

Abstract
Recent studies suggest that glial cells actively participate in the formation, function, maintenance, and repair of the chemical synapse. However, the molecular mechanisms of glia–synapse interactions are largely unknown. We have shown previously that Schwann cell-conditioned medium (SC-CM) promotes synaptogenesis in Xenopus nerve–muscle cocultures. The present study aimed to identify the synaptogenic molecules in SC-CM. Combining biochemical approaches and in vitro bioassays, we found that SC-CM contains transforming growth factor (TGF)-β1, which is expressed in Schwann cells both in vivo and in vitro. Similar to SC-CM, TGF-β1 doubled the size of acetylcholine receptor (AChR) clusters at nerve–muscle contacts and significantly increased the percentage of nerve–muscle contacts that show AChR clusters to ∼60%, compared with ∼20% seen in control cultures. The synaptogenic effects of SC-CM were abolished if SC-CM was immunodepleted of TGF-β1 or if the latency-associated protein or a TGF-β1 receptor kinase inhibitor was added to block the bioactivity of TGF-β1. Similar to frog SC-CM, mammalian SC-CM also showed synaptogenic effects, which were prevented by immunodepletion of TGF-β1. TGF-β1 upregulated agrin expression in spinal neurons, which could explain the increase in AChR clusters in cultures treated with SC-CM. These results suggest that Schwann cells express TGF-β1, which is both sufficient and necessary for mediating the synapse-promoting effects of Schwann cells at the developing neuromuscular junction. Schwann cell-derived TGF-β1 thus joins other astrocyte-derived synaptogenic factors in further strengthening the emerging concept that glial cells contribute to synaptogenesis in both the PNS and the CNS.
Keywords: agrin, glia, neuromuscular junctions, Schwann cell, synaptogenesis, TGF-β1
Introduction
Synapse-associated glial cells, previously considered as passive supporters of the chemical synapse, have recently gained much recognition as an integral and essential component of the tripartite synapse in both the peripheral nervous system and the CNS. A flurry of studies have illustrated that synaptic glia promote synapse formation, maintenance, and repair and are actively involved in modulating synaptic function (Haydon, 2001; Fields and Stevens-Graham, 2002; Newman and Volterra, 2004; Allen and Barres, 2005). However, the molecular mechanisms of synapse–glia interactions are not well understood. It is thus critical to identify glial cell-derived molecules that contribute to synapse formation and function.
In the vertebrate peripheral nervous system, the synapse-associated glial cells are called perisynaptic Schwann cells (PSCs; also known as terminal Schwann cells), which cap the motor nerve terminals at neuromuscular junctions (NMJs). Similar to their counterparts in the CNS, PSCs are also actively involved in multiple aspects of the NMJ (Kang et al., 2003; Koirala et al., 2003; Colomar and Robitaille, 2004; Feng and Ko, 2007; Ko et al., 2007). For example, PSCs extend processes that guide the growth of nerve terminals during development (Herrera et al., 2000; Reddy et al., 2003). These PSC processes are required for the growth and maintenance of developing synapses, because less synaptic growth and more synaptic retraction occur when PSCs are removed by complement-mediated cell lysis (Reddy et al., 2003). In adult muscles, PSCs sense synaptic transmission by increasing intracellular calcium and are capable of modulating synaptic transmission if pharmacologically manipulated (Colomar and Robitaille, 2004; Todd et al., 2006). Similar to their role at the developing NMJ, PSCs also promote the long-term maintenance of synaptic structure and function at the adult NMJ (Reddy et al., 2003). The active role of PSCs has also been demonstrated in muscles after nerve injury, because PSCs sprout and guide nerve terminal regeneration (Reynolds and Woolf, 1992; Son and Thompson, 1995a,b).
Although synapse–glia interactions have been demonstrated in both the PNS and CNS, very few glial cell-derived molecules that contribute to synaptic function and formation have been identified. Recent studies have shown that astrocyte-derived signals, such as thromspondins (TSPs) and cholesterol, promote synapse formation in the CNS (Mauch et al., 2001; Christopherson et al., 2005). Our laboratory has shown that Schwann cell-conditioned medium (SC-CM) contains unidentified small molecules that potentiate spontaneous synaptic transmission at developing Xenopus NMJs (Cao and Ko, 2007). In addition, we have demonstrated that SC-CM increases synaptic numbers in Xenopus nerve–muscle cocultures (Peng et al., 2003). In the present study, we found that both frog and mammalian SC-CM (mSC-CM) contain transforming growth factor (TGF)-β1, which belongs to the TGF-β superfamily involved in various functions, including cell growth and differentiation (Massagué, 2000). TGF-β1 mimics SC-CM and is necessary for promoting synaptogenesis induced by SC-CM. The synaptogenic effect of SC-CM could be attributed to an upregulation of neuronal agrin expression induced by TGF-β1. Our findings demonstrate that TGF-β1 is a Schwann cell-derived synaptogenic molecule and, thus, further advance the molecular understanding of synapse–glia interactions.
Materials and Methods
Xenopus nerve–muscle cocultures.
Spinal neuron and muscle cultures from stage 21–23 Xenopus embryos (Nieuwkoop and Faber, 1994) were prepared as described previously (Peng et al., 1991; Tabti et al., 1998). In brief, neural tubes together with the associated myotomal tissues were collected from the dorsal surface of stage 21–23 Xenopus embryos and dissociated in Ca2+- and Mg2+-free Ringer's solution [containing (in mm): 115 NaCl, 2.6 KCl, 0.4 EDTA, and 10 HEPES, pH 7.6]. The dissociated cells from one embryo were plated on a 35 mm culture dish (VWR) and cultured at room temperature (22–24°C) for 2–4 d before further examination. The basic culture medium consisted of 50% (v/v) Leibovitz's L-15 (Invitrogen) and 50% Ringer's solution [containing (in mm): 115 NaCl, 2 CaCl2, 2.6 KCl, and 10 HEPES, pH 7.6].
Preparation of Xenopus Schwann cell cultures and SC-CM.
To prepare Schwann cell cultures, sciatic nerves were dissected from adult Xenopus as described previously (Peng et al., 2003; Cao and Ko, 2007). After epineurial membranes were removed, nerves were minced and treated with 0.3% collagenase in HBSS without Ca2+ and Mg2+ (Invitrogen), together with 0.25% trypsin-EDTA (Invitrogen) at 37°C for 30 min. Dissociated cells were plated on laminin-1 (Invitrogen)-coated culture dishes and cultured at room temperature in medium containing 45% Ringer's solution, 45% L-15, and 10% fetal bovine serum (FBS) (Invitrogen). Culture medium was changed to serum-free medium (50% Ringer's solution/50% L-15) 1 week after plating. The identity of cultured Schwann cells was confirmed by staining with monoclonal antibody (mAb) 2A12 antibodies, which recognize frog Schwann cells (Astrow et al., 1998). After 2–3 weeks in culture, SC-CM was collected once a week and replenished with fresh serum-free medium. Only cultures in which >90% of the cells were Schwann cells were used to collect SC-CM.
Preparation of mammalian Schwann cell cultures.
Mammalian Schwann cell cultures were prepared according to standard procedures described previously (Cohen and Wilkin, 1995). Briefly, sciatic nerves were collected from neonatal rats (postnatal days 2–7). After removing epineural membranes, nerves were minced and digested in 0.3% collagenase and 0.25% trypsin-EDTA at 37°C for 45 min. After titration and centrifugation, the dissociated cells were plated on poly-l-lysine-coated culture dishes in culture medium containing 90% high-glucose DMEM (Invitrogen) and 10% FBS. The identity of the cultured Schwann cells was confirmed with S-100 antibody (Sigma) staining (Cohen and Wilkin, 1995). Bovine pituitary extract at 10 μg/ml (Sigma) and 2 μm forskolin were used to stimulate Schwann cell proliferation (Chandross et al., 1995). Mammalian SC-CM was collected from confluent Schwann cell cultures every 3 d. Mammalian SC-CM was concentrated 10-fold using centrifuge filter tubes with molecular weight cutoffs at 5 kDa, and was used at a final concentration of 2× in Xenopus nerve–muscle cultures.
Acetylcholine receptor cluster formation assay.
After stimulation under trophic factors plus cAMP elevation for 4 d, Xenopus nerve–muscle cocultures were fixed with 4% paraformaldehyde in Ringer's solution for 30 min at room temperature. The formation of acetylcholine receptor (AChR) clusters was assayed by fluorescence microscopy after labeling nerve–muscle cocultures with Alexa Fluor 594 α-BTX (Invitrogen) for AChR clusters. Nerve–muscle contacts were determined by phase–contrast microscopy. To help identify nerve–muscle contacts, in some experiments neurites were stained with rabbit antibody against Xenopus synapsin-1 (Wang et al., 1995) [G473, a generous gift from Dr. Bai Lu (National Institutes of Health, Bethesda, MD)], followed by FITC-conjugated goat anti-rabbit secondary antibody (Jackson ImmunoResearch Laboratories). A contact made by a growing neurite along the surface of a muscle cell was counted as one nerve–muscle contact. A nerve–muscle contact that was closely associated with BTX-stained spot(s) was counted as one nerve–muscle contact with AChR clusters. The total number of nerve–muscle contacts and the number of those associated with AChR clusters were counted. The size of AChR clusters was measured using NIH ImageJ software.
