

Abstract
Nerve injuries often lead to neuropathic pain syndrome. The mechanisms contributing to this syndrome involve local inflammatory responses, activation of glia cells, and changes in the plasticity of neuronal nociceptive pathways. Cannabinoid CB2 receptors contribute to the local containment of neuropathic pain by modulating glial activation in response to nerve injury. Thus, neuropathic pain spreads in mice lacking CB2 receptors beyond the site of nerve injury. To further investigate the mechanisms leading to the enhanced manifestation of neuropathic pain, we have established expression profiles of spinal cord tissues from wild-type and CB2-deficient mice after nerve injury. An enhanced interferon-γ (IFN-γ) response was revealed in the absence of CB2 signaling. Immunofluorescence stainings demonstrated an IFN-γ production by astrocytes and neurons ispilateral to the nerve injury in wild-type animals. In contrast, CB2-deficient mice showed neuronal and astrocytic IFN-γ immunoreactivity also in the contralateral region, thus matching the pattern of nociceptive hypersensitivity in these animals. Experiments in BV-2 microglia cells revealed that transcriptional changes induced by IFN-γ in two key elements for neuropathic pain development, iNOS (inducible nitric oxide synthase) and CCR2, are modulated by CB2 receptor signaling. The most direct support for a functional involvement of IFN-γ as a mediator of CB2 signaling was obtained with a double knock-out mouse strain deficient in CB2 receptors and IFN-γ. These animals no longer show the enhanced manifestations of neuropathic pain observed in CB2 knock-outs. These data clearly demonstrate that the CB2 receptor-mediated control of neuropathic pain is IFN-γ dependent.
Keywords: interferon-γ, CB2 cannabinoid receptor, microglia, astrocytes, neuropathic pain, cytokine
Introduction
Neuropathic pain refers to pain or increased pain sensitivity caused by nerve injuries. There is now accumulating evidence that inflammatory processes at the site of the nerve injury, the dorsal root ganglia, and the spinal cord projection area contribute to the complex neuropathic pain pathology. Thus, nerve damage stimulates peripheral immune cells including neutrophil granulocytes, macrophages, mast cells, and T-lymphocytes followed by the activation of spinal cord microglia and astrocytes (Scholz and Woolf, 2007). The immune response is orchestrated by inflammatory mediators including cytokines, chemokines, ATP, neuropeptides, prostaglandins, and endocannabinoids (DeLeo and Yezierski, 2001; Clark et al., 2007). Endocannabinoid signaling through CB2 receptors (Zhang et al., 2003) attenuates microglial activation and controls the regional restriction of the neuropathic pain (Ehrhart et al., 2005; Romero-Sandoval and Eisenach, 2007). On sciatic nerve ligation, mice lacking CB2 receptors show an enhanced microglia response (Racz et al., 2008). Thus, activated microglia are found not only in the spinal cord region ipsilateral to the peripheral nerve injury as observed in wild-type controls, but rather throughout the spinal cord. The enhanced microglia response is accompanied by a more widespread neuropathic pain response with enhanced pain sensitivity in the ipsilateral and contralateral sites. Conversely, overexpression of CB2 receptors in neurons and microglia resulted in a significant reduction of neuropathic pain responses. To further elucidate the cellular and molecular mechanism involved in the enhanced neuropathic pain response in the absence of CB2 receptor signaling, we have now established expression profiles from the affected spinal cord tissues. Our analysis provides strong evidence for an involvement of interferon-γ (IFN-γ) as a mediator of CB2 signaling in neuropathic pain.
Materials and Methods
Animal experimental conditions.
Animals were housed in groups of three to five and had ad libitum access to water and food. The housing conditions were maintained at 21 ± 1°C and 55 ± 10% relative humidity in a controlled light/dark cycle (light on between 8:00 A.M. and 8:00 P.M.). All experimental procedures and animal husbandry were conducted according to standard ethical guidelines (European Community Guidelines on the Care and Use of Laboratory Animals 86/609/EEC) and approved by the local ethical committee (Comité Etico Experimental Animal–Instituto Municipal de Asistencia Sanitaria/Universitat Pompeu Fabra, Bezirksregierung Köln). All experiments were performed under blind conditions.
Behavioral experiments.
Hyperalgesia to noxious thermal stimulus (plantar test) and allodynia to mechanical stimuli (von Frey stimulation model) were used as outcome measures of neuropathic pain. In the plantar test, the mean paw withdrawal latencies for the ipsilateral and contralateral hindpaws were determined from the average of three separate trials, taken at 5 min intervals to prevent thermal sensitization and behavioral disturbances using a commercially available apparatus (Ugo Basile Biological Research Apparatus) (Hargreaves et al., 1988). The von Frey filament stimulation experiments were conducted with a Dynamic Aesthesiometer (Ugo Basile Biological Research Apparatus). Clear paw withdrawal, shaking, or licking was considered to be a nociceptive-like response (Chaplan et al., 1994).
IFN-γ−/− (eight males for nerve injury plus six males for sham) and double IFN-γ−/−/CB2 −/− (seven males for nerve injury plus three males for sham) animals were used. Mice were first habituated for 1 h to each different experimental test once daily during 4 d. After the habituation period, baseline responses were established during 2 consecutive days for each paradigm in the following sequence: von Frey model and plantar test (30 min later). All the behavioral tests were performed in the same group of animals. One day after baseline measurements, sciatic nerve injury was induced. A partial ligation of the sciatic nerve at midthigh level just proximal to the trifurcation was performed with one thigh ligature using a 9-0 silk thread to induce neuropathic pain, as previously described (Malmberg and Basbaum, 1998). IFN-γ−/− and IFN-γ−/−/CB2 −/− mice were tested in each paradigm on days 3, 6, 8, 10, and 15 after the surgical procedure using the same sequence as used for baseline responses. Data were compared each experimental day by using a two-way ANOVA (surgery and genotype as between-group factors) followed by corresponding one-way ANOVA when appropriate.
Microarray experiments.
