity in GIRK2−/− mice has been reported previously (Blednov et al., 2002), which could explain this observation. GIRK3−/− animals also showed slightly decreased occurrence of all symptoms with the exception of diarrhea. Interestingly, in GIRK2/3−/− mice, all of the observed withdrawal signs, with the exception of diarrhea, were significantly reduced. Escape jumps and body as well as paw tremor were attenuated most strongly.
Figure 2.
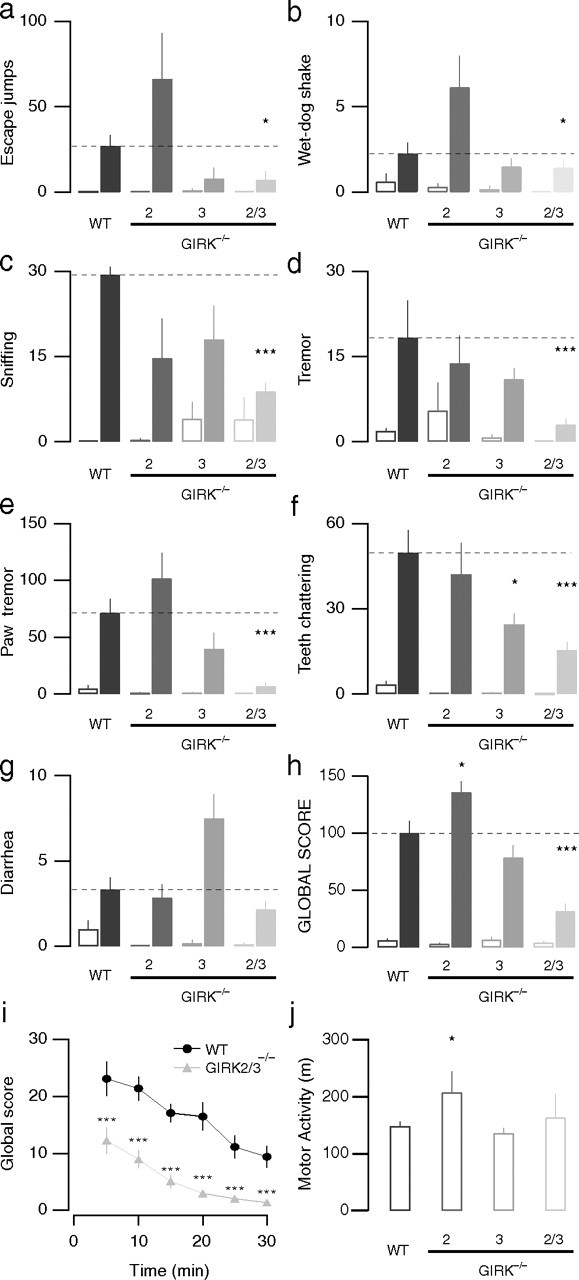
Attenuation of morphine withdrawal syndrome in GIRK2/3−/− mice. a–g, Individual withdrawal signs in GIRK−/− versus WT mice. After five consecutive days of morphine (filled bars; 20–100 mg/kg, i.p.) or saline (open bars) administration, a withdrawal syndrome was precipitated with naloxone (1 mg/kg, i.p.), and the occurrence of each withdrawal sign was monitored for 30 min: a, escape jumps; b, wet-dog shakes; c, sniffing; d, tremor; e, paw tremor; f, teeth chattering; g, diarrhea. h, Global withdrawal scores after naloxone challenge compiled from withdrawal signs above in morphine-treated (filled bars) and saline-treated (open bars) animals. Notice that the global withdrawal score was strongly reduced in GIRK2/3−/− mice but not in the other GIRK−/− lines, whereas saline-treated animals behaved indistinguishably across the genotypes. i, Global withdrawal score as a function of time. Scores in GIRK2/3−/− mice were significantly different from WT mice for all time points. j, Spontaneous locomotor activity (total distance traveled) was greater in GIRK2−/−mice but similar in GIRK3−/− and GIRK2/3−/− mice compared with WT mice. ***p < 0.001; *p < 0.05.
Global withdrawal scores were calculated by compiling all observed signs (Fig. 2h) (see Materials and Methods). Control mice injected with saline had global scores that were not significantly different for WT (6 ± 1), GIRK2−/− (3 ± 1), GIRK3−/− (6 ± 3), and GIRK2/3−/− mice (4 ± 2; n = 6–22; p > 0.05). Although in GIRK2−/− mice, the global withdrawal score (136 ± 10; n = 6) was slightly greater compared with WT mice (100 ± 10; n = 16; p < 0.05), the global withdrawal score in GIRK3−/− mice (79 ± 11; n = 6; p > 0.05) was not significantly different from WT. The global withdrawal score for GIRK2/3−/− mice (32 ± 7; n = 22; p < 0.001), in contrast, was significantly lower than that of WT controls. Breaking down global withdrawal scores into 5 min intervals excluded altered kinetics of the withdrawal response as a cause of the observed differences (Fig. 2i). Furthermore, the lower withdrawal scores of GIRK2/3−/− mice cannot be explained by a general hypoactivity of these animals, because the distances traveled in an open field were similar to those measured in WT mice (WT, 148.5 ± 8.1 m; GIRK2/3−/−, 163.1 ± 41.4 m; n ≥ 6 for each genotype; p > 0.05) (Fig. 2j). Conversely, the higher score in GIRK2−/− mice may be linked to their hyperlocomotor activity (GIRK2−/−, 207.1 ± 37.2 m; n = 12; p < 0.05).