Immunoblotting and immunoprecipitation.
For Western blots, SC-CM was concentrated ∼10-fold using Microcon-3 centrifuge filters (Millipore). The concentrated SC-CM was collected and loaded for SDS-PAGE, and the proteins were transferred to a polyvinylidene fluoride membrane. A polyclonal anti-TGF-β1 antibody (1:200) (Santa Cruz Biotechnology), followed by alkaline phosphatase-conjugated goat anti-rabbit IgG secondary antibody (1:1000) (Santa Cruz Biotechnology) was used to detect TGF-β1 in SC-CM.
For immunoprecipitation, SC-CM was incubated with a monoclonal antibody to TGF-β1 (Sigma) overnight at 4°C. Antigen–antibody complexes were removed by incubation with Protein G-Sepharose beads (Pierce Biotechnology) for 2 h at 4°C and followed by centrifugation to separate the supernatant. Proteins from the protein G-Sepharose beads were eluted and were further analyzed using Western blotting. A sample of the supernatant was saved for Western blotting. The supernatant was examined using the AChR cluster formation assay.
ELISA.
The protein concentration of TGF-β1 in SC-CM was assessed using direct ELISA. A 96-well flat-bottomed microtiter plate (VWR) was coated with SC-CM overnight at 4°C. After washing and blocking, the plate was incubated with a monoclonal antibody to TGF-β1 (1:400) (Sigma) in blocking solution for 2 h at room temperature, followed by a 1 h incubation with an HRP-conjugated anti-mouse secondary antibody (1:1000) (Santa Cruz Biotechnology). 3,3′,5,5′-TMB (Sigma) was used as an HRP substrate. After 30 min of color development in the dark, the reaction was terminated by the addition of 25 μl of 2 M H2SO4 per 100 μl of substrate solution, and the optical densities were measured by a microplate reader at 450 nm. A standard curve using recombinant human TGF-β1 was included in each ELISA experiment. The concentration of TGF-β1 in SC-CM was interpolated from the standard curve.
Reverse transcription PCR.
To test the expression of agrin mRNA, cells from pure neuron cultures were lysed using TRIzol reagent (Invitrogen), and total RNA was isolated after TRIzol reagent protocol. First-strand cDNA was synthesized using an Omniscript RT Kit (Qiagen) and oligo(dT)12–18 primer (Invitrogen) at 37°C for 1 h. cDNA products from the reverse transcription reactions were amplified using the TaqPCR master mix kit (Qiagen). PCRs were performed using the Robocycler Gradient 40 (Stratagene) for 35 cycles of 94°C for 1 min, 55°C for 1.5 min, and 72°C for 1.5 min. PCR products were separated by electrophoresis on a 12% polyacrylamide gel and stained with ethidium bromide. Gels were visualized using a UV transilluminator, and images were taken using a digital camera. For agrin, primers flanking the frog agrin alternative splicing site B were designed based on the GenBank sequence AF096690 (Werle et al., 1999): forward, 5′-TCT CTG GAG GAC AAT GTG AGA AAG C-3′; and reverse, 5′-CCT GAG TGG CTT CAG TCT TTA TGC-3′. The housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used to confirm the equal loading of samples for the reverse transcription and PCRs. The primers for GAPDH were designed on the basis of the GenBank sequence U41573: forward, 5′-GTG TAT GTG GTG GAA TCT-3′; and reverse, 5′-AAG TTG TCG TTG ATG ACC TTT GC-3′.
To examine the expression of TGF-β1 and TGF-β1 type II receptors in Schwann cells, muscle cells, and neurons, total RNA from cultured Schwann cells, cultured muscle cells, and cultured neurons were isolated using the same protocol, respectively, as described above. After reverse transcriptase reactions, the cDNAs were amplified using the TaqPCR master mix kit by different primers. The primers for TGF-β1 were as follows: forward, 5′-GGC GAG TTT GAG TTC CAA AG-3′; and reverse, 5′-TAA CGG GTT CAT CGA TCC TC-3′. The primers for TGF-β1 type II receptors were as follows: forward, 5′-ATG GTG ACG GTA CTG TTC TAC-3′; and reverse, 5′-CCT CAC CTC TCC GGA GGC ATC-3′. The PCR products were separated by 2% agarose gel and visualized with a UV transilluminator.
Statistical analysis.
Duplicate or triplicate cultures with each treatment were prepared for each experiment. The data from at least three independent experiments were pooled together. All data, given as mean ± SEM, were acquired and analyzed blinded to treatment types. Two-tailed and unequal variance Student's t tests were used to determine statistical difference. Significance was defined as p < 0.05.
Results
Schwann cells promote synaptogenesis in vitro
We used Xenopus nerve–muscle cocultures to examine the synaptogenic effect of Schwann cells because Xenopus cultures provide a convenient bioassay to study synaptogenesis. When embryonic Xenopus spinal neurons are cocultured with myotomal muscle cells, neuronal processes make random contacts with muscle cells, followed by the formation of NMJs. One characteristic of vertebrate NMJ formation is the clustering of AChRs on the muscle cell at the nerve–muscle contact, which can be visualized by α-BTX staining (Anderson and Cohen, 1977). The extent of synapse formation can be quantified by counting the percentage of nerve–muscle contacts that are associated with AChR clusters. To avoid the potential variations caused by different batches of SC-CM, the same batch of SC-CM was used when cultures with various treatments were compared with control cultures.
In the absence of trophic support, dissociated Xenopus spinal neurons do not survive long in culture, and the majority of neurons die within 3 d after plating. To increase the survival of Xenopus spinal neurons, we followed the procedures previously described by Peng et al. (2003) using the dissociated Xenopus spinal neurons that could survive for 7–10 d in cultures when treated with a mixture of trophic factors plus elevation of cAMP level (indicated here as trophic stimulation) (see Materials and Methods). As previously shown, although spinal neurons extended neurites profusely after this treatment, NMJ formation between neurons and muscles was reduced, because few AChR clusters were observed at nerve–muscle contacts (Fig. 1 A,B). When SC-CM was added to nerve–muscle cocultures under trophic stimulation, more AChR clusters were formed along nerve–muscle contacts (Fig. 1 C,D). Quantified data are shown in Figure 1 E. Data from sister cultures were compared. Only 20.4 ± 3.5% of the nerve–muscle contacts were associated with AChR clusters in cultures under trophic stimulation alone (625 nerve–muscle contacts counted in four experiments) (duplicate or triplicate cultures were used in each experiment throughout this study; see Materials and Methods). The addition of SC-CM significantly (p < 0.05, Student's t test) increased the formation of AChR clusters to 52.7 ± 3.3% of the nerve–muscle contacts (359 contacts counted in four experiments). These data confirm that, although trophic stimulation promotes neuronal survival and neurite outgrowth, it inhibits synaptogenesis between neurons and muscle cells, as shown previously (Peng et al., 2003). Molecules derived from Schwann cells are able to restore the synaptogenic ability of neurons. Furthermore, because SC-CM is sufficient to promote synapse formation without the presence of Schwann cells themselves, the synaptogenic factors produced by Schwann cells appear to be diffusible molecules.
Figure 1.
