

Abstract
It is becoming increasingly clear that epigenetic modifications are critical factors in the regulation of gene expression. With regard to the nervous system, epigenetic alterations play a role in a diverse set of processes and have been implicated in a variety of disorders. Gaining a more complete understanding of the essential components and underlying mechanisms involved in epigenetic regulation could lead to novel treatments for a number of neurological and psychiatric conditions.
Keywords: epigenetics, chromatin, DNA methylation, histone, transcription, gene
Broadly defined, epigenetics is a type of molecular and cellular “memory” that results in stable changes in gene expression without alterations to the DNA sequence itself. It has long been appreciated that transcription is not occurring on naked DNA, but rather in the context of chromatin which requires the orchestrated effort of not only transcription factors, but also the protein complexes that modify chromatin structure. Currently, commonly studied epigenetic “marks” include DNA methylation and histone modifications, which can include methylation, acetylation, ubiquitination, and phosphorylation, as well as others. Methylation status on any given segment of DNA appears to be controlled in large part by DNA methyltransferases (Ooi and Bestor, 2008). A host of enzymes appear to regulate histone modifications including histone acetyltransferases (HATs) and histone deacetylases (HDACs) as well as methyl-transferases and demethylases (Bhaumik et al., 2007). These epigenetic marks result in alterations to the protein and/or DNA components that make up chromatin structure such that the transcriptional potential of a gene or set of genes near a specific locus is changed. Figure 1 provides an overview of chromatin structure and describes two widely studied epigenetic marks. It is becoming increasingly clear that changes in the chromatin architecture are important factors in gene regulation and understanding these molecular processes and their functional outcomes may give new insight into normal neural function and disease. With regard to brain processes, epigenetic alterations are present and appear to be playing a role in a diverse set of functions including learning and memory processes, drug addiction, neurodegeneration, and circadian rhythms. Epigenetic mechanisms have been implicated in specific human disorders including Fragile X syndrome, Rett syndrome, Huntington's disease, schizophrenia, and bipolar disorder. Understanding the molecular components and environmental conditions that cause or result in epigenetic changes may provide unique opportunities to develop novel interventions and therapies to treat a variety of neurological and psychiatric conditions.
Figure 1.
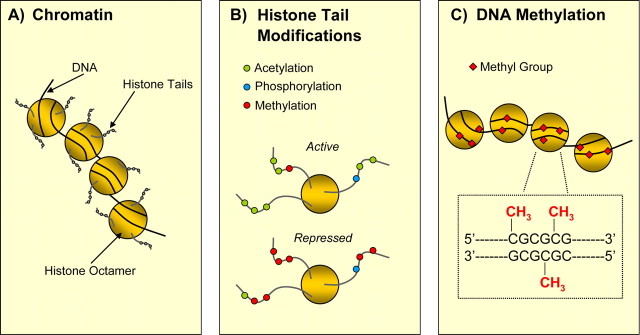
Schematic representation of epigenetic marks. A, DNA is condensed within the nucleus through interactions with histones, and this DNA–protein complex is referred to as chromatin. Two copies each of the histones H2A, H2B, H3, and H4 assemble to form a histone octamer, around which 146 bp of genomic double-stranded DNA are wrapped. B, The N-terminal tail of a histone contains many sites for epigenetic marking via histone acetylation, methylation, and phosphorylation. For example, acetylation of H3, shown as the addition of green circles to the tails, results in a relaxed chromatin state that promotes gene transcription, whereas methylation (shown via red circles) can either promote or repress gene transcription. C, Methylation of DNA is another method of epigenetic marking of the genome, where a methyl group (shown as red diamonds) is transferred to cytosines in genomic regions in and around gene promoters that are rich in cytosine-guanine nucleotides (CpG islands). While the addition of methyl groups at gene promoters is generally linked to transcriptional repression, it is noteworthy that more distal and intragenic portions of many actively transcribed genes are methylated.
The role of chromatin-modifying enzymes in learning and memory processes
The role of transcription in long-lasting forms of synaptic plasticity and memory has been actively investigated since initial experiments showing that transcription is required for long-term memory in goldfish nearly 40 years ago (Agranoff et al., 1967). Recently, it has been reported that epigenetic alterations are involved and, more specifically, it has been demonstrated that the regulation of histone acetylation by HDACs and HATs is pivotal for the transcriptional regulation required for synaptic plasticity and memory processes (for review, see Barrett and Wood, 2008).
To examine the role of HAT activity in synaptic plasticity and memory, genetically modified Cbp [cyclicAMP responsive element-binding (CREB) protein] mutant mice were used. CBP has HAT activity and is also well known as a coactivator recruited by CREB via the interaction between the Ser-133 phosphorylated kinase-inducible domain of CREB and the CBP KIX domain (a large, multihelical region that plays an essential role in binding) (Chrivia et al., 1993; Parker et al., 1996). Cbp mutant mice exhibited significant impairments in long-lasting forms of hippocampal late long-term potentiation (LTP) and long-term memory (Wood et al., 2005, 2006). In particular, it was found that CBP is required for long-term memory for contextual fear and novel object recognition (NOR). These results demonstrate that CBP and histone acetylation are necessary for transcription-dependent forms of synaptic plasticity and long-term memory.
In opposition to HAT activity, HDACs remove acetyl groups and in general are associated with transcriptional repression. Because impairments in CBP activity resulted in memory deficits, it was hypothesized that HDAC inhibition would result in memory enhancements. Indeed, inhibiting HDAC activity with Trichostatin A (TSA) facilitates synaptic plasticity, transforming a transient, transcription-independent form of LTP, into a long-lasting robust form of transcription-dependent LTP (Vecsey et al., 2007). Inhibiting HDAC activity in the hippocampus with TSA led to significant enhancements in long-term memory for contextual fear (Vecsey et al., 2007). Furthermore, using genetically modified Creb and Cbp mutant mice, HDAC inhibition facilitated synaptic plasticity was found to be dependent on CREB and CBP activity. These findings demonstrate that HDACs may potentially function as memory suppressor genes (Abel et al., 1998) and that HDAC inhibition may provide a viable therapeutic strategy. Indeed, HDAC inhibitors (HDACi) are not only currently available as a cancer therapy but are in clinical trials for several neurological disorders.