Absence of a cellular hallmark of withdrawal in GIRK2/3−/− mice
There is general agreement that LC neurons fire excessively during withdrawal. This can be measured in vivo (Aghajanian, 1978), as well as ex vivo in acute brainstem slices (Ivanov and Aston-Jones, 2001). Using the latter model, we performed cell-attached recordings and observed a doubling of the firing frequency in WT mice during withdrawal (0.7 ± 0.2 Hz at baseline vs 1.9 ± 0.4 Hz during withdrawal; n = 6–8; p < 0.05) (Fig. 3a,c). In contrast, LC neurons from GIRK2/3−/− mice tended to have slightly higher basal firing frequencies and did not display increased firing rates during withdrawal (1.1 ± 0.4 Hz at baseline vs 0.6 ± 0.1 Hz during withdrawal; n = 5; p > 0.05) (Fig. 3b,c). These findings are consistent with the behavioral data and suggest that morphine dependence was not induced in GIRK2/3−/− mice.
Figure 3.

Spontaneous firing of LC neurons in GIRK2/3−/− mice. a, b, Representative traces of cell-attached recordings in brainstem slices of WT and GIRK2/3−/− mice at baseline and during morphine withdrawal, after 5 d of morphine treatment (20–100 mg/kg, i.p.) and naloxone administration (1 mg/kg, i.p.). c, Bar graph showing mean firing rates at baseline and during withdrawal. Notice that in WT but not in GIRK2/3−/− mice, there is a significant increase in LC neuron firing rate during withdrawal. **p < 0.01; *p < 0.05.
Effect of morphine on LC neurons in slices from GIRK−/− mice
Given the absence of adaptation in firing rates of LC neurons in morphine-treated GIRK2/3−/− mice, we next examined the effect of morphine on presynaptic and postsynaptic signaling in LC neurons. Opioid-induced presynaptic inhibition of LC neurons can be studied by monitoring GABAergic IPSCs evoked by extracellular stimulation in the proximity of the LC (Williams et al., 2001). Similar to observations in the hippocampus (Lüscher et al., 1997), we found that the presynaptic inhibitory effect of morphine was similar in magnitude for all genotypes tested (WT, 53 ± 1%; GIRK2−/−, 54 ± 4%; GIRK3−/−, 54 ± 9%; GIRK2/3−/−, 53 ± 3%; n ≥ 6 for each genotype; p > 0.05) (Fig. 4a,b).
Figure 4.

Impact of GIRK ablation on presynaptic and postsynaptic inhibition in the LC. a, Morphine inhibited evoked IPSCs to a similar degree in GIRK2−/−, GIRK3−/−, and GIRK2/3−/− mice compared with WT. Morphine, 5 μm; naloxone (Nal), 1 μm. Insets are averages of 15 individual traces at corresponding time points. Calibration: 50 pA, 10 ms. b, Bar graph summarizing the inhibition of evoked IPSCs by morphine. c, Morphine (5 μm) applied for 15 min evoked a sustained outward current at a holding potential of −63 mV in WT neurons that was reduced in GIRK−/− mice. d, Quantification of postsynaptic current amplitude. GIRK2/3−/− mice lack the GIRK-dependent component of the morphine-induced postsynaptic current. The small residual current measured in WT LC neurons in the presence of Ba was blocked by the cAMP analog 8-Br-cAMP (10 mm; 2.4 ± 1.4 pA; n = 6; p < 0.05) (data not shown). Insensitivity to Ba excludes GIRK channels as the underlying conductance, whereas the cAMP sensitivity is compatible with a background K+ channel, similar to the residual currents observed in other neurons (Cruz et al., 2004). **p < 0.01; *p < 0.05.
Although morphine-induced presynaptic inhibition in the LC was unaffected by GIRK ablation, we found that morphine-induced somatodendritic GIRK currents were significantly smaller in GIRK−/− animals compared with WT, with the most prominent effect seen in LC neurons from GIRK2/3−/− mice (WT, 41.8 ± 6.5 pA; GIRK2−/−, 23.4 ± 3.9 pA; GIRK3−/−, 20.9 ± 2.4 pA; GIRK2/3−/−, 6.3 ± 1.2 pA; n > 5; p < 0.05) (Fig. 4c,d). Moreover, the small residual current observed in GIRK2/3−/− mice (8.6 ± 1.1 pA; n = 5) was insensitive to barium (Ba; 1 mm). In the presence of Ba, a current of similar amplitude was also evoked in WT mice (14.3 ± 2.5 pA; p < 0.01; n = 7). Therefore, ablation of both GIRK2 and GIRK3 is necessary and sufficient to eliminate the GIRK-dependent component of the morphine-induced current in LC neurons.