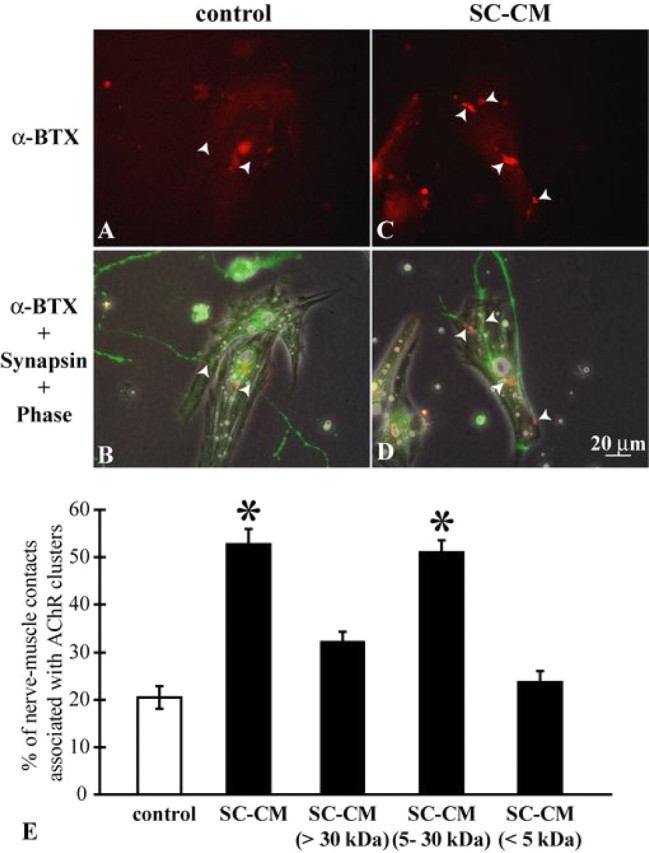
Schwann cell-conditioned medium promotes synapse formation in vitro. A, B, Xenopus nerve–muscle cocultures were treated with neurotrophins plus cAMP elevation (trophic stimulation). In this example, no α-BTX-positive staining was detected at the nerve–muscle contacts (arrowheads). In this figure and the following figures, the top panels depict α-BTX staining for AChRs seen with fluorescence microscopy; the bottom panels combine fluorescence microscopy of α-BTX and synapsin staining for neurites with phase-contrast microscopy. Note that yolk granules, which are autofluorescent, can be seen in some muscle fibers. C, D, Nerve–muscle cocultures with trophic stimulation were also treated with SC-CM. Positive α-BTX staining at nerve–muscle contacts is labeled with arrowheads. Scale bar in D applies to A–C. E, Quantification of AChR cluster formation at nerve–muscle contacts under various treatments. Nerve–muscle cocultures with trophic stimulation alone showed AChR clusters only at 20.4 ± 3.5% of nerve–muscle contacts [control (open bar), 625 contacts]. SC-CM and the fraction of SC-CM containing molecules between 5 and 30 kDa increased the percentage of nerve–muscle contacts associated with AChR clusters to 52.7 ± 3.3% (359 contacts) and 51.1 ± 2.5% (360 contacts), respectively. Neither the fraction of SC-CM containing molecules smaller than 5 kDa (23.7 ± 2.3%; 216 contacts) nor the fraction containing molecules larger than 30 kDa (32.2 ± 2.1%; 397 contacts) increased significantly, compared with control, the percentage of nerve–muscle contacts associated with AChR clusters. Thus, molecules derived from Schwann cells that are active in increasing synaptic number are within the molecular weight range of 5–30 kDa. Data are mean ± SEM. *p < 0.05; two-tailed, unequal variance Student's t test.
To identify the synaptogenic molecules of Schwann cells, we first investigated the ranges of their molecular weights. Using centrifugal filters with different molecular weight cutoffs (5 or 30 kDa), we could separate SC-CM into three fractions: the fraction with molecules smaller than 5 kDa, the fraction with those between 5 kDa and 30 kDa, and the fraction with those larger than 30 kDa. These three fractions of SC-CM were then surveyed for their synaptogenic effects using AChR cluster formation assays. As quantified in Figure 1 E, only the fraction containing molecules between 5 and 30 kDa was effective in increasing the formation of AChR clusters at nerve–muscle contacts [SC-CM (5–30 kDa), 51.1 ± 2.5%; 360 nerve–muscle contacts counted in four experiments]. This fraction of SC-CM that contained molecules between 5 and 30 kDa enhanced the formation of AChR clusters to a degree similar to the same batch of SC-CM without filtering (SC-CM, 52.7 ± 3.3%; 359 contacts counted in four experiments). The other two fractions of SC-CM that contained molecules either smaller than 5 kDa or larger than 30 kDa had no significant effect on the formation of AChR clusters at nerve–muscle contacts [SC-CM (<5 kDa), 23.7 ± 2.3%; 216 contacts counted in four experiments; and SC-CM (>30 kDa), 32.2 ± 2.1%; 397 contacts counted in four experiments]. Together, these results suggest that the synaptogenic molecules derived from Schwann cells are molecules between 5 and 30 kDa.
TGF-β1 mimics the synaptogenic effect of SC-CM
A variety of molecules, including growth factors and extracellular matrix molecules, are secreted by Schwann cells (Mirsky et al., 2002). Some of these molecules, such as BDNF, glial cell line-derived neurotrophic factor, and NT3/4, are already present in our control culture medium without SC-CM and, therefore, unlikely to be responsible for the synaptogenic effect of SC-CM. Using a candidate approach, we identified TGF-β1 as an intriguing possible synaptogenic factor. In Drosophila, BMPs, members of the TGF-β superfamily, promote synaptic growth at NMJs (Marqués et al., 2003; McCabe et al., 2003). Whether TGF-β1 plays a similar role at vertebrate NMJs has been unknown. However, TGF-β receptor type II (TβR-II) is expressed by motor neurons and localized at NMJs (Krieglstein et al., 1998; Jiang et al., 2000), and vertebrate Schwann cells express TGF-β1 (Scherer et al., 1993). More importantly, TGF-β1 presents itself as a homodimer protein of 25 kDa, which is within the molecular weight range of 5–30 kDa. Together, these results led us to explore the possibility that TGF-β1 mediates, at least in part, the synaptogenic effect of Schwann cells.
We first examined whether TGF-β1 could promote synaptogenesis in Xenopus nerve–muscle cultures. Human recombinant TGF-β1 was added to Xenopus nerve–muscle cultures under trophic stimulation. Similar to SC-CM, 2-d treatment of TGF-β1 (10 ng/ml) significantly increased the prevalence of NMJ formation: the percentage of nerve–muscle contacts associated with AChR clusters in TGF-β1-treated cultures was increased to 60.3 ± 3.0% (1069 nerve–muscle contacts counted in 11 experiments), compared with 20.6 ± 1.8% (874 nerve–muscle contacts counted in 11 experiments) in sister cocultures under trophic stimulation alone (p < 0.05, Student's t test) (Fig. 2 A–E). This effect of TGF-β1 was dose-dependent, with the concentration of TGF-β1 ranging from 5 to 20 ng/ml and reaching a plateau at a concentration of 10 ng/ml (Fig. 2 F). Thus, 10 ng/ml TGF-β1 was used in all subsequent experiments. In addition, TGF-β1 enhanced the size of AChR clusters formed along nerve–muscle contacts (Fig. 2 G). The increase of synaptogenesis by TGF-β1 or SC-CM is not attributable to nonspecific effects on neuronal survival or neurite outgrowth, because no significant changes were observed in neuronal survival, neurite numbers, or neurite outgrowth in pure Xenopus neuron cultures under trophic stimulation (supplemental Fig. S1A–C, available at www.jneurosci.org as supplemental material). Additionally, the enhancement of synaptogenesis by TGF-β1 or SC-CM is not a direct effect of AChR cluster formation on muscles, as shown by the lack of changes in the number or the size of spontaneous AChR clusters in pure Xenopus muscle cultures treated with TGF-β1 or SC-CM (supplemental Fig. S1D,E, available at www.jneurosci.org as supplemental material). Altogether, these results demonstrate that TGF-β1, similar to SC-CM, promotes synaptogenesis in Xenopus nerve–muscle cocultures.
Figure 2.

TGF-β1 mimics the synaptogenesis effect of SC-CM. A, B, Control nerve–muscle cocultures were treated with trophic stimulation alone. No α-BTX staining was observed at the nerve–muscle contacts (arrowheads) in this example. C, D, Nerve–muscle cocultures were treated with TGF-β1 (10 ng/ml) in addition to trophic stimulation. Note that α-BTX staining was present at nerve–muscle contacts (arrowheads). Scale bar in D applies to A–C. E, The effect of TGF-β1 (10 ng/ml) on the formation of AChR clusters was quantified. Whereas AChR clusters were formed only at 20.3 ± 1.9% of nerve–muscle contacts (n = 985), in cocultures with trophic stimulation only (control, open bar), both TGF-β1 (10 ng/ml) and SC-CM significantly increased the percentage of nerve–muscle contacts associated with AChR clusters (TGF-β1, 59.8 ± 3.3%, 1013 contacts; and SC-CM, 62.3 ± 2.8%, 901 contacts). Altogether, TGF-β1, similar to SC-CM, increased the formation of AChR clusters in nerve–muscle cocultures. F, The dose–response curve of TGF-β1 in the formation of AChR clusters at nerve–muscle contacts. The synaptogenic effect of TGF-β1 ranged from 38.8 ± 1.8% (n = 300 contacts) at 5 ng/ml to 61.9 ± 1.3% (n = 213 contacts) at 20 ng/ml, and it reached a plateau at the concentration of 10 ng/ml (63.2 ± 2.6%, n = 501 contacts). G, The effects of TGF-β1 and SC-CM on the size of AChR clusters at nerve–muscle contacts in nerve–muscle cocultures were quantified. The average size of each AChR cluster formed along nerve–muscle contacts with trophic stimulation alone was 50.5 ± 5.5 μm2 (n = 40 contacts) (open bar). The addition of TGF-β1 or SC-CM doubled the size of AChR clusters formed per unit length of nerve–muscle contacts (TGF-β1, 95.4 ± 8.6 μm2, n = 121 contacts; and SC-CM, 106.1 ± 7.6 μm2, n = 83 contacts). Data are mean ± SEM. Two-tailed, unequal variance Student's t test was used to determine statistical difference. Significance was defined as *p < 0.05.