Male and female CB2 knock-out (CB2 −/−) mice and wild-type littermates (CB2 +/+) on a C57BL/6J congenic background (Buckley et al., 2000) were used. At the end of the behavioral experiments (Racz et al., 2008), the mice were killed and the lumbar section of the spinal cords was rapidly dissected and fresh-frozen in liquid nitrogen. Tissue was stored at −80°C until RNA isolation. The samples were analyzed by MG_430 2.0 Affymetrix GeneChips in single array. Total RNA was extracted from spinal cord after mechanical homogenization using TRIzol (Invitrogen). mRNA was purified using RNeasy Mini kit (QIAGEN) according to the manufacturer's instructions. The tissue samples from nine animals were pooled. Samples yielded ∼20 μg of total RNA. RNA integrity was assessed using Agilent Bioanalyzer 2100 (Agilent). Five micrograms of total RNA was converted into double-stranded cDNA using T7-oligodT coupled primers per manufacturer's instructions. Ten micrograms of all fragmented cDNA samples were hybridized to MG_430 2.0 Affymetrix GeneChips following the manufacturer's instructions. All samples reported a glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and β-actin average 3′-to-5′ ratio >0.9 on the GeneChips, respectively. All mice reported a comparable number of genes expressed (58.1% in treated and 57.4% in the control groups). Microarray expression values were generated using MAS5 software “ArrayAssist 3.0” (Stratagene). The MAS5-obtained log2-transformed expression values were compared between tissues from sham-operated and sciatic nerve-ligated animals. A total of 490 transcripts were found to be differentially regulated by the nerve ligation at a level of p < 0.05. We further eliminated all transcripts that were upregulated or downregulated by more than twofold in two or more sham-operated tissues, and, furthermore, all those transcripts with absolute expression values <20. This selection yielded 219 transcripts for additional analysis.
Bioinformatics analysis of the promoter regions of the differentially regulated genes reported in the microarray study.
Promoter regions spanning from −600 to +100 bp relative to the transcription start site were downloaded from public domain at University of California Santa Cruz (mouse genome assembly mm9). Duplicated RefSeq identifiers and overlapped regions were discarded to generate a nonredundant sequence dataset. Matches to putative regulatory motifs were obtained with 126 JASPAR transcription factor matrices at 0.90 similarity threshold (Sandelin et al., 2004). Motif overrepresentation was evaluated using 10,000 randomly sampled datasets of the same size taken from the rest of promoter sequences. Empirical p value as (r + 1)/(n − 1) were estimated from occurrences distribution for each specific matrix on randomly sampled datasets. In addition, random expectation (RE) was calculated from 100 datasets of 18,983 synthetic promoters. Each synthetic promoter sequence was generated using order-1 Markov chains from GpC, GC-rich or AT-rich regions obtained on nonredundant and nonoverlapping real promoters (Bellora et al., 2007). RE provides information on the noise of the motif prediction (a high number of hits usually indicates poor matrix quality) and on relative enrichment (observed/expected). A value of p < 0.01 was considered to be significant for all the analyses and motifs with enrichments <2× were excluded.
Validation procedure.
Igtp, Tgtp, and Gbp2 mRNA were quantified by RT-PCR using gene expression assays (Applied Biosystems: Igtp, Mm00497611_m1; Tgtp, Mm00786926_s1; Gbp2, Mm00494575_m1). The real-time PCR amplifications were performed with the Taqman 7900HT from Applied Biosystems in 20 μl reaction volumes. Thermal cycling proceeded with one amplification cycle of denaturation at 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. Results were expressed as the relative amount of Igtp, Tgtp, and Gbp2 normalized by β-actin expression. All reactions were performed in triplicate, and the data were analyzed with SDS analyzing software (Applied Biosystems; SDS 2.2.2) as described previously (Livak and Schmittgen, 2001).
Double immunofluorescence staining.
Five mice per group were anesthetized with ketamine/xylacin (50/10 mg/kg) and then intracardially perfused with heparinized phosphate buffer, followed by 4% paraformaldehyde. The lumbar region of the spinal cord was removed and cryopreserved in 30% sucrose solution at 4°C. The section from L2 to L6–S1 of the spinal cord was selected and then embedded in OCT, sliced in 25 μm sections on a cryostat, and six sections per mice and two mice per slide mounted in Star frost-coated slides.
The slides were incubated in anti iba-1 antibody (1:200; Wako Chemicals) for microglia staining or polyclonal GFAP antibody (1:1000; Dako) for astrocyte staining and monoclonal anti-IFN-γ antibody (1:150; Millipore Bioscience Research Reagents) at the same time at 4°C overnight. The double immunofluorescence staining was followed by incubation with CY3-conjugated anti-rabbit secondary antibody (1:500; Jackson ImmunoResearch) for iba-1 or GFAP, and biotinylated anti-rat secondary antibody (1:500; Vector) for IFN-γ for 1 h. After one wash with phosphate buffer (0.1 m), the slides were incubated with streptavidin Alexa Fluor 488 (1:500; Invitrogen).
For the low-magnification images, the stained sections of the lumbar dorsal horn were viewed at 10× objective and recorded with a Leica DMR microscope equipped with digital camera Leica DFC 300 FX.
Confocal images were obtained using a Leica SP2 confocal microscope, adapted to an inverted Leica DM IRBE microscope. Tissue sections of the lumbar dorsal horn were examined with a 40×, 1.25 numerical aperture oil-immersion in Leica Plan Apochromatic objective at zoom of 2×. Alexa Fluor 488 and CY3 were excited with the 488 nm line of an argon laser and the 543 nm line of a green neon laser, respectively, and double immunofluorescence images of three stained sections were taken for each animal in a sequential mode. From each section were always recorded images of the ipsilateral and contralateral dorsal horns. Fifteen images were analyzed for each experimental group with Image J software and analyzed for the colocalization of GFAP and IFN-γ with the intensity correlation analysis plugin (Li et al., 2004). Data were compared by using one-way ANOVA and least significant difference post hoc analysis.
Results
Transcriptional changes induced by peripheral nerve injury
To evaluate transcriptional events at the level of the spinal cord that may underlie the increased pain sensitivity after nerve injury and the enhanced neuropathic pain phenotype of CB2 −/− animals, we performed a series of microarray experiments in CB2 −/− and CB2 +/+ mice. For this purpose, we dissected lumbar spinal cord segments ipsilateral and contralateral to the nerve ligation from CB2 −/− and CB2 +/+ mice, plus the corresponding sham controls. RNA from these tissues was isolated and analyzed on Affymetrix GeneChip arrays (MG_430 2.0).
We found 490 genes differentially expressed in tissues from animals with nerve injury compared with sham control. Of these transcripts, we excluded from additional analysis all those with expression levels <0.5% of β-actin and those showing differential expression (more than log 1) in more than one of the sham control tissues. We thus obtained a total of 219 transcripts, of which 117 were downregulated and 102 upregulated (supplemental Table S1, available at www.jneurosci.org as supplemental material). These transcripts included a large number of genes with a function in cell signaling, regulation of gene expression and immune responses (Fig. 1), which is consistent with the observed microglia response triggered by the nerve injury (Racz et al., 2008). Most of the suppressed, 93, and one-half, 47, of the induced transcripts were more strongly regulated on the ipsilateral compared with the contralateral side (supplemental Table 1, available at www.jneurosci.org as supplemental material).
