

Abstract
The Cas9 protein from the Streptococcus pyogenes CRISPR-Cas immune system has been adapted for both RNA-guided genome editing and gene regulation in a variety of organisms, but can mediate only a single activity at a time within any given cell. Here we characterize a set of fully orthogonal Cas9 proteins and demonstrate their ability to mediate simultaneous and independently targeted gene regulation and editing in bacteria and in human cells. We find that Cas9 orthologs display consistent patterns in their recognition of target sequences and identify a highly targetable protein from Neisseria meningitidis. Our results provide a basal set of orthogonal RNA-guided proteins for controlling biological systems and establish a general methodology for characterizing additional proteins and adapting them to eukaryotic cells.
Introduction
Clustered, regularly interspaced, short palindromic repeats (CRISPR)/CRISPR-associated (Cas) systems provide bacteria and archaea with acquired immunity by incorporating fragments of viral or plasmid DNA into CRISPR loci and utilizing the transcribed crRNAs to guide the degradation of homologous sequences1, 2. In type II CRISPR systems, a ternary complex of Cas9 nuclease with crRNA and tracrRNA (trans-activating crRNA) binds to and cleaves dsDNA protospacer sequences that match the crRNA spacer and also contain a short protospacer-adjacent motif (PAM)3, 4. Fusing the crRNA and tracrRNA produces a single guide RNA (sgRNA) that is sufficient to target Cas94.
As an RNA-guided nuclease and nickase, Cas9 has been adapted for targeted gene editing5–9 and selection10 in a variety of organisms. While these successes are arguably transformative, nuclease-null Cas9 variants may prove to be at least as useful for regulatory purposes, as the ability to localize proteins and RNA to nearly any set of dsDNA sequences affords tremendous versatility for controlling biological systems11–17. Beginning with targeted gene repression through promoter and 5′-UTR obstruction in bacteria18, Cas9-mediated regulation was recently extended to transcriptional activation by means of VP64 recruitment in human cells19, 20. Looking forward, we anticipate a cornucopia of Cas9-mediated transcriptional activators, repressors, fluorescent protein labels, chromosome tethers, and numerous other tools.
While the Cas9 protein from S. pyogenes can mediate one activity at many different target sites, it cannot concurrently mediate a different activity at other targets. For example, a cell engineered with a Cas9 activator cannot undergo genome editing using a Cas9 nuclease without also cutting the sites being targeted by the activator. Simultaneously employing multiple RNA-guided activities within a single cell will require methods of independently targeting each activity to its own set of target sites. To establish this level of concerted control over cellular behavior21, 22, we developed methods enabling the characterization of orthogonal Cas9 proteins for multiplexed RNA-guided transcriptional activation, repression, and gene editing.
Results
Selecting putatively orthogonal Cas9 proteins
Cas9 RNA binding and sgRNA specificity is primarily determined by the ~36 base pair repeat sequence in pre-crRNA. We began by examining known Cas9 genes for highly divergent repeats in their adjacent CRISPR loci. We chose the well-studied Cas9 protein from Streptococcus pyogenes (SP), the smaller Cas9 proteins from Streptococcus thermophilus CRISPR1 and Neisseria meningitidis (ST1 and NM), and the large Cas9 protein from Treponema denticola (TD). The CRISPR loci associated with these genes harbor repeats that differ by at least 13 nucleotides from one another (Fig. 1a).
Fig. 1.

Comparison and characterization of putatively orthogonal Cas9 proteins. (a) Repeat sequences of SP, ST1, NM, and TD. Bases are colored to indicate the degree of conservation. (b) Plasmids used for characterization of Cas9 proteins in E. coli. All carry compatible replication origins and antibiotic resistance genes. (c) Selection scheme to identify PAMs. Cells expressing a Cas9 protein and one of two spacer-containing targeting plasmids were transformed with one of two PAM libraries with corresponding protospacers and subjected to antibiotic selection. Surviving uncleaved plasmids were subjected to deep sequencing. Cas9-mediated PAM depletion was quantified by comparing the relative abundance of each sequence within the matched versus the mismatched protospacer libraries. (d) Functional PAMs are depleted from the library by Cas9 when the targeting plasmid spacer matches the library plasmid protospacer. (e) Cas9 does not cut when the spacer and protospacer do not match. (f) Nonfunctional PAMs are never cut or depleted.