To determine whether the synaptogenic effect is specific to TGF-β1, we also tested TGF-β2 in Xenopus nerve–muscle cocultures. TGF-β2 was not effective in increasing the number of AChR clusters at nerve–muscle contacts [TGF-β2 (10 ng/ml), 21.0 ± 2.7%; 156 nerve–muscle contacts counted in three experiments, compared with sister control cultures, 22.2 ± 3.3%; 84 contacts counted in three experiments). A recent study has shown that TSPs derived from astrocytes promote synapse formation in retinal ganglia cell cultures (Christopherson et al., 2005). In contrast to the CNS, TSPs did not enhance synapse number in our Xenopus cultures [TSP (100 ng/ml), 22.1 ± 2.6%; 186 nerve–muscle contacts counted in two experiments, compared with sister control cultures, 21.9 ± 3.6%; 167 contacts counted in two experiments]. Therefore, although promoting synaptogenesis is a shared feature among synaptic glial cells, astrocytes and Schwann cells appear to exert their effects via different molecular signaling pathways.
The synaptogenic effect of Schwann cells is mediated by TGF-β1
To explore the possibility that TGF-β1 mediates the synaptogenic effect of SC-CM, we first examined whether TGF-β1 proteins are present in SC-CM. By RT-PCR, we found that TGF-β1 gene was expressed in Schwann cells from Xenopus sciatic nerves, as well as cultured Xenopus Schwann cells, but was not detectable in cultured Xenopus spinal neurons or muscle cells (Fig. 3 A). In addition, TGF-β1 protein was detected in SC-CM by Western blot (Fig. 3 B). The concentration of TGF-β1 in SC-CM (without being concentrated) was ∼20 ng/ml, as assessed by ELISA. Furthermore, as shown by RT-PCR, TβR-II was expressed in cultured Xenopus spinal neurons, but not in cultured Xenopus muscle cells (Fig. 3 C). Hence, the presence of TGF-β1 protein in SC-CM and the expression of TβR-II in cultured Xenopus spinal neurons suggest that Schwann cell-derived TGF-β1 may bind to TβR-II on neurons, which in turn activates a TGF-β signaling pathway in neurons and may play a role in modulating neuronal functions.
Figure 3.

SC-CM contains TGF-β1. A, The expressions of TGF-β1 mRNA were examined using RT-PCR. Both Xenopus Schwann cells from sciatic nerves and cultured Xenopus Schwann cells, but not cultured Xenopus neurons or muscle cells, expressed TGF-β1. B, The presence of TGF-β1 protein in SC-CM was detected by Western blot. TGF-β1 protein, molecular weight of 25 kDa in nonreducing condition, was detected in SC-CM. Human recombinant TGF-β1 was used as a positive control and culture medium was used as a negative control for Western blot. C, The expression of TβR-II in cultured Xenopus neurons and muscle cells. TβR-II mRNA was expressed in cultured neurons but not in muscle cells. The expression of general gene GAPDH was used as a positive control for the sample of total cDNA in RT-PCR (A, C).
To test the potential involvement of TGF-β1 in the synaptogenic effect of SC-CM, we used immunoprecipitation to deplete TGF-β1 protein from SC-CM. The absence of TGF-β1 protein in SC-CM after immunoprecipitation was confirmed by Western blot (Fig. 4 J). Whether SC-CM after immunodepletion of TGF-β1 is capable of increasing synaptic numbers was then examined in nerve–muscle cocultures under trophic stimulation. As shown in Figure 4, whereas few AChR clusters were formed at the nerve–muscle contacts in control cultures (A, B), the treatment of TGF-β1 (C, D) or SC-CM (E, F) substantially increased the number of AChR clusters formed along the nerve–muscle contacts. When TGF-β1 was removed from the same batch of SC-CM by immunoprecipitation, SC-CM was no longer effective in increasing the formation of AChR clusters at the nerve–muscle contacts (G, H). As quantified in Figure 4 I, both TGF-β1 and SC-CM restored the level of synapse formation (TGF-β1, 60.9 ± 3.5%; 809 contacts counted in nine experiments; and SC-CM, 62.9 ± 3.1%; 518 contacts counted in nine experiments), compared with sister control cocultures under only trophic stimulation (19.7 ± 2.1%; 687 contacts counted in nine experiments). In contrast, SC-CM that had been immunodepleted of TGF-β1 failed to increase the percentage of nerve–muscle contacts associated with AChR clusters (SC-CM depleted of TGF-β1, 19.8 ± 2.5%; 742 contacts counted in five experiments), which was a result similar to that found with control cultures.
Figure 4.

TGF-β1 mediates SC-CM-promoted synaptogenesis. A, B, Control nerve–muscle cocultures after trophic stimulation. No AChR clusters, as indicated by the absence of α-BTX staining, were formed along nerve–muscle contacts (arrowheads) in this example. C, D, Nerve–muscle cocultures were treated with TGF-β1 in addition to trophic stimulation. More α-BTX staining was observed at nerve–muscle contacts (arrowheads). E, F, Nerve–muscle cocultures were treated with SC-CM in the presence of trophic stimulation. In this example, note that AChR clusters were formed at nerve–muscle contacts (arrowheads). G, H, One example of nerve–muscle cocultures treated with SC-CM that was immunodepleted of TGF-β1. No α-BTX staining was detected at nerve–muscle contacts when SC-CM was depleted of TGF-β1. Scale bar in H applies to A–G. I, The formation of AChR clusters at nerve–muscle contacts under different treatments was quantified. Data from 5–10 independent experiments were combined. Only 19.7 ± 2.1% of nerve–muscle contacts (n = 687) were associated with AChR clusters in control cultures with trophic stimulation only, whereas both TGF-β1 and SC-CM increased the percentage of nerve–muscle contacts associated with AChR clusters (TGF-β1, 60.9 ± 3.5%, n = 809 contacts; and SC-CM, 62.9 ± 3.1%, n = 519 contacts). SC-CM immunodepleted of TGF-β1 lost its synaptogenic effect: only 19.8 ± 2.5% of contacts (n = 742) were associated with AChR clusters. LAP, which prevents TGF-β1 from binding to its receptors, blocked the synaptogenic effect of TGF-β1 and SC-CM (TGF-β1 + LAP, 17.5 ± 2.6%, n = 556 contacts; SC-CM + LAP, 21.4 ± 2.5%, n = 624 contacts). Additionally, blockade of TβR-I kinase ALK-5 abolished the synaptogenic effect of TGF-β1 and SC-CM (TGF-β1 + TβR-I inhibitor, 20.0 ± 2.3%, n = 251 contacts; SC-CM + TβR-I, 25.8 ± 1.7%, n = 284 contacts). Thus, TGF-β1 is required for the synaptogenic effect of SC-CM. Data are mean ± SEM, *p < 0.05; two-tailed, unequal variance Student's t test. J, TGF-β1 protein was detected in SC-CM (lane 1), as well as in SC-CM with mock immunoprecipitation (IP) (lane 2) (beads without antibodies against TGF-β1, lane 4). After immunoprecipitation with antibodies against TGF-β1, TGF-β1 protein was absent from SC-CM (lane 3) but was associated with protein G-Sepharose beads (lane 5).
It is known that TGF-β1 is secreted as a latent dimeric complex and activated by the release of mature TGF-β1 from its noncovalently associated proproteins, called latency-associated proteins (LAPs) (Massagué, 1998; Hyytiäinen et al., 2004). The association of recombinant LAP prevents TGF-β1 from binding to its cell-surface receptor TβR-II, thereby inhibiting the bioactivity of TGF-β1 (Gentry and Nash, 1990; Böttinger et al., 1996). To test whether LAP blocks the synaptogenic effect of TGF-β1 in our culture system, recombinant LAP (25 ng/ml) was applied together with TGF-β1 to nerve–muscle cocultures. As quantified in Figure 4 I, the synaptogenic effect of TGF-β1 was blocked by LAP (TGF-β1 + LAP, 17.5 ± 2.6%; 556 contacts counted in five experiments, compared with TGF-β1, 60.9 ± 3.5%). To examine whether TGF-β1 is necessary for the effect of SC-CM in synaptogenesis, LAP was added to nerve–muscle cultures together with SC-CM. The synaptogenic effect of SC-CM was also abolished by LAP (SC-CM + LAP, 21.4 ± 2.5%; 624 contacts counted in five experiments, compared with SC-CM, 62.9 ± 3.1%). Together, these findings suggest that TGF-β1 is not only sufficient but also required for the synaptogenic effect of SC-CM.
Having demonstrated the requirement of TGF-β1 in the synaptogenic effect of SC-CM, we next asked whether the typical TGF-β signaling pathway was involved in TGF-β1- or SC-CM-enhanced synaptogenesis. TGF-β1 elicits its bioactivity via a heteromeric receptor complex, consisting of TGF-β receptors type I (TβR-I) and TβR-II (ten Dijke and Hill, 2004). TβR-II is required for the transmembrane signaling of TGF-β1, whereas phosphorylation of TβR-I by TβR-II is required for activating SMAD proteins and propagating the cellular signal downstream of TGF-β1. As shown above, cultured Xenopus spinal neurons express TβR-II (Fig. 3 C), and preventing the binding of TGF-β1 to TβR-II by LAP abolishes the synaptogenic effect of TβR-II (Fig. 4 I). To directly test the involvement of TGF-β receptors, TβR-I kinase inhibitor activin receptor-like kinase-5 (ALK-5) was used together with TGF-β1 in nerve–muscle cocultures. The increase induced by TGF-β1 in the percentage of nerve–muscle contacts associated with AChR clusters was abolished by ALK-5 inhibitor (100 nm) (TGF-β1 + TβR-I inhibitor, 20.0 ± 2.3%; 251 contacts counted in four experiments) (Fig. 4 I). The synaptogenic effect of SC-CM was also impeded by TβR-I kinase inhibitor (SC-CM + TβR-I inhibitor, 28.2 ± 0.9%; 284 contacts counted in four experiments). Therefore, our data suggest that TGF-β1 elicits its synaptogenic effect via the activation of TβR-I. The fact that TβR-I kinase inhibitor also blocks the synaptogenic effect of SC-CM further supports the hypothesis that TGF-β1 mediates Schwann cell-promoted synaptogenesis.