Figure 1.

Classification of genes showing significant changes in expression in the spinal cord after sciatic nerve injury. About one-third of upregulated genes are responsible for the inflammatory/immunological processes, whereas only a small amount of genes belonging to this category was downregulated. A large number of genes involved in signaling and metabolism were upregulated and downregulated. A fewer number of genes regulating cell structure, translation/transcription, and DNA repair functions are in rather equal number upregulated or downregulated. A large number of genes with unknown function have a different expression after sciatic nerve injury.
We considered those transcripts that were more strongly induced or repressed in CB2 −/− animals, or vice versa, as potential candidate mediators of the enhanced neuropathic pain phenotype. Indeed, 54 transcripts were more strongly regulated in CB2 −/− mice and 36 in CB2 +/+ mice (Fig. 2).
Figure 2.

Affymetrix gene array analysis of spinal cord tissues during neuropathic pain development in CB2 −/− mice. A, The list of genes that were more strongly regulated in CB2 −/− animals compared with CB2 +/+ mice. B, Genes that were more strongly regulated in CB2 +/+ animals compared with CB2 −/− mice. The intensity and direction of gene regulation are represented in a heat map (red, downregulated; green, upregulated).
Enhanced upregulation of interferon-γ-inducible transcripts in CB2 knock-out mice
We next performed a bioinformatics analysis to obtain the candidate cis-regulatory motifs and their binding transcription factors in a given dataset of coregulated genes. This method was specifically developed to identify regulatory motifs that are highly specific for a group of genes under study (N. Bellora and M. Albà, unpublished data). In this analysis, we used 72 promoter regions of the genes differentially regulated after nerve injury in both genotypes, as revealed by microarray experiments (supplemental Table 1, available at www.jneurosci.org as supplemental material). We identified a significant overrepresentation of transcription factor interferon regulatory factor-1 (IRF-1) and IRF-2 binding sites (p < 0.001) in 22.2 and 6.9% of the genes analyzed, respectively, thus indicating a high enrichment over synthetic sequences and random promoter datasets (Table 1).
Table 1.
Identification of regulatory motifs in genes upregulated after nerve injury

Transcription factor binding site (TFBS) matrix name indicates the identifier from a JASPAR position weight matrices (PWM) collection. The TFBS matrix logo represents the transcription factor binding site sequences, in which the size of the letter is a measure of nucleotide frequency. Logos of TFBS can be read in forward or reverse, so the complementary logo is also valid for any PWM. The observed sites on the target dataset identify the total number of sites detected. Compared with the expected sites on synthetic sequence datasets or random promotor datasets, IRF-1 and IRF-2 binding sites were enriched in our dataset (number in brackets). Genes with sites represent the number of genes with at least one site. Overrepresentation obs./esp. sites, epval.: epval, empirical p value indicating the probability of obtaining at least the same number of sites on random datasets as the one observed.
These data indicate a strong interferon response after nerve injury. Interestingly, a number of IFN-γ-induced genes were among those transcripts that were more strongly regulated in the absence of CB2 receptors (Fig. 3 A). These transcripts included several members of the 47 kDa and 65 kDa families of GTPases. In CB2 +/+ animals, we observed a 1.7- to 2.4-fold upregulation, with the exception of Tgtp, which was 3.6 times higher, whereas these transcripts were 2.3- to 7-fold upregulated in CB2 −/− mice. Validation of three of these genes using Taqman real-time PCR analysis confirmed their stronger induction in CB2 −/− animals (Fig. 3 B). IFN-inducible GTPases such as Igtp and Gbp2 have been implicated in the regulation of immune responses toward infections (Martens and Howard, 2006; Degrandi et al., 2007). These results suggest an enhanced IFN-γ response in CB2 −/− mice after nerve injury.
Figure 3.

Expression of genes regulated by IFN-γ or IFN-α after partial sciatic nerve ligation (PNL) was upregulated. A, The increase in expression level of IFN-γ-regulated genes was more intensive in CB2 −/− than in CB2 +/+ mice, whereas the upregulation in IFN-α-regulated genes was less intensive in CB2 −/− animals. B, Taqman real-time PCR analysis of the expression of three GTPase regulated genes also showed a stronger expression change in CB2 −/− than in CB2 +/+ mice after neuropathy. wt-i, Ipsilateral paw of CB2 +/+ mice; wt-c, contralateral paw of CB2 +/+ mice; ko-i, ipsilateral paw of CB2 −/− mice; ko-c, contralateral paw of CB2 −/− mice.
The enhanced manifestations of neuropathic pain were not observed in double IFN-γ−/−/CB2 −/− mice
To clarify the involvement of interferon proteins in the enhanced manifestations of neuropathic pain in CB2 −/− mice, we crossed CB2 −/− animals to IFN-γ−/− mice to generate double knock-out mice deficient in both CB2 receptors and IFN-γ. These double knock-out animals showed no gross behavioral phenotypes or any obvious symptoms of a disease.
We therefore subjected these mice to a peripheral nerve ligation and evaluated hyperalgesia to a noxious thermal stimulus (plantar test), and allodynia to a mechanical stimulus (von Frey stimulation model) as outcome behavioral measures of neuropathic pain at various time points after the nerve injury. Baseline nociceptive responses of IFN-γ−/− and double IFN-γ−/−/CB2 −/− mice were similar in the plantar test and von Frey stimulation model. Sham operation also did not produce any modification of these nociceptive thresholds in both genotypes. Furthermore, thermal hyperalgesia and mechanical allodynia induced by sciatic nerve injury in the ipsilateral paw were similar in IFN-γ−/− and double IFN-γ−/−/CB2 −/− mice (Fig. 4) and no mirror image of pain was revealed in any experimental group. Thus, the enhanced manifestations of neuropathic pain in CB2 −/− mice was completely suppressed in the absence of IFN-γ. Therefore, IFN-γ is directly involved in the regulation of neuropathic pain responses by CB2 receptors.
Figure 4.