PAM characterization
Known Cas9 proteins will only target dsDNA sequences flanked by a 3′ PAM sequence specific to the Cas9 of interest. Of the four Cas9 variants, only SP has an experimentally characterized PAM, while the ST1 PAM and, very recently, the NM PAM were deduced bioinformatically. SP is thought to be the most readily targetable due to its short PAM of NGG10, while ST1 and NM targeting are constrained by PAMs of NNAGAAW and NNNNGATT, respectively23, 24. We hypothesized that bioinformatic approaches might infer more stringent PAM requirements for Cas9 activity than are empirically necessary for effector cleavage due to the additional requirement for spacer acquisition in natural systems. Because the PAM sequence is the most frequent target of mutation in escape phages, greater stringency during spacer acquisition might provide redundancy and sometimes preclude resistance. We therefore adopted a library-based approach to comprehensively characterize these sequences in bacteria using high-throughput sequencing.
Genes encoding ST1, NM, and TD were assembled from synthetic fragments and cloned into bacterial expression plasmids with their associated tracrRNAs (Fig. 1b, Supplementary Fig. 1). Prior experience with variably effective spacer sequences using SP (data not shown) led us to select two SP-functional spacers for incorporation into the six targeting plasmids. Each targeting plasmid encodes a constitutively expressed crRNA in which one of the two spacers is followed by the 36 base-pair repeat sequence specific to a Cas9 protein (Fig. 1b). Plasmid libraries containing one of the two protospacers followed by all possible 8 base pair PAM sequences were generated by PCR and assembly25. Future experiments could utilize >8 base pair libraries to account for even longer PAMs. Each library was electroporated into E. coli cells harboring Cas9 expression and targeting plasmids, for a total of 12 combinations of Cas9 protein, spacer, and protospacer (Fig. 1c). Surviving library plasmids were selectively amplified by barcoded PCR and sequenced by MiSeq to distinguish between functional PAM sequences, which are depleted only when the spacer and protospacer match (Fig. 1d–e), from nonfunctional PAMs, which are never depleted (Fig. 1f).
To graphically depict the importance of each nucleotide at every position, we plotted the log relative frequency of each base for matched spacer-protospacer pairs relative to the corresponding mismatched case (Fig. 2). As hypothesized, our results revealed that NM and ST1 recognize PAMs that are less stringent and more complex than earlier bioinformatic predictions, suggesting that requirements for spacer acquisition are indeed more stringent than those for effector cleavage. Most strikingly, NM absolutely requires a single G nucleotide positioned five bases from the 3′ end of the protospacer (Fig. 2a), while ST1 and TD each require at least three specific bases (Fig. 2b–c). Sorting our results by position allowed us to quantify depletion of any PAM sequence from each protospacer library (Fig. 2d–f). All three enzymes cleaved protospacer B more effectively than protospacer A when paired with most PAMs, with ST1 exhibiting the greatest preference (Supplementary Fig. 2a). However, there was also considerable PAM-dependent variation in this interaction. For example, NM cleaved protospacers A and B approximately equally when they were followed by sequences matching TNNNGNNN, but was 10-fold more active in cleaving protospacer B for the set of sequences with PAMs matching ANNNGNNN (Supplementary Fig. 2b).
Fig. 2.

Depletion of functional protospacer-adjacent motifs (PAMs) from libraries by Cas9 proteins. The log frequency of each base at every position for matched spacer-protospacer pairs is plotted relative to control conditions in which spacer and protospacer did not match. Results reflect the mean depletion of libraries by NM (a), ST1 (b), and TD (c) based on two distinct protospacer sequences (d). Depletion of specific sequences for each protospacer are plotted separately for each Cas9 protein (d–f).
Our results highlight the difficulty of defining a single acceptable PAM for a given Cas9. Not only did activity levels depend upon the sequence of the protospacer, but specific combinations of unfavorable PAM bases substantially reduced activity even when the primary base requirements were met. We initially identified PAMs as patterns that underwent >100-fold average depletion with the lower-activity protospacer A and >50-fold depletion of all derivative sequences in which one base unspecified in the parent (e.g. N) is set to A, T, C, or G. (Table 1). While these levels are presumably sufficient to defend against targets in bacteria, we noticed that particular combinations of deleterious mutations dramatically reduced activity. For example, NM depleted sequences matching NCCAGGTN by only 4-fold (PAM matches underlined, Supplementary Fig. 2c). We therefore defined a more stringent threshold requiring >500-fold depletion of matching sequences and >200-fold depletion of one-base derivatives for applications requiring higher affinity (Table 1).
Table 1.