Mammalian Schwann cells promote synaptogenesis in vitro
The synaptogenic effect of Xenopus Schwann cells raises the question of whether mammalian Schwann cells also promote NMJ formation. As demonstrated in Figure 5 A, TGF-β1 mRNA was detected in neonatal rat sciatic nerves and cultured rat Schwann cells. In addition, TGF-β1 proteins were present in mSC-CM as confirmed with Western blot (Fig. 5 B). Thus, mammalian Schwann cells, by releasing TGF-β1, might also enhance synaptogenesis. To test this hypothesis, we first examined the effect of mSC-CM in synapse formation using AChR cluster formation assay in Xenopus nerve–muscle cultures (Fig. 5 C–K). The presence of mSC-CM significantly increased the percentage of nerve–muscle contacts associated with AChR clusters, compared with sister control cultures (mSC-CM, 66.8 ± 4.5%; 247 contacts counted in five experiments, versus control, 18.3 ± 2.5%; 242 contacts counted in five experiments). The enhancement by mSC-CM is similar to that produced by TGF-β1 treatment (68.1 ± 2.5%; 239 contacts counted in five experiments). Furthermore, mSC-CM immunodepleted of TGF-β1 failed to increase the number of synaptic contacts (mSC-CM depleted of TGF-β1, 18.6 ± 2.2%; 311 contacts counted in five experiments, compared with control, 18.3 ± 2.5%), suggesting that the synaptogenic effect of mammalian SC-CM requires TGF-β1. Together, these results show that mammalian Schwann cells, similar to Xenopus Schwann cells, also promote synaptogenesis in vitro. The synaptogenic effect of mammalian Schwann cells is likely mediated by TGF-β1 as well.
Figure 5.

Mammalian Schwann cells promote synapse formation in vitro. A, The expression of TGF-β1 mRNA in mammalian tissues was examined using RT-PCR. Cultured rat Schwann cells, as well as sciatic nerves from neonatal rats, expressed TGF-β1 mRNA. B, The presence of TGF-β1 protein was investigated by Western blot. TGF-β1 protein was detected in mammalian SC-CM. Human recombinant TGF-β1 was used as a positive control and culture medium (DMEM) was used as a negative control for Western blot. C, D, Xenopus nerve–muscle cocultures were treated with trophic stimulation only. The formation of AChR clusters was reduced along the nerve–muscle contacts (arrowheads). E, F, Nerve–muscle cocultures with trophic stimulation were treated with TGF-β1. The formation of AChR clusters was restored at nerve–muscle contacts (arrowheads), as demonstrated by the positive staining of α-BTX. G, H, Nerve–muscle cocultures were treated with mammalian SC-CM in the presence of trophic stimulation. The addition of mammalian SC-CM increased the formation of AChR clusters at nerve–muscle contacts (arrowheads). I, J, Nerve–muscle cocultures were treated with mammalian SC-CM that was immunodepleted of TGF-β1 protein. In this example, note that no α-BTX staining was observed at nerve–muscle contacts (arrowheads). Scale bar in J applies to C–I. K, Quantification of the formation of AChR clusters at nerve–muscle contacts. Data from five independent experiments were combined. In control nerve–muscle cocultures with trophic stimulation alone, only 18.3 ± 2.5% of nerve–muscle contacts (n = 242) were associated with AChR clusters. The addition of mammalian SC-CM increased the percentage of nerve–muscle contacts associated with AChR clusters to 66.8 ± 4.5% (mSC-CM, 247 contacts), which is similar to the effect produced by treatment with TGF-β1 (TGF-β1, 68.1 ± 2.5%, 279 contacts). When TGF-β1 protein was immunodepleted from SC-CM, SC-CM was no longer effective in promoting the formation of AChR clusters at nerve–muscle contacts (mSC-CM after immunoprecipitation with mAb TGF-β1, 18.6 ± 2.2, n = 311 contacts]. Because mammalian Schwann cells were cultured in DMEM, DMEM was used as a control. DMEM alone had no effect on the formation of AChR clusters at nerve–muscle contacts (DMEM, 17.4 ± 1.3%, n = 238 contacts). Data are mean ± SEM, *p < 0.05; two-tailed, unequal variance Student's t test.
TGF-β1 promotes synaptogenesis by upregulating neural agrin expression
How does TGF-β1 promote synaptogenesis? As shown above, TGF-β1 itself does not affect AChR cluster formation in pure muscle cultures (supplemental Fig. S1D, available at www.jneurosci.org as supplemental material). Together with the observation that TβR-II is expressed in spinal neurons but not muscle cells in Xenopus cultures (Fig. 3 C), it is likely that TGF-β1 elicits its synaptogenesis-promoting effect via presynaptic neurons.
It is known that neuronal agrin is required for the development of postsynaptic structures at vertebrate NMJs (Sanes and Lichtman, 2001). There are four isoforms of agrin, which differ in the number (0, 8, 11, or 19) of amino acids inserted at the B site (called Z site in mammalian agrin) near the C terminal (Tsim et al., 1992). B8, B11, and B19, but not B0, agrin isoforms are effective in inducing AChR clusters in muscles (McMahan et al., 1992; Gesemann et al., 1995). Motoneurons, muscles, and Schwann cells all express B0 agrin, whereas only motoneurons (Tsim et al., 1992) and Schwann cells (in the frog) (Yang et al., 2001) express B8, B11, and B19 agrin. We have previously shown that although trophic stimulation promotes neurite outgrowth and neuronal survival, it inhibits the expression of agrin in spinal neurons, thereby preventing the formation of AChR clusters at nerve–muscle contacts (Peng et al., 2003). Schwann cell-derived molecules restore neuronal agrin expression and promote synapse formation. To test whether TGF-β1 acts in a manner similar to SC-CM in regulating agrin expression, RT-PCR was performed to examine agrin mRNA levels in pure spinal neurons under various conditions. As shown in Figure 6 A, a very low level of agrin was expressed in neurons in control cultures under trophic stimulation alone (lane 2). The addition of TGF-β1 (lane 3) or SC-CM (lane 4) restored the expression of all isoforms of agrin in cultured neurons. However, SC-CM that had been immunodepleted of TGF-β1 did not affect the expression of agrin in neurons (lane 5), which showed a level of agrin similar to that in controls (lane 2). Agrin expression levels under various treatments were quantified in Figure 6 B. TGF-β1 and SC-CM each increased the mRNA level of active agrin (B8, B11, and B19) to ∼5-fold compared with control neuron cultures under trophic stimulation alone, whereas SC-CM depleted of TGF-β1 did not affect agrin mRNA levels in neurons. Agrin expression in muscle cells was not affected by TGF-β1 or SC-CM: only B0 agrin was expressed in muscle cells under all conditions (Fig. 6 C). The effect of TGF-β1 on agrin expression is not caused by general upregulation of synaptic proteins because synapsin expression in neurons was not affected by TGF-β1 or SC-CM (Fig. 6 D). Together, these data suggest that TGF-β1 may exert its synaptogenesis-promoting effect by upregulating the expression of neuronal agrin. In addition, the similar effects of TGF-β1 and SC-CM with regard to increasing agrin expression in neurons, together with the fact that TGF-β1-depleted SC-CM has no effect on agrin expression, further supports the theory that TGF-β1 is required for SC-CM-promoted synapse formation.
Figure 6.