Interestingly, every genetically modified Cbp mutant mouse characterized thus far exhibits impaired long-term memory for NOR (Bourtchouladze et al., 2003; Alarcón et al., 2004; Korzus et al., 2004; Wood et al., 2006; Oliveira et al., 2007). These results suggest that long-term memory for NOR is especially sensitive to alterations in CBP activity and levels of histone acetylation. Thus, the question of whether inducing a histone hyperacetylated state, via HDAC inhibition, would enhance long-term memory for NOR was examined. Initial findings show that HDAC inhibition enhances long-term memory for NOR and that enhanced memory following inhibition is persistent across long time intervals. This is particularly interesting because one alluring aspect of examining the role of chromatin modifications in modulating transcription required for long-term memory processes is that these modifications may provide potentially stable epigenetic marks in the service of activating and/or maintaining transcriptional profiles underlying specific cellular states. Together, these findings suggest that HATs (i.e., CBP) and HDACs are pivotal for regulating chromatin-modification mechanisms required for synaptic plasticity and memory processes.
Linking epigenetic gene regulation to fear memory formation
Regulation of the brain-derived neurotrophic factor (bdnf) gene is another fascinating example of how epigenetic modifications control gene expression with measurable changes in electrophysiology and behavior. Investigations into the role of BDNF in the adult CNS have identified this protein as a major regulator of synaptic plasticity and memory formation (for review, see Bramham and Messaoudi, 2005). There are multiple bdnf mRNA transcripts that are all translated into the same BDNF protein. The bdnf gene consists of nine 5′ noncoding exons each linked to individual promoter regions and a 3′ coding exon (IX), which codes for the BDNF preprotein amino acid sequence (Liu et al., 2006; Aid et al., 2007). The activity of specific promoter regions within the bdnf gene dictates the spatial and temporal expression of specific bdnf transcript isoforms (Nanda and Mack, 1998). In addition, neuronal activity has been demonstrated to elicit promoter site-specific transcription initiation from a subset of bdnf promoters in hippocampus (Metsis et al., 1993; Lauterborn et al., 1996; Huang et al., 2002; Rattiner et al., 2004; Tsankova et al., 2004, 2006). However, the significance of this differential regulation of bdnf promoters in the adult brain is not completely clear. The literature suggests that 5′ untranslated regions (5′UTRs) associated with the transcriptional start site of specific genes can control the efficiency of translation of the encoded protein (Gromeier et al., 1999; Jopling and Willis, 2001; Pal et al., 2003; Aranda-Abreu et al., 2005). In addition, 5′ UTRs have been shown to direct the subsequent trafficking and targeting of transcripts to specific cellular compartments of the neuron (Blichenberg et al., 1999). Thus, the unique promoter regions and each noncoding exon are important regulatory sites that may serve to direct differential expression and localization of bdnf exon-specific transcripts.
Initial investigations into the transcriptional mechanisms underlying differential regulation of bdnf transcripts in the brain have focused on the role of transcription factors, such as CREB and nuclear factor-κB (NF-κB), binding to bdnf promoter regions to initiate recruitment and activation of several coactivators of transcription (Shieh and Ghosh, 1999; Lubin et al., 2007). However, recent reports have highlighted another level of transcriptional regulation of genes in the adult nervous system involving alterations in chromatin structure. With regard to bdnf expression, Bredy et al. (2007) suggest that histone modifications around specific bdnf promoters is clearly correlated with memory formation. They demonstrate that extinction of conditioned fear in mice is associated with an increase in histone H4 acetylation around the promoter of bdnf exon IV, as well as increases in bdnf exons I and IV mRNA in prefrontal cortex. Furthermore, several studies support DNA methylation as a provocative epigenetic mechanism for ongoing regulation of bdnf transcription to mediate neuronal functions (Martinowich et al., 2003; Levenson et al., 2006; Brown et al., 2008; Nelson et al., 2008). For example, a recent study examined the intriguing possibility that the adult nervous system may have co-opted DNA methylation as an epigenetic mechanism to mediate bdnf gene expression in neuronal cells during memory formation. Specifically, the study investigated whether DNA methylation regulates bdnf exon-specific transcription within hippocampus in a mammalian contextual conditioned fear memory model. Using direct molecular techniques, they report that differential bdnf DNA methylation at specific promoter and exonic regions is dynamically regulated in the adult hippocampus through NMDA receptor activation during consolidation of fear memories (Lubin et al., 2008). Together, these studies present a clearer understanding of the epigenetic regulation of the bdnf gene in several behaviors and highlight a new activity-dependent transcriptional mechanism for gene expression changes in the adult brain that will have to be incorporated in future molecular studies.
Methyl-cytosine binding protein 2 and its potential role in autism-spectrum disorder and Rett syndrome
It is becoming increasingly clear that epigenetic regulation may be a key component of certain disease processes. To date, one of the best studied disorders that involves epigenetic mechanisms is Rett syndrome, an X-linked neurodevelopmental disorder characterized by severe motor and cognitive impairment as well as autistic symptoms. The syndrome affects females primarily, with early postnatal lethality in males (Amir et al., 1999; Weaving et al., 2005; Chahrour and Zoghbi, 2007). Rett syndrome is caused by mutation in the methyl-cytosine binding protein 2 (MeCP2) gene. MeCP2 is predicted to bind to methylated CpG DNA sequences in specific promoters, which in vitro studies have shown promotes transcriptional silencing at those loci (Jones et al., 1998; Nan et al., 1998; Weaving et al., 2005; Akbarian et al., 2006; Chahrour and Zoghbi, 2007), but transcriptional profiling studies in MeCP2 mutant brain suggest a more complex effect on gene expression (Chahrour et al., 2008). Furthermore, in the first large-scale mapping study, Yasui et al. (2007) found that neuronal MeCP2 binding sites on 26.3 Mb of target imprinted and nonimprinted loci revealed that 59% of MeCP2 binding sites are outside of genes, and only 5.9% are in CpG islands. Furthermore, integrated genome-wide promoter analysis of MeCP2 binding, CpG methylation, and gene expression revealed that 66% of MeCP2-bound promoters are actively expressed and, surprisingly, only 6% are highly methylated. JUNB, an immediate early gene relevant to the pathogenesis of Rett syndrome, is one example of a highly active gene whose expression is modulated by distal and proximal binding to a partially methylated promoter (Yasui et al., 2007). Therefore, these results support a predominant role for MeCP2 as a long-range epigenetic modulator rather than a proximal silencer of gene expression.
