

Significance
In the present work, we show that overexpression of TWINKLE helicase reduces the amount of ROS-induced mtDNA mutations and ameliorates cardiomyopathy in Sod2+/− mice. We demonstrate that increased ROS in mitochondria result in a rise of base transversions and mtDNA rearrangements. Increased TWINKLE availability improves mtDNA integrity and protects cardiomyocytes by inhibiting apoptosis via p21. Our findings offer unique approaches to limit the loss of cardiomyocytes due to oxidative stress, a common problem in various disease conditions and during normal aging.
Keywords: recombination, mtDNA mutations, repair
Abstract
Mitochondrial DNA (mtDNA) in adult human heart is characterized by complex molecular forms held together by junctional molecules of unknown biological significance. These junctions are not present in mouse hearts and emerge in humans during postnatal development, concomitant with increased demand for oxidative metabolism. To analyze the role of mtDNA organization during oxidative stress in cardiomyocytes, we used a mouse model, which recapitulates the complex mtDNA organization of human hearts by overexpression of the mitochondrial helicase, TWINKLE. Overexpression of TWINKLE rescued the oxidative damage induced replication stalling of mtDNA, reduced mtDNA point mutation load, and modified mtDNA rearrangements in heterozygous mitochondrial superoxide dismutase knockout hearts, as well as ameliorated cardiomyopathy in mice superoxide dismutase knockout in a p21-dependent manner. We conclude that mtDNA integrity influences cell survival and reason that tissue specific modes of mtDNA maintenance represent an adaptation to oxidative stress.
Mammalian mitochondrial DNA (mtDNA) is a 16.5-kb circular double-stranded molecule that exists in thousands of copies per cell. It is essential for ATP production in mitochondria because it encodes 13 subunits of the protein complexes required for oxidative phosphorylation, as well as tRNAs and rRNAs necessary for mitochondrial protein biosynthesis. Mitochondria are especially abundant in the heart, the most energy demanding tissue in the mammalian body. Efficient mitochondrial activity is essential for normal heart function and embryonic development (1).
The majority of O2 in mitochondria is consumed by complex IV of the electron transport chain (ETC) during controlled reduction of O2 to water. However, because of electron leaks at complexes I and III, some oxygen molecules are reduced to superoxide anions (O2−), which, in turn, are converted to H2O2 by mitochondrial superoxide dismutase (SOD2) and further into water in a reaction facilitated by catalase (2, 3). Both O2− and H2O2 can react to form highly destructive OH· radicals, and all three are therefore commonly referred to as reactive oxygen species (ROS). Mitochondrial ROS directly damage mtDNA, oxidize disulfides in proteins, and cause peroxidation of membrane fatty acids (4, 5). Oxidative damage has been suggested to be a major source of somatic mtDNA mutations because it cross-links DNA and causes nucleotide modifications as well as single- and double-strand DNA breaks (6). The importance of ROS damage specifically to the heart is particularly evident in Sod2 knockout mice. Complete lack of SOD2 in homozygous knockout mice results in early postnatal lethality, whereas reduction of SOD2 activity in heterozygous (Sod2+/−) mice causes dilated cardiomyopathy during aging (7, 8). Other tissues in Sod2+/− mice are not markedly affected under physiological conditions with the exception of an increased rate of tumor formation in aged mice (9).
Oxidative mtDNA damage increases during postnatal heart development in rats when mitochondrial biogenesis is up-regulated and a metabolic switch from carbohydrate metabolism to β-oxidation of fatty acids occurs (10). In rodents, the developmental increase in ROS exposure temporarily elicits mtDNA repair responses but does not lead to major changes in mtDNA topology or replication during aging (10). In contrast, human heart mtDNA topology changes considerably during postnatal development (10). Although adult human heart mtDNA is organized in complex networks and shows high levels of junctional molecules, the mtDNA organization in the hearts of newborn babies is simple, resembling the situation in rodents (11–13).
Although four-way junctions and complex mtDNA molecules are not present at detectable levels in normal mouse heart, they can be induced by transgenic overexpression of the TWINKLE helicase (12). Besides being necessary for the maintenance of four-way junctions in human heart (10), TWINKLE also possesses strand-annealing activity in vitro, making it an attractive candidate conferring mitochondrial recombination activity (14).
We have hypothesized previously that enhanced recombination protects human heart mtDNA from chronic ROS exposure during long lifetime (11, 15). This view is also supported by the acquisition of complex mtDNA organization in postnatal human hearts concomitant with the increase in oxidative metabolism (12, 13) and ROS-dependent activation of recombination-dependent replication (RDR) in yeast (16). To test whether the mtDNA organization seen in human hearts protects against ROS, we took advantage of TWINKLE overexpressing (Tw+) mice, which recapitulate the structural phenotype of human heart mtDNA and crossed them with heterozygous Sod2+/− mice. We found that TWINKLE overexpression essentially eliminated the elevated mtDNA mutation load in Sod2+/− mouse hearts, changed the type of mtDNA rearrangements, and rescued cardiomyopathy in Sod2+/− mice, most likely by preventing apoptosis of cardiomyocytes via p21-dependent signaling. Our results indicate that TWINKLE maintains mtDNA integrity, hence promoting cardiomyocyte survival.
Results
Sod2+/− Heart mtDNA Shows Oxidative Damage, Replication Stalling, and Topological Changes.
