

Significance
Type IV pili are long, thin fibers, formed mainly of polymers of a single pilin protein, which are displayed on the surfaces of many bacteria, including several human pathogens. Here, we report three-dimensional reconstructions of the PilMNO inner membrane complex, alone and in complex with pilin protein, through a combination of X-ray crystallography and electron microscopy. PilMNO forms a dimeric T-shaped structure, binding two copies of the pilin protein at its extremities. The results provide a structural model for the way in which pilin is harvested from the inner membrane and made available to other components of the type IV pilus biogenesis machinery.
Abstract
Type IV pili are long fibers that are assembled by polymerization of a major pilin protein in the periplasm of a wide range of bacteria and archaea. They play crucial roles in pathogenesis, DNA transformation, and motility, and are capable of rapid retraction, generating powerful motor forces. PilN and PilO are integral inner membrane proteins that are essential for type IV pilus formation. Here, we show that PilN and PilO from Thermus thermophilus can be isolated as a complex with PilM, a cytoplasmic protein with structural similarities to the cytoskeletal protein MreB. The crystal structure of the periplasmic portion of PilN forms a homodimer with an extensive, conserved interaction interface. We conducted serial 3D reconstructions by electron microscopy of PilMN, PilMNO, and PilMNO bound to the major pilin protein PilA4, to chart the assembly of the inner membrane pilus biogenesis platform. PilN drives the dimerization of the PilMN complex with a stoichiometry of 2:2; binding of two PilO monomers then causes the PilN periplasmic domains to dissociate. Finally, two PilA4 monomers bind to the periplasmic domains of PilN and PilO, to generate a T-shaped complex that is primed for addition of the pilin to the nascent pilus fiber. Docking of structures for PilM, PilN, PilO, and PilA4 into the electron density maps of the transmembrane complexes was used to generate a sequence of molecular structures that chart the initial events in type IV pilus formation, and provide structural information on the early events in this important secretion process.
Type IV pili (T4P) constitute a major class of fimbriae; they are widespread and are found mainly in Gram-negative bacteria, including several notable human pathogens, such as Neisseria meningitidis, Pseudomonas aeruginosa, and Vibrio cholerae (1, 2). T4P are typically 1–5 μm long and composed predominantly of polymers of a single, major pilin protein that are linked noncovalently in a helical arrangement (3). They play a key role in mediating bacterial attachment to a variety of surfaces and, through their roles in colonization, are major factors in determining virulence. T4P are also involved in DNA competence and uptake, although the precise relationship of transforming DNA to type IV pilus biogenesis is unclear at present (2). Remarkably, T4P are also capable of rapid depolymerization; this process is apparently catalyzed by a separate ATPase, PilT (4), and is responsible for a phenomenon known as twitching motility (5), which enables bacteria to migrate across solid surfaces (6).
T4P can be categorized into two subtypes, IVa and IVb: this subdivision is based on several structural characteristics, including the length of the signal sequence, which is shorter for type IVa pilins. Pilins from both type IV groups adopt variations based on a similar “core” architecture: an α/β-type fold, consisting of an α-helix packed against a four- or five-stranded β-sheet. Critically, the N-terminal α-helix appears to have a dual function. Models for the construction of T4P place this helix at the center of the helical assembly, such that helices in adjacent subunits interact noncovalently to stabilize the assembly (3). It has also been proposed that the hydrophobic N-terminal helix promotes the insertion of the pilin into the inner membrane in the depolymerized state (3). The inner membrane would therefore act as a reservoir for pilin subunits following rapid depolymerization of the fiber.
Around a dozen different proteins are known, from genetic studies, to be associated with T4P biogenesis (1, 7). Of these, a core set of T4P biogenesis proteins have been identified (7). This core group includes an assembly ATPase, which provides the energy for pilus formation through ATP hydrolysis, and an outer membrane secretin protein, which forms a channel for extrusion of the assembled pilus fiber across the outer membrane (8, 9). In this respect, parallels have been drawn with the type II secretion system (T2SS), which has marked structural and mechanistic similarities with T4P biogenesis (10). Central to both systems is the assembly of a pilus or pseudopilus fiber in the periplasm, leading to suggestions that the mechanisms of the two secretion systems could function in similar ways. A central question, which remains unresolved, is how the energy derived from ATP hydrolysis is coupled to the polymerization of the pilus fiber in the periplasm. There is evidence that, in addition to the assembly ATPase, several proteins associated with the inner membrane also play a critical part in this process. PilM, PilN, and PilO are frequently cotranscribed (1); PilM is a soluble protein and forms an actin-like structure (11). It binds to the N terminus of PilN, an integral membrane protein with a single transmembrane α-helix and a large, periplasmic domain. PilO also has a single transmembrane α-helix at its N terminus and adopts a two-domain structure in the periplasm: a coiled-coil domain, followed by a ferredoxin-like fold (12). Results from studies in P. aeruginosa established that PilN and PilO bind together and can be copurified with PilP, a lipoprotein that is associated with the PilQ outer membrane secretin (13).
Experiments conducted in Thermus thermophilus have shown that pilM, pilN, or pilO mutants are all nonpiliated and exhibit phenotypes characteristic of the absence of T4P (14, 15). Given their location in the inner membrane, it is reasonable to propose that they form a platform for T4P formation, but the details whereby they might contribute to this process are unclear. Here, we report a structural investigation of the PilMNO complex from T. thermophilus (denoted as TtPilMNO; we will use the prefix “Tt” to identify the pilus biogenesis proteins from T. thermophilus). The T4P biogenesis machinery has been well characterized in T. thermophilus, providing an excellent basis for structural investigations (14–16). We show that the complex can be assembled in an incremental manner, starting with association of TtPilM with TtPilN, then TtPilO, and, finally, the major pilin (TtPilA4). We propose that this in vitro reconstitution process effectively mimics the sequence of binding events in vivo and therefore provides a model for the initial steps in the assembly of T4P.
Results
Isolation of the TtPilMN and TtPilMNO Complexes and Binding to the TtPilA437–122 Major Pilin.
Analysis of genome sequences has shown that PilM, PilN, and PilO are frequently found as consecutive genes in many Gram-negative bacteria and may be cotranscribed (1). Given previous evidence that TtPilM binds to an N-terminal TtPilN peptide (11), we attempted to coexpress TtPilM with full-length TtPilN. TtPilM, when expressed alone, is a soluble protein (11), but coexpression with TtPilN resulted in a significant amount of TtPilM being associated with the purified membranes. Solubilization with n-dodecyl β-d-maltopyranoside (DDM), followed by metal chelate affinity and size exclusion chromatography, gave a purified product which contained both proteins (Fig. 1A, lane 4). It was also possible to coexpress TtPilO with TtPilM and TtPilN, and to isolate the TtPilMNO complex in a similar way (Fig. 1A, lane 6).
Fig. 1.

Isolation of TtPilMN and TtPilMNO complexes and binding to TtPilA37–122. (A) Isolation of TtPilMN and TtPilMNO complexes and binding to TtPilA437–122. Samples shown are fractions from size exclusion chromatography run on a Superdex 200 10/300 GL column (GE Healthcare). For each sample, the elution peak volume is given in brackets. Lane 1, molecular mass markers (in kilodaltons); lane 2, TtPilA437–122 elution peak (18.5 mL); lane 3, TtPilA437–122 control (12 mL); lane 4, TtPilMN elution peak (12 mL); lane 5, TtPilMN plus TtPilA437–122 elution peak (12 mL); lane 6, TtPilMNO elution peak (12 mL); lane 7, TtPilMNO plus TtPilA437–122 elution peak (12 mL). (B) Examination of binding of TtPilA437–122, Tt121836–123, and Tt122238–123 to the TtPilMNO complex. Lane 1, molecular mass markers (in kilodaltons); lane 2, TtPilA437–122 elution peak (18.5 mL); lane 3, Tt121836–123 elution peak (18.5 mL); lane 4, Tt122238–123 elution peak (18.5 mL); lane 5, TtPilMNO elution peak (12 mL); lane 6, TtPilMNO plus TtPilA437–122 elution peak (12 mL); lane 7, TtPilMNO plus Tt121836–123 elution peak (12 mL); lane 8, TtPilMNO plus Tt122238–123 elution peak (12 mL); lane 9, TtPilA437–122 control (12 mL); lane 10, Tt121836–123 control (12 mL); lane 11, Tt122238–123 control (12 mL). Other conditions are as for A.