Protospacer-adjacent motifs for each Cas9. We defined two thresholds of activity for PAMs. The moderate threshold required >100-fold depletion of sequences matching the protospacer as well as >50-fold depletion of all sequences with one additional base defined (plain text). The stringent PAM threshold required >500-fold depletion of sequences matching the protospacer as well as >200-fold depletion of all sequences with one additional base defined (bold).
| NM | ST1 | TD |
|---|---|---|
|
| ||
| NNNNGANN | NNAGAA | NAAAAN |
| NNNNGTTN | NNAGGA | NAAANC |
| NNNNGNNT | NNGGAA | NANAAC |
| NNNNGTNN | NNANAA | NNAAAC |
| NNNNGNTN | NNGGGA | |
Orthogonality in bacteria
We originally selected our set of Cas9 proteins for their disparate crRNA repeat sequences. To verify that they are indeed orthogonal, we cotransformed each Cas9 expression plasmid with each of the four targeting plasmids containing spacer B. These cells were challenged by transformation with substrate plasmids containing either protospacer A or protospacer B and a suitable PAM. We observed plasmid depletion exclusively when each Cas9 was paired with its own crRNA, demonstrating that all four constructs are completely orthogonal in bacteria (Fig. 3).
Fig. 3.
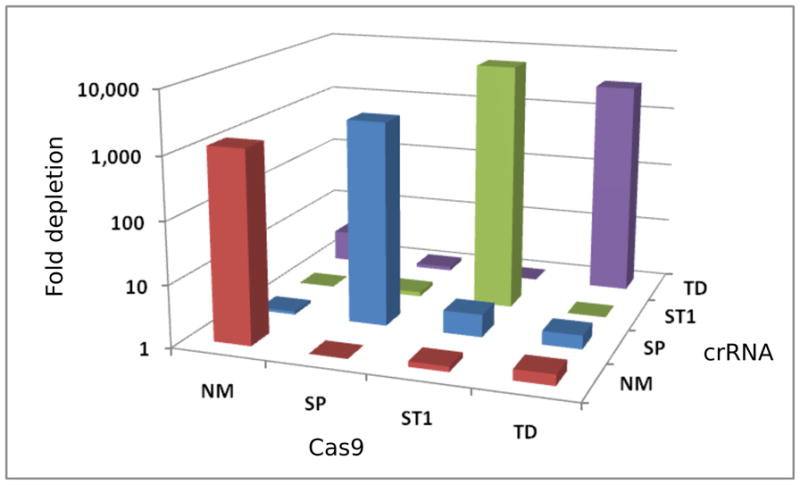
Orthogonal recognition of crRNAs in E. coli. Cells with all 16 combinations of Cas9 and crRNA were challenged with a plasmid bearing a matched or mismatched protospacer and appropriate PAM. Sufficient cells were plated to reliably obtain colonies from matching spacer and protospacer pairings. Total colony counts on the resulting 32 plates were used to calculate the fold depletion. Values less than one were set to one for clarity.
Transcriptional regulation in bacteria
A nuclease-null variant of SP has been demonstrated to repress targeted genes in bacteria with an efficacy dependent upon the position of the targeted protospacer18, 20, 26. Because the diverse PAMs of the new variants allow them to target sites lacking the SP PAM, we wondered whether nuclease-null versions of these proteins might be similarly capable of targeted repression. We identified the catalytic residues of the RuvC and HNH nuclease domains of each ortholog by sequence homology and inactivated them to generate nuclease-null NM, ST1, and TD (Online Methods). To create suitable reporters, we inserted protospacer B with an appropriate PAM for each Cas9 into the non-template strand within the coding sequence of a YFP reporter plasmid (Fig. 4a). We cotransformed each of these constructs into E. coli together with their corresponding targeting plasmids and measured the resulting fluorescence. Cells with matching spacer and protospacer exhibited much weaker fluorescence than the corresponding mismatched case for nuclease-null SP, ST1, and especially NM, but less so for TD. To determine whether this was an artifact of the low basal activity of the TD reporter, we also tested an alternative reporter design in which protospacer A was placed in the 5′ UTR, which confirmed that TD is much less effective as a repressor (Fig. 4b). These results indicate that not all Cas9 proteins are equally suitable for every task and suggest possible differences between larger Cas9 proteins such as TD and smaller members of the family. More practically, our results demonstrate that three of our four orthologs can function as robust RNA-guided repressors in bacteria.
Fig. 4.

