

Significance
This paper describes the mechanism by which copper mediates the interplay between the two energy-producing pathways, respiration and glycolysis. Many tumors produce increased levels of lactate, even when oxygen abounds, reflecting aerobic glycolysis (“Warburg effect”), whereas most normal tissues solely use respiration. We demonstrate that reducing systemic copper with a chelating drug impaired mitochondrial energy metabolism and decreased ATP levels despite induction of glycolysis. We propose that the metabolic phenotype of tumors is modulated in part by variable levels of copper in tumor microenvironment. Our work identifies copper as a tumor promoter by demonstrating that chronic exposure to elevated levels of copper in drinking water—to the maximum allowed in public water supplies—accelerates tumor growth in mice.
Keywords: pancreatic neuroendocrine tumor, cancer metabolism, tumor energetics, mitochondria, Warburg effect
Abstract
Copper is an essential trace element, the imbalances of which are associated with various pathological conditions, including cancer, albeit via largely undefined molecular and cellular mechanisms. Here we provide evidence that levels of bioavailable copper modulate tumor growth. Chronic exposure to elevated levels of copper in drinking water, corresponding to the maximum allowed in public water supplies, stimulated proliferation of cancer cells and de novo pancreatic tumor growth in mice. Conversely, reducing systemic copper levels with a chelating drug, clinically used to treat copper disorders, impaired both. Under such copper limitation, tumors displayed decreased activity of the copper-binding mitochondrial enzyme cytochrome c oxidase and reduced ATP levels, despite enhanced glycolysis, which was not accompanied by increased invasiveness of tumors. The antiproliferative effect of copper chelation was enhanced when combined with inhibitors of glycolysis. Interestingly, larger tumors contained less copper than smaller tumors and exhibited comparatively lower activity of cytochrome c oxidase and increased glucose uptake. These results establish copper as a tumor promoter and reveal that varying levels of copper serves to regulate oxidative phosphorylation in rapidly proliferating cancer cells inside solid tumors. Thus, activation of glycolysis in tumors may in part reflect insufficient copper bioavailability in the tumor microenvironment.
Copper is an essential trace element that is necessary for the activity of a number of metalloenzymes (1). Remarkably, the homeostatic balance of bioavailable copper is metastable and of evident importance, in that a number of tissue abnormalities and disease states in humans have been associated with either reduced or elevated levels of copper (2). Serum copper levels are elevated in cancer patients and correlate with the severity of the disease and response to therapies (3, 4). In animal models, copper-chelating drugs have been reported to have antiangiogenic activity (5–8). The molecular and cellular mechanisms by which copper levels modulate tumor angiogenesis and the generality of its effects on angiogenesis in different cancer types remain unclear, as are its potentially broader effects on tumor growth.
Using a genetically engineered mouse model of human cervical carcinoma, we previously observed and reported that cancer cells express higher levels of the copper transporter Ctr1 and that the tumors were differentially sensitive to reduction in systemic copper levels compared with normal tissues (9), leading us to hypothesize that cancer cells might have a greater demand for and dependence upon copper. To address this hypothesis, we turned to another genetically engineered mouse model of multistage tumorigenesis, the RIP1–Tag2 transgenic mouse line that expresses the simian virus 40 oncogenes under control of a rat insulin gene promoter (10). In this pancreatic neuroendocrine tumor model, islets undergo discrete steps of tumorigenesis, from hyperplasia, induction of angiogenesis, tumor growth, to invasive carcinoma, enabling us to investigate the effects of modulating bioavailable copper at distinct stages of tumor progression.
Here we provide evidence that differential copper intake levels modulate the activity of the copper-binding enzyme cytochrome c oxidase in cancer cells. Pharmacological suppression of systemic copper impairs oxidative phosphorylation and tumor growth, without attendant effects on ongoing tumor angiogenesis beyond a delay in initial angiogenic switching that may be consequent to impaired proliferation of cancer cells in incipient neoplasias. The results reveal that copper accessibility, much like that of oxygen and glucose, can modulate tumor phenotypes, suggesting that bioavailable copper may be amenable to therapeutic targeting of its heretofore-unappreciated role in tumor metabolism.
Results
Copper Is a Limiting Factor for Tumor Growth.