We then examined whether these inner membrane complexes were able to bind to the major type IV pilin from T. thermophilus, TtPilA4 (14). To generate a form of TtPilA4 that would be soluble and tractable to binding studies, we engineered an expression construct that had 36 residues removed from the N terminus of the mature protein (designated TtPilA437–122). In other type IV pilins, these residues form part of the hydrophobic N-terminal helix, which is a well-conserved structural feature that plays an important role in mediating pilus assembly (3). This strategy was successful and resulted in a pilin fragment that was soluble and monomeric (Fig. 1A, lane 2). Premixing of TtPilA437–122 with either TtPilMN or TtPilMNO, followed by fractionation by size exclusion chromatography, showed that the pilin coeluted with both complexes (Fig. 1A, lanes 5 and 7), whereas it did not elute at that position in the chromatogram when applied alone (Fig. 1A, lane 3). To demonstrate the specificity of the interaction, the experiment was repeated using two pilin-like proteins from T. thermophilus, Tt121836–123 and Tt122238–123. These proteins were predicted to form similar structures to TtPilA437–122, with the canonical pilin fold (3); they were also expressed by truncation of their hydrophobic N termini, resulting in soluble proteins that migrated as monomers by size exclusion chromatography, in a similar fashion to TtPilA437–122 (Fig. 1B, lanes 2–4). Repetition of the binding experiment showed that TtPilA437–122 coeluted with the TtPilMNO complex (Fig. 1B, lane 6), but both Tt121836–123 and Tt122238–123 failed to do so (lanes 7 and 8). The results indicate that the TtPilMNO complex appears to specifically bind to TtPilA437–122.
Our strategy to assemble a molecular model for the TtPilMNO–pilin complex required the determination of the structures of TtPilA437–122 and the periplasmic domain of TtPilN, as these have not been reported to date. Crystal structures of both these components were therefore obtained.
Crystal Structure of the TtPilA437–122 Major Pilin.
Crystals of TtPilA437–122 were obtained, which diffracted to 1.7-Å resolution, and the structure was solved by phasing using anomalous diffraction from iodide ions (Table 1). The structure contained two molecules in the asymmetric unit, packed at ∼90° (Fig. 2A). The α/β fold is typical of type IVa pilins, but simplified, reflecting the smaller size of the protein compared with other type IVa pilins whose structures have been determined (2, 3). The single α-helix packs against a four-stranded antiparallel β-sheet (Fig. 2B). The structure overlaid with a core root mean square deviation of 2.9 Å against the PilE pilin from Neisseria gonorrhoeae (17), a fold typical of type IVa pilins (Fig. 2C), but with two important differences. First, the D-region, which forms a defined C-terminal segment in type IVa and IVb pilins (3), is absent from TtPilA437–122. Second, the location of the single disulfide bond within the TtPilA437–122 structure is unusual: it is formed between Cys85 in the first β-strand and Cys71 in the αβ loop, which links the N-terminal helix to β1. Structures of other type IV pilins have located disulfide bonds in a conserved location that spans the D-regions, including PilE from Neisseria gonorrheae (17, 18), TcpA from Vibrio cholera (19), BfpA from Escherichia coli (20), and the minor pilin PilX from Neisseria meningitidis (21). An alternative location for a disulfide bond is found in pilins from Pseudomonas aeruginosa strain K122-4 (22) and FimA from Dichelobacter nodosus (23), with a linkage formed between the αβ-loop and the second β-strand. This serves to further illustrate the structural diversity found within type IV pilus structures.
Table 1.
Crystal structure of TtPilA437–122: Data collection and refinement statistics
| Space group | P 32 2 1 | |
| Unit cell parameters | a = b = 36.8 Å, c = 207.8 Å | a = b = 36.6 Å, c = 208.7 Å |
| Native | Iodine derivative | |
| X-ray source and wavelength, Å | DLS* I04 (0.946) | DLS I24 (1.500) |
| Resolution range, Å | 35–1.70 (1.74–1.70)† | 30–1.87 (1.92–1.87) |
| Multiplicity | 10.1 (10.5) | 7.7 (1.2) |
| Significance (<I>/sd) | 17.1 (3.5) | 16.5 (2.3) |
| No. unique reflections | 19,190 | 12,198 |
| Completeness, % | 99.8 (99.6) | 84.8 (14.8) |
| Rmerge, %‡ | 7.1 (76.0) | 6.8 (15.4) |
| Rmeas, %§ | 7.7 (83.2) | 8.4 (20.8) |
| Refinement statistics | ||
| Rcryst | 21.6 | |
| Rfree, 5% data | 24.1 | |
| Nonhydrogen atoms | ||
| All | 1,361 | |
| Water | 133 | |
| Mean overall B, Å2 | 25.6 | |
| Rmsd from ideal values | ||
| Bond distance, Å | 0.013 | |
| Bond angle, degrees | 1.5 | |
Diamond Light Source.
Values in parentheses refer to the outer resolution shell.
Rmerge = ΣhklΣsym|I − <I>|/ΣhklI.
Ref. 70.
Fig. 2.

Crystal structure of the TtPilA437–122 major pilin. (A) Structure of the TtPilA437–122 dimer in the crystallographic asymmetric unit, showing the approximate 90° angle packing between adjacent subunits. N termini are labeled. (B) Details of secondary structural elements within a TtPilA437–122 monomer. The arrow indicates the disulfide bond between β1 and the αβ loop. N and C termini are labeled. (C) Two orthogonal views of a structural alignment between TtPilA437–122 and the type IV pilin PilE from Neisseria gonorrheae (PDB ID code 2HIL). TtPilA437–122 is shown in gray, and PilE in purple (residues 29–123) and green (124–158). Residues 124–158 comprise the D-region (3), which is absent from TtPilA437–122.
Crystal Structure of the Periplasmic Domain of TtPilN.
A recombinant fragment, corresponding to the periplasmic domain of TtPilN and spanning residues 49–207, was overexpressed and purified (designated TtPilN49–207). TtPilN49–207 formed a dimer in solution, as measured by size exclusion chromatography combined with multiangle laser light scattering analysis (SEC MALLS) (Fig. S1A). The protein was crystallized, native data collected to resolution of 2.05 Å, and the structure solved by Zn phasing (Table 2). TtPilN49–207 forms a dimer in the asymmetric unit, with each individual monomer adopting an α/β sandwich-type fold (Fig. 3A, Left). Secondary structure predictions had previously suggested that the PilN periplasmic domain would form a ferredoxin-like fold, in a similar fashion to PilO (12) and the T2SS protein EpsL (24). A comparison with the structure of the periplasmic domains of PilO from P. aeruginosa established that this was indeed the case, although there are some significant differences in the arrangement of secondary structural elements (Fig. 3A, Right). Both PilO and EpsL form an αββαββ fold, with the two α-helices packed against a four-stranded antiparallel β-sheet. In contrast, TtPilN49–207 has an additional α-helix inserted between β1 and β2 (α2 in Fig. 3A), to give an αβαβαββ order of secondary structure. An alignment of the sequences of PilN from other bacteria suggested that the α2 helix is generally present, although there is less conservation in this region than in other parts of the domain (Fig. S2A). It should also be noted that electron density was only observed starting from residue 94 in the TtPilN49–207 crystal structure; the absence of detectable density for the missing 45 residues at the N terminus may be due to proteolysis during crystallization and/or disorder of these residues in the crystalline state.
Table 2.
Crystal structure of TtPilN49–207: Data collection and refinement statistics
| Space group | I 41 | |
| Unit cell parameters | a = b = 67.8 Å, c = 147.2 Å | |
| Native | Zn derivative | |
| X-ray source and wavelength, Å | DLS* I24 (0.969) | DLS I03 (1.282) |
| Resolution range, Å | 40–2.05 (2.10–2.05)† | 74–2.50 (2.60–2.50) |
| Multiplicity | 3.4 (3.4) | 7.2 (2.6) |
| Significance (<I>/sd) | 12.9 (2.4) | 17.0 (2.4) |
| No. unique reflections | 20,669 | 10,878 |
| Completeness, % | 99.4 (98.8) | 94.6 (66.9) |
| Rmerge, %‡ | 6.8 (56.9) | 8.3 (38.1) |
| Rmeas, %§ | 9.7 (77.2) | 8.8 (46.1) |
| Refinement statistics | ||
| Rcryst | 19.6 | |
| Rfree, 5% data | 22.6 | |
| Nonhydrogen atoms | ||
| All | 1,831 | |
| Water | 142 | |
| Mean overall B, Å2 | 33.4 | |
| Rmsd from ideal values | ||
| Bond distance, Å | 0.022 | |
| Bond angle, degrees | 1.7 | |
Diamond Light Source.