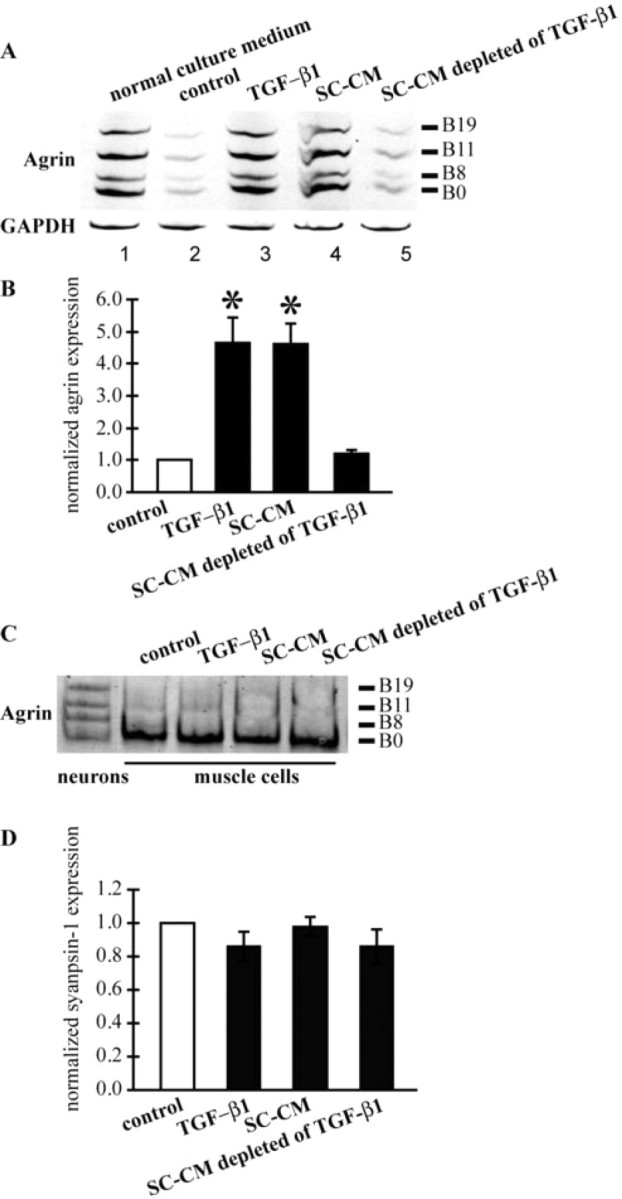
TGF-β1 upregulates neuronal agrin expression. A, Expression of agrin isoforms at C-terminal B splicing site in pure neuron cultures was investigated using RT-PCR. Two-day-old pure neuron cultures without any trophic stimulation expressed all four isoforms of agrin (top, lane 1). Trophic stimulation reduced the expression of all isoforms of agrin (top, lane 2). The addition of TGF-β1 and SC-CM each restored agrin expression in neurons (top, lanes 3 and 4, respectively). Depleting TGF-β1 from SC-CM abolished the effect of SC-CM in promoting agrin expression (top, lane 5). GAPDH gene expression was not affected by any of the treatments (bottom). B, Quantification of active agrin (B8, B11, and B19) expression in pure neurons. Both TGF-β1 and SC-CM were able to significantly (*p < 0.05) increase the expression of active agrin in pure neuron cultures. Additionally, TGF-β1 was required for the upregulation of agrin by SC-CM. C, Only the inactive isoform of agrin (B0) was expressed in muscle cells under the various treatments indicated. D, Quantification of synapsin-1 expression in pure neurons with the different treatments indicated. Similar levels of synapsin-1 were expressed in pure neuron cultures with these treatments.
Discussion
Schwann cell-derived TGF-β1 promotes synaptogenesis at the developing NMJ in vitro
The present study has provided several lines of evidence supporting the novel idea of TGF-β1 as a Schwann cell-derived factor that promotes synaptogenesis. First, the synaptogenic effect (measured as the increase in AChR clusters at nerve–muscle contacts) of Schwann cells is mediated by soluble molecules within the molecular weight range of 5–30 kDa. Second, TGF-β1, a homodimer protein of 25 kDa, is sufficient to increase synaptic numbers in Xenopus nerve–muscle cocultures. Third, Xenopus Schwann cells express TGF-β1, and SC-CM contains TGF-β1 protein as well. Fourth, SC-CM immunodepleted of TGF-β1 fails to promote synaptogenesis, indicating that TGF-β1 is required for the synaptogenic effect of SC-CM. In addition, LAP, which prevents TGF-β1 bioactivity by forming latent complex with TGF-β1, blocks the effects of both TGF-β1 and SC-CM in synapse formation. Finally, ALK-5 kinase inhibitor abolishes the effects of both TGF-β1 and SC-CM in enhancing synaptogenesis. Thus, the synaptogenic effect of TGF-β1 is elicited via activating TβR-I receptor kinase ALK-5. Together, our findings demonstrate that TGF-β1 is necessary and sufficient to mediate the SC-CM-induced synaptogenesis in vitro. To our knowledge, TGF-β1 is the first Schwann cell-derived synaptogenic molecule identified in the PNS.
We have recently shown that SC-CM also enhances spontaneous synaptic transmission at developing NMJs both in cultures and in situ (Cao and Ko, 2007). The potentiation of SC-CM in synaptic transmission is mediated by unknown small molecules between 500 and 5000 Da. Because TGF-β1 itself has no effect on spontaneous transmitter release (data not shown), and TGF-β1 is a homodimer protein of 25 kDa, the potentiation effect of Schwann cells is unlikely to be mediated by TGF-β1. In the CNS, it has been reported that TSPs mediate the synaptogenic effect of astrocytes (Christopherson et al., 2005). However, TSPs did not increase the synaptic numbers in Xenopus nerve–muscle cultures. In addition, TSPs form large (>300 kDa) oligomeric complexes, which are beyond the range of 5–30 kDa. Thus, TSPs themselves unlikely have a direct effect on the synaptogenic effect of Schwann cells.
How does TGF-β1 promote synaptogenesis?
The increase in the number of AChR clusters formed at nerve–muscle contacts indicates that TGF-β1 promotes synaptogenesis. We have previously shown that Schwann cells express both active and inactive forms of agrin (Yang et al., 2001). Thus, it is possible that Schwann cell-released agrin results in a global increase in the formation of AChR clusters on muscle cells, which would result in a higher chance of encountering AChR clusters near neurites. This, however, seems unlikely because SC-CM contains only a small amount of native agrin, probably because of protein degradation in SC-CM collected from the long-term Schwann cell cultures (Yang et al., 2001). In addition, we did not observe any increase in the number of AChR clusters in pure muscle cultures treated with SC-CM or TGF-β1. Furthermore, SC-CM depleted solely of TGF-β1 loses its synaptogenic effect on nerve–muscle cultures. We also excluded the possibility that TGF-β1-induced synaptogenesis might be attributed to an increase in neuronal survival or neurite outgrowth, which could result in the formation of more nerve–muscle contacts. As shown in supplemental Fig S1A–C, available at www.jneurosci.org as supplemental material, TGF-β1 does not affect the survival or the neurite outgrowth of Xenopus spinal neurons in cultures. Although TGF-β1 has been shown to promote neuronal survival in rat motoneuron cultures (Martinou et al., 1990), the trophic stimulation in our culture system might have already elicited the maximum effect in promoting neuronal survival and neurite outgrowth (Hanson et al., 1998). Thus, the increase in the synaptic number is most likely an effect of TGF-β1 on synaptogenesis per se.
In neuron-muscle cocultures, growing neurites randomly contact muscle cells. After the encounter, postsynaptic structures are differentiated at the nerve–muscle contacts, including the clustering of AChRs. Neuronal agrin is important in clustering AChRs because agrin-deficient neurons fail to induce the formation of AChR clusters on muscle cells in vitro (Misgeld et al., 2005). However, relatively little is known about how the expression of agrin in motoneurons is regulated during development. Previously, we have reported that although trophic stimulation promotes neuronal survival and neurite outgrowth, it downregulates neuronal agrin synthesis, which may account for the decreased synaptogenic ability of spinal neurons (Peng et al., 2003). Molecules derived from Schwann cells promote synaptogenesis at the nerve–muscle contact by restoring the expression of neuronal agrin. Our current data demonstrate that Schwann cell-derived TGF-β1 also restores agrin expression in neurons, which could explain the increase of synaptic numbers by TGF-β1. The similar property of SC-CM and TGF-β1 with regard to modulating agrin expression further suggests that TGF-β1 mediates Schwann cell-enhanced synapse formation. Given that fewer and smaller AChR clusters are formed at nerve–muscle contacts in agrin −/− mice (Gautam et al., 1996), restoration of neuronal agrin expression would induce larger and more numerous AChR clusters. Our finding is consistent with this idea, because both the number and the size of AChR clusters at nerve–muscle contacts were increased in TGF-β1-treated cocultures.
Our finding that TGF-β1 upregulates agrin expression in pure neuron cultures indicates that TGF-β1 acts directly on presynaptic neurons rather than through muscle cells. Furthermore, the changes in gene expression of agrin suggest the involvement of the neuronal soma in the synaptogenic effect of TGF-β1. We do not know, however, whether TGF-β1 acts directly on nerve terminals, motor axons, and/or the neuronal soma. TβR-II receptors are localized at mammalian motor nerve terminals and on motoneuron cell bodies (Krieglstein et al., 1998; Jiang et al., 2000). It is thus possible that TGF-β1 binds to TβR-II receptors on the nerve terminal, which phosphorylates TβR-I and in turn activates the signaling pathway in neurons that turns on agrin expression.