To assess a possible link between copper intake levels and tumor development, we asked whether 20 μM copper supplied via drinking water—which is the maximal level of copper permitted in public drinking-water supplies by the Environmental Protection Agency—affects tumor growth using a multistage tumorigenesis mouse model of pancreatic islet cell carcinoma, RIP1–Tag2 (10). In this model, islets undergo discrete steps of tumorigenesis, beginning with hyperplasia/dysplasia (4–5 wk of age), followed by the onset of angiogenesis (8–10 wk) and tumor formation and growth (11–15 wk) until end stage (15–16 wk) (11). Strikingly, when RIP1–Tag2 mice were chronically subjected to 20 μM copper via drinking water from the time of weaning (4 wk of age) to a defined endpoint near end stage (15 wk), the total tumor volume was increased twofold (Fig. 1 A and B), demonstrating that copper is a limiting factor for tumor growth.
Fig. 1.

Copper promotes tumor growth. (A and B) RIP1–Tag2 mice were subjected to 20 μM copper in drinking water or regular water from the time of weaning (4 wk of age). (A) Dissected tumors from 15-wk-old RIP1–Tag2 mice are dissociated and displayed adjacent to the pancreas (whitish tissue), where spleen (dark red, oblong tissue) is shown in the upper right corner. (B) Quantification of angiogenic islets and tumors are shown. Results are means and SEM (n = 15). *P < 0.05. (C) Schematic diagram of stepwise tumorigenesis phases in RIP1–Tag2 mice and pharmacological trial regimens involving the copper chelator TM. (D) RIP1–Tag2 mice were given 1 mg of TM daily from 6 to 9 wk (n = 17) (Left), 9 to 12 wk (n = 8) (Center), or 12 to 15 wk (n = 12) (Right) as diagramed in C and assessed by the same parameters as in B. Mice were euthanized at the end of each treatment period for analyses of angiogenic islets and tumors. Results are means and SEM. *P < 0.05. n.a., not applicable.
We next sought to determine the stage of tumorigenesis at which copper is required, by examining the effects of lowering systemic copper with the copper chelator tetrathiomolybdate (TM) during distinct periods of stepwise tumor progression. TM is a pharmacological agent developed for the treatment of Wilson's disease, a genetic disorder of excess copper accumulation (12). TM mainly acts by forming a complex with dietary copper, preventing its absorption in the intestine and in the blood by binding and sequestering copper. We determined the maximally tolerated dose of TM in these mice to be 1 mg daily for up to 3 wk. When RIP1–Tag2 mice were subjected to this dosage, plasma copper levels dropped to 40% by week 2 (Fig. S1). Using this dosage, we treated a cohort of RIP1–Tag2 mice with TM from 6 to 9 wk, 9 to 12 wk, or 12 to 15 wk of age (Fig. 1C). Administration of TM from 6 to 9 wk resulted in a twofold reduction in the number of angiogenic islets, indicative of a reduction or delay in angiogenic switching in multifocal hyperproliferative islet progenitor lesions; solid tumors were not detected at this stage (Fig. 1D). TM treatment from 9 to 12 wk, when tumors were forming from angiogenic islets and beginning to grow, did not result in significant changes in the numbers of angiogenic islets or tumors or in the total tumor volume (Fig. 1D). However, when mice were given TM from 12 to 15 wk of age, when tumors were growing expansively, tumor growth was markedly reduced (Fig. 1D). The collective tumor burden increased by sevenfold during these 3 wk, whereas in the TM-treated mice, tumors grew by only 1.6-fold. These data demonstrate that systemic reduction in copper levels reduces proliferation of cancer cells in incipient neoplasias and late-stage tumors, delays the onset of tumor angiogenesis (the angiogenic switch) in the premalignant lesions, and impairs late-stage tumor growth.
Copper Stimulates Cancer-Cell Proliferation.