Values in parentheses refer to the outer resolution shell.
Rmerge = ΣhklΣsym|I − <I>|/ΣhklI.
Ref. 70.
Fig. 3.

Crystal structure of the TtPilN49–207 periplasmic domain dimer. (A) Fold of the TtPilN49–207 monomer (orange), with secondary structures labeled (Left) and overlaid with the periplasmic domain of PilO from P. aeruginosa in light blue (Right; PDB ID code 2RJZ). The rmsd of backbone atoms between the two structures was 1.9 Å; the overlay and figure were generated using CCP4MG (69). (B) Two orthogonal views of the TtPilN49–207 dimer. Chain A is shown in orange, and chain B in green. The image was generated using CCP4MG (69). (C) Electrostatic surface of chain A, with chain B in green wire frame superimposed. The images were generated using CCP4MG (69). (D) Two views of an atomic space-filling model of TtPilN49–207 from chain A, colored by sequence conservation using CONSURF (26). Residues colored in yellow are those for which there are insufficient data to determine their degree of sequence conservation. Some selected, highly conserved residues are indicated. Chain B is shown as a green wire frame. The images were generated using Chimera (66).
The most notable feature of the TtPilN49–207 structure is the dimer interface, which is mediated by symmetric contacts between residues in α1, β1, β3, and α3 (Fig. 3B). The parallel alignment of the two copies of the α1 helix leads to a series of molecular contacts along the length of the helix, and could explain why the position of α1 in TtPilN49–207 deviates from that of the equivalent helix in P. aeruginosa PilO (Fig. 3A, Right). Examination of the packing contacts between adjacent TtPilN49–207 monomers in the crystal structure using PISA (25) confirmed that this interface had the largest contact area (1,428 Å2) and, with a complexation significance score of 0.816, was most likely to persist in solution. Calculation of an electrostatic surface demonstrated that the dimer interface was predominantly hydrophobic in character (Fig. 3C). The interface was also well conserved in a comparison of different PilN sequences, conducted using CONSURF (26), notably with the inclusion of some hydrophobic side chains (e.g., Phe175; Fig. 3D, Left, and Fig. S2A). Interestingly, the opposite face of the TtPilN49–207 monomer also contained some well-conserved residues (Glu170, Phe179; Fig. 3D, Right): these may be involved in interaction with TtPilO or TtPilA4.
Structures of TtPilMN, TtPilMNO, and TtPilMNO–TtPilA437–122.
We adopted an incremental approach to the construction of the TtPilMNO–TtPilA437–122 complex, through comparison of electron microscopy-generated volumes of TtPilMN, TtPilMNO, and TtPilMNOA, determined by single-particle averaging. At each stage, structures of the component proteins were docked into the electron density map for each complex. The data presented in Fig. 1 established that TtPilMN can be isolated as an integral membrane protein complex; analysis of the complex by SEC MALLS gave an estimated molecular mass of 120 kDa for the main peak (Fig. S1B). This measurement, combined with the formation of a dimer by TtPilN49–207, suggested that a dimeric organization for TtPilMN was most likely (i.e., two copies of TtPilM and TtPilN per complex). Transmission electron microscopy (TEM) of the isolated TtPilMN complex showed particles of a size that were consistent with dimer formation (Fig. S3). A structure of the TtPilMN complex was then generated by single particle averaging to a resolution of 27 Å, with imposition of C2 symmetry (Fig. 4, Left). The structure is readily interpretable, as it is clearly divided into two unequal halves; we ascribe the upper, small-density feature to TtPilN, as its periplasmic domain is smaller than the mass of TtPilM. The lower half corresponds to TtPilM, with the narrower part of the volume attributable to the transmembrane region and associated detergent. TtPilM, we have previously noted (11), is monomeric; the structure therefore suggests that dimerization of the TtPilMN complex is driven by the TtPilN dimer interface revealed by the crystal structure described above. To generate a molecular model for the complex, the protein–protein docking program SymmDock (27) was used to generate a family of possible TtPilM dimers, which were then screened for optimal fit to the electron density map and consistency with the proposed location of the transmembrane region (for the short, bound TtPilN cytoplasmic peptide). This TtPilM dimer was then docked into the density map. The crystal structure of TtPilN49–207 could then be located in the upper part of the complex, making allowance for the missing coiled-coil region and transmembrane helices (Fig. 4, Right).
Fig. 4.

Structure of the TtPilMN dimer. The left-hand panels (Upper and Lower) show the electron microscopy density map for the TtPilMN complex. (Scale bar: 20 Å and located at approximately the proposed position of the membrane bilayer in the upper panel.) The right-hand panels depict the docked crystal structures of the periplasmic dimer of TtPilN (orange) and TtPilM (blue) with TtPilN peptide bound. The upper panels show a side view, and the lower panels are seen from the top of the complex, along the C2 symmetry axis. The images were generated using Chimera (66).
A similar approach was taken with the TtPilMNO complex, which also gave well-dispersed particles (Fig. S3). The resulting 3D reconstruction of the TtPilMNO complex reveals some significant differences from the TtPilMN volume (Fig. 5). The transmembrane region is expanded: this observation is readily explained by the fact that TtPilO also contains a transmembrane α-helix at its N terminus. In addition, it is likely that this complex binds more detergent than TtPilMN. The second major difference from TtPilMN lies in the periplasmic domains: not only are these larger in volume, to accommodate TtPilO, but the density in this region is clearly divided into two distinct halves. We were unable to convincingly dock the TtPilN49–207 dimer structure into this volume and concluded that binding of TtPilO induces dissociation of the TtPilN dimer. This would also be consistent with the observation of the formation of a PilN–PilO heterodimer in P. aeruginosa (12). To investigate the plausibility of this hypothesis, we first generated a homology model for the periplasmic domains of TtPilO and carried out docking simulations to the TtPilN49–207 monomer structure. The results were then screened to identify the heterodimer most consistent with the electron microscopy density map: a convincing solution was found, which gave excellent agreement with the TtPilMNO volume (Fig. 5, Right). There is little interaction between the TtPilNO heterodimers across the C2 symmetry axis, and the complex appears to be held together by association of the PilN and PilO transmembrane domains.
Fig. 5.

Structure of the TtPilMNO dimer. The left-hand panels (Upper and Lower) show the electron microscopy density map for the TtPilMNO complex. The right-hand panels show the docked crystal structures of TtPilN (orange) and TtPilM (blue), and the homology model of TtPilO (green). Two orthogonal views are depicted, both viewed parallel to the proposed plane of the membrane. Scale bar details are as for Fig. 4. The images were generated using Chimera (66).
The final stage in the assembly sequence was to add TtPilA437–122 to TtPilMNO and repeat the electron microscopy reconstruction of the assembled complex, to identify the location of the pilin (Fig. S3). The resulting volume produced one obvious and striking difference from TtPilMNO, with an additional density feature lying adjacent to the predicted location of TtPilN, on the periplasmic side of the complex (Fig. 6A). The dimensions of the density feature were consistent with that expected for TtPilA437–122, and the pilin crystal structure could be readily docked into place, with the imposition of a constraint that the N-terminal helix in TtPilA437–122 was pointing downward toward the predicted location of the inner membrane. TtPilA437–122 binds to one end of the TtPilN molecule, the main site of interaction on the pilin being the αβ loop between α1 and β1 (Fig. 2B). An attractive feature of this model for the assembly is that the two copies of the bound TtPilA4 lie close to the predicted location of the membrane bilayer. Extension of the N-terminal helix would bring it into contact with the membrane, consistent with current models that place unpolymerized pilin subunits in the membrane (3).
Fig. 6.

Structure of the TtPilMNO–TtPilA437–122 complex. (A) Side view. (Left) Electron microscopy density map for the TtPilMNO–TtPilA437–122 complex. Scale bar details are as for Fig. 4. (Right) Docked structures of TtPilM, N and O (colored as Fig. 5). The TtPilA437–122 crystal structure is shown in pink. (B) Close-up view of the TtPilN:TtPilA4 interaction. Other details are as for A.