The results are significant and represent advances in the autism and Rett syndrome fields in that they demonstrate that MeCP2 modulates the expression level of many genes and does not act exclusively as a transcriptional silencer as has been hypothesized. Furthermore, the results support the concept that epigenetic regulation is complex, and only with continued effort testing existing and forming novel hypotheses will answers become known.
Epigenetic mechanisms in drug addiction
A frontier of epigenomics where the impact is just beginning to be realized is in the pathogenesis and persistence of psychiatric disorders such as drug addiction. People who repeatedly use drugs of abuse often transition to an addicted state characterized by compulsive drug seeking and taking despite severe adverse consequences. While the precise mechanisms underlying this transition remain unclear, changes in gene expression in brain reward regions such as the nucleus accumbens (NAc) are thought to contribute to this process. Indeed, some of these gene expression changes (mediated, for example, by the transcription factors, ΔFosB or CREB) have been directly shown to alter an animal's motivation to self-administer cocaine or other drugs of abuse (Hyman et al., 2006). However, the mechanisms by which chronic drug exposure converges on the genome to alter the activity of specific genes have remained largely unknown. Recent evidence suggests that drug-induced modifications to chromatin structure may contribute to these long-lasting changes in gene expression and ultimately to addictive behaviors.
One of the best studied drug-induced changes in chromatin structure is histone acetylation, which is considered a marker of actively transcribed genes (Fig. 1). Together, several studies have implicated histone acetylation in behavioral responses to cocaine (Kumar et al., 2005; Levine et al., 2005; Renthal et al., 2007, 2008; Freeman et al., 2008) and suggest a role in modulating the saliency of drug experiences. However, these studies, for the most part, investigated the promoters of genes already known to be regulated by drugs of abuse. To gain insight into novel cocaine-induced transcriptional regulation, an unbiased, genome-wide approach coupling chromatin immunoprecipitation with promoter microarrays (ChIP–chip) was used.
Since increases in histone acetylation can both mark actively transcribed genes as well as genes which appear to be primed for later induction (Tsankova et al., 2007; Renthal and Nestler, 2008), ChIP–chip provides exciting new information about transcriptional regulation in response to drugs of abuse beyond that obtained from analyses of steady-state mRNA levels. One new mechanistic insight is that the majority of cocaine-induced histone hyperacetylation occurs on either histone H3 or H4 but only rarely on both histones at the same gene. The mechanisms which determine whether a gene is hyperacetylated at H3 or H4 remain unclear, but these new data suggest that, after chronic cocaine, there may be two distinct signaling pathways that converge on either H3 or H4 to activate gene expression.
In addition to providing novel insight into basic transcriptional mechanisms co-opted by cocaine, these data describe several new signaling pathways that are regulated by cocaine. One example is a family of protein deacetylases, the sirtuins (for review, see Michan and Sinclair, 2007), where it has been found that the promoters of both SIRT1 and SIRT2 are significantly hyperacetylated at histone H3 after chronic cocaine, which is associated with significant upregulation of both genes (W. Renthal, personal communication). This upregulation appears to be physiologically relevant, as pharmacological and genetic manipulation of SIRT1 and SIRT2 activity in the NAc potently regulates cocaine reward and self-administration behavior.
These data provide a wealth of new information about how chronic cocaine exposure alters chromatin structure throughout the genome in the NAc and, most importantly, establish a proof of principle that novel signaling molecules involved in addiction-related behaviors can be identified by studying genome-wide changes in histone acetylation.
Histone modifications and neuropsychiatric disease
Given the data emerging from epigenomic studies using genetic, cellular, and behavioral measures and in light of the role of epigenetics in disease processes, developing novel therapeutics that can ameliorate neurological and neuropsychiatric symptoms would be highly desirable. It is currently recognized that drugs acting as HDACi that presumably promote transcription of specific genes, elicit changes in animal behavior, which is often accompanied by a neurotrophic and/or neuroprotective mechanism. HDACi have shown therapeutic efficacy in many rodent models of neurodegenerative diseases, including Huntington's and other triplet repeat disorders, and also Parkinson's disease; it is now recognized that HDACi are broadly neuroprotective, preventing or delaying neuronal dysfunction and death in vitro and in vivo (Qi et al., 2004; Minamiyama et al., 2004; Camelo et al., 2005; Faraco et al., 2006; Petri et al., 2006; Avila et al., 2007; H. J. Kim et al., 2007; Fischer et al., 2007; Langley et al., 2008). These findings are very promising from a treatment-development perspective, and the underlying molecular mechanisms are thought to involve transcriptional regulation because histone acetylation is typically enriched in euchromatin at sites of active gene promoters.
From a genome-wide perspective, other types of histone modifications show a distribution that is highly complementary to most (histone) acetylation marks. For example, the di- and tri-methylated forms of histone H3-lysine 9 are typically enriched in heterochromatin and, when present at the sites of promoters, these marks are typically involved in transcriptional repression and silencing (Nakayama et al., 2001; Peters et al., 2002; Rice et al., 2003; Guenatri et al., 2004; Appanah et al., 2007; Barski et al., 2007). These mechanisms likely contribute to the neurobiology of disease. For example, dysregulation of H3-methyl-lysine 9 was reported in postmortem brain studies of subjects diagnosed with Huntington's disease (Ryu et al., 2006) or Friedreich's Ataxia (Al-Mahdawi et al., 2008), both of which are triplet repeat disorders.