Transcriptional repression simultaneous with nuclease activity in bacteria. (a) Reporter plasmids for quantification of Cas9 repression contained protospacer B and a suitable PAM in the non-template strand after the YFP start codon. Normalized cellular fluorescence is shown for mismatched and matched spacer-protospacer pairs. PAMs for each Cas9 are shown. (b) Cas9 ortholog repression was verified with a second reporter plasmid containing protospacer A and a PAM in the non-template strand within the 5′ UTR. Error bars represent the standard deviation of eight independently picked cultures for all repression experiments. (c) Cells containing the plasmids used for NM-mediated repression (Fig. 3a) were transformed with a compatible plasmid encoding SP, its tracrRNA, and a 5-spacer CRISPR locus designed to cleave filamentous phage gene III at multiple sites and challenged with M13mp18. The phage defense plasmid completely prevented plaque formation while preserving YFP repression. (d) Cells were transformed with a compatible plasmid encoding carbenicillin resistance and either wild-type gene III or a recoded version lacking protospacers and plated. Plasmids encoding wild-type gene III were perfectly excluded from cells encoding the SP phage defense, which did not interfere with YFP repression. Scale bars, 10 mm.
Simultaneous gene regulation and nuclease activity
Having demonstrated that our orthogonal Cas9 proteins are capable of both nuclease activity and transcriptional repression, we next engineered E. coli to employ both activities simultaneously. We constructed a plasmid encoding SP to defend against filamentous phage infection and utilized our previous constructs encoding nuclease-null NM, the most readily targetable of the orthologs, to repress the YFP reporter. As expected, the resulting cells successfully repressed YFP transcription and cleaved incoming filamentous phage genomes at multiple locations within gene III, completely preventing plaque formation by M13mp18 (Fig. 4c) and precluding transformation with a plasmid containing the targeted gene (Fig. 4d). These results demonstrate the ability of our orthogonal Cas9 proteins to mediate multiple independent activities within a single cell.
Genome editing in human cells
We next sought to apply these Cas9 variants to engineer human cells. We constructed single guide RNAs (sgRNAs) from the corresponding crRNAs and tracrRNAs for NM and ST1, the two smaller and predictably active Cas9 orthologs, by examining complementary regions between crRNA and tracrRNA27 (Supplementary Fig. 2) and fusing the two sequences via a stem-loop at various fusion junctions analogous to those of the sgRNAs created for SP (Supplementary Fig. 3). When the existing sequence might cause problems for our expression system (e.g. due to multiple successive uracils causing Pol III termination), we generated multiple single-base mutants. The complete 3′ tracrRNA sequence was always included, as truncations are known to be detrimental8.
All sgRNAs were assayed for activity along with their corresponding Cas9 protein using our previously described homologous recombination assay in 293 cells8. Briefly, a genomically integrated non-fluorescent GFP reporter line was constructed for each Cas9 protein in which the GFP coding sequence was interrupted by an insert encoding a stop codon and protospacer sequence with functional PAM. Reporter lines were transfected with expression vectors encoding a Cas9 protein and corresponding sgRNA along with a repair donor capable of restoring fluorescence upon nuclease-induced homologous recombination (Fig. 5a). Notably, we observed that full-length crRNA-tracrRNA fusions were active in all instances and therefore represent a reliable method of testing novel Cas9 ortholog activity in eukaryotic cells (Supplementary Figs. 3–5). Some but not all truncated versions are equally active. We selected highly active sgRNA for both NM and ST1 to use in future experiments (Supplementary Figs. 3–5).
Fig. 5.

Cas9-mediated gene editing in human cells. (a) A homologous recombination assay was used to quantify gene editing efficiency. Cas9-mediated double-strand breaks within the protospacer stimulated repair of the interrupted GFP cassette using the donor template, yielding cells with intact GFP. Three different templates were used in order to provide the correct PAM for each Cas9. Fluorescent cells were quantified by flow cytometry. (b) Homologous recombination efficiencies for NM, ST1, and TD in combination with each of their respective sgRNAs. Substrate PAMs are displayed below each Cas9. Data represent mean values ± s.e.m. (n=3).
Cas9 orthogonality in mammalian cells
To verify that none of the three proteins can be guided by the sgRNAs of the others in human cells, we employed the same homologous recombination assay to measure the comparative efficiency of SP, NM, and ST1 in combination with each of the three sgRNAs. Importantly, NM and ST1 induced genome editing at levels comparable to SP (Fig. 5b). Corroborating our findings with crRNAs in bacteria, our results unequivocally show that all three Cas9 proteins are fully orthogonal to one another, demonstrating that they are capable of targeting distinct and non-overlapping sets of sequences within the same cell (Fig. 5b, Supplementary Fig. 6).