Because GIRK channels may be constitutively active (Lüscher et al., 1997; Torrecilla et al., 2002; Chen and Johnston, 2005; Koyrakh et al., 2005), LC neurons observed in GIRK2/3−/− mice may exhibit elevated intrinsic excitability. We therefore monitored the basal firing frequency of LC neurons while blocking synaptic transmission. Under these conditions, the firing frequency of LC neurons from GIRK2/3−/− mice was significantly higher than that measured in LC neurons from WT mice (GIRK2/3−/−, 0.9 ± 0.3 Hz; WT, 0.3 ± 0.1 Hz; n > 5; p < 0.05) (Fig. 5a). Although this difference is evidently partially occluded when synaptic transmission remains intact (compare with Fig. 3), the increased intrinsic excitability of LC neurons from GIRK2/3−/− mice likely explains the elevated NA concentration measured in the cortex of GIRK2/3−/− mice (WT, 275 ± 17 pg/mg; GIRK2/3−/−, 395 ± 36 pg/mg; n ≥ 6; p < 0.05 vs WT) (Fig. 5b). Together, the electrophysiological studies revealed that GIRK channels in LC neurons are postsynaptic effectors of MORs and shape intrinsic excitability.
Figure 5.

Spontaneous firing rate recorded in LC neurons from WT and GIRK2/3−/− mice. a, When synaptic transmission was blocked (2 mm kynurenic acid, 100 μm picrotoxin, and 1 μm strychnine), the spontaneous firing of LC neurons was higher in GIRK2/3−/− mice compared with WT. b, Cortical NA concentration determined in WT and GIRK2/3−/− mice at baseline and after DSP4 treatment. Note that although the baseline NA level was higher in GIRK2/3−/− mice, DSP4 treatment led to a comparable reduction in the cortex of WT and GIRK2/3−/− mice. ***p < 0.001; *p < 0.05.
Rescue of the withdrawal syndrome in GIRK2/3−/− mice
Given the behavioral and electrophysiological observations, we hypothesized that the abolished withdrawal syndrome in GIRK2/3−/− mice may be attributable to the lack of GIRK-dependent postsynaptic inhibition of the LC. In other words, postsynaptic inhibition of LC neurons may be required for the induction of dependence. This hypothesis predicts that GIRK-independent inhibition of the LC should rescue the withdrawal syndrome in GIRK2/3−/− mice. Accordingly, we exposed GIRK2/3−/− mice to DSP4, a toxin that selectively destroys the adrenergic axons originating in the LC, leading to a long-lasting adrenergic depletion (Fritschy and Grzanna, 1992). To assess the integrity of the adrenergic system in GIRK2/3−/− mice and the efficacy of the DSP4 treatment, we measured NA levels in the cortex 4–10 d after the injection of the toxin. At baseline, NA concentrations were higher in GIRK2/3−/− mice than in WT mice (Fig. 5b). After DSP4 treatment, NA concentrations were reduced by 77% in WT mice (67 ± 6 pg/mg; n = 12; p < 0.001) and by 69% in GIRK2/3−/− mice (123 ± 32 pg/mg after treatment; n = 6; p < 0.001), indicating that DSP4 was equally efficacious in both genotypes.
When challenged with naloxone after chronic morphine exposure and in line with previous observations (Chieng and Christie, 1995; Dossin et al., 1996; Caille et al., 1999), global withdrawal scores compiled for seven distinct withdrawal signs (Fig. 6 a–h) were not different between mice treated with a low concentration of DSP4 (50 mg/kg) and untreated WT mice (100 ± 7 vs 100 ± 10; n = 9; p > 0.05) (Fig. 6a,i). Similarly, DSP4 treatment did not have any impact on global withdrawal scores of saline-treated animals of either genotype (WT, 13 ± 6; DSP4-WT, 3 ± 2; GIRK2/3−/−, 3 ± 2; DSP4-GIRK2/3−/−, 15 ± 10; n = 6–8; p > 0.05). However, DSP4 treatment almost completely rescued the withdrawal syndrome in GIRK2/3−/− mice (DSP4-GIRK2/3−/−, 81 ± 7; control-GIRK2/3−/−, 29 ± 9; n ≥ 10; p < 0.001). A sign-by-sign analysis (Fig. 6 b–h) revealed that after DSP4 treatment of GIRK2/3−/− mice, escape jumps, wet-dog shakes, and diarrhea fully recovered, whereas paw tremor showed only a partial recovery. For body tremor and teeth chattering, the increase was not significant.
Figure 6.