If the upregulation of histone acetylation generally seems to be beneficial for neuronal functions and behaviors, then what phenotype would be expected in genetically engineered animals with neuron-specific elevations of repressive chromatin marks such as H3-tri(di-)-methyl-lysine 9? This was examined using transgenic mice overexpressing the H3K9-specific histone methyl-transferase, SET domain bifurcated 1 (Setdb1) (Schultz et al., 2002), also known as Erg-associated protein with SET domain (Yang et al., 2002). When expression in adult brain is upregulated (via transgenes expressed under control of neuron-specific promoters), levels of trimethylated H3 lysine 9 in heterochromatin surrounding pericentromeric repeat DNA became significantly elevated. Preliminary results from ongoing studies indicated that gross neurological function in these mice, as evaluated by rotarod and locomotor assays, body weight, and breeding behaviors were either normal or showed only subtle changes. However, preliminary findings suggest that Setdb1-overexpressing mice outperform their wild-type littermates in the Morris Water Maze and the Object Recognition tests, which are thought to relate to hippocampus- and cortex-related memory functions (Y. Jiang and S. Akbarian, personal communication). These findings were unexpected and suggest that therapeutic benefits in preclinical models of memory disorders are not limited to HDAC-mediated histone acetylation, but the mechanisms by which Setdb1-mediated H3-lysine 9 methylation alters neuronal function and behavior remain to be explored.
The Setdb1 histone methyltransferase is not the only histone modifying enzyme that emerges as an important regulator of neuronal chromatin in postnatal and mature brain. For example, hippocampal synaptic plasticity is altered in mice heterozygous for a truncated, lacZ knock-in allele of Mll1, the founding member of the mixed-lineage leukemia family of histone methyltransferases and essential component of a trithorax chromatin remodeling complex involved in the methylation of the H3-lysine 4 residue (S. Y. Kim et al., 2007). Interestingly, Mll1-mediated H3-lysine 4 methylation might be involved in the molecular pathology of prefrontal dysfunction of schizophrenia and is upregulated at a subset of GABAergic gene promoters in mice treated with the atypical antipsychotic, clozapine (Huang et al., 2007). The potential importance of the fine-tuning of the H3 methylation marks in brain chromatin is further underscored by the discovery that one of the X-linked mental retardation and autism genes, Jumonji AT-rich interactive domain 1C (also termed SMCX), which encodes a histone demethylase that uses H3-tri-methyl-lysine 9 as a docking site for the subsequent demethylation of H3-lysine 4 (Iwase et al., 2007; Adegbola et al., 2008). Therefore, it will be exciting to examine the dynamic interplay of multiple histone lysine methylation marks in the context of neuropsychiatric disorders, including autism, schizophrenia, and some cases of depression, that are thought to involve defects in orderly brain development and maturation.
Histone deacetylase inhibitors as broadly effective neuroprotective agents
In vitro models of neuronal death have allowed for the dissection of some of the molecular players and pathways involved HDAC inhibition mediated neuroprotection. For example, one of the many gene expression changes following HDAC inhibition is an upregulation of the cyclin-dependent kinase inhibitor p21cip1/waf1/sdi1 (p21) (Langley et al., 2008). p21 was independently identified by multiple laboratories based on its interaction with cyclin-dependent kinase (cip1), its role as a p53-inducible protein that can inhibit proliferation (waf1), and its expression in senescent cells (sdi1). Despite knock-out studies suggesting that p21 is not essential for development (Nakayama and Nakayama, 1998), p21 appears to play an essential role in cellular protection in response to a range of stresses (Besson et al., 2008). Investigations into the role p21 plays downstream of HDAC inhibition have revealed that transgenic overexpression of p21 in neurons can mimic the protective effects of HDAC inhibition against both oxidative stress-induced and DNA damage-induced death (Langley et al., 2008). Despite this, p21-deletion studies show that p21 is not strictly necessary for HDAC inhibitor mediated protection against oxidative stress and is only necessary for a portion of HDAC inhibitor mediated protection against DNA damage (Langley et al., 2008). Considering the number of putatively protective genes and pathways induced by HDAC inhibition [for example, p21 (Langley et al., 2008), gelsolin (Meisel et al., 2006), HSP70 (Ren et al., 2004), and peroxiredoxin-1 (Hoshino et al., 2005)], these findings are not surprising. Indeed, it is likely that the ability of HDAC inhibition to induce a number, or “cassette,” of protective genes and pathways is in part responsible for the broad neuroprotection observed in vivo.
Though some traction has been gained with regard to understanding HDAC inhibitor-mediated neuroprotection, a number of critical questions still remain to be resolved including the roles of individual HDAC isoforms as well as which HDACs are bona fide targets for neuroprotection. Another question is whether or not epigenetic modulation of histones per se is even required for neuroprotection. Indeed, despite being called “histone deacetylases,” nonhistone proteins are also substrates for the activities of some HDACs. Some of these include transcription factors, e.g., p53, Sp1, GATA-2, and NF-κB; cytoskeletal proteins, e.g., tubulin; molecular chaperones, e.g., HSP90; and nuclear import factors (Glozak et al., 2005). However, unlike histone modification, the roles these acetylated/deacetylated proteins play in neuroprotection are largely unknown.
Discussion
Epigenetics is a relatively new frontier in neuroscience, especially with regard to regulation of gene expression in the brain. There is still much to learn about how changes in chromatin architecture affect transcriptional regulation and what genes are targeted for regulation. Important questions remain with regard to how environmental conditions impact the epigenome of an organism as well as questions addressing developmental and aging effects. Another intriguing concept is how epigenetic alterations can be modulated to induce long-lasting behavioral changes due to their potential stability; this opens up interesting opportunities regarding intervention therapies. In addition, although progress is being made (Jiang et al., 2008), the field needs to address some important methodological issues because most chromatin and DNA methylation assays lack single cell resolution. These and other technologic improvements should provide unique opportunities to study epigenetic changes in postmitotic neurons across the full life span of the animal, or in humans (Siegmund et al., 2007). Clearly, one should expect that epigenetics-focused research will provide fertile ground for understanding aspects of brain function and behavior, as well as potentially presenting opportunities to develop novel therapies for a wide range of neurological and psychiatric diseases.