Possible roles of TGF-β1 in the development of NMJs in vivo
We have shown that mammalian Schwann cells, by releasing TGF-β1, also increase synapse number in Xenopus cultures. A similar synaptogenic effect of mammalian Schwann cells has been reported in mammalian motoneuron cultures (Ullian et al., 2004), as well as in rat motoneuron and human muscle fiber cocultures (Guettier-Sigrist et al., 2000; Mars et al., 2001). It would be intriguing to test whether these synaptogenic effects of Schwann cells are also mediated by TGF-β1. The in vitro effect of TGF-β1 raises the question whether TGF-β1 plays a similar synaptogenic role in developing NMJs in vivo. Studies from Drosophila have shown that BMPs, members of the TGF-β superfamily, are retrograde signals from muscles to enhance synaptic growth at the NMJ (McCabe et al., 2003). From the invertebrate studies, it is tempting to speculate that TGF-β1 also plays a role in developing vertebrate NMJs in vivo. TGF-β1 is expressed by Schwann cells, and its protein is present in vertebrate NMJs (Scherer et al., 1993). Furthermore, TβR-II receptors are localized at the NMJ (Krieglstein et al., 1998; Jiang et al., 2000). However, direct evidence that TGF-β1 promotes synaptogenesis at vertebrate NMJs is missing. TGF-β1 −/− mice have been generated to study the multiple functions of TGF-β1. Unfortunately, 50% of TGF-β1 −/− mice of a mixed genetic background die before embryonic day 9 because of defects in vasculogenesis and angiogenesis (Shull et al., 1992; Kulkarni et al., 1993). The remaining 50% die at ∼3–4 weeks because of wasting syndrome. Whether NMJ development is impaired in TGF-β1 −/− mice remains to be examined.
Our findings provide additional evidence supporting the concept that Schwann cells are active players in the NMJ. PSCs extend processes that guide nerve terminal growth during development and regeneration. The loss of transiently formed NMJs in mutants lacking Schwann cells suggests the importance of Schwann cells in the maintenance of developing NMJs (Riethmacher et al., 1997; Morris et al., 1999; Woldeyesus et al., 1999; Lin et al., 2000; Wolpowitz et al., 2000). Furthermore, our previous study, which used complement-mediated cell lysis to ablate PSCs in tadpole muscles, further highlighted the essential role of PSCs in promoting and guiding synaptic growth as well as AChR clustering (Reddy et al., 2003). Our present study indicates that Schwann cell-derived TGF-β1 promotes synaptogenesis. Whether TGF-β1 is also involved in the maintenance or guidance roles of PSCs in synaptic growth remains to be investigated. Moreover, our recent finding that Schwann cell-derived molecules potentiate spontaneous synaptic transmission at developing NMJs provides another potential mechanism by which Schwann cells promote the maturation and growth of developing NMJs (Cao and Ko, 2007).
In conclusion, we have demonstrated that TGF-β1 is a Schwann cell-derived molecule that promotes synaptogenesis at nerve–muscle contacts in vitro. Similar glia-enhanced synaptogenesis has also been observed at CNS synapses. Together, these studies further support the concept that glial cells are an essential and integral element of the tripartite synapse. The identification of TGF-β1 as a Schwann cell-derived synaptogenic molecule that complements astrocyte-derived synaptogenic molecules in the CNS should advance the molecular understanding of synapse–glia interactions.
Footnotes
This work was supported by National Institutes of Health Grant NS17954. We thank Dr. S. Koirala and members of the Ko laboratory, S. Lin, K. Ling, and Y. E. Yoo, for their critical comments on this manuscript.
References
- Allen NJ, Barres BA. Signaling between glia and neurons: focus on synaptic plasticity. Curr Opin Neurobiol. 2005;15:542–548. doi: 10.1016/j.conb.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Anderson MJ, Cohen MW. Nerve-induced and spontaneous redistribution of acetylcholine receptors on cultured muscle cells. J Physiol. 1977;268:757–773. doi: 10.1113/jphysiol.1977.sp011880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astrow SH, Qiang H, Ko CP. Perisynaptic Schwann cells at neuromuscular junctions revealed by a novel monoclonal antibody. J Neurocytol. 1998;27:667–681. doi: 10.1023/a:1006916232627. [DOI] [PubMed] [Google Scholar]
- Böttinger EP, Factor VM, Tsang ML, Weatherbee JA, Kopp JB, Qian SW, Wakefield LM, Roberts AB, Thorgeirsson SS, Sporn MB. The recombinant proregion of transforming growth factor beta1 (latency-associated peptide) inhibits active transforming growth factor beta1 in transgenic mice. Proc Natl Acad Sci U S A. 1996;93:5877–5882. doi: 10.1073/pnas.93.12.5877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao G, Ko CP. Schwann cell-derived factors modulate synaptic activities at developing neuromuscular synapses. J Neurosci. 2007;27:6712–6722. doi: 10.1523/JNEUROSCI.1329-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandross KJ, Chanson M, Spray DC, Kessler JA. Transforming growth factor-beta 1 and forskolin modulate gap junctional communication and cellular phenotype of cultured Schwann cells. J Neurosci. 1995;15:262–273. doi: 10.1523/JNEUROSCI.15-01-00262.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120:421–433. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Cohen J, Wilkin GP. Neural cell culture: a practical approach. New York: Oxford UP; 1995. [Google Scholar]
- Colomar A, Robitaille R. Glial modulation of synaptic transmission at the neuromuscular junction. Glia. 2004;47:284–289. doi: 10.1002/glia.20086. [DOI] [PubMed] [Google Scholar]
- Feng Z, Ko CP. Neuronal glia interactions at the vertebrate neuromuscular junction. Curr Opin Pharmacol. 2007;7:316–324. doi: 10.1016/j.coph.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Fields RD, Stevens-Graham B. New insights into neuron-glia communication. Science. 2002;298:556–562. doi: 10.1126/science.298.5593.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam M, Noakes PG, Moscoso L, Rupp F, Scheller RH, Merlie JP, Sanes JR. Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell. 1996;85:525–535. doi: 10.1016/s0092-8674(00)81253-2. [DOI] [PubMed] [Google Scholar]
- Gentry LE, Nash BW. The pro domain of pre-pro-transforming growth factor beta 1 when independently expressed is a functional binding protein for the mature growth factor. Biochemistry. 1990;29:6851–6857. doi: 10.1021/bi00481a014. [DOI] [PubMed] [Google Scholar]
- Gesemann M, Denzer AJ, Ruegg MA. Acetylcholine receptor-aggregating activity of agrin isoforms and mapping of the active site. J Cell Biol. 1995;128:625–636. doi: 10.1083/jcb.128.4.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guettier-Sigrist S, Coupin G, Warter JM, Poindron P. Cell types required to efficiently innervate human muscle cells in vitro. Exp Cell Res. 2000;259:204–212. doi: 10.1006/excr.2000.4968. [DOI] [PubMed] [Google Scholar]
- Hanson MG, Jr, Shen S, Wiemelt AP, McMorris FA, Barres BA. Cyclic AMP elevation is sufficient to promote the survival of spinal motor neurons in vitro. J Neurosci. 1998;18:7361–7371. doi: 10.1523/JNEUROSCI.18-18-07361.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haydon PG. GLIA: listening and talking to the synapse. Nat Rev Neurosci. 2001;2:185–193. doi: 10.1038/35058528. [DOI] [PubMed] [Google Scholar]
- Herrera AA, Qiang H, Ko CP. The role of perisynaptic Schwann cells in development of neuromuscular junctions in the frog (Xenopus laevis) J Neurobiol. 2000;45:237–254. doi: 10.1002/1097-4695(200012)45:4<237::aid-neu5>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Hyytiäinen M, Penttinen C, Keski-Oja J. Latent TGF-beta binding proteins: extracellular matrix association and roles in TGF-beta activation. Crit Rev Clin Lab Sci. 2004;41:233–264. doi: 10.1080/10408360490460933. [DOI] [PubMed] [Google Scholar]
- Jiang Y, McLennan IS, Koishi K, Hendry IA. Transforming growth factor-beta 2 is anterogradely and retrogradely transported in motoneurons and up-regulated after nerve injury. Neuroscience. 2000;97:735–742. doi: 10.1016/s0306-4522(00)00084-1. [DOI] [PubMed] [Google Scholar]
- Kang H, Tian L, Thompson W. Terminal Schwann cells guide the reinnervation of muscle after nerve injury. J Neurocytol. 2003;32:975–985. doi: 10.1023/B:NEUR.0000020636.27222.2d. [DOI] [PubMed] [Google Scholar]
- Ko CP, Sugiura Y, Feng Z. The biology of perisynaptic (terminal) Schwann cells. In: Armati P, editor. The biology of schwann cells: development, differentiation and immunomodulation. New York: Cambridge UP; 2007. pp. 72–99. [Google Scholar]