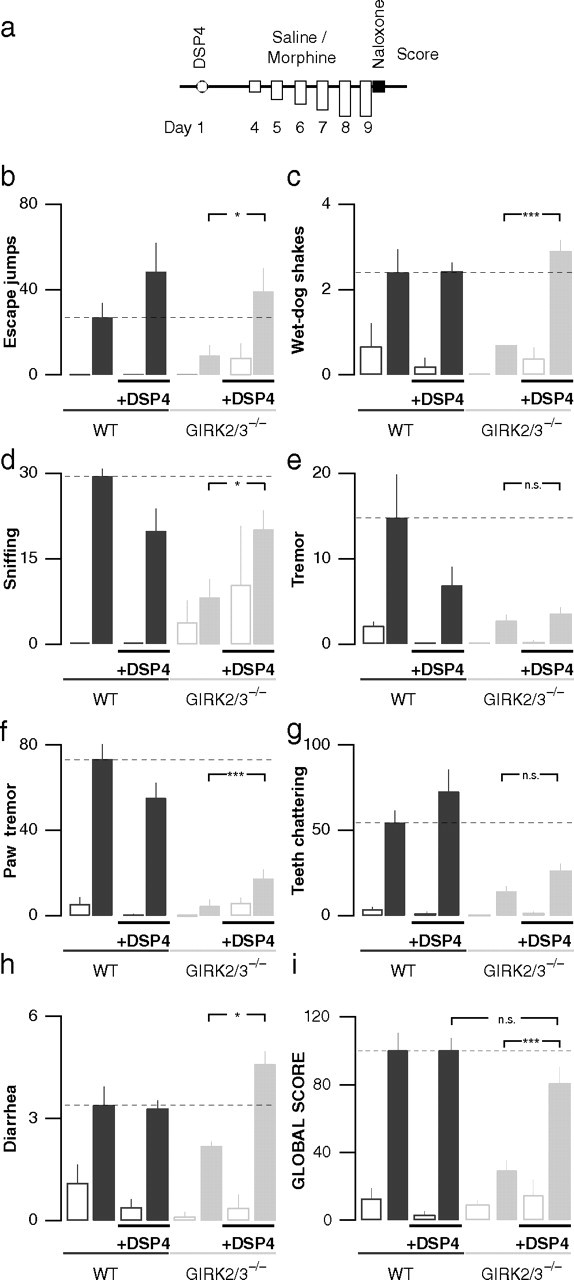
Individual withdrawal signs in GIRK2/3−/− animals treated with DSP4. a, WT and GIRK2/3−/− mice were injected either with DSP4 (50 mg/kg, i.p.) or with saline 3–5 d before starting the chronic morphine administration procedure. After 5 d of chronic morphine (filled bars) or saline (open bars) administration, a withdrawal syndrome was precipitated with naloxone (1 mg/kg, i.p.), and each withdrawal sign was counted for 30 min: b, escape jumps; c, wet-dog shakes; d, sniffing; e, tremor; f, paw tremor; g, teeth chattering; h, diarrhea. i, Global withdrawal score showing that DSP4 treatment did not have any significant influence on withdrawal in WT animals but did rescue withdrawal in GIRK2/3−/− mice. ***p < 0.001; *p < 0.05.
We also tested whether DSP4 treatment affected baseline nociception and morphine analgesia and found that in the hot-plate test, there was no significant effect of DSP4 on baseline latency in WT mice, nor did DSP4 influence morphine analgesia (supplemental Fig. S3a,b, available at www.jneurosci.org as supplemental material). These observations suggest that the adrenergic output of the LC exerts little impact on systemic morphine analgesia.
Discussion
Here, we present evidence that the morphine withdrawal syndrome is severely attenuated in GIRK2/3−/− mice, whereas morphine analgesia is mostly preserved. GIRK2/3−/− mice also failed to display an increase in LC neuron firing rate, an established electrophysiological hallmark of withdrawal. Furthermore, the GIRK-dependent component of the morphine-induced postsynaptic current was absent in LC neurons from GIRK2/3−/− mice. Altogether, the behavioral and electrophysiological data suggested that inhibition of adrenergic output from the LC is required for induction of dependence, a contention supported by experiments using the neurotoxin DSP4. Indeed, DSP4-treated GIRK2/3−/− mice show a near-normal morphine withdrawal syndrome, arguing that the attenuated withdrawal syndrome in GIRK2/3−/− mice is not caused by an inability to express morphine dependence.
Among the brain regions implicated in opioid dependence and withdrawal, the LC has generated great interest. There is general consensus that LC neurons are hyperactive during withdrawal (Aghajanian, 1978). Enhanced synaptic drive (e.g., strengthening of excitatory afferents onto LC neurons (Johnson et al., 2002) and intrinsic upregulation of neuronal excitability (Lane-Ladd et al., 1997; Han et al., 2006) have been implicated in the hyperactivity of the LC. Nevertheless, the relevance of this hyperactivity to withdrawal behavior remains controversial (Christie et al., 1997). A large body of evidence implicates the LC in the expression of the opiate withdrawal syndrome (Maldonado, 1997; Nestler, 2004). For example, surgical lesions of the LC abolished the withdrawal syndrome (Maldonado and Koob, 1993). In addition, studies involving molecular and pharmacological interventions aimed at disrupting MOR-mediated inhibition of adenylyl cyclase, the cAMP-responsive element-binding protein function (Maldonado et al., 1996), phosphodiesterases (Mamiya et al., 2001), or the cAMP system were all supportive of a key role played by the LC in the expression of morphine withdrawal (Lane-Ladd et al., 1997; Han et al., 2006). Nevertheless, pharmacological ablation of LC function failed to attenuate morphine withdrawal (Chieng and Christie, 1995; Caille et al., 1999).