First, we studied whether increased oxidative stress alters mtDNA maintenance by analyzing mtDNA from Sod2+/− hearts. We detected high levels of mitochondrial 8-oxoguanine (8-oxoG), a hallmark of oxidative DNA damage (9), in mtDNA of Sod2+/− hearts but not in wild-type (Wt) or Tw+ littermates. Overexpression of TWINKLE did not influence the 8-oxoG levels in compound Sod2+/−;Tw+ mice (Fig. 1A). Sod2+/− mice also showed evidence of replication stalling (increased levels of y forms; see ref. 17) in the noncoding region (NCR). X forms, suggestive for recombination junctions, were readily detectable in Tw+ and Sod2+/−;Tw+ mice, but absent in Sod2+/− mice (Fig. 1 B and C). Sod2+/− mice also exhibited altered heart mtDNA topology. Instead of single, monomeric circles, Sod2+/− mice carried a distinct high-molecular weight mtDNA structure, which differed from the more diffuse forms seen in Tw+ and Sod2+/−;Tw+ mice (Fig. 1 C and D). The high-molecular weight mtDNA structure found in Sod2+/− hearts resembled unicircular mtDNA dimers characteristic for the human heart (11, 18), because it formed a well-defined band resistant to the decatenating enzyme Escherichia coli Topoisomerase IV (Topo IV) (Fig. 1D). The relative mtDNA content in Sod2+/− hearts was similar to Wt, whereas the mtDNA content in Sod2+/−;Tw+ mice was comparable to the content in Tw+ mice (Fig. 1D).
Fig. 1.

Oxidative damage causes changes in mouse heart mtDNA replication and topological organization. (A) Mouse heart mtDNA was cut with MluI and separated over a 0.4% agarose gel, stained with EtBr, and subsequently blotted for detection of 8-oxoG modifications. (B) Two-dimensional AGE of ClaI-digested mouse heart mtDNA. Mouse hearts have few replication intermediates compared with liver or kidney (11). Sod2+/− mouse mtDNA shows distinct accumulation of y forms (Y), indicative of stalled replication. Molecules with recombination junctions (X) were detected only in Twinkle overexpressing mice (Tw+ and Sod2+/−;Tw+). (C) Undigested heart mtDNA with (+) or without (−) decatenating Topoisomerase IV treatment. Catenated dimers (open arrowhead) are sensitive to the enzyme, whereas unicircular dimers (filled arrowhead) are not. Topo IV relaxes supercoils (fastest moving band and asterisk). (D) Relative mtDNA content in the mouse hearts. a and b denote statistically significant differences between groups (P < 0.05, one-way ANOVA). Two to three males and 2–4 females were used per group.
TWINKLE Reverses the Increase in mtDNA Mutations in Sod2+/− Heart.
ROS has been suggested to be a major source of mtDNA mutations driving somatic aging (6), although this hypothesis has not been proven experimentally (19). Therefore, we analyzed whether the elevated ROS in Sod2+/− mice increases the mutation load of mtDNA. Single-nucleotide variants (SNVs) from mouse heart mtDNA were identified by using MitoSeq, a deep sequencing approach developed for enriched mtDNA (20). Although in next generation sequencing (NGS) the true mutation load might be partially masked by the relatively high false call rate for transversions compared with transitions (21), we can assume a consistent error rate in all samples, because they were sequenced together and the high sequence coverage allowed reliable determination of differences in SNV loads between samples. Our data revealed that overexpression of TWINKLE did not lead to significantly increased SNV loads compared with Wt mice (Fig. 2 and Fig. S1). In contrast, we observed significantly higher levels of SNVs in Sod2+/− mice, including G > C or C > A transversions, potentially caused by polymerase errors at 8-oxoG. We also noticed a nonsignificant increase in the frequency of base-pair transitions, the predominant type of mtDNA mutations. Surprisingly, overexpression of TWINKLE rescued the elevated SNV load in Sod2+/− mtDNA, and levels of transitions and G > C or C > A transversions returned to normal (Fig. 2). Baseline levels for error-corrected mtDNA mutation loads in the human brain are approximately 3.5 × 10−5 (21), indicating a 2- to 3-fold increase in total SNV load in Sod2+/− mice relative to Wt. Taken together, the relative increase of 1.6 SNVs per mtDNA in Sod2+/− mouse hearts was efficiently rescued by overexpression of TWINKLE.
Fig. 2.
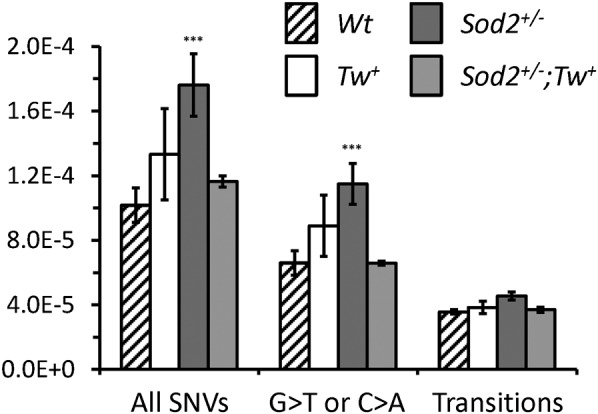
ROS increases the incidence of mtDNA point mutations. Frequencies of single-nucleotide variants (SNVs) per base for Wt, Tw+, Sod2+/−, and Sod2+/−;Tw+ mice. Shown are All SNVs (Left), G > T or C > A transversions at G or C bases (Center), and all transitions (G > A, A > G, C > T, or T > C; Right). Error bars indicate range by rank of frequency in littermates with different genotypes. ***P < 0.01, one-way ANOVA.