Discussion
The results presented here have provided a framework for the initial events in type IV pilus biogenesis. Recent work in the field has identified a limited number of core components of the T4P biogenesis machinery, which are absolutely required for pilus assembly in most organisms. These comprise an assembly ATPase, an outer membrane secretin, and the PilMNOP proteins, in addition to the major type IV pilin. On the basis of our results, we propose a sequence of events that would prime T4P assembly (Fig. 7). The formation of the PilMN binary complex starts the process, establishing a transmembrane platform for the assembly. The dimerization of PilN is the main driver for this stage of the assembly process. PilO then docks on to PilN, opens up the structure, and primes it for pilin binding. Finally, the major pilin, which has been processed by the prepilin peptidase PilD and migrates within the inner membrane, can diffuse laterally and bind to PilMNO. The result is a “T”-shaped complex, with two pilin copies at its outermost extremities (Fig. 7). Given we observed that the TtPilMN complex is also capable of binding TtPilA437–122 (Fig. 1), we suggest that the function of PilO lies in modifying the complex by splitting the PilN dimer and setting up the pilin binding site in the correct orientation to capture pilin subunits from the inner membrane. Our observation that TtPilA437–122 binds to the TtPilMNO complex is in agreement with other evidence for association of T4P and T2SS pilins with their respective inner membrane complexes (28–30). Clearly, recruitment of the major pilin subunit could be the first “priming” step in T4P assembly.
Fig. 7.
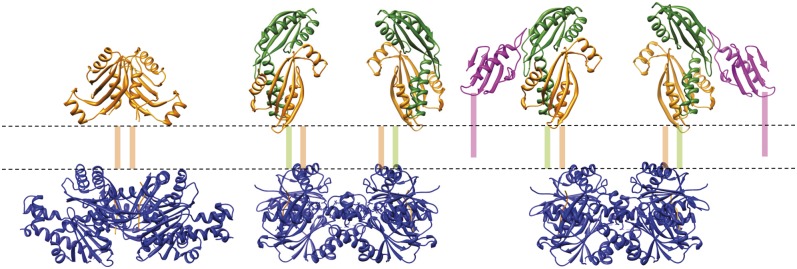
Assembly of the core type IV pilus biogenesis platform in the inner membrane. The figure shows, from Left to Right, TtPilMN, TtPilMNO, and TtPilMNO–TtPilA437–122 complexes. Approximate locations of the inner membrane (dotted lines) and transmembrane helices are shown. Color coding of the components of the complexes is as for Figs. 4–6.
How would a TtPilMNO–TtPilA4 complex participate in the next stage of T4P assembly? An important clue comes from work on the role of secretin-binding proteins in T4P biogenesis and the T2SS in other bacteria. Secretins form channels in the outer membrane for the passage of secreted proteins and assembled T4P (31). The T2SS secretin GspD and the T4P secretin PilQ form dodecameric assemblies enclosing chambers through which the secreted proteins pass (8, 32). Recently, it has been noted that the lipoprotein PilP, which is located in the inner membrane and binds to PilQ (33), adopts a similar β structure to GspC, a component of the T2SS that has a related function in binding to its cognate secretin GspD (34). Moreover, there are also similarities in the ways the two secretin-binding proteins recognize the secretin α/β domains in PilQ and GspD (8, 34). The PilP lipoprotein is formed from a long, flexible N terminus, which contains the lipid attachment site, and a globular C-terminal domain (35). The C-terminal domain binds to PilQ (8) and the N terminus recognizes PilN as part of the PilMNO complex (13, 36). It therefore forms a crucial link between the dodecameric secretin assembly and the PilMNO inner membrane platform. It is reasonable to infer that the formation of secretin dodecamers in the outer membrane could act as a driver for oligomerization of the PilMNO inner membrane components. Indeed, similar suggestions have been made in the case of the T2SS (34). The consequence would be that, when a major pilin binds to this assembly, pilins from adjacent PilMNO complexes are brought into close spatial proximity, where they could associate and thus be immediately available for incorporation into a pilus fiber. We should note that there is no equivalent of the PilP lipoprotein in T. thermophilus, although there is a separate protein, PilW, which has no orthologs in proteobacteria but may carry out a similar function (14).
An obvious question, which leads on from our structural characterization of an inner membrane platform for T4P biogenesis, is how ATP hydrolysis actively promotes pilus formation. Although our structural model of the TtPilMNO–TtPilA437–122 complex shows a well-defined platform linking the cytoplasmic TtPilM protein to pilus subunits in the periplasm, it is unclear how ATP hydrolysis would influence the assembly process. TtPilM binds ATP, but we failed to record any ATP hydrolase activity associated the protein (11). Current opinion suggests that the energy for secretion is provided by the assembly ATPases: these are members of the AAA+ superfamily that are generally associated with mechanical processes, such as protein folding (37). Our recent electron microscopy structural work on the T4P assembly ATPase from T. thermophilus, PilF (16), has shown that it forms a hexameric assembly with an unusual “dumbbell”-like structure (38). The C-terminal half, which forms the ATP-binding part of the complex, is located at one end of the structure. Structural changes that occur on hydrolysis of ATP are transmitted through a narrower stem structure to the N-terminal half of the complex, which is predicted to harbor three GSPII domains. GSPII is a structural fold that is found in ATPases associated with the T2SS, as well as T4P biogenesis (16, 39). GSPII domains are known to be capable of large structural rearrangements (39), suggesting a mechanism for energization of the secretion process, whereby the GSPII domains would transmit conformational changes to integral inner membrane components.
There is evidence for binding of secretion ATPases to PilM-like proteins from studies on other systems. Abendroth et al. determined the crystal structure of a complex between the N-terminal domain of the T2SS ATPase EpsE and the cytoplasmic domain of EpsL (40), which is similar in structure to PilM (11). In addition, BfpC, a key protein in the biogenesis of type IVb bundle forming pili, shares structural similarity with EpsL and PilM (41) and binds to its cognate assembly ATPase, BfpD (40, 42). Interestingly, both the cytoplasmic and periplasmic domains of EpsL form dimers (24, 43), raising the question of how this is consistent with binding to the hexameric EpsE ATPase (44). A solution has been proposed in the form of an assembly in which three EpsL dimers bind to a single EpsE hexamer (24). It is possible, therefore, that a similar arrangement could pertain in T. thermophilus, with three TtPilMNO dimers binding to the PilF ATPase. At present, however, we have no direct evidence for such an assembly, and other arrangements remain possible.
A second question that emerges from our observations is how the TtPilA4 subunit would dissociate from the TtPilMNO complex and incorporate into the pilus fiber. Of relevance here are recent studies conducted on the PuII/J/K pseudopilins, which form part of the T2SS assembly for pullulanase secretion (45). Formation of the PulI/J/K heterotrimer effectively displaces the PulK pseudopilin from the membrane, thus forming a structure that is an effective nucleus for initiation of pilus formation. We speculate that, in general, the PilMNO platform could act as an intermediary between the cytoplasmic ATPase and mechanical displacement of pilin subunits out of the membrane and into the growing pilus fiber. It is also interesting to note that any model for T4P assembly or disassembly would require an “anchor point” at the base of the fiber as an intermediate during the extension/retraction cycle. The PilMNO platform could provide such a function. Clearly, further work is required to establish with greater precision the precise role of the PilMNO assembly in T4P formation.
Materials and Methods
Expression and Purification of TtPilMNO, TtPilMN, TtPilN47–207, TtPilA37–122, TTHA1218, and TTHA1222.
The genome sequence of the type IV pilus operon in Thermus thermophilus HB8 indicated that pilM, pilN, and pilO are cotranscribed, which suggested a straightforward strategy for coexpression, through amplification of the pilM/pilN or pilM/pilN/pilO genes, including the intragenic regions. DNA containing the full-length pilMNO and pilMN genes was amplified by PCR from Thermus thermophilus HB8 genomic DNA using the primer sets CGCGCGGCAGCCATATGGTGTTCAAAAGCCTTAGCCAG and GTTAGCAGCCGGATCCTCATGGGGTGCTCCCTCC, and GTATAAGGAGGGCATATGTTCAAAAGCCTTAGCCAG and CTCCCGCTGTGAATTCCTAGCGAGCACCGCTTTC.
