

Significance
Calcium-activated chloride channels play key roles in physiology, from mediating sensory transduction and nociception to regulating mucine secretion in airway epithelia and controlling excitability of smooth muscle fibers. Recently, TMEM16A was identified as the pore-forming subunit of these channels. It remains unclear, however, whether this protein is sufficient to form calcium-activated chloride channels, or whether association with other subunits is required for function. Recently, association with calmodulin has been proposed to be required for the calcium-dependent activation and ion selectivity of these channels. Here we show that purified and reconstituted TMEM16A is necessary and sufficient to recapitulate the properties of native and heterologously expressed calcium-activated chloride currents. Thus, association of TMEM16A with other proteins is not required for function.
Keywords: ion channels, anoctamin, reconstitution, membrane protein
Abstract
Ca2+-activated Cl− channels (CaCCs) are key regulators of numerous physiological functions, ranging from electrolyte secretion in airway epithelia to cellular excitability in sensory neurons and muscle fibers. Recently, TMEM16A (ANO1) and -B were shown to be critical components of CaCCs. It is still unknown whether they are also sufficient to form functional CaCCs, or whether association with other subunits is required. Recent reports suggest that the Ca2+ sensitivity of TMEM16A is mediated by its association with calmodulin, suggesting that functional CaCCs are heteromultimers. To test whether TMEM16A is necessary and sufficient to form functional CaCCs, we expressed, purified, and reconstituted human TMEM16A. The purified protein mediates Ca2+-dependent Cl− transport with submicromolar sensitivity to Ca2+, consistent with what is seen in patch–clamp experiments. The channel is synergistically gated by Ca2+ and voltage, so that opening is promoted by depolarizing potentials. Mutating two conserved glutamates in the TM6-7 intracellular loop selectively abolishes the Ca2+ dependence of reconstituted TMEM16A, in a manner similar to what was reported for the heterologously expressed channel. Well-characterized CaCC blockers inhibit Cl− transport with Kis comparable to those measured for native and heterologously expressed CaCCs. Finally, direct physical interactions between calmodulin and TMEM16A could not be detected in copurification experiments or in functional assays. Our results demonstrate that purified TMEM16A is necessary and sufficient to recapitulate the biophysical and pharmacological properties of native and heterologously expressed CaCCs. Our results also show that association of TMEM16A with other proteins, such as calmodulin, is not required for function.
Ca2+-activated Cl− channels (CaCCs) regulate a number of physiological functions including epithelial electrolyte secretion in cardiac muscle contractility, preventing polyspermy in oocytes, olfactory responses, and regulating the membrane potential in the nervous system (1, 2). The initial discovery of these channels dates to over 30 y ago in salamander photoreceptors (3) and was followed by numerous reports of Ca2+-activated Cl− currents in salivary glands, airway epithelia, Xenopus oocytes, and smooth muscle cells (1). The molecular identity of CaCCs remained unknown until recently, when TMEM16A and -B (also known as Anoctamin1 and 2) were identified as pore-forming components of Ca2+-activated Cl− channels (4–6). This seminal discovery allowed researchers to identify the novel roles played by these channels in a variety of physiological processes (2) including mediating salt secretion in kidneys, lungs, and airway epithelia, controlling the excitability of cardiac and smooth muscle cells (7, 8), nociception in sensory neurons (9, 10), and cell proliferation (11–14).
Despite these breakthroughs, the TMEM16A channels remain poorly understood at the molecular level, even for their most basic features. Strikingly, two fundamental questions that remain are as follows. (i) Is TMEM16A sufficient to form a functional Ca2+-activated channel? Or, is association with ancillary subunits required? (ii) Does Ca2+ activate TMEM16A directly or indirectly? Recent work has shown that TMEM16A forms dimers in cells (15–17), associates with members of the ezrin–moesin network of cytoskeletal proteins (18), and that channel activation and ion selectivity require an obligatory association with calmodulin (CaM) (19, 20). It remains unclear, however, whether these proteins are integral components of the functional CaCC complex or whether they are dispensable for function. The basis of the Ca2+ sensitivity of TMEM16A is controversial: Several reports suggest that Ca2+ modulates TMEM16A indirectly, through association of the channel with calmodulin (19–21). Indeed, the TMEM16A primary sequence lacks recognizable Ca2+-binding motifs (22), whereas it possesses at least two putative calmodulin-binding domains (20). In contrast, a conserved pair of acidic residues in the intracellular TM6-7 loop was recently shown to play a key role in the Ca2+ sensitivity of TMEM16A and -F (23, 24), suggesting that Ca2+ might directly interact with the channel.
To address these two questions, we purified the TMEM16A channel and tested its function in vitro. We found that the purified human (h)TMEM16A protein alone recapitulates all of the fundamental biophysical and pharmacological properties of native and heterologously expressed CaCCs. We could not detect a direct interaction between purified TMEM16A and calmodulin. Our results demonstrate that TMEM16A is necessary and sufficient to form functional Ca2+-activated Cl− channels and that association with other proteins, such as calmodulin, is not required for their activity.
Results
Purified hTMEM16A Forms Dimers.