To disentangle the comparative contributions of sgRNA and PAM to orthogonal targeting, we tested a variety of downstream PAM sequences with SP and ST1 and their respective sgRNAs. Certain PAMs were acceptable to both SP and ST1, enabling both enzymes to target the exact same sequence, but cutting occurred only when each enzyme was paired with its corresponding sgRNA. These results highlight the importance of both sgRNA and PAM for Cas9 activity, but also emphasize that the specific affinity of each Cas9 for its corresponding sgRNA is sufficient for orthogonality (Supplementary Fig. 7).
Transcriptional activation in human cells
We next investigated the ability of NM and ST1 to mediate transcriptional activation in human cells. Nuclease-null NM and ST1 genes were fused to the VP64 activator domain at their C-termini to yield putative RNA-guided activators modeled after our SP activator19. Reporter constructs for activation consisted of a protospacer with an appropriate PAM inserted upstream of the tdTomato coding region. Vectors expressing an RNA-guided transcriptional activator, an sgRNA, and an appropriate reporter were cotransfected and the extent of transcriptional activation measured by FACS (Fig. 6). In each case, we observed robust transcriptional activation by all three Cas9 variants, similar to a corresponding TAL-VP64 activator (Fig. 6). Each Cas9 activator also stimulated transcription only when paired with its corresponding sgRNA, confirming orthogonal genome regulation by the three Cas9 proteins.
Fig. 6.

Transcriptional activation in human cells. (a) Reporter constructs for transcriptional activation featured a minimal promoter driving tdTomato. Nuclease-null Cas9-VP64 fusion proteins binding to upstream protospacers resulted in transcriptional activation and enhanced fluorescence. (b) Cells were transfected with all combinations of Cas9 activators and sgRNAs and tdTomato fluorescence visualized by microscopy. Transcriptional activation occurred only when each Cas9 was paired with its own sgRNA. Scale bars, 100 μm. (c) Activation was quantified by flow cytometry along with a TAL-VP64 effector targeting an upstream sequence for comparison. Data represent mean values ± s.e.m. (n=3).
Discussion
By experimentally characterizing and demonstrating orthogonality between multiple Cas9 proteins in bacteria and human cells, we have substantially expanded the repertoire of orthogonal RNA-guided DNA-binding elements and constructed a pipeline for characterizing additional examples. Together, these proteins constitute the basics of a platform enabling simultaneous transcriptional regulation, labeling, and gene editing within individual cells.
Our results illustrate the remarkable diversity of proteins within a single family of CRISPR systems. Though clearly related, the Cas9 proteins from S. pyogenes, N. meningitidis, S. thermophilus, and T. denticola range from 3.25 to 4.6 kb in length and recognize completely different PAM sequences. These findings are in keeping with the strongly diversifying selective pressures facing defense systems engaged in molecular arms races28 and suggest that many other Cas9 proteins may be equally orthogonal.
Using two distinct protospacers for comprehensive PAM characterization allowed us a glimpse of the complexities governing protospacer and PAM recognition. Differential protospacer cleavage efficiencies exhibited a consistent trend across diverse Cas9 proteins, although the magnitude of the disparity varied considerably between orthologs. This pattern suggests that sequence-dependent differences in D-loop formation or stabilization determine the basal targeting efficiency for each protospacer, but that additional Cas9 or repeat-dependent factors also play a role. Similarly, numerous factors preclude efforts to describe PAM recognition with a single sequence motif. Individual bases adjacent to the primary PAM recognition determinants can combine to dramatically decrease overall affinity. Indeed, certain PAMs appear to interact nonlinearly with the spacer or protospacer to determine the overall activity. Moreover, different affinity levels may be required for distinct activities across disparate cell types. Finally, we observed that our experimentally identified PAMs required fewer bases than those inferred from bioinformatic analyses, suggesting that spacer acquisition requirements differ from those for effector cleavage.
This difference is most notable for the Cas9 protein from Neisseria meningitidis, which has fewer PAM requirements when paired with our spacers than either its bioinformatic prediction or the currently popular Cas9 from S. pyogenes, and considerably fewer than either ST1 or TD. It would be interesting to determine whether the total protospacer+PAM specificity of these four proteins is related to organismal genome size, a relationship that could point towards more specific Cas9 orthologs. More immediately, the characterization of NM considerably expands the number of sequences that can be readily targeted with a Cas9 protein. At 3.25 kbp in length, the NM gene is also 850 bp smaller than the SP gene; both the NM and ST1 genes are small enough to fit into standard viral vectors for in vivo delivery. NM may represent a more suitable starting point for directed evolution efforts designed to alter PAM recognition or specificity. We expect future experiments aimed at characterizing additional Cas9 orthologs to further improve our mechanistic understanding and expand our engineering capabilities.