Footnotes
This work was supported by the National Institute of Child Health and Human Development, the National Institute on Drug Abuse, the National Institute of Mental Health, the National Institute of Neurological Disorders and Stroke, the Epilepsy Foundation, the Evelyn F. McKnight Brain Research Foundation (F.D.L.), University of Texas Southwestern Medical Scientist Training Program (W.R.), the Whitehall Foundation (M.A.W.), the Adelson Program for Neural Repair and Rehabilitation, and the New York State Spinal Cord Injury Research Board CoRE (B.L.). We would like to recognize and apologize to colleagues whose work could not be acknowledged due to space limitations. We thank Michele Pearson for her assistance preparing this manuscript and Tania Roth for additional helpful input.
References
- Abel T, Kandel E. Positive and negative regulatory mechanisms that mediate long-term memory storage. Brain Res Brain Res Rev. 1998;26:360–378. doi: 10.1016/s0165-0173(97)00050-7. [DOI] [PubMed] [Google Scholar]
- Adegbola A, Gao H, Sommer S, Browning M. A novel mutation in JARID1C/SMCX in a patient with autism spectrum disorder (ASD) Am J Med Genet A. 2008;146A:505–511. doi: 10.1002/ajmg.a.32142. [DOI] [PubMed] [Google Scholar]
- Agranoff BW, Davis RE, Casola L, Lim R. Actinomycin D blocks formation of memory of shock-avoidance in goldfish. Science. 1967;158:1600–1601. doi: 10.1126/science.158.3808.1600. [DOI] [PubMed] [Google Scholar]
- Aid T, Kazantseva A, Piirsoo M, Palm K, Timmusk T. Mouse and rat BDNF gene structure and expression revisited. J Neurosci Res. 2007;85:525–535. doi: 10.1002/jnr.21139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbarian S, Jiang Y, Laforet G. The molecular pathology of Rett syndrome: synopsis and update. Neuromolecular Med. 2006;8:485–494. doi: 10.1385/NMM:8:4:485. [DOI] [PubMed] [Google Scholar]
- Alarcón JM, Malleret G, Touzani K, Vronskaya S, Ishii S, Kandel ER, Barco A. Chromatin acetylation, memory, and LTP are impaired in CBP+/- mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron. 2004;42:947–959. doi: 10.1016/j.neuron.2004.05.021. [DOI] [PubMed] [Google Scholar]
- Al-Mahdawi S, Pinto RM, Ismail O, Varshney D, Lymperi S, Sandi C, Trabzuni D, Pook M. The Friedreich ataxia GAA repeat expansion mutation induces comparable epigenetic changes in human and transgenic mouse brain and heart tissues. Hum Mol Genet. 2008;17:735–746. doi: 10.1093/hmg/ddm346. [DOI] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Appanah R, Dickerson DR, Goyal P, Groudine M, Lorincz MC. An unmethylated 3′ promoter-proximal region is required for efficient transcription initiation. PLoS Genet. 2007;3 doi: 10.1371/journal.pgen.0030027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranda-Abreu GE, Hernández ME, Soto A, Manzo J. Possible Cis-acting signal that could be involved in the localization of different mRNAs in neuronal axons. Theor Biol Med Model. 2005;2:33. doi: 10.1186/1742-4682-2-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avila AM, Burnett BG, Taye AA, Gabanella F, Knight MA, Hartenstein P, Cizman Z, Di Prospero NA, Pellizzoni L, Fischbeck KH, Sumner CJ. Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy. J Clin Invest. 2007;117:659–671. doi: 10.1172/JCI29562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett RM, Wood MA. Beyond transcription factors: the role of chromatin modifying enzymes in regulating transcription required for memory. Learn Mem. 2008;15:460–467. doi: 10.1101/lm.917508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Besson A, Dowdy SF, Roberts JM. CDK inhibitors: cell cycle regulators and beyond. Dev Cell. 2008;14:159–169. doi: 10.1016/j.devcel.2008.01.013. [DOI] [PubMed] [Google Scholar]
- Bhaumik SR, Smith E, Shilatifard A. Covalent modifications of histones during development and disease pathogenesis. Nat Struct Mol Biol. 2007;14:1008–1016. doi: 10.1038/nsmb1337. [DOI] [PubMed] [Google Scholar]
- Blichenberg A, Schwanke B, Rehbein M, Garner CC, Richter D, Kindler S. Identification of a cis-acting dendritic targeting element in MAP2 mRNAs. J Neurosci. 1999;19:8818–8829. doi: 10.1523/JNEUROSCI.19-20-08818.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourtchouladze R, Lidge R, Catapano R, Stanley J, Gossweiler S, Romashko D, Scott R, Tully T. A mouse model of Rubinstein-Taybi syndrome: defective long-term memory is ameliorated by inhibitors of phosphodiesterase 4. Proc Natl Acad Sci U S A. 2003;100:10518–10522. doi: 10.1073/pnas.1834280100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramham CR, Messaoudi E. BDNF function in adult synaptic plasticity: the synaptic consolidation hypothesis. Prog Neurobiol. 2005;76:99–125. doi: 10.1016/j.pneurobio.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Bredy TW, Wu H, Crego C, Zellhoefer J, Sun YE, Barad M. Histone modifications around individual BDNF gene promoters in prefrontal cortex are associated with extinction of conditioned fear. Learn Mem. 2007;14:268–276. doi: 10.1101/lm.500907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SE, Weaver IC, Meaney MJ, Szyf M. Regional-specific global cytosine methylation and DNA methyltransferase expression in the adult rat hippocampus. Neurosci Lett. 2008;440:49–53. doi: 10.1016/j.neulet.2008.05.028. [DOI] [PubMed] [Google Scholar]