The abc splice variant of hTMEM16A (4, 25) was expressed in Sf9 insect cells and purified using metal-affinity chromatography. The purified protein ran on a gel-filtration column as one main peak eluting at ∼10.4 mL (Fig. 1A). Of the two minor peaks in the elution profile, the first was at ∼8 mL whereas the second eluted at ∼14 mL and was a truncated TMEM16A that lacks at least its N-terminal domain, as verified by an antihistidine Western blot (Fig. S1A), and migrates as a monomer (Fig. 1B), consistent with the observation that the N terminus is necessary for dimerization (17). The identity of both bands was confirmed using mass spectrometry. Rerunning the main peak through gel filtration after 12–24 h yielded a single, monodisperse peak at the same position (Fig. 1A), indicating that the purified hTMEM16Aabc protein is stable. The elution volume of the hTMEM16A channel complex is comparable to that of the purified MthK K+ channel tetramer (Fig. S1B), suggesting that the molecular mass of TMEM16A is ∼250 kDa, consistent with the expectation that this Cl− channel forms dimers (15–17). Further support for this conclusion comes from experiments in which we used glutaraldehyde to cross-link the channel complex (26, 27). Whereas the nonspecific nature of this cross-linking reagent can lead to overestimating the size of a molecular complex, in the case of purified TMEM16A the results are clean (Fig. 1C): Upon glutaraldehyde treatment, the TMEM16A protein band quantitatively shifts from its monomer position (∼120 kDa) to one consistent with a dimer (∼250 kDa) (Fig. 1C), with no higher molecular mass bands appearing. The same treatment under denaturing conditions yielded a single band at the monomer position, confirming that the dimer is disrupted in SDS (Fig. 1C). In all cases, glutaraldehyde-treated samples ran as poorly defined bands, consistent with the heterogeneous products of this reaction (28).
Fig. 1.

Purification of TMEM16A. (A) Size-exclusion chromatography profile of purified hTMEM16Aabc (black). The numbers refer to the fractions collected, with peak II being the main peak where hTMEM16Aabc elutes. The red trace corresponds to the rerun of peak II after 16 h at 4 °C. (B) SDS/PAGE of purified hTMEM16Aabc. Lane 1, marker; lane 2, after nickel column; lanes 3–5, peaks I/II/III from A. (C) Glutaraldehyde (GA) cross-linking. Lanes 1–6, protein with and without Ca2+ in the absence of GA; lane 7, marker; lanes 8–11, cross-linking induced by 0.125% GA in the presence of 1 mM Ca2+ (lane 8) or 1 mM EGTA (lane 10) is prevented by incubation with 2% SDS (lanes 9 and 11); lanes 12 and 13, cross-linking induced by 0.5% GA in the presence of 1 mM Ca2+ (lane 12) or 1 mM EGTA (lane 13). Arrowheads indicate the position of hTMEM16Aabc without GA treatment (red), with GA treatment (green), and with GA treatment after incubation with SDS (blue). The yellow arrowhead indicates the presence of a GA-induced large molecular mass aggregate induced by high concentrations of GA.
Purified hTMEM16A Is a Ca2+-Dependent and Cl−-Selective Channel.
We assayed the functionality of purified hTMEM16A by reconstituting it in proteoliposomes and testing whether it mediates passive Cl− efflux using a Cl−-selective electrode (29) (Fig. 2A). Proteoliposomes were reconstituted in the presence of a 300-fold KCl gradient, and Cl− efflux was initiated by the addition of the K+ ionophore valinomycin, which plays a dual role: It shunts the voltage established by the Cl− gradient and sets the membrane potential to the K+ reversal potential (30). Protein-free liposomes reconstituted under these conditions showed no significant Cl− leak (Fig. 2B). In contrast, addition of valinomycin induced Cl− efflux from hTMEM16A-containing liposomes (Fig. 2B). Consistent with expectations, the rate of Cl− transport was enhanced by the presence of 1 mM Ca2+ (Fig. 2B). These experiments also demonstrate that TMEM16A forms Cl−-selective channels with minimal permeability to cations. If the channels had a finite permeability to K+, then the liposomes would lose their Cl− content before the addition of valinomycin, as seen for the poorly selective TMEM16 channel afTMEM16 (31). Thus, purified hTMEM16A is a Cl−-selective channel that is activated by Ca2+.
Fig. 2.

Purified hTMEM16Aabc mediates Ca2+-dependent Cl− transport. (A) Schematic representation of the Cl− efflux assay. (B) Time course of valinomycin-induced Cl− efflux from protein-free liposomes in the presence (blue) or absence of extraliposomal Ca2+ (cyan). The time course is normalized to the value of Cl− after the addition of detergent (*) to dissolve all liposomes. Cl− efflux from liposomes reconstituted with 5 µg TMEM16A/mg lipid in the presence of 1 mM (red) and absence of Ca2+ (purple). In all cases, efflux was initiated at t = 0 by addition of a 1-μL solution of 2 mg/mL of valinomycin. The asterisk denotes the addition of 40 μL of 1.5 M n-octyl-β-D-glucopyranoside to dissolve all liposomes. (C) Protein dependence of Cl− efflux. Liposomes were reconstituted at 0 (black), 0.25 (red), 0.5 (green), 1 (yellow), 2.5 (blue), 5 (purple), and 10 (cyan) µg protein/mg lipid. (D) Fraction of inactive liposomes as a function of protein concentration. The solid line represents the fit to Eq. 1 with θ = 0.40 ± 0.03, φ = 0.41 ± 0.03, and p0 = 1.13 ± 0.15. Each data point represents the average of n = 3–6 independent experiments, and the error is the SEM. The error on the fit parameters θ, φ, and p0 is the uncertainty on the fit.
Estimating the Fraction of Active TMEM16A Channels.