To dissect the mechanisms by which the copper chelator TM retards tumorigenesis at the early (6–9 wk) and late (12–15 wk) stages, we analyzed the percentage of cancer cells undergoing apoptosis or proliferation in tumors harvested at 9 or 15 wk of age following a 3-wk treatment with TM. TM treatment in the early stage did not affect apoptosis of cancer cells, but reduced the percentage of proliferating cells by twofold (Fig. 2A). TM treatment in the late stage also reduced the rate of cancer cell proliferation, by more than threefold (Fig. 2B). Conversely, tumors of mice that were chronically exposed to 20 μM copper via drinking water showed a 40% increase in the percentage of proliferating cells (Fig. 2B), indicating that differential levels of copper intake affect the rate of cancer cell proliferation. Unrestricted growth of tumors depends on formation of new blood vessels, and copper has been shown to be involved in this process (5–8). However, vascular density in tumors was not affected by copper status under our experimental conditions (Fig. 2C), although angiogenic switching was delayed (Fig. 1D). The distinctive effects of TM on the tumor angiogenic phenotype comparing our results with previous studies (5–8) may reflect differences in strain background and/or cancer/tissue types (Fig. S2); in humans, serum copper levels are influenced by multiple genetic and environmental factors (13), consistent with this supposition.
Fig. 2.

Copper stimulates cancer cell proliferation. (A) RIP1–Tag2 mice were euthanized at 9 wk of age following a 3-wk treatment with the copper chelator TM. BrdU was injected 2 h before euthanization. The percentage of proliferating cells was calculated by counting BrdU-positive and -negative cancer cells. Apoptosis was detected by TUNEL staining and by counting TUNEL-positive and -negative cancer cells. More than 20 tumors from five mice per treatment arm were analyzed. Results are means and SEM. *P < 0.05. (B and C) RIP1–Tag2 mice were treated with the copper chelator TM for 3 wk (12–15 wk) or chronically exposed to 20 μM copper via drinking water (4–15 wk) before being euthanized at 15 wk of age. (B) The percentage of proliferating and apoptotic cells was quantified as described in A. (C) Vascular density is presented as the percentage of area covered with Meca-32–positive tube structures. More than 20 tumors from three mice per treatment arm were analyzed. Results are means and SEM. *P < 0.05. (D) βTC3 cells derived from RIP1–Tag2 tumors (Left) and the human ovarian carcinoma cells A2780 (Right) were treated with 10 μM TM and varying amounts of copper (0, 2, and 5 μM). The number of cells was counted at the time of treatment and 48 h later. Data are means and SEM (n = 3). *P < 0.05.
We further assessed the effect of copper chelation on cancer cell proliferation in vitro using βTC3 cells derived from RIP1–Tag2 tumors (14). Copper levels modulated the rate of cell proliferation in culture: 10 μM TM—which is within the range of plasma TM concentrations in mice given 1 mg of TM orally (9)—suppressed proliferation of βTC3 cells, an effect that was reversed by additional copper (Fig. 2D, Left). This trend was also observed in human ovarian carcinoma cells A2780 (Fig. 2D, Right). These results demonstrate that the proliferation rate of cancer cells can be accelerated or slowed down both in vivo and in vitro by increasing or decreasing copper levels, respectively.
Copper Regulates Cancer Cell Proliferation and Oxidative Phosphorylation.
To understand the role of copper in cell proliferation, we examined the effects of copper depletion on cell-cycle progression. Treatment of βTC3 cells with 10 μM TM resulted in accumulation of cells in the G2 phase of the cell cycle: the percentage of cells in G2 was 9% in untreated controls vs. 22% in TM-treated cells after 24 h (Fig. 3A). During mitosis, ATP is required for chromosome condensation, cohesion, and separation (15–17), and severe ATP reduction induced by a mitochondrial inhibitor has been shown to increase the proportion of cells in the G2 phase of the cell cycle (18). The major supply of ATP in mammalian cells is from mitochondrial oxidative phosphorylation in the presence of oxygen. Copper plays an essential role in this process by forming a catalytic core of cytochrome c oxidase (19), the terminal enzyme complex of the electron transport chain that produces ATP in the mitochondria (oxidative phosphorylation). The effects of systemic copper depletion on the activity of cytochrome c oxidase have been shown to be tissue specific: inhibition of the enzyme is most prominent in the heart and muscle (20). The effects on tumors had not been examined. We hypothesized that the reduced proliferation rate of cancer cells under copper chelation and accumulation of cells in G2 might be due to decreased activity of cytochrome c oxidase and concomitant decrease in ATP levels. Indeed, TM treatment reduced the cytochrome c oxidase activity by twofold in βTC3 cells (Fig. 3B). We also observed a twofold decrease in the mitochondrial membrane potential in TM-treated βTC3 cells (Fig. 3C), indicating diminished activity of the electron transport chain. Together these results imply that copper limitation reduces oxidative phosphorylation in cancer cells.