Analysis of deletion breakpoints by mapping the 3′ and 5′ positions of aligned segments of chimeric reads revealed increased levels of recombined molecules in Tw+ and Sod2+/− mouse hearts (Fig. 3 and Figs. S2 and S3). We found comparable numbers of rearrangements irrespective of inclusion or exclusion of replication origins in the NCR. The most striking difference between Sod2+/−and Tw+ mice was the overrepresentation (coverage) of certain sequences adjacent to common breakpoint regions (Fig. S2). The results from Sod2+/− mice resembled to some extent the increase in control region coverage generated by control region multimers (CRMs) occurring in polymerase γ 3′-5′ exonuclease-deficient mtDNA mutator mice (20). However, overrepresented sequences in Sod2+/− mice were spread around 6–7 hotspots throughout the mitochondrial genome and not restricted to the control region. Interestingly, these same regions have a higher H-strand G-content relative to other regions (Fig. S2D). The strand bias indicates that the generation of these rearrangements was not due to difficulty of DNA polymerases to replicate GC-rich regions, either in vivo or in vitro, but rather caused by 8-oxoG lesions during lagging strand synthesis. Overexpression of TWINKLE reduced the overrepresentation of specific short sequence stretches adjacent to abundant breakpoint regions in Sod2+/− mice, but had little effect on breakpoint frequency.
Fig. 3.

Rearrangement hotspots in mouse heart mtDNA correspond to regions of homology. (A) Scatter plots showing nucleotide coordinates of 3′- (y axis) and 5′-ends (x axis) of recombined molecules sequenced from the different mice. Gene loci are shown next to each axis. Black squares indicate tRNAs, red bars rRNA, and blue bars polypeptide encoding loci. Canonical deletions or molecules still retaining the 16299/1 numbering origin and, hence, the NCR are marked with red dots; noncanonical deletions, which lack 16299/1 are marked in black. Color intensity indicates rearrangement frequency. The similarity in the distribution of breakpoints arises from the regions of homology on mouse mtDNA (Fig. S3). rRNA and tRNA loci show lower levels of recombination. (B) An example of a rearrangement hotspot at the ND2 gene. The end of the ND2 sequence shows high levels of recombination with the ND6 gene locus but also with ND4, ND4L, and ND5. The few breakpoints at the NCR region are close to the replication origin (15). Sequence comparisons of homologies at the common breakpoints 4,521:13,695 and 4,821:13,948 are shown.
mtDNA Rearrangement Break Points in Sod2+/− Mice Correspond to Regions of Homology.
Further analysis of the rearranged mtDNA molecules revealed that 85% of sequence breakpoints occurred at regions sharing homology of at least three nucleotides with a median sequence homology of five nucleotides (Fig. 3 and Fig. S3). The most common class (14%) were rearrangements containing a section that had lost more than 16 kb of the genome, where the rearranged molecules retained only a <300-bp fragment of one parental molecule (Fig. S3C). It should be noted that determining whether a rearrangement is a deletion or duplication on a circular genome is not possible with the used deep sequencing platform.
TWINKLE Overexpression Ameliorates the Hypertrophic Cardiomyopathy in Sod2+/− Mice.
Next, we investigated whether the reduced rate of mtDNA mutations in Sod2+/−;Tw+ compound mice was functionally relevant and improved the cardiac phenotype of Sod2+/− mice. MRI and morphological analysis was performed with 8-mo-old Sod2+/− mice, in which cardiomyopathy manifests by gradual loss of cardiomyocytes and subsequent replacement fibrosis (8). One of the hallmarks of the Sod2+/− phenotype is the continuous thickening of the left ventricle, leading to increased end diastolic mass (Fig. 4 A and C and Fig. S4). This pathological phenotype was absent in aged Sod2+/−;Tw+ mice (Fig. 4 A and C and Fig. S4). Furthermore, morphological analysis revealed a significantly reduced amount of fibrosis in Sod2+/−;Tw+ compared with Sod2+/− hearts, although collagen III deposition remained increased compared with Wt mice (Fig. 4 B and D). These results indicate that TWINKLE overexpression significantly improves the cardiomyopathy in Sod2+/− mice.
Fig. 4.

Twinkle overexpression ameliorates the heart phenotype in the aged Sod2+/− mice. (A) End systole MRI of Wt, Tw+, Sod2+/−, and Sod2+/−/;Tw+ hearts. The cardiomyopathy in Sod2+/− mice is indicated by the thickened left ventricle and reduced lumen size. TWINKLE overexpression attenuated the phenotype. (B) Immunofluorescence staining for collagen type III reveals high level of replacement fibrosis in Sod2+/− mouse hearts, which is partially rescued by TWINKLE overexpression. (Scale bar: 50 μm.) (C) Quantitative assessment of cardiac function. (D) Quantification of collagen III levels. ED, end diastolic; EDV, end diastolic volume; EF, ejection fraction; ES, end systolic; ESV, end systolic volume; SV, systolic volume. Results are presented as mean ± SD. P values were calculated by using one-way ANOVA with Tukey´s multiple comparison test.
Up-Regulation of the DNA Damage Response Gene p21 Corresponds to Improvement of Cardiomyopathy in Sod2+/−;Tw+ Mice.
It seemed unlikely that normalization of SNV accumulation and a change in the organization of mtDNA rearrangements directly improve the cardiomyopathy in Sod2+/−;Tw+ mice. Rather, improved mtDNA integrity might affect retrograde cellular survival pathways that prevent the gradual loss of cardiomyocytes. Therefore, we analyzed transcriptional changes evoked in Tw+ and Sod2+/−;Tw+ compared with Wt and Sod2+/− mice. For these experiments, we used 14-wk-old mice to avoid indirect effects caused by the emerging cardiomyopathy.