To determine the fraction of active purified channels, we performed the reconstitutions at various protein-to-lipid ratios. Assuming that the reconstitution process randomly inserts proteins into liposomes, the fraction of liposomes containing zero copies of a protein, f0, is given by a modified Poisson distribution (29, 32),
where θ is the fraction of liposomes that is refractory to protein incorporation, p is the protein density, 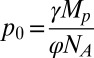 is the protein density at which the expected occupancy of the liposomes by the protein complex is 1, γ is the number of liposomes per mass of lipid, Mp is ∼228 kDa (the mass of the TMEM16A dimer), φ is the unknown fraction of active proteins, and NA is Avogadro’s number. By fitting the experimentally determined f0s at various protein densities (Fig. 2C), we found that p0 ∼ 1.1 and φ ∼ 0.41. The high refractory pool of liposomes, θ ∼ 0.4, leads to an underestimation of the fraction of active proteins and is likely due to the reconstitution method. Indeed, the f0 of liposomes containing high copy numbers of CLC-ec1, a well-characterized H+/Cl− exchanger that has ∼100% activity (29), is low when prepared via dialysis and high when reconstituted via Bio-Beads (Fig. S2). The idea that a large fraction of liposomes is refractory to TMEM16A incorporation is also consistent with the observation that f0 is constant when the protein density increases from 1 to 10 µg protein/mg lipid (Fig. 2D), indicating that at 1 µg protein/mg lipid all accessible liposomes contain at least one active protein.
is the protein density at which the expected occupancy of the liposomes by the protein complex is 1, γ is the number of liposomes per mass of lipid, Mp is ∼228 kDa (the mass of the TMEM16A dimer), φ is the unknown fraction of active proteins, and NA is Avogadro’s number. By fitting the experimentally determined f0s at various protein densities (Fig. 2C), we found that p0 ∼ 1.1 and φ ∼ 0.41. The high refractory pool of liposomes, θ ∼ 0.4, leads to an underestimation of the fraction of active proteins and is likely due to the reconstitution method. Indeed, the f0 of liposomes containing high copy numbers of CLC-ec1, a well-characterized H+/Cl− exchanger that has ∼100% activity (29), is low when prepared via dialysis and high when reconstituted via Bio-Beads (Fig. S2). The idea that a large fraction of liposomes is refractory to TMEM16A incorporation is also consistent with the observation that f0 is constant when the protein density increases from 1 to 10 µg protein/mg lipid (Fig. 2D), indicating that at 1 µg protein/mg lipid all accessible liposomes contain at least one active protein.
Ca2+ Directly Activates TMEM16A.
We then determined whether activation of purified TMEM16A by Ca2+ mirrors that of native CaCC currents in cells. We prepared liposomes in 0 Ca2+ and varied the external Ca2+ concentration (Ca2+ex). As expected, the rate of Cl− efflux mediated by TMEM16A increased as a function of Ca2+ex (Fig. 3A), with a midpoint of activation of ∼210 nM (Fig. 3B), a value close to that found in electrophysiological measurements (25). The curve is well-fit with a Hill coefficient of ∼2 (Fig. 3B), suggesting that binding of two Ca2+ ions might be necessary to open the channel.
Fig. 3.

Ca2+ dependence of TMEM16A. (A) Cl− efflux mediated by TMEM16A in the presence of 0 (black), 100 nM (red), 150 nM (brown), 200 nM (pink), 300 nM (yellow), 400 nM (green), 1 µM (cyan), and 1 mM (blue) external Ca2+ and 0 internal Ca2+. (B) Ca2+ dependence of the normalized Cl− efflux velocity of WT (black symbols) and E724Q/E727Q (red symbols). The velocity of the mutant was normalized to the maximal efflux velocity of the WT channel. The solid line is the fit to a Hill equation with K1/2 = 210 ± 3 nM and n = 2.2 ± 0.7. Each data point represents the average of n = 3–6 independent experiments, and the error is the SEM. The error on the fit parameters K1/2 and n is the uncertainty on the fit. (C) Sequence alignment of the TM6-7 intracellular loop of TMEM16A, -B, -E, and -F. The two conserved glutamates are highlighted in red. (D) Cl− efflux mediated by purified E724Q/E727Q mutant of TMEM16A in 0 (black), 1 µM (red), 10 µM (green), 100 µM (yellow), 1 mM (blue), and 2 mM (pink) Ca2+. The asterisk denotes the addition of 40 μL of 1.5 M β-OG to dissolve all liposomes.
Recently, a conserved diacidic motif in the intracellular TM6-7 loop was shown to be important for the Ca2+ sensitivity of TMEM16A (23) (Fig. 3C). We therefore purified and reconstituted the E724Q/E727Q double mutant of TMEM16A and tested its sensitivity to Ca2+. As expected, this mutant mediates slow and Ca2+-independent Cl− fluxes: An ∼105-fold increase in free Ca2+, from ∼8 nM to 2 mM, failed to activate Cl− transport (Fig. 3 B and D).
Synergistic Activation of TMEM16A by Ca2+ and Voltage.
One of the defining characteristics of native CaCCs and of TMEM16A is that Ca2+ and voltage synergistically gate the channel (22). To test whether the purified TMEM16A protein also recapitulates this key property, we oriented our system by reconstituting the proteoliposomes in 0 internal Ca2+, Ca2+in , and 0 or 1 mM external Ca2+, Ca2+ex. In this way, we could selectively activate the channels oriented with the C terminus to the external solution (Fig. 4A) and apply different voltages using valinomycn in the presence of opportune K+ gradients. In the absence of voltage (V = 0 mV) and Ca2+, TMEM16A is inactive (Fig. 4B, blue trace). Application of a high transmembrane voltage, V ∼ +140 mV (Fig. 4B, green trace), or the addition of saturating Ca2+ex elicits robust efflux (Fig. 4B, red trace), consistent with the idea that voltage or Ca2+ are sufficient to promote channel opening. The rate of Cl− efflux is maximal in the presence of both Ca2+ and voltage (Fig. 4B, black trace), indicating that the two synergistically gate the purified TMEM16A channels. By systematically varying the applied potential between 0 and +137 mV, we found that the voltage dependence of TMEM16A can be well-described by a modified Boltzmann function (Fig. 4 C and D), with a V1/2 of ∼100 mV. The presence of Ca2+ on both sides of the membrane does not appreciably shift this voltage dependence (Fig. 4D). Finally, the Ca2+-insensitive E724Q/E727Q mutant remains voltage-dependent, as Cl− efflux mediated by this mutant is negligible at 0 mV and maximal at +137 mV (Fig. S3 A and B). This suggests that the mutation selectively ablates the Ca2+ sensitivity but not the voltage dependence of TMEM16A, as previously reported (23). Thus, Ca2+ and voltage act synergistically to open purified TMEM16A, as they do on the channel in live cells.