Fig. 3.
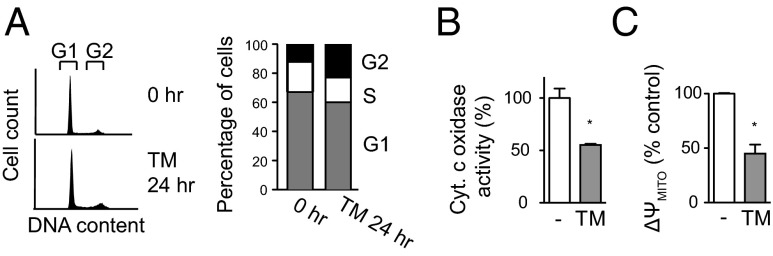
Copper-deficient cancer cells accumulate in G2 with decreased cytochrome c oxidase activity. (A, Left) Representative histograms of DNA content in βTC3 cells treated with 10 μM TM. (Right) TM treatment shifts cell-cycle distribution toward G2 phase. n = 3. P < 0.01. (B and C) Effect of TM on cytochrome c oxidase activity (B) and mitochondrial membrane potential (C). βTC3 cells were treated with 10 μM TM for 24 h. Data are means and SEM (n = 3). *P < 0.05.
Copper-Deficient Cancer Cells Have Increased Dependency on Glycolysis.
We observed a transient decline in cellular ATP levels in TM-treated βTC3 cells (Fig. 4A, 12 h), further supporting the hypothesis that mitochondrial ATP production is impaired in cancer cells that are under copper limitation. However, the ATP level recovered subsequently (Fig. 4A, 24 h), suggesting a compensatory mechanism. Another pathway that produces ATP is glycolysis, in which glucose, NAD+, and ADP are converted to pyruvate, NADH, and ATP through multistep enzymatic reactions. When cells induce glycolysis for ATP production, most often under anaerobic conditions, the glycolysis end-product pyruvate is reduced to lactate instead of being converted to acetyl-CoA that enters the tricarboxylic acid (TCA) cycle and fuels oxidative phosphorylation. Reduction of pyruvate to lactate is accompanied by oxidation of NADH to NAD+, which feeds into glycolysis for further ATP production. This higher rate of lactate production reflects the increased contribution of glycolysis to total ATP production. We found that TM treatment increased glucose uptake by 30% (Fig. 4B) and cellular lactate levels by 47% (Fig. 4C) in βTC3 cells, indicative of enhanced glycolysis for ATP production. Metabolic stresses that result in decreased cellular ATP levels trigger phosphorylation of 5′ AMP-activated protein kinase (AMPK), which increases cellular ATP levels by inhibiting energy-consuming biosynthetic pathways and stimulating energy-producing catabolic pathways (21). We observed increased phosphorylation both of the α subunit of AMPK (at Thr-172) and of its downstream target, acetyl-CoA carboxylase (ACC), in TM-treated βTC3 cells (Fig. 4D), indicating involvement of AMPK in the TM-induced bioenergetic reprogramming in cancer cells. However, proliferation is still reduced despite such reprogramming and ATP recovery, implying that ATP turnover rate is low.
Fig. 4.

Copper chelation activates AMPK and enhances glycolysis in cancer cells. (A–C) βTC3 cells were analyzed for the effects of TM on ATP (A), glucose consumption (B), and lactate (C). Data are means and SEM (n = 3). *P < 0.05. (D) Immunoblot detecting phosphorylation of AMPKα (Thr-172) and of ACC in TM-treated βTC3 cells. Data are means and SEM (n = 3). *P < 0.05. (E) Effect of TM on NADP+/NADPH ratio. Data are means and SEM (n = 3). *P < 0.05. (F) Effect of TM on lactate levels in human cancer cell lines from primary pancreatic adenocarcinoma (BxPC3), metastatic pancreatic adenocarcinoma (SUIT2), breast carcinoma (MDAMB157), and ovarian carcinoma (SKOV3). Cells were treated with 10 μM TM for 24 h. Data are means and SEM (n = 3). *P < 0.05. (G) Effect of TM and oxamate on cancer cell proliferation. βTC3 (Left) and SKOV3 (Right) cells were treated with either 10 μM TM and/or 50 mM oxamate for 48 h. Data are means and SEM (n = 3). *P < 0.05.