Development of neuropathic pain in male IFN-γ−/− and IFN-γ−/−/CB2 −/− knock-out animals. A single tight ligature around one-third or one-half of sciatic nerve was made to induce the neuropathic pain. Mice were tested in the ipsilateral and contralateral paw for evaluating mechanical allodynia (von Frey model) and thermal hyperalgesia (plantar model) on days 3, 6, 8, 10, and 15 after surgery. Mechanical allodynia data are expressed as mean ± SEM percentage values of basal responses of sham-operated mice. Thermal hyperalgesia data are expressed as mean ± SEM values of withdrawal latencies. The black stars represent comparisons between time points in IFN-γ−/− animals (n = 8 for nerve injury, n = 6 for sham). The white stars represent comparison between time points in IFN-γ−/−/CB2 −/− mice (n = 7 for nerve injury; n = 3 for sham). One star, p < 0.05; two stars, p < 0.01; three stars, p < 0.001.
Immunofluorescence analysis of IFN-γ expression in microglial cells and astrocytes
To further study IFN-γ in activated microglia and astrocytes under these experimental conditions, double immunofluorescence analysis was performed using antibodies against iba-1 or GFAP and IFN-γ in CB2 −/− and wild type mice exposed to sciatic nerve injury. IFN-γ signals were detected in the dorsal horn of lumbar spinal cord mainly in the ipsilateral side of CB2 +/+ mice, as well as in the ipsilateral and contralateral sides of CB2 −/− mice. IFN-γ immunoreactivity could be detected in a minor subset of GFAP+ astrocytes but did not colocalize with iba+ microglial cells. Using the intensity correlation analysis, we observe that the colocalization of IFN-γ signal with the GFAP signal was significantly enhanced in the ipsilateral and contralateral dorsal horns of the CB2 −/− mice, whereas in CB+/+ this effect was only revealed in the ipsilateral dorsal horn. The expression of IFN-γ in astrocytes matched the pattern of nociceptive hypersensitivity (see Fig. 6). IFN-γ protein has also been reported to be expressed in neurons (Neumann et al., 1997). Indeed, a positive IFN-γ staining was observed in the cytoplasm of cells recognized as neurons by their morphology (Fig. 5) identifying spinal cord neurons as a potential source for IFN-γ after nerve injury.
Figure 6.
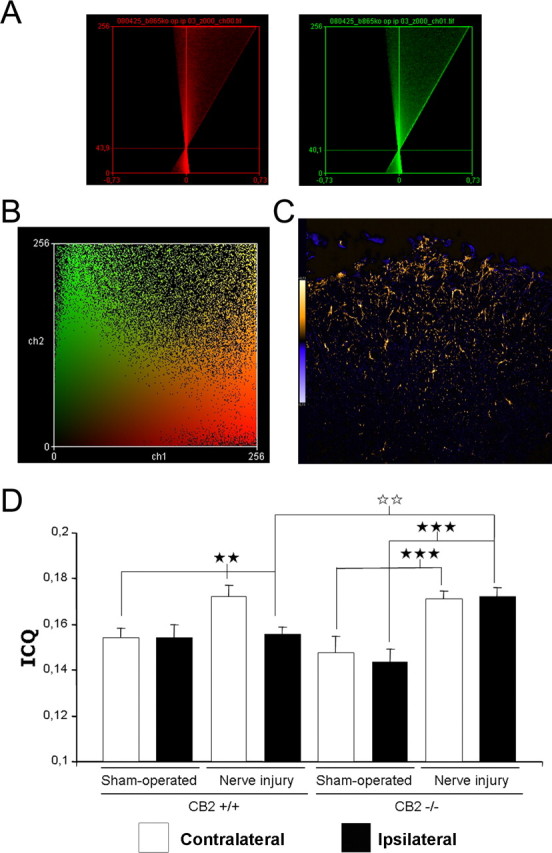
Intensity correlation analysis for the colocalization of GFAP with IFN-γ in CB2 +/+ and CB2 −/− mice after sciatic nerve injury. A, Representative intensity correlation plots of GFAP (astrocyte) (red channel) and IFN-γ (green channel) in the ipsilateral dorsal horn of a CB2 −/− mice after nerve injury. GFAP was visualized by staining of cryostat sections with rabbit anti-GFAP antibody followed by CY3-conjugated secondary anti-rabbit antibody (red). IFN-γ expression was visualized by staining of cryostat sections with rat anti-IFN-γ antibody and a biotinylated anti-rat secondary antibody followed by streptavidin Alexa Fluor 488 (green). B, Scatter plot of pixel staining intensities of the same image. C, Image of the product of the differences of the mean values (PDM) of the same image. D, Intensity correlation quotients for the different experimental groups (n = 5). The white bars represent the contralateral dorsal horn, and the black bars represent the ipsilateral dorsal horn. Error bars indicate SEM. The black stars represent comparisons between sham-operated and nerve injury, or between ipsilateral and contralateral paw. The white stars represent comparisons between genotypes. Two stars, p < 0.01; three stars, p < 0.001.
Figure 5.

IFN-γ expression by astrocytes and neuronal cells in the spinal cord of CB2 +/+ and CB2 −/− mice after sciatic nerve injury. A, Representative low-magnification images of double immunofluorescence staining with GFAP (astrocyte: red, CY3-conjugated secondary anti-rabbit Ab) and IFN-γ (green, streptavidin Alexa Fluor 488) recorded with 10× objective in the lumbar dorsal horn of sciatic nerve injury CB2 +/+ and CB2 −/− mice. B, Confocal microscopy of representative spinal cord sections after double immunofluorescence staining for microglial marker iba-1 or astrocyte marker GFAP (red, CY3-conjugated secondary anti rabbit Ab) and for IFN-γ (green, streptavidin Alexa Fluor 488). IFN-γ expression in GFAP+ astrocytes and not iba-1 microglial cells in the lumbar dorsal horn of CB2 +/+ and CB2 −/− mice. C, IFN-γ immunoreactivity in neuron-like cells as evidenced by neuronal morphology in the ventral horn of spinal cords of neuropathic WT mice. IFN-γ expression was visualized by staining of cryostat sections with rat anti-IFN-γ antibody and a biotinylated anti-rat secondary antibody followed by streptavidin Alexa Fluor 488 (green). D, Colocalization of GFAP with IFN-γ in the ipsilateral dorsal horn of neuropathic CB2 −/− mice. GFAP was visualized by staining of cryostat sections with rabbit anti-GFAP antibody followed by CY3-conjugated secondary anti-rabbit antibody (red).