The attenuated withdrawal syndrome seen in GIRK2/3−/− mice may not reflect a selective deficit of the LC. Indeed, it is possible that the observed phenotype is influenced by developmental alterations linked to constitutive gene ablation and/or the absence of GIRK2 and/or GIRK3 from neuron populations that normally modulate withdrawal behavior. But does the rescue of the withdrawal syndrome by DSP4 implicate the LC? DSP4 binds to the adrenergic transporters located on adrenergic neuron terminals (Lee et al., 1982). DSP4 shows strong selectivity for adrenergic terminals arising from the LC, as evidenced by immunohistochemical analysis of dopamine β-hydroxylase (a marker of adrenergic terminals) (Fritschy and Grzanna, 1991) and biochemical measurements of NA that showed abolished levels only in regions targeted by the LC (Prieto and Giralt, 2001). It is important to note that the molecular basis for this selectivity remains elusive. Furthermore, recent studies indicate that the selectivity of DSP4 for LC projections is relative, not absolute (Dailly et al., 2006).
Our experience with DSP4 confirmed that withdrawal can be observed in WT mice when the LC output is abolished (Chieng and Christie, 1995; Caille et al., 1999) and in turn suggested a novel role for the central adrenergic system in opiate dependence (Fig. 7). Our hypothesis is that NA normally prevents the induction of dependence, and that the morphine-dependent activation of GIRK channels effectively suppresses NA release, perhaps by its action on the LC, permitting the induction of dependence. Although our experience with DSP4 certainly implicates the LC, it is appropriate to consider other adrenergic projections given questions concerning the anatomic selectivity of this toxin. Indeed, structures neighboring the LC, including afferents to the periaqueductal gray, have been implicated in morphine withdrawal (Maldonado et al., 1995; Chieng and Christie, 1996). The adrenergic fibers arising from medullary A1–A2 neuron groups projecting to the bed nucleus of the stria terminalis may also contribute to the opioid withdrawal syndrome (Delfs et al., 2000). Rebound of adenylyl cyclase activity in these parts of the brain (Punch et al., 1997; Williams et al., 2001; McClung et al., 2005) may constitute the molecular correlate of withdrawal.
Figure 7.

Proposed model. MOR mediates analgesia mainly via presynaptic effectors such as voltage-gated calcium channels or voltage-sensitive K channels (Kv). Chronic exposure to morphine leads to a counter-adaptation of adenylyl cyclase signaling and downstream events that are responsible for the expression of dependence. Top left, In WT mice, this pathway is normally gated by the afferents from adrenergic nuclei such as the LC. Morphine, through activation of MORs and postsynaptic GIRK channels, inhibits adrenergic neurons to allow the induction of dependence. Top right, In GIRK2/3−/− mice, where the potency but not the efficacy of morphine analgesia is affected, dependence is not induced because the adrenergic tone is persistently/constitutively high. Bottom, When GIRK2/3−/− mice are exposed to DSP4, the adrenergic output is abolished, and withdrawal is rescued.
Studies involving mice with mutant Girk genes have implicated GIRK channels in nociception and the acute analgesic effect of morphine (Ikeda et al., 2000; Blednov et al., 2003; Mitrovic et al., 2003; Marker et al., 2004, 2006). For example, weaver mice, which harbor a mutation in the Girk2 gene, exhibited blunted responses to systemic morphine analgesia as measured by the hot-plate test (Ikeda et al., 2000). Similarly, GIRK2−/− mice exhibited hyperalgesia and blunted systemic morphine analgesia in both thermal and chemical tests of nociception (Mitrovic et al., 2003). Notably, this study demonstrated that analgesic efficacy of systemic morphine was preserved in GIRK2−/− mice, whereas potency was reduced. Therefore, GIRK channels contribute to systemic morphine analgesia but are not required for a maximal analgesic response.
Therefore, given the results from this and published studies, it seems reasonable to suggest that the analgesic effect of systemic morphine primarily involves presynaptic inhibition. Activation of MORs on presynaptic terminals is known to modulate calcium channels, the neurotransmitter release machinery, and voltage-gated potassium channels, leading to decreased neurotransmitter release (Miller, 1998). The importance of presynaptic mechanisms to opioid analgesia is supported by observations in β-arrestin2−/− mice, which display an enhanced analgesic response to morphine (Bohn et al., 1999). In slices of the periaqueductal gray and the LC from these mice, morphine causes an augmented presynaptic inhibition, whereas postsynaptic hyperpolarizing currents are normal (Bradaia et al., 2005).
In summary, we describe a dissociation of systemic morphine analgesia and physical dependence. Because this is achieved with the ablation of postsynaptic effectors, we conclude that morphine analgesia is primarily mediated by presynaptic mechanisms. Furthermore, we argue that GIRK channels containing GIRK2 and GIRK3 subunits present in the LC or other adrenergic nuclei inhibit adrenergic output during morphine exposure, thereby gating the induction of dependence. Our model predicts that the selective activation of MORs expressed on presynaptic terminals would have strong analgesic effects without inducing dependence.