Upon import into a cell, glucose is immediately phosphorylated. The resultant glucose-6-phosphate serves as a precursor for several metabolic pathways, including glycolysis, generating pyruvate accompanied by ATP production, and the pentose phosphate pathway, which produces NADPH and pentoses used for reductive reactions and the synthesis of fatty acids, nucleotides, and amino acids. Avid glucose uptake by cancers has been proposed to reflect biosynthetic activities of multiplying cancer cells involving the pentose phosphate pathway (22). To determine whether the increased import of glucose in TM-treated cancer cells feeds into the pentose phosphate pathway, we measured the NADP+/NADPH ratio in TM-treated cells and untreated controls. TM treatment of βTC3 cells increased the NADP+/NADPH ratio by 80% (Fig. 4E), implying inhibition of the biosynthetic and reducing activities of the pentose phosphate pathway. Thus, when copper is limiting, glucose is primarily consumed for ATP production in cancer cells. Lactate production was also induced by TM in a set of human cancer cell lines derived from a primary (BxPC3) and a metastatic (SUIT2) human pancreatic adenocarcinoma, a breast carcinoma (MDAMB157), and an ovarian carcinoma (SKOV3) (Fig. 4F).
These observations collectively imply that copper-starved cancer cells may rely on glycolysis for supplementary energy production and thus might be hypersensitive to glycolysis inhibition. To test this idea, we treated copper-deficient cancer cells with oxamate, an inhibitor of lactate dehydrogenase that converts the glycolytic end-product pyruvate into lactate and provides NAD+ to glycolysis. We found that oxamate, dosed at a concentration that does not suppress cell proliferation, enhanced the inhibitory effect of TM on βTC3 cells (Fig. 4G, Left), as well as on human ovarian carcinoma cells SKOV3 (Fig. 4G, Right). We observed a similar combinatorial effect with another glycolysis inhibitor, 2-deoxyglucose (Fig. S3), which inhibits the hexokinase that catalyzes the first step of glycolysis. Together these results support our hypothesis that copper-deficient cancer cells depend on glycolysis for viability and proliferation.
ATP Levels Are Reduced in Copper-Deficient Tumors Despite Enhanced Glycolysis.
We extended our metabolic analyses to tumors in vivo. The activity of cytochrome c oxidase was lower in tumors from TM-treated mice and higher in those from mice that were on copper water (Fig. 5A). Positron emission tomography (PET) using 18F-labeled deoxyglucose (18FDG), whose uptake into cells mimics that of glucose, revealed increased accumulation of 18FDG in TM-treated tumors compared with untreated controls: the median 18FDG level per tumor volume in TM-treated mice was four times that of control animals (Fig. 5B). Tumor lactate levels were also higher in TM-treated mice (Fig. 5C), indicating increased ATP production through glycolysis. Interestingly, we observed lower copper concentrations in larger tumors (Fig. 5D), accompanied by decreased cytochrome c oxidase activity (Fig. 5E) and increased 18FDG uptake (Fig. 5F), suggesting that the copper deficiency elicited by TM can also occur naturally in certain large tumors. These in vivo data collectively indicate decreased oxidative phosphorylation and enhanced glycolysis in copper-deficient tumors, consistent with our in vitro data with cancer cells.
Fig. 5.

ATP levels are reduced in copper-deficient tumors despite enhanced glycolysis. (A) Cytochrome c oxidase activity of tumors following TM or copper treatment. Data are means and SEM (n > 30). *P < 0.05. (B) PET imaging of RIP1–Tag2 mice using 18FDG. Horizontal bars in the graph represent median. Representative images are shown from three mice per treatment group. 18FDG-positive tumors appear as green ∼ yellow signals. K, kidney. *P < 0.05. (C) Lactate levels in tumors treated with TM or copper. Data are means and SEM (n = 5). *P < 0.05. (D) Copper concentration in tumors according to tumor weight. (E) Cytochrome c oxidase activity in small (<2 mm) vs. large (>3 mm) tumors. Data are means and SEM (n > 20). *P < 0.05. (F) 18FDG concentrations in tumors according to tumor volume. (G) ATP levels in tumors treated with TM or copper. Data are means and SEM (n = 5). *P < 0.05.