Online Methods
Vector and Strain Construction
Cas9 sequences from S. thermophilus, N. meningitidis, and T. denticola were obtained from NCBI and human codon optimized using JCAT (www.jcat.de)29 and modified to facilitate DNA synthesis and expression in E. coli. 500 bp gBlocks (Integrated DNA Technologies) were joined by hierarchical overlap PCR and isothermal assembly25. The resulting full-length products were subcloned into bacterial and human expression vectors. Nuclease-null Cas9 cassettes (NM: D16A D587A H588A N611A, SP: D10A D839A H840A N863A, ST1: D9A D598A H599A N622A, TD: D13A D878A H879A N902A) were constructed from these templates by standard methods.
Bacterial plasmids
Cas9 was expressed in bacteria from a cloDF13-aadA plasmid backbone using the medium-strength proC constitutive promoter. All tracrRNA cassettes, including promoters and terminators from the native bacterial loci, were synthesized as gBlocks and inserted downstream of the Cas9 coding sequence for each vector for robust tracrRNA production. When the tracrRNA cassette was expected to additionally contain a promoter in the opposite orientation, the lambda t1 terminator was inserted to prevent interference with cas9 transcription. Bacterial targeting plasmids were based on a p15A-cat backbone with the strong J23100 promoter followed by one of two 20 base pair spacer sequences (Fig. 2D) previously determined to function using SP. Substrate plasmids for orthogonality testing in bacteria were identical to library plasmids (see below) but with the following PAMs: GAAGGGTT (NM), GGGAGGTT (SP), GAAGAATT (ST1), AAAAAGGG (TD). Spacer sequences were immediately followed by one of the three 36 base pair repeat sequences depicted in Fig. 1A. YFP reporter vectors were based on a pSC101-kan backbone with the pR promoter driving GFP and the T7 g10 RBS preceding the EYFP coding sequence. Two types were created: one with protospacer B + PAM inserted into the non-template strand just after the start codon of YFP, and one with protospacer A + PAM inserted into the non-template strand in the 5′-UTR. PAMs are as listed in Fig. 4A and 4B. The plasmid conferring immunity to filamentous phages via SP features a colE1-erm backbone, the SP cas9 gene and tracrRNA exactly as in the standard cloDF13-aadA plasmids, and the J23100 promoter driving a CRISPR locus targeting five sites within M13 gene III. Transformed plasmids carry the bla gene for carbenicillin resistance and either wild-type gene III or a recoded gene III. The CRISPR locus and recoded gene III were synthesized by Genewiz. All vectors and sequences are available through Addgene.
Mammalian vectors
Mammalian Cas9 expression vectors were based on pcDNA3.3-TOPO with C-terminal SV40 NLSs. sgRNAs for each Cas9 were designed by aligning crRNA repeats with tracrRNAs and fusing the 5′ crRNA repeat to the 3′ tracrRNA so as to leave a stable stem for Cas9 interaction27. sgRNA expression constructs were generated by cloning the U6-sgRNA expressing fragments synthesized as gBlocks into the pCR-BluntII-TOPO vector backbone. Spacers were identical to those used in previous work8. Lentivectors for the broken-GFP HR reporter assay were modified from those previously described to include appropriate PAM sequences for each Cas9 and used to establish the stable GFP reporter lines.
RNA-guided transcriptional activators consisted of nuclease-null Cas9 proteins fused to the VP64 activator and corresponding reporter constucts bearing a tdTomato driven by a minimal promoter were constructed as previously described20. All vectors and sequences are available through Addgene.
Library construction and transformation
Protospacer libraries were constructed by amplifying the pZE21 vector (ExpressSys, Ruelzheim, Germany) using primers (IDT, Coralville, IA) encoding one of the two protospacer sequences followed by 8 random bases and assembled by standard isothermal methods25. Library assemblies were initially transformed into NEBTurbo cells (New England Biolabs, Ipswich MA), yielding >1E8 clones per library according to dilution plating, and purified by Midiprep (Qiagen, Carlsbad CA). Electrocompetent NEBTurbo cells containing a Cas9 expression plasmid (DS-NMcas, DS-ST1cas, or DS-TDcas) and a targeting plasmid (PM-NM!sp1, PM-NM!sp2, PM-ST1!sp1, PM-ST1!sp2, PM-TD!sp1, or PM-TD!sp2) were transformed with 200 ng of each library and recovered for 2 hours at 37C prior to dilution with media containing spectinomycin (50 μg/mL), chloramphenicol (30 μg/mL), and kanamycin (50 μg/mL). Serial dilutions were plated to estimate post-transformation library size. All libraries exceeded ~1E7 clones, indicative of complete coverage of the 65,536 random PAM sequences.