Role of the CB2 receptor in IFN-γ-inducible inducible nitric oxide synthase mRNA and CCR2 mRNA expression in microglial cells
Peripheral nerve injury induced inflammation is associated with enhanced gene transcription of inducible nitric oxide synthase (iNOS) and nitric oxide (NO) release from locally recruited macrophages and microglial cells (Martucci et al., 2008). Recent studies demonstrated that spinal microglial infiltration in neuropathic pain is critically dependent on binding of chemokine monocyte chemoattractant protein-1 [MCP-1; chemokine ligand 2 (CCL2)] to its receptor CCR2 expressed on infiltrated microglial cells (Zhang et al., 2007). To test the hypothesis that IFN-γ-inducible iNOS and CCR2 gene expression is influenced by CB2 signaling in microglial cells, we stimulated BV-2 microglial cells with IFN-γ in the presence or absence of the CB2 agonist (6aR,10aR)-3-(1,1-dimethylbutyl)-6a,7,10,10a-tetrahydro-6,6,9-trimethyl-6H-dibenzo[b,d]pyran (JWH-133) (Fig. 7). mRNA expression of CB2 is upregulated in IFN-γ stimulated BV-2 cells and unaffected by treatment with JWH-133 and the CB2-selective antagonist N-[(1S)-endo-1,3,3-trimethyl bicyclo[2.2.1]heptan-2-yl]-5-(4-chloro-3-methylphenyl)-1-(4-methylbenzyl)-pyrazole-3-carboxamide (SR144528) (SR2). However, exposure of IFN-γ-stimulated microglial cells to JWH-133 inhibited iNOS gene expression, whereas the CB2 antagonist SR144528 was able to reverse this effect (Fig. 7). This demonstrates that this inhibitory effect of the JWH-133 is indeed mediated by the CB2 receptors. Next, we tested the influence of CB2 receptor signaling on CCR2 mRNA expression in BV-2 microglial cells after stimulation with IFN-γ. Treatment of BV-2 cells with JWH-133 significantly diminishes IFN-γ dependent CCR2 mRNA upregulation in BV-2 cells. This inhibitory effect was fully reversed in the presence of the CB2 antagonist SR144528. These findings suggest that CB2 signaling significantly downregulates IFN-γ-dependent iNOS and CCR2 gene expression in microglial cells, the gene products of which are critically involved in activation and chemotaxis of microglial cells after peripheral nerve injury.
Figure 7.

The CB2 agonist JWH-133 reduces IFN-γ-inducible iNOS and CCR2 mRNA expression in microglial cells. Relative expression levels of CB2, iNOS, and CCR2 mRNA were determined by quantitative real-time PCR. BV-2 microglial cells were stimulated either with 50 U/ml IFN-γ for 15 h alone, or in the presence of JWH-133 (5 μm) with or without CB2 receptor antagonist SR144528 (1 μm). cDNA was generated from BV-2 cells and expression of CB2, iNOS, and CCR2 mRNA was evaluated by real-time RT-PCR. Quantitative RT-PCR results are expressed as a ratio of average copies per copy of GAPDH for mean values ± SEM of n = 6 for all conditions. *p < 0,05; **p < 0,005 (BV-2 cells stimulated with IFN-γ vs with IFN-γ in the presence of 5 μm JWH-133).
Discussion
The present results revealed the crucial role of CB2 cannabinoid receptors in the development of neuropathic pain through an immune mechanism linked to modified IFN-γ activity. Hyperalgesia and allodynia induced by sciatic nerve injury were enhanced in CB2 −/− mice, as revealed by a mirror image of pain in the contralateral side. These behavioral manifestations of neuropathic pain matched the changes induced in microglial and astrocyte activation, astrocytic IFN-γ expression, and other biochemical parameters related to the immune response.
Glial activators include chemokines that enhance pain sensation and are under the control of immune mediators, as well as several neuromodulators released by nearby neurons, such as prostaglandins (Tanga et al., 2006). Interferons represent crucial modulators of the central and peripheral immune responses (Bach et al., 1997) and the enhanced induction of IFN-γ genes in CB2 −/− mice could participate in their nociceptive hypersensitivity. Indeed, a prolonged spinal increase in IFN-γ levels in inflammatory responses in diseases such as viral infections and multiple sclerosis, is thought to contribute to the associated persistent pain states (Miyazaki et al., 2008). Furthermore, IFN-γ treatments in cancer therapy can result in spontaneous pain in humans (Quesada et al., 1986; Mahmoud et al., 1992). Intrathecal IFN-γ administration in mice can also cause pain-related behaviors in normal, but not in IFN-γ receptor knock-out mice (Robertson et al., 1997). Although the molecular and cellular mechanism of interferon-induced pain remains mostly unclear, it has been demonstrated that IFN-γ can cause spontaneous firing of dorsal horn neurons in vitro and in vivo, as well as enhanced wind-up responses to electrical stimulation (Vikman et al., 2003, 2005, 2007). Interestingly, the pharmacological activation of CB2 cannabinoid receptors suppresses wind-up responses of spinal nociceptive neurons and this effect was more pronounced in the presence of pathological pain (Nackley et al., 2004).
The enhanced IFN-γ response revealed by microarray experiments in CB2 −/− mice exposed to nerve injury has an important functional relevance in vivo. Thus, a direct relationship between the enhanced IFN-γ response and the neuropathic pain manifestations of CB2 −/− mice was demonstrated by using double knock-out animals deficient in CB2 receptors and IFN-γ. The behavioral phenotype of CB2 −/− mice showing an enhanced neuropathic pain was completely abolished in these double knock-out animals. IFN-γ is a crucial modulator of the central and peripheral immune responses suggesting that an immune alteration seems to underline the neuropathic pain responses in CB2 −/− mice.
The manifestations of neuropathic pain observed in CB2 −/− mice and double knock-out mice deficient in CB2 and IFN-γ suggest that endocannabinoids play an important role in the control of the immune responses leading to the development of neuropathic pain. In support of this hypothesis, an enhancement in the levels of the two main endocannabinoids, anandamide and 2-arachidonoyl-glycerol, was revealed after sciatic nerve injury in the spinal cord and several brain areas involved in pain, such as the periaqueductal gray matter and the rostral ventral medulla (Petrosino et al., 2007). The endocannabinoid levels were also enhanced after sciatic nerve injury in the dorsal root ganglia (Mitrirattanakul et al., 2006) and the section of the sciatic nerve proximal to the lesion (Agarwal et al., 2007). These increased endocannabinoid levels are likely related to enhanced biosynthesis or decreased catabolism and transport because endocannabinoids are produced on demand without any substantial storage (Di Marzo, 1998). Therefore, endocannabinoids could produce a tonic activation of CB2 receptors after sciatic nerve injury that would limit the immune responses leading to the development of neuropathic pain. In agreement, both mechanical and thermal hyperalgesia produced after sciatic nerve injury were attenuated by the administration of N-arachidonoyl-serotonin, an inhibitor of fatty acid amide hydrolase, the enzyme responsible for the degradation of anandamide (Maione et al., 2007), which further support the role of endocannabinoids in the modulation of neuropathic pain.