Surprisingly, despite enhanced glycolysis, ATP levels in tumors remained low following a 3-wk TM treatment (Fig. 5G), which is in contrast to the rapid recovery of ATP levels observed in TM-treated cancer cells in vitro (Fig. 4A). This discrepancy may reflect physiological and microenvironmental differences between cultured cancer cells and those inside solid tumors. These results support the interpretation that copper-starved tumors attempt to overcome the mitochondrial ATP shortage by up-regulating glycolysis and the TCA cycle, but nevertheless fail to fully recover from the ATP deficiency—perhaps due to insufficient nutrient supply in the tumor microenvironment—resulting in suppression of tumor growth.
Copper-Chelator Treatment Does Not Promote Tumor Invasion or Increase Reactive Oxygen Species.
Enhanced glucose uptake, reflected by increased 18FDG-PET signals, is clinically associated with aggressiveness of tumors (23). Although we observed increased 18FDG accumulation in TM-treated tumors, they were not more invasive: in fact, the percentage of invasive tumors was slightly decreased (Fig. 6A), consistent with previous reports showing inhibition of invasion and metastasis by TM (24, 25). It is important to note that 18FDG-PET measures glucose entry and phosphorylation, but does not provide information on how glucose is being used. The pentose phosphate pathway, which diverts glucose for duplication of biomass after the first enzymatic step of glycolysis, is up-regulated in tumors (22), which explains in part their enhanced 18FDG-PET uptake. The mechanism for the TM-induced increase in 18FDG uptake appears different, not involving the pentose phosphate pathway (Fig. 4E); rather, the elevated 18FDG import and lactate secretion are suggestive of an attempt to restore ATP through augmented glycolysis (Fig. 5C).
Fig. 6.

Copper chelator treatment does not promote tumor invasion. (A) Effect of TM on tumor invasiveness. Invasiveness was determined by scoring the percentage of invasive fronts occupying the tumor margin in H&E-stained sections. Noninvasive, 0%; grade I, <50%; grade II, >50% (n = 20). (B) Effect of TM on ROS-induced lipid damage in tumors. Lipid peroxidation levels were determined by measuring the major toxic product of lipid peroxidation, HNE. n = 6. (C) Effect of TM on prototypical HIF-1α target gene transcripts. RNA was purified from βTC3 cells treated with 10 μM TM for 24 h and analyzed by TaqMan using L19 as an internal control. n = 3.
We also examined the effects of copper chelation on reactive oxygen species (ROS) in tumors, because systemic copper deficiency has been shown to inhibit the activity of copper–zinc superoxide dismutase 1 (SOD1) in normal tissues, especially in the liver (20). SOD1 catalyzes dismutation of superoxides, thereby reducing ROS (26). ROS not only causes damages to DNA, lipids, and proteins, but also can promote invasion by stabilizing hypoxia inducible factor 1α (HIF-1α) (27), a transcription factor whose target genes include a number of genes involved in invasion as well as glycolysis (28). ROS-induced lipid damage, 4-hydroxy-2-nonenal (HNE), was not increased in tumors by TM treatment, but was actually decreased (Fig. 6B). The lower mitochondrial membrane potential (Fig. 3C) that results from TM treatment is also consistent with reduced production of ROS (29). Furthermore, a few signature HIF-1α target genes were not induced by TM treatment of βTC3 cells (Fig. 6C). Collectively, the results suggest that ROS levels are not elevated in tumors by the degree of copper deficiency imposed under our experimental conditions, which are nevertheless sufficient to functionally disrupt another copper-dependent enzyme, cytochrome c oxidase.
Discussion
Our work provides direct evidence that varying levels of copper can modulate the proliferation of cancer cells and associated tumor growth, indicating that copper can be a rate-limiting nutrient for tumors, much like oxygen and glucose. We do not think, however, that copper is a carcinogen: exposure of wild-type mice to 20 μM copper in drinking water for up to 2 y did not result in increased cancer incidence. We demonstrate herein that the activity of cytochrome c oxidase, a key enzyme in oxidative phosphorylation, in tumors is affected by copper levels. Additional bioavailable copper evidently facilitates increased production of ATP, which is consumed to fuel rapid proliferation of cancer cells. Thus, copper may not initiate transformation, but may stimulate proliferation of transformed cells by providing energy needed for cell-cycle progression.