Fig. 4.

Synergistic activation of reconstituted TMEM16A by Ca2+ and voltage. (A) Schematic representation of the experimental setup. (B) Time course of Cl− efflux from TMEM16A proteoliposomes in 0 Ca2+ex at 0 (blue) or 137 mV (red) and 1 mM Ca2+ex at 0 (green) or 137 mV (black). (C) Cl− efflux of TMEM16A in 0 Ca2+in/1 mM Ca2+ex and V = 0 (pink), 47 (blue), 68 (yellow), 98 (green), 116 (red), and 137 mV (black). (D) Normalized velocities of Cl− transport as a function of membrane potential in 0 Ca2+in/1 mM Ca2+ex (black) and symmetrical 1 mM Ca2+ (red). Solid lines correspond to a fit to a modified Boltzmann function with v(0) = 0.12 ± 0.005, z = 0.6 ± 0.2, and V1/2 = 100 ± 5 (black trace) and v(0) = 0.27 ± 0.005, z = 0.4 ± 0.2, and V1/2 = 90 ± 5 (red trace). Each data point represents the average of n = 3–6 independent experiments, and the error is the SEM. The error on the fit parameters v(0), V1/2, and n is the uncertainty on the fit.
Pharmacological Profile of Purified TMEM16A.
We tested whether the purified channel is also sufficient to recapitulate the pore properties of the CaCC currents recorded in live cells. To this end, we tested whether addition of known CaCC blockers, such as NPPB [5-nitro-2-(3-phenylpropylamino)-benzoate], NFA (niflumic acid), NPA (n-phenylanthranilic acid), and DIDS (4,4′-di-isothiocyano-2,2′-stilbenedisulfonic acid), inhibit TMEM16A-mediated Cl− efflux. Consistent with work on CaCCs in native and heterologous systems (1, 5, 6), all tested compounds inhibited TMEM16A to varying degrees (Fig. 5 A and B). In the presence of the blockers, the time course of Cl− efflux becomes a double exponential (Fig. 5 A and C), with a fast component, with kinetics similar to those seen in unblocked channels, and a slow component, which reflects the reduced transport by the blocked channels. For this reason, we focused our analysis on the total Cl− efflux rather than on the initial velocity. We found that 50 µM external NPA and DIDS inhibits ∼20% of Cl− efflux whereas NFA and NPPB are more potent, with 50–60% block (Fig. 5B). The inhibition constant of NPPB for the reconstituted hTMEM16A is Ki ∼ 15 µM (Fig. 5 C and D), a value comparable to the ∼22 μM reported for native CaCCs (33).
Fig. 5.

Inhibition of purified TMEM16A. (A) Time course of Cl− efflux from TMEM16A proteoliposomes with saturating Ca2+ (black), with DMSO (red), and in the presence of 50 µM inhibitor: NPA (green), DIDS (yellow), NFA (blue), and NPPB (pink). (B) Steady-state normalized activity in the presence of inhibitors. The activity, A/A0 = (1 − f*0)/(1 − f0), where f*0 is the fraction of inactive liposomes under each condition and f0 is the fraction under control conditions. (C) Time course of Cl− efflux from TMEM16A proteoliposomes in the presence of varying amounts of NPPB: 0 (black), 1 µM (cyan), 5 µM (pink), 10 µM (blue), 25 µM NPPB (yellow), 50 µM (green), or 80 µM (red). (D) Steady-state normalized activity as a function of [NPPB]. The solid line is a fit to a Hill equation with K1/2 = 15 ± 5 µM, n = 1.2 ± 0.4, and Amin = 0.3 ± 0.1. In all cases data points represent the average of n = 3–6 independent experiments, and the error is the SEM. The error on the fit parameters Amin, K1/2, and n is the uncertainty on the fit.
Calmodulin and TMEM16Aabc Do Not Form a Stable Complex.
Our results show that Ca2+ directly binds to and activates purified TMEM16A. However, it was suggested that calmodulin forms an obligatory and permanent complex with TMEM16A (19, 20), and that this interaction is required for channel activation. To address this discrepancy, we tested whether we could detect the formation of a TMEM16A–calmodulin complex in vitro. First, we mixed the two proteins at a 1 TMEM16A:4 calmodulin molar ratio in the presence of 0.5 mM Ca2+ and ran the mixture on a size-exclusion column (Fig. 6A). The two proteins ran independently: The peak at ∼10.4 mL contained only TMEM16A and the one at ∼16.2 mL contained only calmodulin (Fig. 6 A and B), suggesting that the two proteins do not form a stable complex. However, this experiment might not detect a weak interaction between calmodulin and the channel, as the different migration speeds of the two proteins over the size-exclusion chromatography column might lead to their eventual separation. We therefore tested whether TMEM16A bound to Ni-NTA beads through its His tag can pull down calmodulin. We tested this by premixing the two proteins and then loading them onto the beads or by binding TMEM16A to the beads and then adding calmodulin (Fig. 6C). In both cases, calmodulin and TMEM16A elute separately, consistent with the idea that they do not form a stable complex. To place an upper limit on the amount of calmodulin that might be copurifying with TMEM16A in these experiments, we loaded on an SDS gel predefined mixtures of calmodulin and TMEM16A at various molar ratios (Fig. 6D). The calmodulin band is visible even at a ratio of 0.1 calmodulin:TMEM16A, indicating that the calmodulin contamination in our experiments is <<10%.