In parallel, DNA encoding the periplasmic fragment of the pilN gene (spanning residues 49–207) was amplified using the primer pair CGCGCGGCAGCCATATGGCCAAGGCGGAGCGGGAC and GTTAGCAGCCGGATCCCTAGCGAGCACCGCTTTCAC.
The amplified pilMN product and the pET28a vector (Novagen) were treated with restriction enzymes NdeI and EcoRI, purified and ligated to generate the pilMN_28a expression construct. The pilN and pilMNO DNA fragments were cloned into the pET15b vector, which had been predigested with restriction enzymes NdeI and BamHI, using the In-Fusion HD cloning system (Clontech); this generated the pilN_15b and pilMNO_15b expression constructs. pilN_15b encoded the predicted periplasmic domain of TtPilN with a 6×-His tag followed by a thrombin cleavage site at the N terminus; the expressed protein was designated TtPilN49–207. pilMN_28a and pilMNO_15b encoded TtPilM with a 6×-His tag followed by a thrombin cleavage site at the N terminus, and full-length, native TtPilN and TtPilO proteins, without tags. Isolation of the TtPilMN and TtPilMNO complexes by metal chelate affinity chromatography thus demonstrated noncovalent association of TtPilN and TtPilO with TtPilM.
TTHA1221 in the T. thermophilus HB8 genome sequence was identified as having the highest sequence similarity to PilA4 from T. thermophilus HB27, which is the major pilin protein in that strain (14). TTHA1221 was therefore designated as TtPilA4, to ensure consistency. To generate an expression vector for TtPilA437–122, a fragment of TTHA1221 from T. thermophilus HB8, spanning residues 37–122, was prepared by custom synthesis (Life Technologies), excised from the manufacturer’s standard vector using the restriction enzymes NcoI and XhoI, and ligated into the pET22b vector. The pilA4_22b expression construct encoded TtPilA4 residues 37–122, thus omitting part of the predicted N-terminal α-helix, replacing it at the N terminus with a pelB leader peptide sequence and a 6×-His tag at the C terminus. Expression vectors for the minor pilins TTHA1218 and TTHA1222 were generated in a similar fashion, by custom gene synthesis and cloning into pET22b. The 1218_22b construct encoded residues 36–123 and 1222_22b encoded residues 38–123; the resulting proteins were designated Tt121836–123 and Tt122238–123.
Each expression vector was transformed into T7 Express cells (New England Biolabs) and two to three colonies were inoculated into 100 mL of 2X-YT medium containing the appropriate antibiotics (100 μg/mL ampicillin for pilMNO_15b, pilN_15b, pilA_22b, 1218_22b, and 1222_22b; 40 μg/mL kanamycin for pilMN_28a) and allowed to grow for 3–4 h at 37°C. The 100-mL culture was diluted into 4 L of fresh 2X-YT medium and the cells were grown at 37 °C until the absorbance at 600 nm reached 0.8–1.0. At this stage, the temperature was reduced to 16 °C and the cells were induced by the addition of isopropyl β-d-1-thiogalactopyranoside to a final concentration of 0.1 mM. The cells were harvested after 16 h by centrifugation at 6,000 × g.
E. coli cells containing the overexpressed membrane protein complexes TtPilMN or TtPilMNO were resuspended in 100 mL of buffer A [25 mM Tris⋅HCl, pH 8.0, 100 mM NaCl, 5% (vol/vol) glycerol, 10 mM MgCl2], and 1× EDTA-free protease inhibitor mixture (Roche), 0.4 mg of DNase, and 40 mg of lysozyme were added. The resuspended cells were lysed using a sonication probe (TT13/FZ; Bandelin Sonopuls HD3200) at 30% amplitude for 8 min with pulses of 10 s on and 10 s off. The lysate was centrifuged for 30 min at 18,000 × g and the supernatant was passed through a 0.45-μm filter. The membranes were separated by centrifugation of the filtered supernatant at 130,000 × g for 2 h, and the membrane pellet was washed by resuspending in buffer A containing 1× protease inhibitor mixture and centrifuged again at 130,000 × g. The membranes were solubilized by resuspending in buffer A supplemented with 1% (wt/vol) DDM and 1× protease inhibitor mixture, and incubated for 1 h at 4 °C. Unsolubilized membranes were removed by centrifugation at 130,000 × g for 45 min, and the supernatant was mixed with 4 mL of Ni-NTA resin (Qiagen) and incubated for 3 h at 4 °C. The resin was packed in a gravity flow column (Pierce) and washed with 20 column volumes of buffer A supplemented with 0.015% (wt/vol) DDM and 10–50 mM imidazole. Increasing the imidazole concentration to 200–500 mM eluted the protein complex. The TtPilMN and TtPilMNO complexes were concentrated using 50- and 100-kDa cutoff centrifugal concentrators (Sartorius), respectively. The protein complexes were further purified by gel filtration using Superdex 200 (10/300) GL column (GE Healthcare) equilibrated with buffer A containing 1% (wt/vol) n-octyl β-d-glucoside. The peak fractions were pooled and concentrated and finally spun at 200,000 × g to remove any aggregates.
E. coli cells expressing the truncated pilin proteins (TtPilA437–122, Tt121836–123, Tt122238–123) were resuspended in buffer B (50 mM Tris⋅HCl, pH 8.0, 200 mM NaCl), lysed, and filtered as detailed for the TtPilMN/TtPilMNO complexes. The filtered supernatant was mixed with 4 mL of Ni-NTA resin and incubated for 1 h at 4 °C. The resin was packed and washed, and the protein was eluted in buffer B plus 200–500 mM imidazole, with other details as described above for the TtPilMN/TtPilMNO complexes. Pooled fractions were concentrated using a 5-kDa cutoff centrifugal concentrator (Sartorius) and further purified by size exclusion chromatography using a Superdex 75 (10/300) GL column (GE Healthcare).
For TtPilN49–207, cells were resuspended in buffer C [25 mM Hepes, pH 7.0, 100 mM NaCl, 5% (vol/vol) glycerol], lysed, and filtered as described for TtPilA437–122. TtPilN49–207 was purified by metal chelate affinity chromatography, also as described for TtPilA437–122, except that buffer C was used instead of buffer B. Purified TtPilN49–207 was treated with thrombin (GE Healthcare) for 16 h at 4 °C, while dialyzing in buffer C, for cleavage of the 6×-His tag. The protein was then concentrated using a 10-kDa cutoff centrifugal concentrator (Sartorius) and purified by size exclusion chromatography as described for TtPilA437–122.
Crystallization of TtPilA437–122 and TtPilN49–207.
For crystallization of TtPilA437–122, the protein was preequilibrated in 50 mM Tris⋅HCl (pH 8.0) and 200 mM NaCl. A volume of 500 nL of TtPilA437–122 (7 mg/mL) was mixed with 500 nL of well solution containing 150 mM Mops/NaOH (pH 7.0), 6.0% (vol/vol) ethylene glycol, and 1.8 M ammonium sulfate. Crystals appeared within 2 d and were cryoprotected by washing in well solution, to which glycerol was added to give a final concentration of 20% (vol/vol). For collection of the iodine anomalous data, the crystal was washed in 0.5 M KI plus 20% (vol/vol) glycerol before freezing.
For crystallization of TtPilN49–207, the protein was preequilibrated in 25 mM Hepes/NaOH (pH 7.0), 100 mM NaCl, and 5% (vol/vol) glycerol. A volume of 400 nL of TtPilN49–207 (5 mg/mL) was mixed with 400 nL of well solution containing 0.1 M Hepes/NaOH (pH 7.0), 0.2 M MgCl2, 20% (wt/vol) PEG 6000, and 10 mM ZnCl2. Cryoprotection of the native crystal was achieved by washing in the well solution, to which glycerol was added to give a final concentration of 20% (vol/vol). For formation of the zinc derivative, the crystal was washed in 0.1 M Hepes/NaOH (pH 7.0), 0.2 M MgCl2, 20% (wt/vol) PEG 6000, 40 mM ZnCl2, and 20% (vol/vol) glycerol.
Structure Determination.