- Camelo S, Iglesias AH, Hwang D, Due B, Ryu H, Smith K, Gray SG, Imitola J, Duran G, Assaf B, Langley B, Khoury SJ, Stephanopoulos G, De Girolami U, Ratan RR, Ferrante RJ, Dangond F. Transcriptional therapy with the histone deacetylase inhibitor trichostatin A ameliorates experimental autoimmune encephalomyelitis. J Neuroimmunol. 2005;164:10–21. doi: 10.1016/j.jneuroim.2005.02.022. [DOI] [PubMed] [Google Scholar]
- Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56:422–437. doi: 10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrivia JC, Kwok RP, Lamb N, Hagiwara M, Montminy MR, Goodman RH. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature. 1993;365:855–859. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- Faraco G, Pancani T, Formentini L, Mascagni P, Fossati G, Leoni F, Moroni F, Chiarugi A. Pharmacological inhibition of histone deacetylases by suberoylanilide hydroxamic acid specifically alters gene expression and reduces ischemic injury in the mouse brain. Mol Pharmacol. 2006;70:1876–1884. doi: 10.1124/mol.106.027912. [DOI] [PubMed] [Google Scholar]
- Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai LH. Recovery of learning and memory is associated with chromatin remodelling. Nature. 2007;447:178–182. doi: 10.1038/nature05772. [DOI] [PubMed] [Google Scholar]
- Freeman WM, Patel KM, Brucklacher RM, Lull ME, Erwin M, Morgan D, Roberts DC, Vrana KE. Persistent alterations in mesolimbic gene expression with abstinence from cocaine self-administration. Neuropsychopharmacology. 2008;33:1807–1817. doi: 10.1038/sj.npp.1301577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glozak MA, Sengupta N, Zhang X, Seto E. Acetylation and deacetylation of non-histone proteins. Gene. 2005;363:15–23. doi: 10.1016/j.gene.2005.09.010. [DOI] [PubMed] [Google Scholar]
- Gromeier M, Bossert B, Arita M, Nomoto A, Wimmer E. Dual stem loops within the poliovirus internal ribosomal entry site control neurovirulence. J Virol. 1999;73:958–964. doi: 10.1128/jvi.73.2.958-964.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenatri M, Bailly D, Maison C, Almouzni G. Mouse centric and pericentric satellite repeats form distinct functional heterochromatin. J Cell Biol. 2004;166:493–505. doi: 10.1083/jcb.200403109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino I, Matsubara H, Hanari N, Mori M, Nishimori T, Yoneyama Y, Akutsu Y, Sakata H, Matsushita K, Seki N, Ochiai T. Histone deacetylase inhibitor FK228 activates tumor suppressor Prdx1 with apoptosis induction in esophageal cancer cells. Clin Cancer Res. 2005;11:7945–7952. doi: 10.1158/1078-0432.CCR-05-0840. [DOI] [PubMed] [Google Scholar]
- Huang HS, Matevossian A, Whittle C, Kim SY, Schumacher A, Baker SP, Akbarian S. Prefrontal dysfunction in schizophrenia involves mixed-lineage leukemia 1-regulated histone methylation at GABAergic gene promoters. J Neurosci. 2007;27:11254–11262. doi: 10.1523/JNEUROSCI.3272-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Doherty JJ, Dingledine R. Altered histone acetylation at glutamate receptor 2 and brain-derived neurotrophic factor genes is an early event triggered by status epilepticus. J Neurosci. 2002;22:8422–8428. doi: 10.1523/JNEUROSCI.22-19-08422.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- Iwase S, Lan F, Bayliss P, de la Torre-Ubieta L, Huarte M, Qi HH, Whetstine JR, Bonni A, Roberts TM, Shi Y. The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell. 2007;128:1077–1088. doi: 10.1016/j.cell.2007.02.017. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Matevossian A, Huang HS, Straubhaar J, Akbarian S. Isolation of neuronal chromatin from brain tissue. BMC Neurosci. 2008;9:42. doi: 10.1186/1471-2202-9-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;19:187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- Jopling CL, Willis AE. N-myc translation is initiated via an internal ribosome entry segment that displays enhanced activity in neuronal cells. Oncogene. 2001;20:2664–2670. doi: 10.1038/sj.onc.1204404. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Rowe M, Ren M, Hong JS, Chen PS, Chuang DM. Histone deacetylase inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: multiple mechanisms of action. J Pharmacol Exp Ther. 2007;321:892–901. doi: 10.1124/jpet.107.120188. [DOI] [PubMed] [Google Scholar]
- Kim SY, Levenson JM, Korsmeyer S, Sweatt JD, Schumacher A. Developmental regulation of Eed complex composition governs a switch in global histone modification in brain. J Biol Chem. 2007;282:9962–9972. doi: 10.1074/jbc.M608722200. [DOI] [PubMed] [Google Scholar]
- Korzus E, Rosenfeld MG, Mayford M. CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron. 2004;42:961–972. doi: 10.1016/j.neuron.2004.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Choi KH, Renthal W, Tsankova NM, Theobald DE, Truong HT, Russo SJ, Laplant Q, Sasaki TS, Whistler KN, Neve RL, Self DW, Nestler EJ. Chromatin remodeling is a key mechanism underlying cocaine-induced plasticity in striatum. Neuron. 2005;48:303–314. doi: 10.1016/j.neuron.2005.09.023. [DOI] [PubMed] [Google Scholar]