In most eukaryotes, oxidative phosphorylation is the predominant source of ATP; glycolysis is used in an adaptive response to oxygen limitation. Tumors, however, produce more lactate than normal tissues, even in the presence of ample oxygen, indicative of increased ATP generation from glycolysis (30). A number of oncogenic pathways have been reported to contribute to such metabolic changes (31). Our findings suggest that an environmental factor may also play a critical role in shifting the balance between the two bioenergetic pathways: enhanced glycolysis of tumors may in part reflect insufficient copper bioavailability in the tumor microenvironment. Varying copper levels may have other effects on tumor growth, such as consequent changes in systemic iron distribution or altered function of other copper-dependent factors and pathways (1). Nevertheless, our demonstration that oxidative phosphorylation of tumors can be manipulated by copper levels has important implications for understanding tumor metabolism and for therapeutic strategies.
Our studies indicate that the metabolic changes induced by copper limitation involve a mechanism distinct from the hypoxia-driven glycolytic shift. In hypoxia, HIF-1α is stabilized, resulting in transcriptional activation of a large set of genes, including those involved in oxygen delivery, glycolysis, invasion, and metastasis (28). In contrast, our data indicate that copper limitation does not activate HIF-1α or promote invasion. Notably, however, AMPK was activated by copper chelation. Activation of AMPK can also induce increased glycolysis, independent of the HIF-1α regulatory pathway (21), much as we observed in TM-treated tumors. Thus, copper chelation therapy may up-regulate glycolysis in an attempt to restore ATP in tumors but without necessarily involving activation of HIF-1α, which would likely have made cancers more invasive.
Oxygen consumption is not quantitatively diminished in many tumors despite increased glycolysis, indicative of concurrent oxidative phosphorylation (32). The ATP contribution from oxidative phosphorylation accounts for 70–90% of total ATP produced in the majority of cancer cells, with glycolysis providing the remainder (33). Rapidly proliferating cancer cells exhibit increased sensitivity to respiratory inhibitors (34), and the tumor-forming capacity of transformed cells requires robust oxidative phosphorylation (35, 36). Our findings further support a continuing role for oxidative phosphorylation, despite the induction of aerobic glycolysis in tumor growth, and suggest that this process could be an important target in cancer therapy.
Oxidative phosphorylation is essential for all cells in our body, and, as such, systemic inhibition of this pathway would be highly toxic unless tumors could be selectively targeted. Metformin, which is prescribed to type 2 diabetes patients, exerts its effects in part by inhibiting complex I of the electron transport chain (37, 38), and a number of retrospective clinical studies have found that metformin may offer a benefit in cancer prevention (39, 40). However, inhibition of complex I has been reported to induce ROS (41, 42), and cancer cells treated with metformin or its analog phenformin manifest elevated levels of ROS (43), which could potentially make them more invasive by activating HIF-1α. Copper chelators may offer several advantages over biguanides such as metformin and phenformin. First, copper chelators target complex IV, the rate-limiting enzyme of the mitochondrial respiratory chain and, as such, a key regulator of oxidative phosphorylation and ATP production (44). Second, in our study copper chelation did not increase ROS in tumors or invasiveness, consistent with the notion that complex IV is not a major site for ROS release (29). Third, cancers appear to have an increased need for copper compared with normal tissues (45, 46), and we have observed increased levels of the copper transporter Ctr1 protein in mouse cervical carcinoma (9) and increased Ctr1 mRNA levels in islets undergoing tumorigenesis (Fig. S4).
Although TM exhibits cytostatic antitumor activity when used as a monotherapy both in the mouse model of pancreatic neuroendocrine tumor (this work) and in a mouse model of human papillomavirus 16-driven cervical carcinoma (9), we envision that this class of drug will have a greater therapeutic impact when used in rational combinations that warrant future investigation first in preclinical cancer models and then in focused, proof-of-concept clinical trials. One approach would involve combinations of TM or another copper chelator with platinum-based chemotherapy, wherein TM is predicted to have dual effects: (i) up-regulating the Ctr1 copper transporter in tumors in response to insufficient copper supply so as to increase the Ctr1-mediated import of the cytotoxic platinum drugs (9, 47), and (ii) directly impairing oxidative phosphorylation, ATP production, and consequent cancer cell proliferation and tumor growth (this work). An alternative therapeutic strategy, encouraged by the results of the cell-based combinatorial assays presented in this work, would involve the combination of a copper chelator with a glycolysis inhibitor, thereby simultaneously blocking copper-dependent respiration and glycolysis, the two major pathways for ATP production in highly ATP-demanding, hyperproliferative cancer cells.