Fig. 6.

Calmodulin and TMEM16Aabc do not form a stable complex. (A) Size-exclusion chromatography profiles of TMEM16Aabc (black) and CaM (green) alone and after a 20-min incubation together in a 1:4 molar ratio (red) in the presence of 0.5 mM Ca2+. Arrows denote elution times of TMEM16A (black) and CaM (green). (B) Coomassie-stained SDS gels of the peaks from the elution profile in A. Lane 1, marker; lane 2, TMEM16A alone; lane 3, calmodulin; lane 4, 4:1 mixture of calmodulin and TMEM16A loaded on the size-exclusion chromatography column; lane 5, shoulder eluting at ∼8 mL; lane 6, peak at 10.4 mL; lane 7, peak at 16.2 mL. (C) Elution fractions from the pull-down assay. Lane 1, marker; lane 2, premix of 7 μM TMEM16A, 30 μM calmodulin, and 0.5 mM Ca2+; lane 3, wash; lane 4, elution with 300 mM imidazole; lane 5, beads incubated first with TMEM16A and then calmodulin in the presence of 0.5 mM Ca2+; lane 6, wash; lane 7, elution with 300 mM imidazole. (D) Coomassie-stained SDS gels of mixtures of calmodulin and TMEM16A. Lane 1, marker; lane 2, TMEM16A alone; lanes 3–7, mixtures of calmodulin and TMEM16A at the indicated molar ratios. Arrowheads indicate the position of TMEM16A (black) and calmodulin (green). (E) Time course of Cl− efflux from TMEM16A proteoliposomes at +80 mV, with 1 mM Ca2+ex and 0 mM Ca2+in in the presence of 30 μM external calmodulin (red) or in its absence (black). (F) Efflux velocity of TMEM16A alone (black; n = 4) and TMEM16A with 30 μM calmodulin (red; n = 5).
Finally, we tested whether calmodulin modulates the function of TMEM16A reconstituted in proteoliposomes (Fig. 6 E and F). We prepared proteoliposomes in 0 Ca2+in and 1 mM Ca2+ex to selectively activate the channels with a cytosolic domain facing the external solution. The rates of Cl− efflux mediated by TMEM16A in the presence of 30 μM external calmodulin (red trace) or without calmodulin (black trace) are indistinguishable, v(−Calm) = 2.9 ± 0.5 nmol Cl− s−1 and v(+Calm) = 2.8 ± 0.3 nmol Cl− s−1. These experiments show that calmodulin does not modulate the activity of TMEM16A.
Discussion
Recent work from multiple independent laboratories has demonstrated that TMEM16A is a necessary component of the ubiquitous Ca2+-activated Cl− channels (4–6). However, the question of whether TMEM16A is sufficient to form CaCCs remains open. Several reports have suggested that functional CaCCs are complexes in which TMEM16A associates with other proteins, such as calmodulin, and that these interactions are required for the Ca2+-dependent gating of the channel (19–21). To address this question, we purified and functionally reconstituted hTMEM16A. We found that the isolated protein recapitulates the essential biochemical, biophysical, and pharmacological characteristics of native and heterologously expressed CaCCs. Our data also suggest that TMEM16A is very selective for Cl− over cations. This conclusion is in slight contrast with the nonideal selectivity reported for TMEM16A using macroscopic current recordings in cells (23, 24). However, it is possible that a small cationic current, endogenous to the cells, might be contaminating those measurements. Additional experiments, such as single-channel recordings, will be needed to resolve this issue.
Our results indicate that the TMEM16A protein contains all of the necessary machinery for Ca2+- and voltage-dependent gating of these channels, and that association with other proteins is not required. This conclusion is also consistent with the idea that a conserved pair of acidic residues in the TM6-7 intracellular loop is essential for Ca2+ sensitivity of TMEM16A (13), strengthening our conclusion that Ca2+ binds to TMEM16A directly rather than indirectly. Similarly, known CaCC inhibitors block reconstituted TMEM16A, suggesting that the pore properties of the channel are also preserved in the purified protein. Taken together, our results demonstrate that TMEM16A is necessary and sufficient to form functional Cl− channels that are directly activated by Ca2+ and whose properties closely resemble those seen in native systems and in cells heterologously expressing TMEM16A (1, 5, 6, 33).
This conclusion is in contrast with reports that suggested that calmodulin binds to and modulates the function of TMEM16A, and that the two proteins form an obligatory complex and their interaction is required for channel activation (19, 20). Although our results cannot rule out the possibility that calmodulin may, under specific conditions, bind to and modulate the function of TMEM16A, our results indicate that these two proteins interact weakly, if at all. It is also possible that the formation of a TMEM16A–calmodulin complex might require the presence of additional, but still unknown, partner proteins that act as intermediaries between the two and/or of a specific lipid or ionic environment. The tissue specificity of the putative interactions between TMEM16A and calmodulin would be consistent with this hypothesis. Finally, in our experience, TMEM16A is extremely sensitive to the detergent used for extraction from cellular membranes. Out of >20 different detergents screened, digitonin was the only one that successfully preserved the structural integrity and functional properties of the purified TMEM16A channel. It is therefore possible that the detergent used to extract the protein in pull-down assays might adversely affect the stability of the channel. In turn, this might lead to the exposure of domains that are inaccessible to calmodulin in a correctly folded TMEM16A protein.
The availability of the purified protein will ease the way toward more detailed mechanistic and structural investigations of TMEM16A. The comparison of the properties of these channels studied in a reduced and biochemically defined system with those seen in live cells will be able, for example, to elucidate the mechanistic bases for their modulation by key parameters such as [Ca2+] and lipid composition and to determine the role of partner proteins that might associate with TMEM16A to modulate its function in physiologically meaningful ways.