In contrast with the results here obtained in CB2 −/− mice, the genetic disruption of the CB1 receptor had no major consequences on the development of neuropathic pain (Castañé et al., 2006) despite the high expression of these receptors in the CNS (Tsou et al., 1998). However, CB1 receptors expressed in peripheral nociceptors but not in the CNS seem to be involved in the manifestations of neuropathic pain (Agarwal et al., 2007). In agreement, pharmacological activation of CB1 receptors have also been reported to reduce pain sensitivity in a variety of neuropathic pain models (Pertwee, 2005).
Subsequent to nerve injury, gradients formed by CCL2 and CCL3 orchestrate the recruitment and activation of resident and monocyte-derived microglia via signaling through their respective receptors CCR2, CCR1, and CCR5 (Scholz and Woolf, 2007). In particular, CCR2 expression in either resident microglia or bone marrow-derived macrophages may be sufficient for the development of mechanical allodynia in a murine neuropathic pain model (Zhang et al., 2007). CB2 cannabinoid receptor activity may critically influence the induction of CCR2 expression by monocytes and thus inhibit their chemotaxis (Steffens et al., 2005). Moreover, endocannabinoids were found to abolish microglia activation by inhibiting NO release through a mechanism linked to the MAPK (mitogen-activated protein kinase) pathway (Eljaschewitsch et al., 2006). Our data revealed that IFN-γ treatment of mouse BV-2 microglial cells evoked marked microglial activation as indicated by induction of iNOS and CCR2 gene expression. CB2 signaling, however, interfered with the expression of these two IFN-γ-inducible genes in BV-2 cells. These data suggest that CB2 receptor signaling exerts antiinflammatory effects in the neuropathic response by controlling IFN-γ-mediated microglial activation and recruitment. Therefore, these data complement our in vivo observations in the neuropathic response in CB2 −/− mice.
Mice genetically modified either by increasing or eliminating specific gene may be limited by the fact that this genetic change may be affecting not only the target gene but also other biological components, perhaps participating in the effects evaluated in these genetic models. Therefore, pharmacological studies using selective ligands of CB2 receptors would be useful to confirm the relevance of the present results. Nevertheless, these genetic manipulations have been considered a key approach to the identification of alterations associated to different pathological conditions and to the discovering of new potential therapeutic targets in a variety of neuropsychiatric disorders. In this study, the according results obtained in CB2 −/− mice and double knock-out mice deficient in CB2 and IFN-γ together with the pharmacological studies performed in vitro further support the relevance of the findings.
Our immunofluorescence analysis revealed that IFN-γ was mainly expressed in neurons after sciatic nerve injury and this expression of IFN-γ matched the pattern of nociceptive hypersensitivity in all experiments. IFN-γ expression was also present in astrocytes, but absent in microglia. Previous studies have reported the presence of CB2 receptors in microglial cells (Romero-Sandoval et al., 2008) and neurons (Van Sickle et al., 2005). However, the possible presence of CB2 receptors in neurons and its possible functional role is still a controversial issue that requires additional investigation. Taken all these data into consideration, we can postulate a mechanism to explain the modulation of neuropathic pain through CB2 receptor activation (Fig. 8). Thus, the neuroinflammatory process leading to the development of neuropathic pain seems to be initiated by the microglial activation produced after nerve injury (Scholz and Woolf, 2007). This process requires a coactivation of astrocytes, which, together with neurons, release IFN-γ and promote consolidation and progression of the neuropathic pain state (Zhang et al., 2007). IFN-γ promotes microglia activation by the induction of several inflammatory pathways, including an enhancement in iNOS and CCR2 activity. Interestingly, previous studies have reported the expression of IFN-γ receptors in microglial cells and its modulation under pathological conditions (Cannella and Raine, 2004). CB2 receptors on microglial cells would play a crucial role to control and limit the spreading of this neuroinflammatory process. Thus, the activity of CB2 receptors in microglial cells would reduce the activation of these cells during neuropathic pain by regulating the expression of iNOS and CCR2. In the absence of CB2 receptors, IFN-γ would produce a more widespread activation of microglial cells, which would enhance the manifestations of neuropathic pain and would be responsible for the presence of a mirror image of pain in the contralateral side.
Figure 8.

Hypothetical mechanism to explain the modulation of neuropathic pain through CB2 receptor activation. The release of IFN-γ by activated astrocytes and neurons plays an important role in the neuroinflammatory process leading to the development of neuropathic pain. IFN-γ promotes microglia activation by the induction of several inflammatory pathways, including an enhancement in iNOS and CCR2 activity. The activated microglia promote consolidation and progression of the neuropathic pain state. CB2 receptors on microglial cells would control and limit the spreading of this neuroinflammatory process. Thus, the activity of CB2 receptors in microglial cells would reduce the activation of these cells during neuropathic pain by regulating the expression of iNOS and CCR2. CB2 receptors located in neurons could also participate in the neuropathic pain response by decreasing the production of IFN-γ. These inhibitory effects would restrict the activation of microglial cells and attenuate the development of neuropathic pain. In the absence of CB2 receptors, IFN-γ would produce a more widespread activation of microglial cells, which would enhance the manifestations of neuropathic pain and would be responsible for the presence of a mirror image of pain in the contralateral side.
Footnotes
This work was supported by grants from the National Institute on Drug Abuse (1R01-DA016768-0111) (R.M., A.Z.), European Commission [Framework VI, GENADDICT OJ 2004/C164, 005166 (R.M., A.Z.); PHECOM, LSHM-CT-2007-037669 (R.M.)], Ministerio de Educación y Ciencia (BFU2004-00920/BFI; SAF2007-64062) (R.M.), Generalitat de Catalunya (R.M.), Deutsche Forschungsgemeinschaft (SFB645; FOR926) (A.Z.), and Bundesministerium für Bildung und Forschung (NGFN2) (A.Z.). We thank Dr. Patricia Murtra for her contribution in the histological experiments, Dr. Bernardo Castellano for helping in the microglia protocols, Dr. Patricia Robledo and Dr. Miguel Angel Serra for critical comments on this manuscript, and Edda Erxlebe, Julia Essig, Astrid Markert, Daniela Mauer, Dr. David Otte, Karola Poppensieker, Friederike Stammer, and Öznur Yilmaz for expert help.