We observed no significant transcriptional changes by DNA microarray analysis in the Tw+ mouse hearts compared with controls, with the exception of an eightfold increase in TWINKLE (Peo1), confirming overexpression of the transgene (Fig. S5). In contrast, we detected significant changes in the expression of nearly 5,000 genes (>1.5-fold change) in Sod2+/− compared with Wt hearts (Fig. S5A). Importantly, 60 genes were differentially regulated between Sod2+/− and Sod2+/−;Tw+ mouse hearts, with fold change of ≥1.5 (P < 0.05), including an eightfold increase of TWINKLE expression. In addition, we detected a significant up-regulation of Cdkn1a (p21) (Fig. 5 A and B). p21 is a key regulator of cellular responses under stress conditions, controlling cell cycle and inhibiting apoptosis to facilitate repair of DNA damage or to promote cellular senescence (22). Moreover, we observed an up-regulation of p53 (Trp53), one of the main regulators of p21, in Sod2+/−;Tw+ and, to a lesser degree, in Sod2+/− mouse hearts. In contrast, other genes, which mediated p53-dependent apoptosis such as Trp63, Trp73, Bax, and Bad, were either down-regulated or not altered in Sod2+/−;Tw+ compared with Sod2+/− mice (Fig. 5 A and B) (23). Similar results were obtained for other apoptosis-inducing factors such as E2f1, which is activated by mitochondrial ROS stress in cultured cells (24) and extrinsic apoptosis factors like Fas and Tnfrsf10b (or DR5/KILLER). To test whether TWINKLE overexpressing cells are more resistant to ROS-induced apoptosis, we exposed isolated cardiomyocytes to 100 nM H2O2 for 6 h. TWINKLE overexpressing cardiomyocytes showed increased p21 and Bcl-2 expression, but diminished PARP and Caspase 3 activation in contrast to control cells (Fig. 5C).
Fig. 5.

Overexpression of Twinkle in Sod2+/− mice increases p21 and elicits antiapoptotic signaling. (A) Expression levels of selected transcripts involved in protective responses or apoptosis in Sod2+/− and Tw+ mice. p21 (Cdkn1a) expression was significantly higher in Sod2+/−;Tw+ mice compared with other groups, whereas expression of p53 (Trp53), Akt (Akt3), TGFβ (Tgfb2), and Bcl2 did not differ statistically between Sod2+/− and Sod2+/−;Tw+. Expression of the proapoptotic factors Bax and Bad were lower in Sod2+/− and Sod2+/−;Tw+ hearts compared with controls. (B) Western blot analysis to confirm p21 and Bcl-2 up-regulation in Sod2+/−;Tw+ hearts as detected by the transcriptome analysis. Actinin was used as loading control. (C) Isolated adult mouse cardiomyocytes were exposed to 100 nM H2O2 for 6h. Tw+ cardiomyocytes showed significant increase in p21 and antiapoptotic Bcl-2 levels but lack PARP and caspase 3 activation as seen in the wild-type cells. Values are normalized against untreated samples of the same genotype, except for the cleaved PARP and caspase, which are normalized against treated wild-type. The cleaved caspase 3 is marked by an asterisk. P values were calculated by using one-way ANOVA with Tukey´s multiple comparison test. Two to three male and 2–4 female mice were used per group.
To gain further insight into the role of TWINKLE in the prevention of cardiomyopathy, we studied the p21-related cellular pathways controlling stress responses and looked for signs of cellular senescence. We observed a significant increase in the expression of Tgfb2 and Akt3 and of the cellular senescence marker β-galactosidase 1 (Glb1), in both Sod2+/− and Sod2+/−;Tw+ mouse hearts, although the response was augmented in the latter (Fig. 5A, Fig. S5B, and SI Results and Discussion).
Taken together, our results indicate that overexpression of TWINKLE adds very little on the cellular stress responses activated in the Sod2+/− mouse heart. More likely, TWINKLE-dependent changes in mtDNA seem to restrict via p21 some p53-dependent responses and activate cell survival factors, such as Bcl-2, improving the capability of Sod2+/− ;Tw+ hearts to deal with increased oxidative stress.
Discussion
In the present study, we mimicked the organization of mtDNA in human heart in mice by transgenic overexpression of the helicase TWINKLE and found that the altered mtDNA maintenance mode protected the heart from increased oxidative stress. Compound Sod2+/−;Tw+ hearts showed improvement of cardiomyopathy, associated with decreased mtDNA point mutation load, alterations in the type of mtDNA rearrangements, and activation of a cell survival involving p21.
Maintenance of mtDNA Under Enhanced Oxidative Stress Is Improved by Increased Levels of TWINKLE.