Footnotes
This work was supported by the Roche Research Foundation (M.S.), the Swiss National Science Foundation (C.L.), and National Institutes of Health Grants MH61933 and DA011806 (K.W.). We thank Howard Fields, Matt Frerking, Cheryl Marker, Mauro Serafin, and the members of the Lüscher laboratory for many helpful discussions and Françoise Loctin and Cyril Creton for technical assistance. We thank Markus Stoffel for providing the GIRK2−/− mice and Isabelle Decosterd for providing us with the apparatus used for analgesia testing. We thank E. Grouzman for help with the measurements of noradrenaline.
References
- Aghajanian GK. Tolerance of locus coeruleus neurones to morphine and suppression of withdrawal response by clonidine. Nature. 1978;276:186–188. doi: 10.1038/276186a0. [DOI] [PubMed] [Google Scholar]
- Blednov YA, Stoffel M, Cooper R, Wallace D, Mane N, Harris RA. Hyperactivity and dopamine D1 receptor activation in mice lacking girk2 channels. Psychopharmacology (Berl) 2002;159:370–378. doi: 10.1007/s00213-001-0937-6. [DOI] [PubMed] [Google Scholar]
- Blednov YA, Stoffel M, Alva H, Harris RA. A pervasive mechanism for analgesia: activation of GIRK2 channels. Proc Natl Acad Sci USA. 2003;100:277–282. doi: 10.1073/pnas.012682399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT. Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science. 1999;286:2495–2498. doi: 10.1126/science.286.5449.2495. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG. Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature. 2000;408:720–723. doi: 10.1038/35047086. [DOI] [PubMed] [Google Scholar]
- Borgland SL. Acute opioid receptor desensitization and tolerance: is there a link? Clin Exp Pharmacol Physiol. 2001;28:147–154. doi: 10.1046/j.1440-1681.2001.03418.x. [DOI] [PubMed] [Google Scholar]
- Bradaia A, Berton F, Ferrari S, Lüscher C. beta-Arrestin2, interacting with phosphodiesterase 4, regulates synaptic release probability and presynaptic inhibition by opioids. Proc Natl Acad Sci USA. 2005;102:3034–3039. doi: 10.1073/pnas.0406632102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caille S, Espejo EF, Reneric JP, Cador M, Koob GF, Stinus L. Total neurochemical lesion of noradrenergic neurons of the locus ceruleus does not alter either naloxone-precipitated or spontaneous opiate withdrawal nor does it influence ability of clonidine to reverse opiate withdrawal. J Pharmacol Exp Ther. 1999;290:881–892. [PubMed] [Google Scholar]
- Chen X, Johnston D. Constitutively active G-protein-gated inwardly rectifying K+ channels in dendrites of hippocampal CA1 pyramidal neurons. J Neurosci. 2005;25:3787–3792. doi: 10.1523/JNEUROSCI.5312-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chieng B, Christie MJ. Lesions to terminals of noradrenergic locus coeruleus neurones do not inhibit opiate withdrawal behaviour in rats. Neurosci Lett. 1995;186:37–40. doi: 10.1016/0304-3940(95)11276-3. [DOI] [PubMed] [Google Scholar]
- Chieng B, Christie MD. Local opioid withdrawal in rat single periaqueductal gray neurons in vitro. J Neurosci. 1996;16:7128–7136. doi: 10.1523/JNEUROSCI.16-22-07128.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie MJ, Williams JT, Osborne PB, Bellchambers CE. Where is the locus in opioid withdrawal? Trends Pharmacol Sci. 1997;18:134–140. doi: 10.1016/s0165-6147(97)01045-6. [DOI] [PubMed] [Google Scholar]
- Cruz HG, Ivanova T, Lunn ML, Stoffel M, Slesinger PA, Lüscher C. Bi-directional effects of GABA(B) receptor agonists on the mesolimbic dopamine system. Nat Neurosci. 2004;7:153–159. doi: 10.1038/nn1181. [DOI] [PubMed] [Google Scholar]
- Dailly E, Chenu F, Petit-Demouliere B, Bourin M. Specificity and efficacy of noradrenaline, serotonin depletion in discrete brain areas of Swiss mice by neurotoxins. J Neurosci Methods. 2006;150:111–115. doi: 10.1016/j.jneumeth.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Delfs JM, Zhu Y, Druhan JP, Aston-Jones G. Noradrenaline in the ventral forebrain is critical for opiate withdrawal-induced aversion. Nature. 2000;403:430–444. doi: 10.1038/35000212. [DOI] [PubMed] [Google Scholar]
- Dossin O, Hanoun N, Zajac JM. Involvement of locus coeruleus projections in opiate withdrawal but not in opiate tolerance in mice. Eur J Pharmacol. 1996;308:271–274. doi: 10.1016/0014-2999(96)00318-4. [DOI] [PubMed] [Google Scholar]