- Koirala S, Reddy LV, Ko CP. Roles of glial cells in the formation, function, and maintenance of the neuromuscular junction. J Neurocytol. 2003;32:987–1002. doi: 10.1023/B:NEUR.0000020637.71452.3c. [DOI] [PubMed] [Google Scholar]
- Krieglstein K, Henheik P, Farkas L, Jaszai J, Galter D, Krohn K, Unsicker K. Glial cell line-derived neurotrophic factor requires transforming growth factor-beta for exerting its full neurotrophic potential on peripheral and CNS neurons. J Neurosci. 1998;18:9822–9834. doi: 10.1523/JNEUROSCI.18-23-09822.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, Roberts AB, Sporn MB, Ward JM, Karlsson S. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci U S A. 1993;90:770–774. doi: 10.1073/pnas.90.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Sanchez HB, Deerinck T, Morris JK, Ellisman M, Lee KF. Aberrant development of motor axons and neuromuscular synapses in erbB2-deficient mice. Proc Natl Acad Sci U S A. 2000;97:1299–1304. doi: 10.1073/pnas.97.3.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marqués G, Haerry TE, Crotty ML, Xue M, Zhang B, O'Connor MB. Retrograde Gbb signaling through the Bmp type 2 receptor wishful thinking regulates systemic FMRFa expression in Drosophila. Development. 2003;130:5457–5470. doi: 10.1242/dev.00772. [DOI] [PubMed] [Google Scholar]
- Mars T, Yu KJ, Tang XM, Miranda AF, Grubic Z, Cambi F, King MP. Differentiation of glial cells and motor neurons during the formation of neuromuscular junctions in cocultures of rat spinal cord explant and human muscle. J Comp Neurol. 2001;438:239–251. doi: 10.1002/cne.1312. [DOI] [PubMed] [Google Scholar]
- Martinou JC, Le Van Thai A, Valette A, Weber MJ. Transforming growth factor beta 1 is a potent survival factor for rat embryo motoneurons in culture. Brain Res Dev Brain Res. 1990;52:175–181. doi: 10.1016/0165-3806(90)90233-o. [DOI] [PubMed] [Google Scholar]
- Massagué J. TGF-beta signal transduction. Annu Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- Massagué J. How cells read TGF-beta signals. Nat Rev Mol Cell Biol. 2000;1:169–178. doi: 10.1038/35043051. [DOI] [PubMed] [Google Scholar]
- Mauch DH, Nägler K, Schumacher S, Göritz C, Müller EC, Otto A, Pfrieger FW. CNS synaptogenesis promoted by glia-derived cholesterol. Science. 2001;294:1354–1357. doi: 10.1126/science.294.5545.1354. [DOI] [PubMed] [Google Scholar]
- McCabe BD, Marqués G, Haghighi AP, Fetter RD, Crotty ML, Haerry TE, Goodman CS, O'Connor MB. The BMP homolog Gbb provides a retrograde signal that regulates synaptic growth at the Drosophila neuromuscular junction. Neuron. 2003;39:241–254. doi: 10.1016/s0896-6273(03)00426-4. [DOI] [PubMed] [Google Scholar]
- McMahan UJ, Horton SE, Werle MJ, Honig LS, Kröger S, Ruegg MA, Escher G. Agrin isoforms and their role in synaptogenesis. Curr Opin Cell Biol. 1992;4:869–874. doi: 10.1016/0955-0674(92)90113-q. [DOI] [PubMed] [Google Scholar]
- Mirsky R, Jessen KR, Brennan A, Parkinson D, Dong Z, Meier C, Parmantier E, Lawson D. Schwann cells as regulators of nerve development. J Physiol Paris. 2002;96:17–24. doi: 10.1016/s0928-4257(01)00076-6. [DOI] [PubMed] [Google Scholar]
- Misgeld T, Kummer TT, Lichtman JW, Sanes JR. Agrin promotes synaptic differentiation by counteracting an inhibitory effect of neurotransmitter. Proc Natl Acad Sci U S A. 2005;102:11088–11093. doi: 10.1073/pnas.0504806102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JK, Lin W, Hauser C, Marchuk Y, Getman D, Lee KF. Rescue of the cardiac defect in ErbB2 mutant mice reveals essential roles of ErbB2 in peripheral nervous system development. Neuron. 1999;23:273–283. doi: 10.1016/s0896-6273(00)80779-5. [DOI] [PubMed] [Google Scholar]
- Newman EA, Volterra A. Glial control of synaptic function. Glia. 2004;47:207–208. doi: 10.1002/glia.20085. [DOI] [PubMed] [Google Scholar]
- Nieuwkoop PD, Faber J. Normal table of Xenopus laevis (Daudin): a systematical and chronological survey of development from the fertilized egg till the end of metamorphosis. Ed 2. New York: Garland; 1994. [Google Scholar]
- Peng HB, Baker LP, Chen Q. Tissue culture of Xenopus neurons and muscle cells as a model for studying synaptic induction. Methods Cell Biol. 1991;36:511–526. doi: 10.1016/s0091-679x(08)60294-0. [DOI] [PubMed] [Google Scholar]
- Peng HB, Yang JF, Dai Z, Lee CW, Hung HW, Feng ZH, Ko CP. Differential effects of neurotrophins and Schwann cell-derived signals on neuronal survival/growth and synaptogenesis. J Neurosci. 2003;23:5050–5060. doi: 10.1523/JNEUROSCI.23-12-05050.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy LV, Koirala S, Sugiura Y, Herrera AA, Ko CP. Glial cells maintain synaptic structure and function and promote development of the neuromuscular junction in vivo. Neuron. 2003;40:563–580. doi: 10.1016/s0896-6273(03)00682-2. [DOI] [PubMed] [Google Scholar]
- Reynolds ML, Woolf CJ. Terminal Schwann cells elaborate extensive processes following denervation of the motor endplate. J Neurocytol. 1992;21:50–66. doi: 10.1007/BF01206897. [DOI] [PubMed] [Google Scholar]
- Riethmacher D, Sonnenberg-Riethmacher E, Brinkmann V, Yamaai T, Lewin GR, Birchmeier C. Severe neuropathies in mice with targeted mutations in the ErbB3 receptor. Nature. 1997;389:725–730. doi: 10.1038/39593. [DOI] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW. Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat Rev Neurosci. 2001;2:791–805. doi: 10.1038/35097557. [DOI] [PubMed] [Google Scholar]
- Scherer SS, Kamholz J, Jakowlew SB. Axons modulate the expression of transforming growth factor-betas in Schwann cells. Glia. 1993;8:265–276. doi: 10.1002/glia.440080407. [DOI] [PubMed] [Google Scholar]
- Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D, Annunziata N, Doetschman T. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son YJ, Thompson WJ. Schwann cell processes guide regeneration of peripheral axons. Neuron. 1995a;14:125–132. doi: 10.1016/0896-6273(95)90246-5. [DOI] [PubMed] [Google Scholar]
- Son YJ, Thompson WJ. Nerve sprouting in muscle is induced and guided by processes extended by Schwann cells. Neuron. 1995b;14:133–141. doi: 10.1016/0896-6273(95)90247-3. [DOI] [PubMed] [Google Scholar]
- Tabti N, Alder J, Poo MM. Culturing spinal neurons and muscle cells from Xenopus embryos. In: Banker G, Goslin K, editors. Culturing nerve cells. Ed 2. Cambridge, MA: MIT; 1998. pp. 237–259. [Google Scholar]
- ten Dijke P, Hill CS. New insights into TGF-beta-Smad signalling. Trends Biochem Sci. 2004;29:265–273. doi: 10.1016/j.tibs.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Todd KJ, Serrano A, Lacaille JC, Robitaille R. Glial cells in synaptic plasticity. J Physiol Paris. 2006;99:75–83. doi: 10.1016/j.jphysparis.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Tsim KW, Ruegg MA, Escher G, Kröger S, McMahan UJ. cDNA that encodes active agrin. Neuron. 1992;8:677–689. doi: 10.1016/0896-6273(92)90089-v. [DOI] [PubMed] [Google Scholar]
- Ullian EM, Christopherson KS, Barres BA. Role for glia in synaptogenesis. Glia. 2004;47:209–216. doi: 10.1002/glia.20082. [DOI] [PubMed] [Google Scholar]
- Wang T, Xie K, Lu B. Neurotrophins promote maturation of developing neuromuscular synapses. J Neurosci. 1995;15:4796–4805. doi: 10.1523/JNEUROSCI.15-07-04796.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werle MJ, Jones MA, Stanco AM. Aggregates of acetylcholine receptors are not observed under anti-agrin staining Schwann cell processes at the frog neuromuscular junction. J Neurobiol. 1999;40:45–54. [PubMed] [Google Scholar]
- Woldeyesus MT, Britsch S, Riethmacher D, Xu L, Sonnenberg-Riethmacher E, Abou-Rebyeh F, Harvey R, Caroni P, Birchmeier C. Peripheral nervous system defects in erbB2 mutants following genetic rescue of heart development. Genes Dev. 1999;13:2538–2548. doi: 10.1101/gad.13.19.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolpowitz D, Mason TB, Dietrich P, Mendelsohn M, Talmage DA, Role LW. Cysteine-rich domain isoforms of the neuregulin-1 gene are required for maintenance of peripheral synapses. Neuron. 2000;25:79–91. doi: 10.1016/s0896-6273(00)80873-9. [DOI] [PubMed] [Google Scholar]
- Yang JF, Cao G, Koirala S, Reddy LV, Ko CP. Schwann cells express active agrin and enhance aggregation of acetylcholine receptors on muscle fibers. J Neurosci. 2001;21:9572–9584. doi: 10.1523/JNEUROSCI.21-24-09572.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]