- Langley B, D'Annibale MA, Suh K, Ayoub I, Tolhurst A, Bastan B, Yang L, Ko B, Fisher M, Cho S, Beal MF, Ratan RR. Pulse inhibition of histone deacetylases induces complete resistance to oxidative death in cortical neurons without toxicity and reveals a role for cytoplasmic p21(waf1/cip1) in cell cycle-independent neuroprotection. J Neurosci. 2008;28:163–176. doi: 10.1523/JNEUROSCI.3200-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauterborn JC, Rivera S, Stinis CT, Hayes VY, Isackson PJ, Gall CM. Differential effects of protein synthesis inhibition on the activity-dependent expression of BDNF transcripts: evidence for immediate-early gene responses from specific promoters. J Neurosci. 1996;16:7428–7436. doi: 10.1523/JNEUROSCI.16-23-07428.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenson JM, Roth TL, Lubin FD, Miller CA, Huang IC, Desai P, Malone LM, Sweatt JD. Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J Biol Chem. 2006;281:15763–15773. doi: 10.1074/jbc.M511767200. [DOI] [PubMed] [Google Scholar]
- Levine AA, Guan Z, Barco A, Xu S, Kandel ER, Schwartz JH. CREB-binding protein controls response to cocaine by acetylating histones at the fosB promoter in the mouse striatum. Proc Natl Acad Sci U S A. 2005;102:19186–19191. doi: 10.1073/pnas.0509735102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu QR, Lu L, Zhu XG, Gong JP, Shaham Y, Uhl GR. Rodent BDNF genes, novel promoters, novel splice variants, and regulation by cocaine. Brain Res. 2006;1067:1–12. doi: 10.1016/j.brainres.2005.10.004. [DOI] [PubMed] [Google Scholar]
- Lubin FD, Ren Y, Xu X, Anderson AE. Nuclear factor-kappaB regulates seizure threshold and gene transcription following convulsant stimulation. J Neurochem. 2007;103:1381–1395. doi: 10.1111/j.1471-4159.2007.04863.x. [DOI] [PubMed] [Google Scholar]
- Lubin FD, Roth TL, Sweatt JD. Epigenetic regulation of BDNF gene transcription in the consolidation of fear memory. J Neurosci. 2008;28:10576–10586. doi: 10.1523/JNEUROSCI.1786-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, Fan G, Sun YE. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- Meisel A, Harms C, Yildirim F, Bösel J, Kronenberg G, Harms U, Fink KB, Endres M. Inhibition of histone deacetylation protects wild-type but not gelsolin-deficient neurons from oxygen/glucose deprivation. J Neurochem. 2006;98:1019–1031. doi: 10.1111/j.1471-4159.2006.04016.x. [DOI] [PubMed] [Google Scholar]
- Metsis M, Timmusk T, Arenas E, Persson H. Differential usage of multiple brain-derived neurotrophic factor promoters in the rat brain following neuronal activation. Proc Natl Acad Sci U S A. 1993;90:8802–8806. doi: 10.1073/pnas.90.19.8802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michan S, Sinclair D. Sirtuins in mammals: insights into their biological function. Biochem J. 2007;404:1–13. doi: 10.1042/BJ20070140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamiyama M, Katsuno M, Adachi H, Waza M, Sang C, Kobayashi Y, Tanaka F, Doyu M, Inukai A, Sobue G. Sodium butyrate ameliorates phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Hum Mol Genet. 2004;13:1183–1192. doi: 10.1093/hmg/ddh131. [DOI] [PubMed] [Google Scholar]
- Nakayama J, Rice JC, Strahl BD, Allis CD, Grewal SI. Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science. 2001;292:110–113. doi: 10.1126/science.1060118. [DOI] [PubMed] [Google Scholar]
- Nakayama K, Nakayama K. Cip/Kip cyclin-dependent kinase inhibitors: brakes of the cell cycle engine during development. Bioessays. 1998;20:1020–1029. doi: 10.1002/(SICI)1521-1878(199812)20:12<1020::AID-BIES8>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Nan X, Cross S, Bird A. Gene silencing by methyl-CpG-binding proteins. Novartis Found Symp. 1998;214:6–16. doi: 10.1002/9780470515501.ch2. discussion 16–21, 46–50. [DOI] [PubMed] [Google Scholar]
- Nanda S, Mack KJ. Multiple promoters direct stimulus and temporal specific expression of brain-derived neurotrophic factor in the somatosensory cortex. Brain Res Mol Brain Res. 1998;62:216–219. doi: 10.1016/s0169-328x(98)00242-3. [DOI] [PubMed] [Google Scholar]
- Nelson ED, Kavalali ET, Monteggia LM. Activity-dependent suppression of miniature neurotransmission through the regulation of DNA methylation. J Neurosci. 2008;28:395–406. doi: 10.1523/JNEUROSCI.3796-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira AM, Wood MA, McDonough CB, Abel T. Transgenic mice expressing an inhibitory truncated form of p300 exhibit long-term memory deficits. Learn Mem. 2007;14:564–572. doi: 10.1101/lm.656907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooi SK, Bestor TH. The colorful history of active DNA demethylation. Cell. 2008;133:1145–1148. doi: 10.1016/j.cell.2008.06.009. [DOI] [PubMed] [Google Scholar]
- Pal R, Agbas A, Bao X, Hui D, Leary C, Hunt J, Naniwadekar A, Michaelis ML, Kumar KN, Michaelis EK. Selective dendrite-targeting of mRNAs of NR1 splice variants without exon 5: identification of a cis-acting sequence and isolation of sequence-binding proteins. Brain Res. 2003;994:1–18. doi: 10.1016/j.brainres.2003.08.046. [DOI] [PubMed] [Google Scholar]
- Parker D, Ferreri K, Nakajima T, LaMorte VJ, Evans R, Koerber SC, Hoeger C, Montminy MR. Phosphorylation of CREB at Ser-133 induces complex formation with CREB-binding protein via a direct mechanism. Mol Cell Biol. 1996;16:694–703. doi: 10.1128/mcb.16.2.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters AH, Mermoud JE, O'Carroll D, Pagani M, Schweizer D, Brockdorff N, Jenuwein T. Histone H3 lysine 9 methylation is an epigenetic imprint of facultative heterochromatin. Nat Genet. 2002;30:77–80. doi: 10.1038/ng789. [DOI] [PubMed] [Google Scholar]