- Fritschy JM, Grzanna R. Selective effects of DSP-4 on locus coeruleus axons: are there pharmacologically different types of noradrenergic axons in the central nervous system? Prog Brain Res. 1991;88:257–268. doi: 10.1016/s0079-6123(08)63815-7. [DOI] [PubMed] [Google Scholar]
- Fritschy JM, Grzanna R. Restoration of ascending noradrenergic projections by residual locus coeruleus neurons: compensatory response to neurotoxin-induced cell death in the adult rat brain. J Comp Neurol. 1992;321:421–441. doi: 10.1002/cne.903210309. [DOI] [PubMed] [Google Scholar]
- Grouzmann E, Cavadas C, Grand D, Moratel M, Aubert JF, Brunner HR, Mazzolai L. Blood sampling methodology is crucial for precise measurement of plasma catecholamines concentrations in mice. Pflügers Arch. 2003;447:254–258. doi: 10.1007/s00424-003-1140-x. [DOI] [PubMed] [Google Scholar]
- Han MH, Bolanos CA, Green TA, Olson VG, Neve RL, Liu RJ, Aghajanian GK, Nestler EJ. Role of cAMP response element-binding protein in the rat locus ceruleus: regulation of neuronal activity and opiate withdrawal behaviors. J Neurosci. 2006;26:4624–4629. doi: 10.1523/JNEUROSCI.4701-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda K, Kobayashi T, Kumanishi T, Niki H, Yano R. Involvement of G-protein-activated inwardly rectifying K (GIRK) channels in opioid-induced analgesia. Neurosci Res. 2000;38:113–116. doi: 10.1016/s0168-0102(00)00144-9. [DOI] [PubMed] [Google Scholar]
- Inanobe A, Yoshimoto Y, Horio Y, Morishige KI, Hibino H, Matsumoto S, Tokunaga Y, Maeda T, Hata Y, Takai Y, Kurachi Y. Characterization of G-protein-gated K+ channels composed of Kir3.2 subunits in dopaminergic neurons of the substantia nigra. J Neurosci. 1999;19:1006–1017. doi: 10.1523/JNEUROSCI.19-03-01006.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov A, Aston-Jones G. Local opiate withdrawal in locus coeruleus neurons in vitro. J Neurophysiol. 2001;85:2388–2397. doi: 10.1152/jn.2001.85.6.2388. [DOI] [PubMed] [Google Scholar]
- Johnson AD, Peoples J, Stornetta RL, Van Bockstaele EJ. Opioid circuits originating from the nucleus paragigantocellularis and their potential role in opiate withdrawal. Brain Res. 2002;955:72–84. doi: 10.1016/s0006-8993(02)03367-x. [DOI] [PubMed] [Google Scholar]
- Koyrakh L, Lujan R, Colon J, Karschin C, Kurachi Y, Karschin A, Wickman K. Molecular and cellular diversity of neuronal G-protein-gated potassium channels. J Neurosci. 2005;25:11468–11478. doi: 10.1523/JNEUROSCI.3484-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labouebe G, Lomazzi M, Cruz HG, Creton C, Lujan R, Li M, Yanagawa Y, Obata K, Watanabe M, Wickman K, Boyer SB, Slesinger PA, Lüscher C. RGS2 modulates coupling between GABA(B) receptors and GIRK channels in dopamine neurons of the ventral tegmental area. Nat Neurosci. 2007;10:1559–1568. doi: 10.1038/nn2006. [DOI] [PubMed] [Google Scholar]
- Lane-Ladd SB, Pineda J, Boundy VA, Pfeuffer T, Krupinski J, Aghajanian GK, Nestler EJ. CREB (cAMP response element-binding protein) in the locus ceruleus: biochemical, physiological, and behavioral evidence for a role in opiate dependence. J Neurosci. 1997;17:7890–7901. doi: 10.1523/JNEUROSCI.17-20-07890.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CM, Javitch JA, Snyder SH. Characterization of [3H]desipramine binding associated with neuronal norepinephrine uptake sites in rat brain membranes. J Neurosci. 1982;2:1515–1525. doi: 10.1523/JNEUROSCI.02-10-01515.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao YJ, Jan YN, Jan LY. Heteromultimerization of G-protein-gated inwardly rectifying K+ channel proteins GIRK1 and GIRK2 and their altered expression in weaver brain. J Neurosci. 1996;16:7137–7150. doi: 10.1523/JNEUROSCI.16-22-07137.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C, Jan LY, Stoffel M, Malenka RC, Nicoll RA. G protein-coupled inwardly rectifying K+ channels (GIRKs) mediate postsynaptic but not presynaptic transmitter actions in hippocampal neurons. Neuron. 1997;19:687–695. doi: 10.1016/s0896-6273(00)80381-5. [DOI] [PubMed] [Google Scholar]
- Maldonado R. Participation of noradrenergic pathways in the expression of opiate withdrawal: biochemical and pharmacological evidence. Neurosci Biobehav Rev. 1997;21:91–104. doi: 10.1016/0149-7634(95)00061-5. [DOI] [PubMed] [Google Scholar]
- Maldonado R, Koob GF. Destruction of the locus coeruleus decreases physical signs of opiate withdrawal. Brain Res. 1993;605:128–138. doi: 10.1016/0006-8993(93)91364-x. [DOI] [PubMed] [Google Scholar]