Materials and Methods
Molecular Biology.
The hTMEM16Aabc gene was inserted into pFastBacHTa (Invitrogen). Mutagenesis was carried out using QuikChange (Agilent Technologies) and the plasmid was fully sequenced.
Protein Expression and Purification.
Protein was expressed in baculovirus-infected Sf9 cells using the Bac-to-Bac system (Invitrogen). Infected cells were incubated at 27 °C for 60–72 h, collected by centrifugation at 800 × g for 15 min, and then suspended in buffer A [20 mM Hepes⋅NaOH, 300 mM KCl, 10% (wt/vol) glycerol, pH 7.4] with the addition of a protease inhibitor cOmplete tablet (Roche). Cells were disrupted via sonication for 2 min and centrifuged at 2,000 rpm for 15 min. The membrane fraction was isolated via ultracentrifugation (150,000 × g for 1 h), and the pellet was resuspended in buffer A with 1% (wt/vol) digitonin (EMD Biochemicals) and extracted with gentle stirring for 1 h at 4 °C. Insoluble material was removed via centrifugation (40,000 × g for 45 min) and the cleared supernatant was loaded onto an Ni-NTA resin (Qiagen) column preequilibrated in buffer A with 0.12% digitonin. Nonspecifically bound proteins were removed with 40 mM imidazole, and hTMEM16Aabc was eluted in 300 mM imidazole. After elution, the protein was run on a size-exclusion chromatography column (Superdex200 10/30GL; GE Healthcare) in buffer A with 0.12% digitonin. In some preparations, an aggregate peak was visible (eluting at ∼8 mL); in these cases, the protein was rerun through gel filtration to separate the functional protein from the aggregate. In some cases, the His tag was removed via a 3-h incubation at room temperature (RT) with tobacco etch virus protease followed by gel-filtration chromatography. Because the presence of the His tag did not alter the function of reconstituted hTMEM16A (Fig. S4), most of the experiments were carried out in the construct still bearing the His tag.
Glutaraldehyde Cross-Linking.
The protein was concentrated to 0.25 mg/mL in the presence of 1 mM Ca(NO3)2 or 1 mM EGTA. Glutaraldehyde was added to a final concentration of 0.125% or 0.5% and incubated at room temperature for 3 min, and the reaction was stopped by the addition of 50 mM Tris in SDS loading buffer. In the control samples, 2% SDS was added to unfold the protein before the addition of glutaraldehyde. The samples were run on 6% (wt/vol) SDS/PAGE.
Liposome Reconstitution.
Liposomes were prepared from a 3:1 mixture of Escherichia coli polar lipids and l-α-phosphatidylcholine Egg PC (60%) (Avanti Polar Lipids). Lipids were dried under N2, dissolved in pentane, dried again, and resuspended in buffer B (300 mM KCl, 20 mM Hepes⋅NaOH, pH 7.4) by sonication. The Ca2+ concentration was adjusted to the desired values (see below) and freeze–thawed three times. Liposomes were destabilized by adding 35 mM CHAPS for 15 min at RT, after which the protein was added at the desired concentration. The lipid-detergent-protein mixture was incubated for 15 min at RT and then added to preequilibrated Bio-Beads SM2 (Bio-Rad) at 80 mg beads per 500 μL suspension and incubated at RT for 2 h under constant rotation. Detergent was removed by three successive additions of Bio-Beads at 4 °C for a total incubation time of ∼24 h.
Preparation of Ca2+ Solutions.
The free [Ca2+] present in our standard solution (300 mM KCl, 20 mM Hepes, pH 7.4) was measured with a Ca-sensitive electrode (Orion Research) and found to be 55 ± 5 μM. The amounts of Ca2+ and EGTA to generate the desired free Ca2+ concentrations were calculated using the program Ca-EGTA Calculator v1.3 (www.stanford.edu/∼cpatton/CaEGTA-TS.htm), and the values were verified (for [Ca2+]free > 1 μM) using a Ca-sensitive electrode. For solutions containing less than 1 μM [Ca2+]free, the predicted value was used. Our 0 Ca2+ solution was obtained by adding 1 mM EGTA to our solution for a predicted [Ca2+]free of ∼8 nM.
Cl− Flux Assay.
Cl− fluxes were assayed as described previously (29). Briefly, liposomes were extruded through a 400-nm membrane filter and spun through a Sephadex G-50 column (Sigma-Aldrich) preequilibrated in the external buffer (1 mM KCl, 300 mM Na glutamate/K glutamate, 20 mM Hepes, pH 7.4) and [Ca2+]free was adjusted as described. Two hundred microliters of liposomes was diluted in 1.8 mL of buffer and the Cl− time course was monitored with an Ag:AgCl electrode. After baseline stabilization, 1 µL of the K+ ionophore valinomycin (2 mg/mL) was added to initiate Cl− efflux. The experiment was terminated by addition of 40 µL of 1.5 M n-octyl-β-d-glucopyranoside (Affymetrix) to dissolve liposomes. The velocity of Cl− transport, v, was determined as 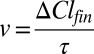 (29), where ΔClfin is the total amount of Cl− released at steady state and τ is the time constant of Cl− efflux and is derived by fitting the data to a single exponential. The velocity of non–channel-mediated Cl− leak was determined by measuring Cl− efflux from protein-free liposomes and subtracted from v.
(29), where ΔClfin is the total amount of Cl− released at steady state and τ is the time constant of Cl− efflux and is derived by fitting the data to a single exponential. The velocity of non–channel-mediated Cl− leak was determined by measuring Cl− efflux from protein-free liposomes and subtracted from v.