High-throughput sequencing
Library DNA was harvested by spin columns (Qiagen, Carlsbad CA) after 12 hours of antibiotic selection. Intact PAMs were amplified with barcoded primers (Supplementary Data) and sequences obtained from overlapping 25bp paired-end reads on an Illumina MiSeq. MiSeq yielded 18,411,704 total reads or 9,205,852 paired-end reads with an average quality score >34 for each library. Paired end reads were merged and filtered for perfect alignment to each other, their protospacer, and the plasmid backbone. The remaining 7,652,454 merged filtered reads were trimmed to remove plasmid backbone and protospacer sequences and then used to generate position weight matrices for each PAM library. Each library combination received at least 450,000 high-quality reads.
Sequence processing
To calculate the fold depletion for each candidate PAM, we employed two scripts to filter the data (Supplementary Data). patternProp (usage: python patternProp.py [PAM] file.fastq) returns the number and fraction of reads matching each 1-base derivative of the indicated PAM. 1-base derivatives are defined as the set of all sequences in which one additional base that was not specified in the parent (i.e. N) is set to A, C, G, or T. patternProp3 returns the fraction of reads matching each 1-base derivative relative to the total number of reads for the library. Spreadsheets detailing depletion ratios for each calculated PAM were used to identify the minimal fold depletion among all 1-base derivatives and thereby classify PAMs (Supplementary Data).
Repression and orthogonality assays in bacteria
Cas9-mediated repression was assayed by transforming the NM expression plasmid and the YFP reporter plasmid with each of the two corresponding targeting plasmids. Colonies with matching or mismatched spacer and protospacer were picked and grown in 96-well plates. Fluorescence at 495–528 nm and absorbance at 600nm were measured using a Synergy Neo microplate reader (BioTek, Winooski VT).
Orthogonality tests were performed by preparing electrocompetent NEBTurbo cells bearing all combinations of Cas9 and targeting plasmids and transforming them with matched or mismatched substrate plasmids bearing appropriate PAMs for each Cas9. Sufficient cells and dilutions were plated to ensure that at least some colonies appeared even for correct Cas9 + targeting + matching protospacer combinations, which typically arise due to mutational inactivation of the Cas9 or the crRNA. Colonies were counted and fold depletion calculated for each.
For the simultaneous nuclease and repression assays, cells were first rendered electrocompetent, transformed with the SP phage defense plasmid, and plated with erythromycin, kanamycin, chloramphenicol, and spectinomycin. Plaque assays were performed by mixing dilutions of M13mp18 phage (New England Biolabs, Ipswich, MA) with 75 uL cells (NEBTurbo, containing the F plasmid), combining with 1 mL soft agar, and plating onto 60 mm LB plates with 50 μg/mL IPTG and 200 μg/mL X-Gal. For the plasmid transformation assay, cells were rendered electrocompetent by standard methods, transformed with plasmids bearing wild-type or recoded gene III, and plated with carbenicillin, erythromycin, kanamycin, chloramphenicol, and spectinomycin. Plaque assays were imaged using a Typhoon FLA 9000 (GE, Fairfield, CT), and the contrast adjusted by setting the maximum saturation to 1.0% using ImageJ.
Cell culture and transfections
HEK 293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen) high glucose supplemented with 10% fetal bovine serum (FBS, Invitrogen), penicillin/streptomycin (pen/strep, Invitrogen), and non-essential amino acids (NEAA, Invitrogen). Cells were maintained at 37°C and 5% CO2 in a humidified incubator.
Transfections involving nuclease assays were as follows: 0.4×106cells were transfected with 2μg Cas9 plasmid, 2μg gRNA and/or 2μg DNA donor plasmid using Lipofectamine 2000 as per the manufacturer’s protocols. Cells were harvested 3 days after transfection and either analyzed by FACS, or for direct assay of genomic cuts the genomic DNA of ~1 × 106 cells was extracted using DNAeasy kit (Qiagen).
For transfections involving transcriptional activation assays: 0.4×106cells were transfected with 2μg Cas9N-VP64 plasmid, 2μg gRNA and/or 0.25μg of reporter construct. Cells were harvested 24–48hrs post transfection and assayed using FACS or immunofluorescence methods, or their total RNA was extracted and these were subsequently analyzed by RT-PCR.
Statistical Analyses
No samples were excluded from any experiments.