Our results demonstrate that the reduced expression of Sod2 expression causes increased oxidative damage, resulting in a striking mtDNA phenotype characterized by replication stalling and formation of unicircular dimers (Fig. 1). We thereby confirm that mtDNA is an important target for ROS, underscoring the importance of SOD2 in mtDNA maintenance and supporting its role in the mitochondrial nucleoid (25). Sod2+/− mice showed increased levels of overall SNVs, G > A and C > T transversions, and transitions in heart mtDNA, which confirms that ROS generated in mitochondria are a major source of mtDNA mutations. The SNV mutation loads in Sod2+/− mice are approximately 10-fold lower than in the PolG mtDNA mutator mouse expressing an error-prone mtDNA polymerase (20). Surprisingly, TWINKLE overexpression abolished the increase in mutation load caused by reduced SOD2 levels, indicating improved mtDNA repair. A possible mechanism is recombination-mediated repair, based on the assumption that the abundant mtDNA X forms in the TWINKLE mice reflect recombination, as they do in many other systems (26–28). In the nucleus, such forms strictly depend on recombinase activity (29), although further mechanistic studies are clearly necessary to unveil the exact function of TWINKLE for mtDNA reorganization. To further demonstrate this point, we also provide evidence of human heart mtDNA recombination, detectable by using rare molecular markers (Fig. S6). Previous evolutionary studies have also suggested that recombination is required for purging de novo mtDNA mutations in different species (30). Alternatively, increased dNTP incorporation into mtDNA, which has been seen in the skeletal muscles of Tw+ mice (31), might indicate that increased mtDNA replication and turnover overcomes the ROS-induced mutations. However, because mtDNA in Sod2+/−;Tw+ mice is equally exposed to ROS at all times, turnover alone seems insufficient (Fig. 1A). Increased replication, without enhanced replication fidelity, should result in a higher chance of introducing mutations due to replication errors, especially when maintaining a constant mtDNA copy number. This effect has been observed to a limited extent in mtDNA mutator mice overexpressing PGC1α, which have an increased mtDNA mutation load despite enhanced mitochondrial biogenesis (32).
Sod2+/− mice also exhibited an impressive amount of rearranged molecules (Fig. 3 and Figs. S2 and S3). More than 85% of all rearrangements occurred between homologous sequences, suggesting that illicit intramolecular or intermolecular recombination events were involved in the generation of rearranged mtDNA molecules. As a result, the breakpoints in rearranged mtDNA correlated with the overall sequence identity of mouse mtDNA (Fig. S3).
A major difference between mtDNA rearrangements in Tw+ mice and Sod2+/− mice was indicated by the differential sequence coverage coinciding with rearrangement breakpoints (Fig. S2A). In comparison with Tw+, Sod2+/− mice showed higher sequence coverage adjacent to the breakpoints. Peaks and breakpoint hotspots correlated with the H-strand G content (Fig. S2D), indicating that 8-oxoG lesions induced replication stalling and repair at the lagging strand, which requires the nascent H strand as a template. In contrast, PolG mutator mice display an increase in the mtDNA sequence coverage between NCR and OL clusters (33) caused by linear deletions without recombination breakpoints (34). The sharp peaks in sequence coverage of Sod2+/− mtDNA assemblies might represent a signature of small linear fragments. Such fragments most likely arise from stalled replication intermediates or turnover of damaged mtDNA.
Death Before Dishonor: mtDNA Integrity Influences Cell Survival.
TWINKLE overexpression did not only influence the type of mtDNA rearrangements and reduce the load of point mutations, but also ameliorated cardiomyopathy in Sod2+/− mice. However, the early postnatal lethality of Sod2−/− mice was not rescued (n = 8 litters). Absence of rescue is not surprising because the high concentrations of ROS in Sod2−/− mice, resulting in multisystemic pathologies including growth failure, metabolic abnormalities, and central nervous system damage (7, 35), cannot be influenced by mtDNA maintenance factors such as TWINKLE.
Our genome-wide transcriptional profiling analysis revealed that TWINKLE overexpression alone, despite the changes in mtDNA maintenance (Fig. 1), had little effect on global gene expression and no effect on gene expression related to the mitochondrial compartment (Fig. S5). In contrast, numerous transcriptional changes were found both in Sod2+/− and Sod2+/−;Tw+ mice, apparently caused by the increased oxidative stress.
Interestingly, senescence marker Glb1 (22) was more pronounced in TWINKLE overexpressing hearts (Fig. S4B). This observation can be explained by up-regulation of p21, which can inhibit apoptosis via factors such as Bcl-2 and induce cellular senescence by a stress response program (22).
The increase of p21 expression in Sod2+/−;Tw+ mice is most likely a consequence of altered mtDNA maintenance, such as efficient rescue of stalled replication intermediates, because TWINKLE itself is unlikely to have independent functions outside of mitochondria. The implicated link between mtDNA integrity and apoptotic signaling might also explain the increased rate of apoptosis in PolG mutator mice, which show high levels of linear mtDNA deletions without increased ROS production (36, 37). Sod2+/− cardiomyocytes are more sensitive to apoptosis caused by cellular stress during myocardial infarction (38) or other adverse conditions (8, 39), although it is difficult to score a measurable increase of apoptotic cardiomyocytes because of the gradual development of the age-dependent cardiomyopathy in Sod2+/− mice. Furthermore, aged Tw+ mouse hearts accumulate cytochrome oxidase negative cardiomyocytes (Fig. S7), indicating that cardiomyocytes with impaired mitochondrial function are maintained in aging mutant mice. Interestingly, a recent study (40) described that TWINKLE protects against cardiomyopathy caused by pressure overload, which is well in line with our findings.
Our study provides direct experimental evidence that ROS cause mtDNA mutations. It also demonstrates that increased helicase availability improves mtDNA integrity and protects cardiomyocytes from chronic oxidative stress. Although our model offers an explanation for the physiological relevance of the unusual mechanism of mtDNA maintenance in adult human hearts, the mechanistic role of TWINKLE in mtDNA modification and the retrograde mtDNA damage signaling pathways remain a challenge.
Materials and Methods
Animals.
Sod2 knockout (8) and TWINKLE transgenic (41) mice have been described. All animal experiments were performed in accordance with Max Planck institutional and German national ethical regulations (permission no. V54-19c20/15-B2/250) and with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health. Both male and female mice were used as indicated in the results.
Cardiomyocyte Isolation, Culture, and Exposure to ROS.