Transmembrane voltage was applied to the liposomes by varying the external K+ concentration from 300 to 1 mM. The velocity of Cl− efflux at different voltages was then fit to a Boltzmann function with an offset of the form 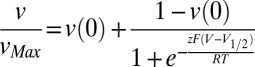 , where vMax and v(0) are the maximal and minimal efflux velocities, respectively, V1/2 is the half-activation potential, z is the apparent gating charge, and the other symbols have their usual meaning.
, where vMax and v(0) are the maximal and minimal efflux velocities, respectively, V1/2 is the half-activation potential, z is the apparent gating charge, and the other symbols have their usual meaning.
Calmodulin Overexpression and Purification.
DNA encoding rat calmodulin CaM1–148 was cloned into a pT7-7 bacterial vector and overexpressed in E. coli following published protocols (34). Briefly, cells were grown to an OD600 of 0.4–0.6 and induced with 0.4 mM Isopropyl β-D-thiogalactopyranoside for 4 h, after which cells were pelleted and resuspended in buffer (20 mM Tris, 1 mM EDTA, pH 7.2). Cells were lysed by sonication and debris was removed by ultracentrifugation at 35,000 rpm at 4 °C for 45 min. Supernatant was then loaded onto a phenyl Sepharose column (Amersham Pharmacia Biotech) preequilibrated in buffer C (20 mM Tris, 10 mM CaCl2, pH 7.4), washed first with buffer C, then with buffer C with 0.5 M NaCl, again with buffer C, and finally eluted with 20 mM Tris, 1 mM EDTA (pH 7.4). Purified rat CaM was dialyzed into 50 mM Hepes, 100 mM KCl, 50 μM EGTA (pH 7.4).
Pull-Down Experiments.
Pull-down experiments were performed by premixing 7 μM TMEM16A with 30 μM calmodulin in the presence of 0.5 mM CaCl2 for 20 min. The mixture was then loaded onto a 0.4-mL Ni-NTA column preequilibrated in buffer A and washed with 20 mL of buffer, and TMEM16A was eluted with buffer A with 300 mM imidazole. Alternatively, purified TMEM16A was first loaded onto a 0.4-mL Ni-NTA column preequilibrated in buffer A, and 30 μM calmodulin was loaded onto the column. After 20 min of incubation the beads were washed with buffer A and TMEM16A was eluted with 300 mM imidazole. Each experiment was performed three or four times with comparable results.
Data Analysis and Statistics.
Data were analyzed using Ana software (by M. Pusch, Istituto di Biofisica, Genoa, Italy) and SigmaPlot 10.0 (Systat Software). The experiments were repeated 3–20 times; exact numbers are reported in the text or in the figure legends. Data points are reported as mean ± SEM. The error reported for parameters derived from fits is the uncertainty of the fit given by the analysis programs, rather than the SEM.
Supplementary Material
Acknowledgments
We thank Dr. Galietta (University of Genoa) for the generous gift of the hTMEM16Aabc clone, Dr. Madeline Shea (University of Iowa) for the generous gift of the rat calmodulin clone, and members of the Accardi laboratory for support and discussion. This work was supported by National Institutes of Health Grant GM085232 and an Irma T. Hirschl/Monique Weill-Caulier Scholar Award (to A.A.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
See Commentary on page 19185.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1312014110/-/DCSupplemental.
References
- 1.Hartzell C, Putzier I, Arreola J. Calcium-activated chloride channels. Annu Rev Physiol. 2005;67:719–758. doi: 10.1146/annurev.physiol.67.032003.154341. [DOI] [PubMed] [Google Scholar]
- 2.Duran C, Hartzell HC. Physiological roles and diseases of Tmem16/Anoctamin proteins: Are they all chloride channels? Acta Pharmacol Sin. 2011;32(6):685–692. doi: 10.1038/aps.2011.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bader CR, Bertrand D, Schwartz EA. Voltage-activated and calcium-activated currents studied in solitary rod inner segments from the salamander retina. J Physiol. 1982;331:253–284. doi: 10.1113/jphysiol.1982.sp014372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caputo A, et al. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science. 2008;322(5901):590–594. doi: 10.1126/science.1163518. [DOI] [PubMed] [Google Scholar]
- 5.Schroeder BC, Cheng T, Jan YN, Jan LY. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell. 2008;134(6):1019–1029. doi: 10.1016/j.cell.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang YD, et al. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature. 2008;455(7217):1210–1215. doi: 10.1038/nature07313. [DOI] [PubMed] [Google Scholar]
- 7.Thomas-Gatewood C, et al. TMEM16A channels generate Ca2+-activated Cl− currents in cerebral artery smooth muscle cells. Am J Heart Circ Physiol. 2011;301(5):H1819–H1827. doi: 10.1152/ajpheart.00404.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang F, et al. Calcium-activated chloride channel TMEM16A modulates mucin secretion and airway smooth muscle contraction. Proc Natl Acad Sci USA. 2012;109(40):16354–16359. doi: 10.1073/pnas.1214596109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu B, et al. The acute nociceptive signals induced by bradykinin in rat sensory neurons are mediated by inhibition of M-type K+ channels and activation of Ca2+-activated Cl− channels. J Clin Invest. 2010;120(4):1240–1252. doi: 10.1172/JCI41084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cho H, et al. The calcium-activated chloride channel anoctamin 1 acts as a heat sensor in nociceptive neurons. Nat Neurosci. 2012;15(7):1015–1021. doi: 10.1038/nn.3111. [DOI] [PubMed] [Google Scholar]