Native crystals of TtPilA437–122 were grown in the presence of ammonium sulfate (see above); data were collected to a resolution of 1.7 Å (Table 1) and processed using XDS (46), as implemented by Xia2 (47). Space group identification was assisted by use of the software package POINTLESS (48). Soaking with a variety of heavy-atom compounds failed to deliver a suitable derivative, possibly due to the high salt conditions under which they were grown. Phase information was eventually extracted by the collection of anomalous data from a crystal soaked in a high concentration of KI, as described above; data were collected at a wavelength of 1.5 Å, to a resolution of 1.87 Å (Table 1). Thirteen iodine sites were found using AUTOSOL (49), as implemented from PHENIX (50), with a figure of merit (FOM) of 0.39. A model, which was >90% complete, was built using AUTOBUILD (51) and used as a search model for molecular replacement of the native data (Table 1), implemented with PHASER (52), from within PHENIX (50). Clear and unambiguous solutions were found to the rotation and translation functions, and the resulting model used for automated building by AUTOBUILD, followed by several cycles of refinement, using phenix.refine (53) combined with manual building using the molecular graphics package COOT (54). The final model comprised two molecules in the asymmetric unit.
Native data were collected from a single TtPilN49–207 crystal (Table 2) and processed using XDS (46) and Xia2 (47). The native crystals were grown in the presence of 10 mM ZnCl2; to extract phase measurements, two SAD datasets were collected from a single crystal that has been soaked in 40 mM ZnCl2 and data subsequently processed using MOSFLM (55), then merged and scaled using AIMLESS from the CCP4 suite (56). Zinc sites were identified using ShelXC/D (57), phases calculated by SHARP (58), density modification by PARROT (59), and the model was built using BUCCANEER (60) in a pipeline implemented in AUTOSHARP (61). At this stage, it was clear that there was a dimer in the asymmetric unit, and that electron density for residues 49–93 was absent. This structure was then used as a molecular replacement model for the 2.05-Å resolution dataset, which was completed using PHASER (52) and AUTOBUILD (51) from within PHENIX (50). Model building was completed manually using COOT (54) and refinement using phenix.refine (53).
Stereochemical parameters for both structures were examined using MOLPROBITY (62) and PROCHECK (63), and were within or better than the tolerance limits expected for each structure at the resolution limits given in Tables 1 and 2. Coordinates and structure factors were deposited in the Protein Data Bank with ID codes 4BHR (TtPilA37–122) and 4BHQ (TtPilN49–207).
TEM and 3D Reconstruction of TtPilMN, TtPilMNO, and TtPilMNO–TtPilA437–122 Complexes by Single-Particle Averaging.
The relatively small size of the TtPilMN/TtPilMNO/TtPilMNO–TtPilA437–122 complexes (<250 kDa) and their low C2 symmetry meant that processing cryo–electron-microscopic data of unstained, low-contrast protein in vitreous ice would be problematic and not necessarily lead to higher resolution data. The TEM structural studies we performed in this project were designed to augment the crystallographic data and provide us with an accurate complex envelope to perform protein docking, and so we opted to prestain the samples in uranyl acetate before rapid freezing and collection of data under cryogenic low-dose conditions. This provided the advantage of improved sample preservation with a higher signal–noise ratio than would have been the case using unstained samples, but limited the attainable resolution to 20–30 Å. For each of the complexes, samples were diluted to a final protein concentration of 10–15 μg/mL in buffer A plus 1% (wt/vol) β-d-octyl glucoside and applied to a freshly glow discharged carbon-coated 400-mesh copper grid (Agar Scientific). After incubation on a 10-µL droplet for 30 s, grids were briefly washed in water and then negatively stained in 1% (wt/vol) uranyl acetate for 1 min. The grids were continuously blotted for 4–5 s in a 90% humidity chamber before plunge-freezing into liquid ethane. Data were then recorded on a Polara FEG operating at 200 kV on a 4K Gatan Ultrascan CCD in low-dose mode (FEI). Full experimental, image recording, and single-particle averaging details for each complex are provided in Table 3. All image processing was performed using EMAN2 (64), applied to contrast transfer function (CTF)-corrected data, using strategies previously described (8, 38). For each complex, particles were selected using stringent selective swarm parameters in E2Boxer and overlapping data were discarded. Data were CTF-corrected and, following class averaging, preliminary models were generated to assess complex heterogeneity and symmetry: the complexes all had clear C2 symmetry. Structures were then generated following five rounds of iterative refinement. C2 symmetry was verified in a separate calculation, where the symmetry assignment was changed from C2 to C1 and the structure refined for a further four to five rounds. In each case, the structures and classes were preserved, demonstrating that imposition of C2 symmetry was correct.
Table 3.
TEM and single-particle averaging: Experimental details
| Category | TtPilMN | TtPilMNO | TtPilMNO–TtPilA437–122 |
| Electron dose, e/Å2 | 20–60 | 20–60 | 20–60 |
| Defocus range, −µm | 0.7–2.1 | 1.1–2.9 | 0.9–2.8 |
| Acceleration voltage, kV | 200 | 200 | 200 |
| Particle numbers | 12,074 | 15,486 | 15,726 |
| Å/pixel | 3.5 | 3.5 | 3.5 |
| Selection box, pix | 48 | 64 | 64 |
| Classes in volume | 120 | 180 | 180 |
| Alignment comp | Fourier ring correlation | ||
| Symmetry | C2 | C2 | C2 |
| Resolution, Å | 27 | 23 | 25 |
Resolution was estimated using the same method as applied previously (9), where each dataset is split into two halves, and the estimated resolution is set at the point at which the Fourier shell correlation of one half with the other reaches 0.5 (Fig. S4 and Table 3). To form the TtPilMNO–TtPilA437–122 complex, TtPilA437–122 was added in a twofold molar excess over TtPilMNO before application to the grid. It should be noted that each 3D structural refinement was performed independently and unique start models were created for each dataset to prevent any initial model bias. Although the differences between the TtPilMN and the TtPilMNO were large and straightforward to interpret, the 3D reconstruction of TtPilMNO–TtPilA437–122 produced an additional small density, which we attributed to TtPilA437–122. This density was verified using a variety of processing criteria: by refining the structure with auto-mask (on or off), changing the number of particle classes, the number of particles included in the averages and the 3D structure, and the σ value to determine the class-keep. Following calculations, including changes to all these parameters, the additional density was still present and appeared after the first round of refinement (Fig. S4).
Construction of Molecular Models for the TtPilMN, TtPilMNO, and TtPilMNO–TtPilA437–122 Complexes.
For the TtPilMN complex, the crystal structure of the TtPilM:TtPilN peptide complex (PDB ID code 2YCH) was analyzed for formation of a homodimer using SymmDock (27, 65), generating 100 different homodimeric complexes. The top three solutions, as scored by SymmDock, were essentially identical, and involved an interface at the 1A and 1C domains (11). The remaining seven of the top 10 solutions formed a separate class, with dimerization at the 2A and 2B domain interface. Docking of both sets of solutions into the electron density map for TtPilMN was then conducted manually in Chimera (66), placing the TtPilM dimer and bound TtPilN peptide into the larger of the two density features (as the mass of TtPilM is larger than the periplasmic domain of TtPilN). Both sets of solutions gave similar correlation coefficients of about 0.93 but the first set of solutions was selected in preference, as the alignment of the C terminus of the bound TtPilN peptide was closer to the predicted location of the transmembrane region, whereas in the second group of dimer models it was located at the outside of the complex, pointing outward, which was implausible. Following location of the TtPilM dimer, the TtPilN periplasmic dimer was manually docked into the second density feature, making allowance for the absence of the short coiled-coil domain from the structure.
To build a molecular model for the TtPilMNO complex, a homology model was constructed for the periplasmic domains of TtPilO, comprising the short coiled-coil region followed by the ferredoxin-like fold (12) (PDB ID code 2RJZ), using SWISS-MODEL (67). The sequence alignment of TtPilO against PilO from P. aeruginosa, which was used to generate the model, is given in Fig. S2B. PatchDock (27) was used to construct 100 separate models, docking a single TtPilO subunit onto the crystal structure of the TtPilN49–207 monomer. Each model of the TtPilO: TtPilN49–207 heterodimer was then refined in FireDock (68). The top 10 energetically favored models were then fitted into the electron density map for the TtPilMNO complex using Chimera (66) and the best-fitting model selected, based on correlation coefficient, with the imposition of two constraints: (i) the periplasmic TtPilN and TtPilO domains needed to be orientated such that their N termini were pointing toward the predicted location of the inner membrane (and hence could plausibly be joined to transmembrane helices), and (ii) the density attributed to the pilin domain (see below) made a majority contact with TtPilN, rather than TtPilO. This latter constraint was imposed because the data in Fig. 1 show TtPilN alone is able to mediate binding to the TtPilA437–122. The TtPilN–TtPilO complex that optimally satisfied these criteria (with a correlation coefficient of 1.606) was also the highest energetically favored model following refinement by FireDock. The position of TtPilM was manually adjusted in Chimera (66) to optimize its fit to the electron density map. For both the TtPilMN and TtPilMNO complexes, the proportions of mass contributed by the molecular models to the whole calculated mass were broadly in agreement with the ratios of the predicted volume from the models to the total volumes of the 3D reconstructions.