Supplementary Material
Acknowledgments
We thank Ben Stranges for protein alignments and W.L. Chew for helpful discussions. This work was supported by US National Institutes of Health NHGRI grant P50 HG005550, Department of Energy grant DE-FG02-02ER63445, and the Wyss Institute for Biologically Inspired Engineering.
Footnotes
Author Contributions
K.M.E. and P.M. conceived the study; K.M.E. and P.M. designed the experiments; K.M.E., J.L.B., and S.J.Y. performed experiments in E. coli, J.L.B. wrote analysis software, P.M. and M.M. performed experiments in human cells, K.M.E. and P.M. analyzed results, and K.M.E. and P.M. wrote the manuscript with input from G.M.C.
Competing Financial Interests
The authors have filed for patents concerning the use of Cas9 proteins for gene targeting and regulation.
References
- 1.Bhaya D, Davison M, Barrangou R. CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annual review of genetics. 2011;45:273–297. doi: 10.1146/annurev-genet-110410-132430. [DOI] [PubMed] [Google Scholar]
- 2.Wiedenheft B, Sternberg SH, Doudna JA. RNA-guided genetic silencing systems in bacteria and archaea. Nature. 2012;482:331–338. doi: 10.1038/nature10886. [DOI] [PubMed] [Google Scholar]
- 3.Gasiunas G, Barrangou R, Horvath P, Siksnys V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E2579–2586. doi: 10.1073/pnas.1208507109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jinek M, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cho SW, Kim S, Kim JM, Kim JS. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nature biotechnology. 2013;31:230–232. doi: 10.1038/nbt.2507. [DOI] [PubMed] [Google Scholar]
- 6.Cong L, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ding Q, et al. Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell stem cell. 2013;12:393–394. doi: 10.1016/j.stem.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mali P, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang H, et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153:910–918. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nature biotechnology. 2013;31:233–239. doi: 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boch J, et al. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 2009;326:1509–1512. doi: 10.1126/science.1178811. [DOI] [PubMed] [Google Scholar]
- 12.Gaj T, Gersbach CA, Barbas CF., 3rd ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends in biotechnology. 2013;31:397–405. doi: 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hockemeyer D, et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nature biotechnology. 2009;27:851–857. doi: 10.1038/nbt.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim YG, Cha J, Chandrasegaran S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:1156–1160. doi: 10.1073/pnas.93.3.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moscou MJ, Bogdanove AJ. A simple cipher governs DNA recognition by TAL effectors. Science. 2009;326:1501. doi: 10.1126/science.1178817. [DOI] [PubMed] [Google Scholar]
- 16.Porteus MH, Carroll D. Gene targeting using zinc finger nucleases. Nature biotechnology. 2005;23:967–973. doi: 10.1038/nbt1125. [DOI] [PubMed] [Google Scholar]
- 17.Urnov FD, et al. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature. 2005;435:646–651. doi: 10.1038/nature03556. [DOI] [PubMed] [Google Scholar]
- 18.Qi LS, et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gilbert LA, et al. CRISPR-Mediated Modular RNA-Guided Regulation of Transcription in Eukaryotes. Cell. 2013;154:442–451. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mali P, et al. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nature biotechnology. doi: 10.1038/nbt.2675. advance online publication, Aug 1 (2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Podgornaia AI, Laub MT. Determinants of specificity in two-component signal transduction. Current opinion in microbiology. 2013;16:156–162. doi: 10.1016/j.mib.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 22.Purnick PE, Weiss R. The second wave of synthetic biology: from modules to systems. Nature reviews molecular cell biology. 2009;10:410–422. doi: 10.1038/nrm2698. [DOI] [PubMed] [Google Scholar]
- 23.Horvath P, et al. Diversity, activity, and evolution of CRISPR loci in Streptococcus thermophilus. Journal of bacteriology. 2008;190:1401–1412. doi: 10.1128/JB.01415-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y, et al. Processing-Independent CRISPR RNAs Limit Natural Transformation in Neisseria meningitidis. Molecular cell. 2013;50:488–503. doi: 10.1016/j.molcel.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gibson DG, et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 26.Bikard D, et al. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic acids research. 2013;41:7429–7437. doi: 10.1093/nar/gkt520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deltcheva E, et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471:602–607. doi: 10.1038/nature09886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bondy-Denomy J, Pawluk A, Maxwell KL, Davidson AR. Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature. 2013;493:429–432. doi: 10.1038/nature11723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grote A, et al. JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic acids research. 2005;33:W526–531. doi: 10.1093/nar/gki376. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.