Isolation and the culture of adult mouse cardiomyocytes were performed by using standard procedures (42). Isolated cardiomyocytes are highly intolerant to H2O2 treatment. We found a concentration of 100 nM (using PBS as vehicle) to be optimal in our hands, enabling cardiomyocytes survival during a 6-h experiment, which is in line with other published studies (43).
mtDNA Analyses.
Fourteen-week-old mice were used for the analyses. Total heart DNA and enriched mtDNA were isolated as described (11). Relative mouse heart mtDNA copy number was determined by Southern blot analysis using MluI digested total DNA (cleaves mouse mtDNA once at nucleotide 1771) separated on 0.4% 1× Tris-Borate-EDTA agarose gels and quantified with a probe covering nucleotides 14,783–15,333 while using the nuclear 18S rDNA as an internal loading control (see SI Materials and Methods for details). Two-dimensional agarose gel electrophoresis, mtDNA topology analysis, and Southern blotting were performed by following standard procedures (44). Oxidative mtDNA damage was estimated by Southwestern blot analysis by using an antibody against 8-oxoG as described (10).
Deep Sequencing Analysis.
Analysis was done by using mtDNA from 14-wk-old Wt, Tw+, Sod2+/−, and Sod2+/−;Tw+ mouse hearts. mtDNA from two mice of each group was sequenced. Deep sequencing was carried out by using the MitoSeq workflow (20) and as indicated in SI Materials and Methods.
Transcriptome Analysis.
Three 14-wk-old female mice per each group were used for transcriptome analyses. Hearts were collected immediately after killing the animals, and total RNA was isolated from left ventricle sections with TRIzol (Invitrogen). Analyses were performed by using Affymetrix GeneChip Mouse Genome 430 2.0 Array following the manufacturer's protocol and scanned by using standard procedures (45). The microarray data have been submitted to ArrayExpress Archive (www.ebi.ac.uk/arrayexpress, accession no. E-MEXP-3983).
SDS/PAGE and Western Blotting.
SDS/PAGE (Laemmli) gels [7.5 and 12% (wt/vol)] as well as NuPAGE 3–12% (wt/vol) Bis-Tris and Tris-Acetate gels (Invitrogen) were used by using standard conditions. Heart muscle samples were homogenized in 4% (wt/vol) SDS and 10 mM Tris at pH 7.4 by using an Ultra-Turrax and sonicated to reduce viscosity. DTT was added to a final sample concentration of 1 mM, and samples were boiled for 5 min after measurement of protein content. Western blotting and immunodetection were carried out by using standard procedures and peroxidase-coupled secondary antibodies.
The primary antibodies used were as follows: Rabbit anti–pan-actin (4968), rabbit anti–α-actinin (3134), rabbit anti-p53 (9289), mouse anti-p21 (2946), rabbit anti-caspase 3 (9662), rabbit anti-cleaved caspase 3 (9661), rabbit anti-cleaved PARP (5625) from Cell Signaling Technology. Mouse anti–Bcl-2 (610538) was purchased from BD Transduction Laboratories.
Mouse Heart Magnetic Resonance Imaging and Immunohistochemistry.
MRI was performed on 32-wk-old male mice essentially as described (46). Six mice were included in each group (Wt, Tw+, Sod2+/−, and Sod2+/−;Tw+). The same animals were used for immunohistochemical analysis of replacement fibrosis. Briefly, the heart was removed immediately after sacrificing and frozen in Tissue-Tek (Sakura) on dry ice. Hearts were cut into 10-μm sections by using a Leica CM 1950 cryostat and fixed in acetone for 10 min. Sections were washed twice in PBS and incubated overnight at 4 °C with 1:200 dilution of rabbit anti-Collagen Type III antibody (Rockland; 600-401-105-0.1). After repeated washing in PBS, sections were incubated at room temperature for 1 h in PBS with a 1:500 dilution of goat anti-rabbit cy2 (Dianova) labeled secondary antibody and 2 ng/μL DAPI (Pierce). After repeated washing in PBS, the coverslip was mounted by using Mowiol 4-88 (10% in 0,1 M Tris⋅HCl at pH 8.5; Roth).
Supplementary Material
Acknowledgments
We thank Dr. Astrid Wietelmann and Ursula Hoffmann for performing the MRI and assisting with the data analyses; Mrs. Marion Wiesnet for isolating adult mouse cardiomyocytes; Mrs. Sylvia Thomas for her superb technical assistance; Mrs. Jennifer Norris for the excellent work with the mouse colony; Dr. Marten Szibor for the discussions regarding ROS in hearts and Dr. James Stewart (Max Planck Institute for the Biology of Ageing, Cologne) for the useful discussions on NGS data; Prof. Douglas Turnbull and Prof. Robert Taylor (University of Newcastle) for providing the KSS patient samples for the recombination analysis presented in Fig. S6; and Prof. Howy Jacobs, because some of the ideas presented in this paper originated from work performed several years ago at the University of Tampere, Finland. This work was supported by the European Molecular Biology Organization, Academy of Finland, Jane and Aatos Erkko Foundation, James and Esther King Biomedical Research Program (3KN09), the Max Planck Society, the Excellence Cluster Cardiopulmonary System, and Deutsche Forschungsgemeinschaft Grant SFB TRR81.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. D.P.K. is a guest editor invited by the Editorial Board.
Data deposition: The microarray datasets reported in this paper have been deposited in the ArrayExpress Archive, http://www.ebi.ac.uk/arrayexpress (accession nos. E-MEXP-3983).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1303046110/-/DCSupplemental.