For the TtPilMNO–TtPilA437–122 complex, the model of the TtPilMNO dimer generated from the previous structure could be docked into the electron density map with minimal adjustment. The additional electron density for the two copies of the TtPilA437–122 pilin was readily apparent at the periphery of the complex, and the TtPilA437–122 crystal structure was manually docked into the density using Chimera (66), with the imposed constraint that the N-terminal helix was pointing toward the inner membrane.
Supplementary Material
Acknowledgments
We thank The Wellcome Trust for grant funding through Project Grant 093388.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org [PDB ID codes 4BHR (TtPilA37–122) and 4BHQ (TtPilN49–207)].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1312313110/-/DCSupplemental.
References
- 1.Pelicic V. Type IV pili: E pluribus unum? Mol Microbiol. 2008;68(4):827–837. doi: 10.1111/j.1365-2958.2008.06197.x. [DOI] [PubMed] [Google Scholar]
- 2.Giltner CL, Nguyen Y, Burrows LL. Type IV pilin proteins: Versatile molecular modules. Microbiol Mol Biol Rev. 2012;76(4):740–772. doi: 10.1128/MMBR.00035-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Craig L, Li J. Type IV pili: Paradoxes in form and function. Curr Opin Struct Biol. 2008;18(2):267–277. doi: 10.1016/j.sbi.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wolfgang M, van Putten JPM, Hayes SF, Dorward D, Koomey M. Components and dynamics of fiber formation define a ubiquitous biogenesis pathway for bacterial pili. EMBO J. 2000;19(23):6408–6418. doi: 10.1093/emboj/19.23.6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mattick JS. Type IV pili and twitching motility. Annu Rev Microbiol. 2002;56:289–314. doi: 10.1146/annurev.micro.56.012302.160938. [DOI] [PubMed] [Google Scholar]
- 6.Burrows LL. Weapons of mass retraction. Mol Microbiol. 2005;57(4):878–888. doi: 10.1111/j.1365-2958.2005.04703.x. [DOI] [PubMed] [Google Scholar]
- 7.Carbonnelle E, Helaine S, Nassif X, Pelicic V. A systematic genetic analysis in Neisseria meningitidis defines the Pil proteins required for assembly, functionality, stabilization and export of type IV pili. Mol Microbiol. 2006;61(6):1510–1522. doi: 10.1111/j.1365-2958.2006.05341.x. [DOI] [PubMed] [Google Scholar]
- 8.Berry J-L, et al. Structure and assembly of a trans-periplasmic channel for type IV pili in Neisseria meningitidis. PLoS Pathog. 2012;8(9):e1002923. doi: 10.1371/journal.ppat.1002923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Collins RF, et al. Structure of the Neisseria meningitidis outer membrane PilQ secretin complex at 12 A resolution. J Biol Chem. 2004;279(38):39750–39756. doi: 10.1074/jbc.M405971200. [DOI] [PubMed] [Google Scholar]
- 10.McLaughlin LS, Haft RJF, Forest KT. Structural insights into the Type II secretion nanomachine. Curr Opin Struct Biol. 2012;22(2):208–216. doi: 10.1016/j.sbi.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karuppiah V, Derrick JP. Structure of the PilM-PilN inner membrane type IV pilus biogenesis complex from Thermus thermophilus. J Biol Chem. 2011;286(27):24434–24442. doi: 10.1074/jbc.M111.243535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sampaleanu LM, et al. Periplasmic domains of Pseudomonas aeruginosa PilN and PilO form a stable heterodimeric complex. J Mol Biol. 2009;394(1):143–159. doi: 10.1016/j.jmb.2009.09.037. [DOI] [PubMed] [Google Scholar]
- 13.Tammam S, et al. Characterization of the PilN, PilO and PilP type IVa pilus subcomplex. Mol Microbiol. 2011;82(6):1496–1514. doi: 10.1111/j.1365-2958.2011.07903.x. [DOI] [PubMed] [Google Scholar]
- 14.Rumszauer J, Schwarzenlander C, Averhoff B. Identification, subcellular localization and functional interactions of PilMNOWQ and PilA4 involved in transformation competency and pilus biogenesis in the thermophilic bacterium Thermus thermophilus HB27. FEBS J. 2006;273(14):3261–3272. doi: 10.1111/j.1742-4658.2006.05335.x. [DOI] [PubMed] [Google Scholar]
- 15.Friedrich A, Prust C, Hartsch T, Henne A, Averhoff B. Molecular analyses of the natural transformation machinery and identification of pilus structures in the extremely thermophilic bacterium Thermus thermophilus strain HB27. Appl Environ Microbiol. 2002;68(2):745–755. doi: 10.1128/AEM.68.2.745-755.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rose I, et al. Identification and characterization of a unique, zinc-containing transport ATPase essential for natural transformation in Thermus thermophilus HB27. Extremophiles. 2011;15(2):191–202. doi: 10.1007/s00792-010-0343-2. [DOI] [PubMed] [Google Scholar]
- 17.Craig L, et al. Type IV pilus structure by cryo-electron microscopy and crystallography: Implications for pilus assembly and functions. Mol Cell. 2006;23(5):651–662. doi: 10.1016/j.molcel.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 18.Parge HE, et al. Structure of the fibre-forming protein pilin at 2.6 A resolution. Nature. 1995;378(6552):32–38. doi: 10.1038/378032a0. [DOI] [PubMed] [Google Scholar]
- 19.Craig L, et al. Type IV pilin structure and assembly: X-ray and EM analyses of Vibrio cholerae toxin-coregulated pilus and Pseudomonas aeruginosa PAK pilin. Mol Cell. 2003;11(5):1139–1150. doi: 10.1016/s1097-2765(03)00170-9. [DOI] [PubMed] [Google Scholar]
- 20.Ramboarina S, et al. Structure of the bundle-forming pilus from enteropathogenic Escherichia coli. J Biol Chem. 2005;280(48):40252–40260. doi: 10.1074/jbc.M508099200. [DOI] [PubMed] [Google Scholar]
- 21.Helaine S, Dyer DH, Nassif X, Pelicic V, Forest KT. 3D structure/function analysis of PilX reveals how minor pilins can modulate the virulence properties of type IV pili. Proc Natl Acad Sci USA. 2007;104(40):15888–15893. doi: 10.1073/pnas.0707581104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Audette GF, Irvin RT, Hazes B. Crystallographic analysis of the Pseudomonas aeruginosa strain K122-4 monomeric pilin reveals a conserved receptor-binding architecture. Biochemistry. 2004;43(36):11427–11435. doi: 10.1021/bi048957s. [DOI] [PubMed] [Google Scholar]
- 23.Hartung S, et al. Ultrahigh resolution and full-length pilin structures with insights for filament assembly, pathogenic functions, and vaccine potential. J Biol Chem. 2011;286(51):44254–44265. doi: 10.1074/jbc.M111.297242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abendroth J, Kreger AC, Hol WGJ. The dimer formed by the periplasmic domain of EpsL from the type 2 secretion system of Vibrio parahaemolyticus. J Struct Biol. 2009;168(2):313–322. doi: 10.1016/j.jsb.2009.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372(3):774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 26.Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N. ConSurf 2010: Calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010;38(Web Server issue):W529–W533. doi: 10.1093/nar/gkq399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005;33(Web Server issue):W363–W367. doi: 10.1093/nar/gki481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gray MD, Bagdasarian M, Hol WGJ, Sandkvist M. In vivo cross-linking of EpsG to EpsL suggests a role for EpsL as an ATPase-pseudopilin coupling protein in the type II secretion system of Vibrio cholerae. Mol Microbiol. 2011;79(3):786–798. doi: 10.1111/j.1365-2958.2010.07487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tammam S, et al. PilMNOPQ from the Pseudomonas aeruginosa type IV pilus system form a transenvelope protein interaction network that interacts with PilA. J Bacteriol. 2013;195(10):2126–2135. doi: 10.1128/JB.00032-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Georgiadou M, Castagnini M, Karimova G, Ladant D, Pelicic V. Large-scale study of the interactions between proteins involved in type IV pilus biology in Neisseria meningitidis: Characterization of a subcomplex involved in pilus assembly. Mol Microbiol. 2012;84(5):857–873. doi: 10.1111/j.1365-2958.2012.08062.x. [DOI] [PubMed] [Google Scholar]