References
- Agarwal N, Pacher P, Tegeder I, Amaya F, Constantin CE, Brenner GJ, Rubino T, Michalski CW, Marsicano G, Monory K, Mackie K, Marian C, Batkai S, Parolaro D, Fischer MJ, Reeh P, Kunos G, Kress M, Lutz B, Woolf CJ, et al. Cannabinoids mediate analgesia largely via peripheral type 1 cannabinoid receptors in nociceptors. Nat Neurosci. 2007;10:870–879. doi: 10.1038/nn1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach EA, Aguet M, Schreiber RD. The IFN gamma receptor: a paradigm for cytokine receptor signaling. Annu Rev Immunol. 1997;15:563–591. doi: 10.1146/annurev.immunol.15.1.563. [DOI] [PubMed] [Google Scholar]
- Bellora N, Farré D, Albà MM. Positional bias of general and tissue-specific regulatory motifs in mouse gene promoters. BMC Genomics. 2007;8:459. doi: 10.1186/1471-2164-8-459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley NE, McCoy KL, Mezey E, Bonner T, Zimmer A, Felder CC, Glass M, Zimmer A. Immunomodulation by cannabinoids is absent in mice deficient for the cannabinoid CB(2) receptor. Eur J Pharmacol. 2000;396:141–149. doi: 10.1016/s0014-2999(00)00211-9. [DOI] [PubMed] [Google Scholar]
- Cannella B, Raine CS. Multiple sclerosis: cytokine receptors on oligodendrocytes predict innate regulation. Ann Neurol. 2004;55:46–57. doi: 10.1002/ana.10764. [DOI] [PubMed] [Google Scholar]
- Castañé A, Célérier E, Martín M, Ledent C, Parmentier M, Maldonado R, Valverde O. Development and expression of neuropathic pain in CB1 knockout mice. Neuropharmacology. 2006;50:111–122. doi: 10.1016/j.neuropharm.2005.07.022. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Clark AK, Yip PK, Grist J, Gentry C, Staniland AA, Marchand F, Dehvari M, Wotherspoon G, Winter J, Ullah J, Bevan S, Malcangio M. Inhibition of spinal microglial cathepsin S for the reversal of neuropathic pain. Proc Natl Acad Sci U S A. 2007;104:10655–10660. doi: 10.1073/pnas.0610811104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degrandi D, Konermann C, Beuter-Gunia C, Kresse A, Würthner J, Kurig S, Beer S, Pfeffer K. Extensive characterization of IFN-induced GTPases mGBP1 to mGBP10 involved in host defense. J Immunol. 2007;179:7729–7740. doi: 10.4049/jimmunol.179.11.7729. [DOI] [PubMed] [Google Scholar]
- DeLeo JA, Yezierski RP. The role of neuroinflammation and neuroimmune activation in persistent pain. Pain. 2001;90:1–6. doi: 10.1016/s0304-3959(00)00490-5. [DOI] [PubMed] [Google Scholar]
- Di Marzo V. Endocannabinoids and other fatty acid derivates with cannabimimetic properties: biochemistry and possible physiopathological relevance. Biochim Biophys Acta. 1998;1392:153–175. doi: 10.1016/s0005-2760(98)00042-3. [DOI] [PubMed] [Google Scholar]
- Ehrhart J, Obregon D, Mori T, Hou H, Sun N, Bai Y, Klein T, Fernandez F, Tan J, Shytle RD. Stimulation of cannabinoid receptor 2 (CB2) suppresses microglial activation. J Neuroinflammation. 2005;2:29. doi: 10.1186/1742-2094-2-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eljaschewitsch E, Witting A, Mawrin C, Lee T, Schmidt PM, Wolf S, Hoertnagl H, Raine CS, Schneider-Stock R, Nitsch R, Ullrich O. The endocannabinoid anandamide protects neurons during CNS inflammation by induction of MKP-1 in microglial cells. Neuron. 2006;49:67–79. doi: 10.1016/j.neuron.2005.11.027. [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Li Q, Lau A, Morris TJ, Guo L, Fordyce CB, Stanley EF. A syntaxin 1, Gαo, and N-type calcium channel complex at a presynaptic nerve terminal: analysis by quantitative immunocolocalization. J Neurosci. 2004;24:4070–4081. doi: 10.1523/JNEUROSCI.0346-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Mahmoud HH, Pui CH, Kennedy W, Jaffe HS, Crist WM, Murphy SB. Phase I study of recombinant human interferon gamma in children with relapsed acute leukemia. Leukemia. 1992;6:1181–1184. [PubMed] [Google Scholar]
- Maione S, De Petrocellis I, de Novellis V, Schiano Moriello A, Petrosino S, Palazzo E, Sca Rossi F, Woodward DF, Di Marzo V. Analgesic actions of N-arachidonoyl-serotonin, a fatty acid amide hydrolase inhibitor with antagonistic activity at vanilloid TRPV1 receptors. Br J Pharmacol. 2007;150:766–781. doi: 10.1038/sj.bjp.0707145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmberg AB, Basbaum AI. Partial sciatic nerve injury in the mouse as a model of neuropathic pain: behavioral and neuroanatomical correlates. Pain. 1998;76:215–222. doi: 10.1016/s0304-3959(98)00045-1. [DOI] [PubMed] [Google Scholar]
- Martens S, Howard J. The interferon-inducible GTPases. Annu Rev Cell Dev Biol. 2006;22:559–589. doi: 10.1146/annurev.cellbio.22.010305.104619. [DOI] [PubMed] [Google Scholar]
- Martucci C, Trovato AE, Costa B, Borsani E, Franchi S, Magnaghi V, Panerai AE, Rodella LF, Valsecchi AE, Sacerdote P, Colleoni M. The purinergic antagonist PPADS reduces pain related behaviours and interleukin-1beta, interleukin-6, iNOS and nNOS overproduction in central and peripheral nervous system after peripheral neuropathy in mice. Pain. 2008;137:81–95. doi: 10.1016/j.pain.2007.08.017. [DOI] [PubMed] [Google Scholar]
- Mitrirattanakul S, Ramakul N, Guerrero AV, Matsuka Y, Ono T, Iwase H, Mackie K, Faull KF, Spigelman I. Site-specific increases in peripheral cannabinoid receptors and their endogenous ligands in a model of neuropathic pain. Pain. 2006;126:102–114. doi: 10.1016/j.pain.2006.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki Y, Iwabuchi K, Kikuchi S, Fukazawa T, Niino M, Hirotani M, Sasaki H, Onoe K. Expansion of CD4+CD28− T cells producing high levels of interferon-γ in peripheral blood of patients with multiple sclerosis. Mult Scler. 2008;14:1044–1055. doi: 10.1177/1352458508092809. [DOI] [PubMed] [Google Scholar]