References
- 1.Goffart S, von Kleist-Retzow JC, Wiesner RJ. Regulation of mitochondrial proliferation in the heart: Power-plant failure contributes to cardiac failure in hypertrophy. Cardiovasc Res. 2004;64(2):198–207. doi: 10.1016/j.cardiores.2004.06.030. [DOI] [PubMed] [Google Scholar]
- 2.Grivennikova VG, Vinogradov AD. Generation of superoxide by the mitochondrial Complex I. Biochim Biophys Acta. 2006;1757(5-6):553–561. doi: 10.1016/j.bbabio.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 3.Andreyev AY, Kushnareva YE, Starkov AA. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc) 2005;70(2):200–214. doi: 10.1007/s10541-005-0102-7. [DOI] [PubMed] [Google Scholar]
- 4.Liu P, Demple B. DNA repair in mammalian mitochondria: Much more than we thought? Environ Mol Mutagen. 2010;51(5):417–426. doi: 10.1002/em.20576. [DOI] [PubMed] [Google Scholar]
- 5.Stefanatos R, Sanz A. Mitochondrial complex I: A central regulator of the aging process. Cell Cycle. 2011;10(10):1528–1532. doi: 10.4161/cc.10.10.15496. [DOI] [PubMed] [Google Scholar]
- 6.Beckman KB, Ames BN. The free radical theory of aging matures. Physiol Rev. 1998;78(2):547–581. doi: 10.1152/physrev.1998.78.2.547. [DOI] [PubMed] [Google Scholar]
- 7.Li Y, et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet. 1995;11(4):376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- 8.Loch T, et al. Different extent of cardiac malfunction and resistance to oxidative stress in heterozygous and homozygous manganese-dependent superoxide dismutase-mutant mice. Cardiovasc Res. 2009;82(3):448–457. doi: 10.1093/cvr/cvp092. [DOI] [PubMed] [Google Scholar]
- 9.Jang YC, Remmen VH. The mitochondrial theory of aging: Insight from transgenic and knockout mouse models. Exp Gerontol. 2009;44(4):256–260. doi: 10.1016/j.exger.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 10.Pohjoismäki JL, et al. Oxidative stress during mitochondrial biogenesis compromises mtDNA integrity in growing hearts and induces a global DNA repair response. Nucleic Acids Res. 2012;40(14):6595–6607. doi: 10.1093/nar/gks301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pohjoismäki JL, et al. Human heart mitochondrial DNA is organized in complex catenated networks containing abundant four-way junctions and replication forks. J Biol Chem. 2009;284(32):21446–21457. doi: 10.1074/jbc.M109.016600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pohjoismäki JL, et al. Developmental and pathological changes in the human cardiac muscle mitochondrial DNA organization, replication and copy number. PLoS ONE. 2010;5(5):e10426. doi: 10.1371/journal.pone.0010426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pohjoismäki JL, et al. Postnatal cardiomyocyte growth and mitochondrial reorganization cause multiple changes in the proteome of human cardiomyocytes. Mol Biosyst. 2013;9(6):1210–1219. doi: 10.1039/c3mb25556e. [DOI] [PubMed] [Google Scholar]
- 14.Sen D, Nandakumar D, Tang GQ, Patel SS. Human mitochondrial DNA helicase TWINKLE is both an unwinding and annealing helicase. J Biol Chem. 2012;287(18):14545–14556. doi: 10.1074/jbc.M111.309468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pohjoismäki JL, Goffart S. Of circles, forks and humanity: Topological organisation and replication of mammalian mitochondrial DNA. Bioessays. 2011;33(4):290–299. doi: 10.1002/bies.201000137. [DOI] [PubMed] [Google Scholar]
- 16.Hori A, Yoshida M, Shibata T, Ling F. Reactive oxygen species regulate DNA copy number in isolated yeast mitochondria by triggering recombination-mediated replication. Nucleic Acids Res. 2009;37(3):749–761. doi: 10.1093/nar/gkn993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wanrooij S, Goffart S, Pohjoismäki JL, Yasukawa T, Spelbrink JN. Expression of catalytic mutants of the mtDNA helicase Twinkle and polymerase POLG causes distinct replication stalling phenotypes. Nucleic Acids Res. 2007;35(10):3238–3251. doi: 10.1093/nar/gkm215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kolesar JE, Wang CY, Taguchi YV, Chou SH, Kaufman BA. Two-dimensional intact mitochondrial DNA agarose electrophoresis reveals the structural complexity of the mammalian mitochondrial genome. Nucleic Acids Res. 2013;41(4):e58. doi: 10.1093/nar/gks1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Larsson NG. Somatic mitochondrial DNA mutations in mammalian aging. Annu Rev Biochem. 2010;79:683–706. doi: 10.1146/annurev-biochem-060408-093701. [DOI] [PubMed] [Google Scholar]
- 20.Williams SL, et al. The mtDNA mutation spectrum of the progeroid Polg mutator mouse includes abundant control region multimers. Cell Metab. 2010;12(6):675–682. doi: 10.1016/j.cmet.2010.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schmitt MW, et al. Detection of ultra-rare mutations by next-generation sequencing. Proc Natl Acad Sci USA. 2012;109(36):14508–14513. doi: 10.1073/pnas.1208715109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Erol A. Deciphering the intricate regulatory mechanisms for the cellular choice between cell repair, apoptosis or senescence in response to damaging signals. Cell Signal. 2011;23(7):1076–1081. doi: 10.1016/j.cellsig.2010.11.023. [DOI] [PubMed] [Google Scholar]
- 23.Haupt S, Berger M, Goldberg Z, Haupt Y. Apoptosis - the p53 network. J Cell Sci. 2003;116(Pt 20):4077–4085. doi: 10.1242/jcs.00739. [DOI] [PubMed] [Google Scholar]