- 31.Korotkov KV, Gonen T, Hol WGJ. Secretins: Dynamic channels for protein transport across membranes. Trends Biochem Sci. 2011;36(8):433–443. doi: 10.1016/j.tibs.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reichow SL, Korotkov KV, Hol WGJ, Gonen T. Structure of the cholera toxin secretion channel in its closed state. Nat Struct Mol Biol. 2010;17(10):1226–1232. doi: 10.1038/nsmb.1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Balasingham SV, et al. Interactions between the lipoprotein PilP and the secretin PilQ in Neisseria meningitidis. J Bacteriol. 2007;189(15):5716–5727. doi: 10.1128/JB.00060-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Korotkov KV, et al. Structural and functional studies on the interaction of GspC and GspD in the type II secretion system. PLoS Pathog. 2011;7(9):e1002228. doi: 10.1371/journal.ppat.1002228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Golovanov AP, et al. The solution structure of a domain from the Neisseria meningitidis lipoprotein PilP reveals a new beta-sandwich fold. J Mol Biol. 2006;364(2):186–195. doi: 10.1016/j.jmb.2006.08.078. [DOI] [PubMed] [Google Scholar]
- 36.Ayers M, et al. PilM/N/O/P proteins form an inner membrane complex that affects the stability of the Pseudomonas aeruginosa type IV pilus secretin. J Mol Biol. 2009;394(1):128–142. doi: 10.1016/j.jmb.2009.09.034. [DOI] [PubMed] [Google Scholar]
- 37.Wendler P, Ciniawsky S, Kock M, Kube S. Structure and function of the AAA+ nucleotide binding pocket. Biochim Biophys Acta. 2012;1823(1):2–14. doi: 10.1016/j.bbamcr.2011.06.014. [DOI] [PubMed] [Google Scholar]
- 38.Collins RF, Hassan D, Karuppiah V, Thistlethwaite A, Derrick JP. Structure and mechanism of the PilF DNA transformation ATPase from Thermus thermophilus. Biochem J. 2013;450(2):417–425. doi: 10.1042/BJ20121599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen Y, et al. Structure and function of the XpsE N-terminal domain, an essential component of the Xanthomonas campestris type II secretion system. J Biol Chem. 2005;280(51):42356–42363. doi: 10.1074/jbc.M506843200. [DOI] [PubMed] [Google Scholar]
- 40.Abendroth J, Murphy P, Sandkvist M, Bagdasarian M, Hol WG. The X-ray structure of the type II secretion system complex formed by the N-terminal domain of EpsE and the cytoplasmic domain of EpsL of Vibrio cholerae. J Mol Biol. 2005;348(4):845–855. doi: 10.1016/j.jmb.2005.02.061. [DOI] [PubMed] [Google Scholar]
- 41.Yamagata A, et al. Structure of an essential type IV pilus biogenesis protein provides insights into pilus and type II secretion systems. J Mol Biol. 2012;419(1-2):110–124. doi: 10.1016/j.jmb.2012.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Milgotina EI, Lieberman JA, Donnenberg MS. The inner membrane subassembly of the enteropathogenic Escherichia coli bundle-forming pilus machine. Mol Microbiol. 2011;81(4):1125–1127. doi: 10.1111/j.1365-2958.2011.07771.x. [DOI] [PubMed] [Google Scholar]
- 43.Abendroth J, Bagdasarian M, Sandkvist M, Hol WGJ. The structure of the cytoplasmic domain of EpsL, an inner membrane component of the type II secretion system of Vibrio cholerae: An unusual member of the actin-like ATPase superfamily. J Mol Biol. 2004;344(3):619–633. doi: 10.1016/j.jmb.2004.09.062. [DOI] [PubMed] [Google Scholar]
- 44.Lu C, et al. Hexamers of the type II secretion ATPase GspE from Vibrio cholerae with increased ATPase activity. Structure. 2013;21(9):1707–1717. doi: 10.1016/j.str.2013.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cisneros DA, Bond PJ, Pugsley AP, Campos M, Francetic O. Minor pseudopilin self-assembly primes type II secretion pseudopilus elongation. EMBO J. 2012;31(4):1041–1053. doi: 10.1038/emboj.2011.454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Winter G. xia2: An expert system for macromolecular crystallography data reduction. J Appl Cryst. 2010;43:186–190. [Google Scholar]
- 48.Evans PR. An introduction to data reduction: Space-group determination, scaling and intensity statistics. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):282–292. doi: 10.1107/S090744491003982X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Terwilliger TC, et al. Decision-making in structure solution using Bayesian estimates of map quality: The PHENIX AutoSol wizard. Acta Crystallogr D Biol Crystallogr. 2009;65(Pt 6):582–601. doi: 10.1107/S0907444909012098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adams PD, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Terwilliger TC, et al. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr D Biol Crystallogr. 2008;64(Pt 1):61–69. doi: 10.1107/S090744490705024X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McCoy AJ, et al. Phaser crystallographic software. J Appl Cryst. 2007;40(Pt 4):658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Afonine PV, et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr. 2012;68(Pt 4):352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 4):486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leslie AGW, Powell HR. Processing diffraction data with MOSFLM. NATO Sci Ser II Math. 2007;245:41–51. [Google Scholar]
- 56.Winn MD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sheldrick GM. A short history of SHELX. Acta Crystallogr A. 2008;64(Pt 1):112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 58.Bricogne G, Vonrhein C, Flensburg C, Schiltz M, Paciorek W. Generation, representation and flow of phase information in structure determination: Recent developments in and around SHARP 2.0. Acta Crystallogr D Biol Crystallogr. 2003;59(Pt 11):2023–2030. doi: 10.1107/s0907444903017694. [DOI] [PubMed] [Google Scholar]
- 59.Cowtan K. Recent developments in classical density modification. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 4):470–478. doi: 10.1107/S090744490903947X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cowtan K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr D Biol Crystallogr. 2006;62(Pt 9):1002–1011. doi: 10.1107/S0907444906022116. [DOI] [PubMed] [Google Scholar]
- 61.Vonrhein C, Blanc E, Roversi P, Bricogne G. Automated structure solution with autoSHARP. Methods Mol Biol. 2007;364:215–230. doi: 10.1385/1-59745-266-1:215. [DOI] [PubMed] [Google Scholar]
- 62.Chen VB, et al. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 1):12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Laskowski RA, McArthur MW, Moss DS, Thornton JM. PROCHECK: A program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;24:946–950. [Google Scholar]
- 64.Tang G, et al. EMAN2: An extensible image processing suite for electron microscopy. J Struct Biol. 2007;157(1):38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 65.Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. Geometry-based flexible and symmetric protein docking. Proteins. 2005;60(2):224–231. doi: 10.1002/prot.20562. [DOI] [PubMed] [Google Scholar]
- 66.Pettersen EF, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 67.Bordoli L, et al. Protein structure homology modeling using SWISS-MODEL workspace. Nat Protoc. 2009;4(1):1–13. doi: 10.1038/nprot.2008.197. [DOI] [PubMed] [Google Scholar]
- 68.Mashiach E, Schneidman-Duhovny D, Andrusier N, Nussinov R, Wolfson HJ. FireDock: A web server for fast interaction refinement in molecular docking. Nucleic Acids Res. 2008;36(Web Server issue):W229–W232. doi: 10.1093/nar/gkn186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Potterton L, et al. Developments in the CCP4 molecular-graphics project. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2288–2294. doi: 10.1107/S0907444904023716. [DOI] [PubMed] [Google Scholar]
- 70.Diederichs K, Karplus PA. Improved R-factors for diffraction data analysis in macromolecular crystallography. Nat Struct Biol. 1997;4(4):269–275. doi: 10.1038/nsb0497-269. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.