- 11.Stanich JE, et al. Ano1 as a regulator of proliferation. Am J Physiol Gastrointest Liver Physiol. 2011;301(6):G1044–G1051. doi: 10.1152/ajpgi.00196.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duvvuri U, et al. TMEM16A induces MAPK and contributes directly to tumorigenesis and cancer progression. Cancer Res. 2012;72(13):3270–3281. doi: 10.1158/0008-5472.CAN-12-0475-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu W, Lu M, Liu B, Huang Y, Wang K. Inhibition of Ca(2+)-activated Cl(−) channel ANO1/TMEM16A expression suppresses tumor growth and invasiveness in human prostate carcinoma. Cancer Lett. 2012;326(1):41–51. doi: 10.1016/j.canlet.2012.07.015. [DOI] [PubMed] [Google Scholar]
- 14.Mazzone A, et al. Inhibition of cell proliferation by a selective inhibitor of the Ca(2+)-activated Cl(−) channel, Ano1. Biochem Biophys Res Commun. 2012;427(2):248–253. doi: 10.1016/j.bbrc.2012.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fallah G, et al. TMEM16A(a)/anoctamin-1 shares a homodimeric architecture with CLC chloride channels. Mol Cell Proteomics. 2011;10(2) doi: 10.1074/mcp.M110.004697. M110.004697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sheridan JT, et al. Characterization of the oligomeric structure of the Ca2+-activated Cl− channel Ano1/TMEM16A. J Biol Chem. 2011;286(2):1381–1388. doi: 10.1074/jbc.M110.174847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tien J, Lee HY, Minor DLJ, Jr, Jan YN, Jan LY. Identification of a dimerization domain in the TMEM16A calcium-activated chloride channel (CaCC) Proc Natl Acad Sci USA. 2013;110(16):6352–6357. doi: 10.1073/pnas.1303672110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perez-Cornejo P, et al. Anoctamin 1 (Tmem16A) Ca2+-activated chloride channel stoichiometrically interacts with an ezrin-radixin-moesin network. Proc Natl Acad Sci USA. 2012;109(26):10376–10381. doi: 10.1073/pnas.1200174109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jung J, et al. Dynamic modulation of ANO1/TMEM16A HCO3− permeability by Ca2+/calmodulin. Proc Natl Acad Sci USA. 2013;110(1):360–365. doi: 10.1073/pnas.1211594110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tian Y, et al. Calmodulin-dependent activation of the epithelial calcium-dependent chloride channel TMEM16A. FASEB J. 2011;25(3):1058–1068. doi: 10.1096/fj.10-166884. [DOI] [PubMed] [Google Scholar]
- 21.Wang M, et al. Downregulation of TMEM16A calcium-activated chloride channel contributes to cerebrovascular remodeling during hypertension by promoting basilar smooth muscle cell proliferation. Circulation. 2012;125(5):697–707. doi: 10.1161/CIRCULATIONAHA.111.041806. [DOI] [PubMed] [Google Scholar]
- 22.Xiao Q, et al. Voltage- and calcium-dependent gating of TMEM16A/Ano1 chloride channels are physically coupled by the first intracellular loop. Proc Natl Acad Sci USA. 2011;108(21):8891–8896. doi: 10.1073/pnas.1102147108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu K, Duran C, Qu Z, Cui YY, Hartzell HC. Explaining calcium-dependent gating of anoctamin-1 chloride channels requires a revised topology. Circ Res. 2012;110(7):990–999. doi: 10.1161/CIRCRESAHA.112.264440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang H, et al. TMEM16F forms a Ca2+-activated cation channel required for lipid scrambling in platelets during blood coagulation. Cell. 2012;151(1):111–122. doi: 10.1016/j.cell.2012.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferrera L, et al. Regulation of TMEM16A chloride channel properties by alternative splicing. J Biol Chem. 2009;284(48):33360–33368. doi: 10.1074/jbc.M109.046607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maduke M, Pheasant DJ, Miller C. High-level expression, functional reconstitution, and quaternary structure of a prokaryotic ClC-type chloride channel. J Gen Physiol. 1999;114(5):713–722. doi: 10.1085/jgp.114.5.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fang Y, Kolmakova-Partensky L, Miller C. A bacterial arginine-agmatine exchange transporter involved in extreme acid resistance. J Biol Chem. 2007;282(1):176–182. doi: 10.1074/jbc.M610075200. [DOI] [PubMed] [Google Scholar]
- 28. Migneault I, Dartiguenave C, Bertrand MJ, Waldron KC (2004) Glutaraldehyde: Behavior in aqueous solution, reaction with proteins, and application to enzyme crosslinking. Biotechniques 37(5):790–796, 798–802. [DOI] [PubMed]
- 29.Walden M, et al. Uncoupling and turnover in a Cl−/H+ exchange transporter. J Gen Physiol. 2007;129(4):317–329. doi: 10.1085/jgp.200709756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nguitragool W, Miller C. Uncoupling of a CLC Cl−/H+ exchange transporter by polyatomic anions. J Mol Biol. 2006;362(4):682–690. doi: 10.1016/j.jmb.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 31.Malvezzi M, et al. Ca2+-dependent phospholipid scrambling by a reconstituted TMEM16 ion channel. Nat Commun. 2013;4:2367. doi: 10.1038/ncomms3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee SY, Letts JA, MacKinnon R. Functional reconstitution of purified human Hv1 H+ channels. J Mol Biol. 2009;387(5):1055–1060. doi: 10.1016/j.jmb.2009.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu G, Hamill OP. NPPB block of Ca++-activated Cl− currents in Xenopus oocytes. Pflugers Arch. 1992;420(2):227–229. doi: 10.1007/BF00374996. [DOI] [PubMed] [Google Scholar]
- 34.Pedigo S, Shea MA. Quantitative endoproteinase GluC footprinting of cooperative Ca2+ binding to calmodulin: Proteolytic susceptibility of E31 and E87 indicates interdomain interactions. Biochemistry. 1995;34(4):1179–1196. doi: 10.1021/bi00004a011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.