- 24.Raimundo N, et al. Mitochondrial stress engages E2F1 apoptotic signaling to cause deafness. Cell. 2012;148(4):716–726. doi: 10.1016/j.cell.2011.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kienhöfer J, et al. Association of mitochondrial antioxidant enzymes with mitochondrial DNA as integral nucleoid constituents. FASEB J. 2009;23(7):2034–2044. doi: 10.1096/fj.08-113571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Webb MR, Plank JL, Long DT, Hsieh TS, Kreuzer KN. The phage T4 protein UvsW drives Holliday junction branch migration. J Biol Chem. 2007;282(47):34401–34411. doi: 10.1074/jbc.M705913200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Branzei D, et al. Ubc9- and mms21-mediated sumoylation counteracts recombinogenic events at damaged replication forks. Cell. 2006;127(3):509–522. doi: 10.1016/j.cell.2006.08.050. [DOI] [PubMed] [Google Scholar]
- 28.Mankouri HW, Ashton TM, Hickson ID. Holliday junction-containing DNA structures persist in cells lacking Sgs1 or Top3 following exposure to DNA damage. Proc Natl Acad Sci USA. 2011;108(12):4944–4949. doi: 10.1073/pnas.1014240108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vanoli F, Fumasoni M, Szakal B, Maloisel L, Branzei D. Replication and recombination factors contributing to recombination-dependent bypass of DNA lesions by template switch. PLoS Genet. 2010;6(11):e1001205. doi: 10.1371/journal.pgen.1001205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsaousis AD, Martin DP, Ladoukakis ED, Posada D, Zouros E. Widespread recombination in published animal mtDNA sequences. Mol Biol Evol. 2005;22(4):925–933. doi: 10.1093/molbev/msi084. [DOI] [PubMed] [Google Scholar]
- 31.Ylikallio E, Tyynismaa H, Tsutsui H, Ide T, Suomalainen A. High mitochondrial DNA copy number has detrimental effects in mice. Hum Mol Genet. 2010;19(13):2695–2705. doi: 10.1093/hmg/ddq163. [DOI] [PubMed] [Google Scholar]
- 32.Dillon LM, et al. Increased mitochondrial biogenesis in muscle improves aging phenotypes in the mtDNA mutator mouse. Hum Mol Genet. 2012;21(10):2288–2297. doi: 10.1093/hmg/dds049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ameur A, et al. Ultra-deep sequencing of mouse mitochondrial DNA: Mutational patterns and their origins. PLoS Genet. 2011;7(3):e1002028. doi: 10.1371/journal.pgen.1002028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bailey LJ, et al. Mice expressing an error-prone DNA polymerase in mitochondria display elevated replication pausing and chromosomal breakage at fragile sites of mitochondrial DNA. Nucleic Acids Res. 2009;37(7):2327–2335. doi: 10.1093/nar/gkp091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lebovitz RM, et al. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc Natl Acad Sci USA. 1996;93(18):9782–9787. doi: 10.1073/pnas.93.18.9782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kujoth GC, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309(5733):481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 37.Trifunovic A, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429(6990):417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 38.Van Remmen H, et al. Multiple deficiencies in antioxidant enzymes in mice result in a compound increase in sensitivity to oxidative stress. Free Radic Biol Med. 2004;36(12):1625–1634. doi: 10.1016/j.freeradbiomed.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 39.Van Remmen H, et al. Knockout mice heterozygous for Sod2 show alterations in cardiac mitochondrial function and apoptosis. Am J Physiol Heart Circ Physiol. 2001;281(3):H1422–H1432. doi: 10.1152/ajpheart.2001.281.3.H1422. [DOI] [PubMed] [Google Scholar]
- 40.Tanaka A, et al. The overexpression of Twinkle helicase ameliorates the progression of cardiac fibrosis and heart failure in pressure overload model in mice. PLoS ONE. 2013;8(6):e67642. doi: 10.1371/journal.pone.0067642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tyynismaa H, et al. Twinkle helicase is essential for mtDNA maintenance and regulates mtDNA copy number. Hum Mol Genet. 2004;13(24):3219–3227. doi: 10.1093/hmg/ddh342. [DOI] [PubMed] [Google Scholar]
- 42.O’Connell TD, Rodrigo MC, Simpson PC. Isolation and culture of adult mouse cardiac myocytes. Methods Mol Biol. 2007;357:271–296. doi: 10.1385/1-59745-214-9:271. [DOI] [PubMed] [Google Scholar]
- 43.Goto K, et al. Unique mode of cell death in freshly isolated adult rat ventricular cardiomyocytes exposed to hydrogen peroxide. Med Mol Morphol. 2009;42(2):92–101. doi: 10.1007/s00795-009-0439-x. [DOI] [PubMed] [Google Scholar]
- 44.Pohjoismäki JL, et al. Alterations to the expression level of mitochondrial transcription factor A, TFAM, modify the mode of mitochondrial DNA replication in cultured human cells. Nucleic Acids Res. 2006;34(20):5815–5828. doi: 10.1093/nar/gkl703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lipshutz RJ, Fodor SP, Gingeras TR, Lockhart DJ. High density synthetic oligonucleotide arrays. Nat Genet. 1999;21(1) Suppl:20–24. doi: 10.1038/4447. [DOI] [PubMed] [Google Scholar]
- 46.Ziebart T, et al. Sustained persistence of transplanted proangiogenic cells contributes to neovascularization and cardiac function after ischemia. Circ Res. 2008;103(11):1327–1334. doi: 10.1161/CIRCRESAHA.108.180463. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.