- Petri S, Kiaei M, Kipiani K, Chen J, Calingasan NY, Crow JP, Beal MF. Additive neuroprotective effects of a histone deacetylase inhibitor and a catalytic antioxidant in a transgenic mouse model of amyotrophic lateral sclerosis. Neurobiol Dis. 2006;22:40–49. doi: 10.1016/j.nbd.2005.09.013. [DOI] [PubMed] [Google Scholar]
- Qi X, Hosoi T, Okuma Y, Kaneko M, Nomura Y. Sodium 4-phenylbutyrate protects against cerebral ischemic injury. Mol Pharmacol. 2004;66:899–908. doi: 10.1124/mol.104.001339. [DOI] [PubMed] [Google Scholar]
- Rattiner LM, Davis M, Ressler KJ. Differential regulation of brain-derived neurotrophic factor transcripts during the consolidation of fear learning. Learn Mem. 2004;11:727–731. doi: 10.1101/lm.83304. [DOI] [PubMed] [Google Scholar]
- Ren M, Leng Y, Jeong M, Leeds PR, Chuang DM. Valproic acid reduces brain damage induced by transient focal cerebral ischemia in rats: potential roles of histone deacetylase inhibition and heat shock protein induction. J Neurochem. 2004;89:1358–1367. doi: 10.1111/j.1471-4159.2004.02406.x. [DOI] [PubMed] [Google Scholar]
- Renthal W, Nestler EJ. Epigenetic mechanisms in drug addiction. Trends Mol Med. 2008;14:341–350. doi: 10.1016/j.molmed.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renthal W, Maze I, Krishnan V, Covington HE, 3rd, Xiao G, Kumar A, Russo SJ, Graham A, Tsankova N, Kippin TE, Kerstetter KA, Neve RL, Haggarty SJ, McKinsey TA, Bassel-Duby R, Olson EN, Nestler EJ. Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron. 2007;56:517–529. doi: 10.1016/j.neuron.2007.09.032. [DOI] [PubMed] [Google Scholar]
- Renthal W, Carle TL, Maze I, Covington HE, 3rd, Truong HT, Alibhai I, Kumar A, Montgomery RL, Olson EN, Nestler EJ. DeltaFosB mediates epigenetic desensitization of the c-fos gene after chronic amphetamine exposure. J Neurosci. 2008;28:7344–7349. doi: 10.1523/JNEUROSCI.1043-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice JC, Briggs SD, Ueberheide B, Barber CM, Shabanowitz J, Hunt DF, Shinkai Y, Allis CD. Histone methyltransferases direct different degrees of methylation to define distinct chromatin domains. Mol Cell. 2003;12:1591–1598. doi: 10.1016/s1097-2765(03)00479-9. [DOI] [PubMed] [Google Scholar]
- Ryu H, Lee J, Hagerty SW, Soh BY, McAlpin SE, Cormier KA, Smith KM, Ferrante RJ. ESET/SETDB1 gene expression and histone H3 (K9) trimethylation in Huntington's disease. Proc Natl Acad Sci U S A. 2006;103:19176–19181. doi: 10.1073/pnas.0606373103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz DC, Ayyanathan K, Negorev D, Maul GG, Rauscher FJ., 3rd SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002;16:919–932. doi: 10.1101/gad.973302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh PB, Ghosh A. Molecular mechanisms underlying activity-dependent regulation of BDNF expression. J Neurobiol. 1999;41:127–134. [PubMed] [Google Scholar]
- Siegmund KD, Connor CM, Campan M, Long TI, Weisenberger DJ, Biniszkiewicz D, Jaenisch R, Laird PW, Akbarian S. DNA methylation in the human cerebral cortex is dynamically regulated throughout the life span and involves differentiated neurons. PLoS ONE. 2007;2:e895. doi: 10.1371/journal.pone.0000895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsankova NM, Kumar A, Nestler EJ. Histone modifications at gene promoter regions in rat hippocampus after acute and chronic electroconvulsive seizures. J Neurosci. 2004;24:5603–5610. doi: 10.1523/JNEUROSCI.0589-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci. 2006;9:519–525. doi: 10.1038/nn1659. [DOI] [PubMed] [Google Scholar]
- Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci. 2007;8:355–367. doi: 10.1038/nrn2132. [DOI] [PubMed] [Google Scholar]
- Vecsey CG, Hawk JD, Lattal KM, Stein JM, Fabian SA, Attner MA, Cabrera SM, McDonough CB, Brindle PK, Abel T, Wood MA. Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP-dependent transcriptional activation. J Neurosci. 2007;27:6128–6140. doi: 10.1523/JNEUROSCI.0296-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaving LS, Ellaway CJ, Gecz J, Christodoulou J. Rett syndrome: clinical review and genetic update. J Med Genet. 2005;42:1–7. doi: 10.1136/jmg.2004.027730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood MA, Kaplan MP, Park A, Blanchard EJ, Oliveira AM, Lombardi TL, Abel T. Transgenic mice expressing a truncated form of CREB-binding protein (CBP) exhibit deficits in hippocampal synaptic plasticity and memory storage. Learn Mem. 2005;12:111–119. doi: 10.1101/lm.86605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood MA, Attner MA, Oliveira AM, Brindle PK, Abel T. A transcription factor-binding domain of the coactivator CBP is essential for long-term memory and the expression of specific target genes. Learn Mem. 2006;13:609–617. doi: 10.1101/lm.213906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Xia L, Wu DY, Wang H, Chansky HA, Schubach WH, Hickstein DD, Zhang Y. Molecular cloning of ESET, a novel histone H3-specific methyltransferase that interacts with ERG transcription factor. Oncogene. 2002;21:148–152. doi: 10.1038/sj.onc.1204998. [DOI] [PubMed] [Google Scholar]
- Yasui DH, Peddada S, Bieda MC, Vallero RO, Hogart A, Nagarajan RP, Thatcher KN, Farnham PJ, Lasalle JM. Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes. Proc Natl Acad Sci U S A. 2007;104:19416–19421. doi: 10.1073/pnas.0707442104. [DOI] [PMC free article] [PubMed] [Google Scholar]