- Maldonado R, Valverde O, Garbay C, Roques BP. Protein kinases in the locus coeruleus and periaqueductal gray matter are involved in the expression of opiate withdrawal. Naunyn Schmiedebergs Arch Pharmacol. 1995;352:565–575. doi: 10.1007/BF00169392. [DOI] [PubMed] [Google Scholar]
- Maldonado R, Blendy JA, Tzavara E, Gass P, Roques BP, Hanoune J, Schutz G. Reduction of morphine abstinence in mice with a mutation in the gene encoding CREB. Science. 1996;273:657–659. doi: 10.1126/science.273.5275.657. [DOI] [PubMed] [Google Scholar]
- Mamiya T, Noda Y, Ren X, Hamdy M, Furukawa S, Kameyama T, Yamada K, Nabeshima T. Involvement of cyclic AMP systems in morphine physical dependence in mice: prevention of development of morphine dependence by rolipram, a phosphodiesterase 4 inhibitor. Br J Pharmacol. 2001;132:1111–1117. doi: 10.1038/sj.bjp.0703912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marker CL, Cintora SC, Roman MI, Stoffel M, Wickman K. Hyperalgesia and blunted morphine analgesia in G protein-gated potassium channel subunit knockout mice. NeuroReport. 2002;13:2509–2513. doi: 10.1097/00001756-200212200-00026. [DOI] [PubMed] [Google Scholar]
- Marker CL, Stoffel M, Wickman K. Spinal G-protein-gated K+ channels formed by GIRK1 and GIRK2 subunits modulate thermal nociception and contribute to morphine analgesia. J Neurosci. 2004;24:2806–2812. doi: 10.1523/JNEUROSCI.5251-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marker CL, Lujan R, Colon J, Wickman K. Distinct populations of spinal cord lamina II interneurons expressing G-protein-gated potassium channels. J Neurosci. 2006;26:12251–12259. doi: 10.1523/JNEUROSCI.3693-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClung CA, Nestler EJ, Zachariou V. Regulation of gene expression by chronic morphine and morphine withdrawal in the locus ceruleus and ventral tegmental area. J Neurosci. 2005;25:6005–6015. doi: 10.1523/JNEUROSCI.0062-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RJ. Presynaptic receptors. Annu Rev Pharmacol Toxicol. 1998;38:201–227. doi: 10.1146/annurev.pharmtox.38.1.201. [DOI] [PubMed] [Google Scholar]
- Mitrovic I, Margeta-Mitrovic M, Bader S, Stoffel M, Jan LY, Basbaum AI. Contribution of GIRK2-mediated postsynaptic signaling to opiate and alpha 2-adrenergic analgesia and analgesic sex differences. Proc Natl Acad Sci USA. 2003;100:271–276. doi: 10.1073/pnas.0136822100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ. Historical review: molecular and cellular mechanisms of opiate and cocaine addiction. Trends Pharmacol Sci. 2004;25:210–218. doi: 10.1016/j.tips.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Pravetoni M, Wickman K. Behavioral characterization of mice lacking GIRK/Kir3 channel subunits. Genes Brain Behav. 2008 doi: 10.1111/j.1601-183X.2008.00388.x. in press. [DOI] [PubMed] [Google Scholar]
- Prieto M, Giralt MT. Effects of N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine (DSP4) on alpha2-adrenoceptors which regulate the synthesis and release of noradrenaline in the rat brain. Pharmacol Toxicol. 2001;88:152–158. doi: 10.1034/j.1600-0773.2001.d01-97.x. [DOI] [PubMed] [Google Scholar]
- Punch LJ, Self DW, Nestler EJ, Taylor JR. Opposite modulation of opiate withdrawal behaviors on microinfusion of a protein kinase A inhibitor versus activator into the locus ceruleus or periaqueductal gray. J Neurosci. 1997;17:8520–8527. doi: 10.1523/JNEUROSCI.17-21-08520.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Signorini S, Liao YJ, Duncan SA, Jan LY, Stoffel M. Normal cerebellar development but susceptibility to seizures in mice lacking G protein-coupled, inwardly rectifying K+ channel GIRK2. Proc Natl Acad Sci USA. 1997;94:923–927. doi: 10.1073/pnas.94.3.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrecilla M, Marker CL, Cintora SC, Stoffel M, Williams JT, Wickman K. G-protein-gated potassium channels containing Kir3.2 and Kir3.3 subunits mediate the acute inhibitory effects of opioids on locus ceruleus neurons. J Neurosci. 2002;22:4328–4334. doi: 10.1523/JNEUROSCI.22-11-04328.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickman K, Pu WT, Clapham DE. Structural characterization of the mouse Girk genes. Gene. 2002;284:241–250. doi: 10.1016/s0378-1119(01)00884-8. [DOI] [PubMed] [Google Scholar]
- Williams JT, Christie MJ, Manzoni O. Cellular and synaptic adaptations mediating opioid dependence. Physiol Rev. 2001;81:299–343. doi: 10.1152/physrev.2001.81.1.299. [DOI] [PubMed] [Google Scholar]