- Nackley AG, Zvonok AM, Makriyannis A, Hohmann AG. Activation of cannabinoid CB2 receptors suppresses C-fiber responses and windup in spinal wide dynamic range neurons in the absence and presence of inflammation. J Neurophysiol. 2004;92:3562–3574. doi: 10.1152/jn.00886.2003. [DOI] [PubMed] [Google Scholar]
- Neumann H, Schmidt H, Wilharm E, Behrens L, Wekerle H. Interferon gamma gene expression in sensory neurons: evidence for autocrine gene regulation. J Exp Med. 1997;186:2023–2031. doi: 10.1084/jem.186.12.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG. Pharmacological actions of cannabinoids. Handb Exp Pharmacol. 2005;168:1–51. doi: 10.1007/3-540-26573-2_1. [DOI] [PubMed] [Google Scholar]
- Petrosino S, Palazzo E, de Novellis V, Bisogno T, Rossi F, Maione S, Di Marzo V. Changes in spinal and supraspinal endocannabinoid levels in neuropathic rats. Neuropharmacology. 2007;52:415–422. doi: 10.1016/j.neuropharm.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Quesada JR, Talpaz M, Rios A, Kurzrock R, Gutterman JU. Clinical toxicity of interferons in cancer patients: a review. J Clin Oncol. 1986;4:234–243. doi: 10.1200/JCO.1986.4.2.234. [DOI] [PubMed] [Google Scholar]
- Racz I, Nadal X, Alferink J, Baños JE, Rehnelt J, Martín M, Pintado B, Gutierrez-Adan A, Sanguino E, Manzanares J, Zimmer A, Maldonado R. Crucial role of CB2 cannabinoid receptor in the regulation of central immune responses during neuropathic pain. J Neurosci. 2008;28:12125–12135. doi: 10.1523/JNEUROSCI.3400-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson B, Xu XJ, Hao JX, Wiesenfeld-Hallin Z, Mhlanga J, Grant G, Kristensson K. Interferon-gamma receptors in nociceptive pathways: role in neuropathic pain-related behaviour. Neuroreport. 1997;8:1311–1316. doi: 10.1097/00001756-199703240-00050. [DOI] [PubMed] [Google Scholar]
- Romero-Sandoval A, Eisenach JC. Spinal cannabinoid receptor type 2 activation reduces hypersensitivity and spinal cord glial activation after paw incision. Anesthesiology. 2007;106:787–794. doi: 10.1097/01.anes.0000264765.33673.6c. [DOI] [PubMed] [Google Scholar]
- Romero-Sandoval A, Nutile-McMenemy N, DeLeo JA. Spinal microglial and perivascular cell cannabinoid receptor type 2 activation reduces behavioral hypersensitivity without tolerance after peripheral nerve injury. Anesthesiology. 2008;108:722–734. doi: 10.1097/ALN.0b013e318167af74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandelin A, Alkema W, Engström P, Wasserman WW, Lenhard B. JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 2004;32:D91–D94. doi: 10.1093/nar/gkh012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci. 2007;10:1361–1368. doi: 10.1038/nn1992. [DOI] [PubMed] [Google Scholar]
- Steffens S, Veillard NR, Arnaud C, Pelli G, Burger F, Staub C, Karsak M, Zimmer A, Frossard JL, Mach F. Low dose oral cannabinoid therapy reduces progression of atherosclerosis in mice. Nature. 2005;434:782–786. doi: 10.1038/nature03389. [DOI] [PubMed] [Google Scholar]
- Tang SC, Arumugam TV, Xu X, Cheng A, Mughal MR, Jo DG, Lathia JD, Siler DA, Chigurupati S, Ouyang X, Magnus T, Camandola S, Mattson MP. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci U S A. 2007;104:13798–13803. doi: 10.1073/pnas.0702553104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanga FY, Raghavendra V, Nutile-McMenemy N, Marks A, Deleo JA. Role of astrocytic S100beta in behavioral hypersensitivity in rodent models of neuropathic pain. Neuroscience. 2006;140:1003–1010. doi: 10.1016/j.neuroscience.2006.02.070. [DOI] [PubMed] [Google Scholar]
- Tsou K, Brown S, Sañudo-Peña MC, Mackie K, Walker JM. Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience. 1998;83:393–411. doi: 10.1016/s0306-4522(97)00436-3. [DOI] [PubMed] [Google Scholar]
- Van Sickle MD, Duncan M, Kingsley PJ, Mouihate A, Urbani P, Mackie K, Stella N, Makriyannis A, Piomelli D, Davison JS, Marnett LJ, Di Marzo V, Pittman QJ, Patel KD, Sharkey KA. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science. 2005;310:329–332. doi: 10.1126/science.1115740. [DOI] [PubMed] [Google Scholar]
- Vikman KS, Hill RH, Backström E, Robertson B, Kristensson K. Interferon-gamma induces characteristics of central sensitization in spinal dorsal horn neurons in vitro. Pain. 2003;106:241–251. doi: 10.1016/S0304-3959(03)00262-8. [DOI] [PubMed] [Google Scholar]
- Vikman KS, Siddall PJ, Duggan AW. Increased responsiveness of rat dorsal horn neurons in vivo following prolonged intrathecal exposure to interferon-gamma. Neuroscience. 2005;135:969–977. doi: 10.1016/j.neuroscience.2005.06.059. [DOI] [PubMed] [Google Scholar]
- Vikman KS, Duggan AW, Siddall PJ. Interferon-gamma induced disruption of GABAergic inhibition in the spinal dorsal horn in vivo. Pain. 2007;133:18–28. doi: 10.1016/j.pain.2007.02.010. [DOI] [PubMed] [Google Scholar]
- Zhang J, Hoffert C, Vu HK, Groblewski T, Ahmad S, O'Donnell D. Induction of CB2 receptor expression in the rat spinal cord of neuropathic but not inflammatory chronic pain models. Eur J Neurosci. 2003;17:2750–2754. doi: 10.1046/j.1460-9568.2003.02704.x. [DOI] [PubMed] [Google Scholar]
- Zhang J, Shi XQ, Echeverry S, Mogil JS, De Koninck Y, Rivest S. Expression of CCR2 in both resident and bone marrow-derived microglia plays a critical role in neuropathic pain. J Neurosci. 2007;27:12396–12406. doi: 10.1523/JNEUROSCI.3016-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]