Materials and Methods
Mice, Tumor Analysis, and Cell Culture.
RIP1–Tag2 mice have been described (10, 11). For TM treatment, 10 mg of ammonium tetrathiomolybdate (Sigma) was dissolved in 1 mL of H2O, and 100 μL of the solution was orally given to each mouse every 24 h with a gavage needle. For copper water treatment, 1 mL of 8 mM CuSO4 was added to water bottles containing 400 mL of drinking water (final Cu concentration ∼20 μM; Cu concentration in drinking water without additional copper was 0.15–0.3 μM). Water was prepared fresh every week. βTC3 cells were derived from RIP1–Tag2 tumors (14). A2780, SKOV3, BxPC3, SUIT2, and MDAMB157 were from ATCC. Methods for tumor analysis and cell culture conditions are described in SI Materials and Methods.
Cytochrome c Oxidase Activity, Metabolites, Mitochondrial Membrane Potential, and Lipid Peroxidation.
Details of enzyme activity measurements and metabolites are described in SI Materials and Methods.
Western Blotting and Quantitative PCR.
Protocols and antibodies used for Western blotting and primers used for quantitative PCR are described in SI Materials and Methods.
PET.
See SI Materials and Methods for PET procedures.
Tissue Copper Measurement.
Tissues were weighed and digested in 10 μL of nitric acid per mg of tumor tissue for 2 h at 75 °C. Twenty-five microliters of digest and 25 μL of analytical grade H2O were mixed with 450 μL of 100 parts per billion cobalt, which served as an internal control for inductively coupled plasma mass spectrometry (ICP-MS) analysis. Fifty parts per billion copper was used as a standard.
Supplementary Material
Acknowledgments
We thank R. Franks (University of California, Santa Cruz) for technical support with ICP-MS; F. Schaufele (University of California, San Francisco) for assistance with microscopy and image analysis; and F. McCormick (University of California, San Francisco) and members of our laboratories for comments and feedback. This study was supported by a Flight Attendant Medical Research Institute Clinical Innovator Award (D.H. and S.I.); grants from the Swiss Cancer League (D.H. and S.I.), National Cancer Institute (D.H.), the Swiss Federal Institute of Technology Lausanne (D.H. and J.A.), the European Research Commission Advanced Grant (J.A.), and the Swiss National Science Foundation (J.A.); and an award from the William K. Bowes, Jr. Charitable Foundation (D.H.). The work was supported in part by Biomedical Imaging Research Center of the Swiss Federal Institute of Technology Lausanne, University of Lausanne, University of Geneva, University Hospital of Geneva, Central University Hospital of Vaudois, and the Leenaards and Jeantet Foundations (C.P.-Y.). J.A. is the Nestlé Chair in Energy Metabolism at the Swiss Federal Institute of Technology Lausanne.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1318431110/-/DCSupplemental.
References
- 1.Kim BE, Nevitt T, Thiele DJ. Mechanisms for copper acquisition, distribution and regulation. Nat Chem Biol. 2008;4(3):176–185. doi: 10.1038/nchembio.72. [DOI] [PubMed] [Google Scholar]
- 2.Adelstein SJ, Vallee BL. Copper metabolism in man. N Engl J Med. 1961;265:892–897. doi: 10.1056/NEJM196111022651806. [DOI] [PubMed] [Google Scholar]
- 3.Linder MC, Moor JR, Wright K. Ceruloplasmin assays in diagnosis and treatment of human lung, breast, and gastrointestinal cancers. J Natl Cancer Inst. 1981;67(2):263–275. [PubMed] [Google Scholar]
- 4.Pagliardi E, Giangrandi E. Clinical significance of the blood copper in Hodgkin’s disease. Acta Haematol. 1960;24:201–212. doi: 10.1159/000206463. [DOI] [PubMed] [Google Scholar]
- 5.Brem SS, et al. Inhibition of angiogenesis and tumor growth in the brain. Suppression of endothelial cell turnover by penicillamine and the depletion of copper, an angiogenic cofactor. Am J Pathol. 1990;137(5):1121–1142. [PMC free article] [PubMed] [Google Scholar]
- 6.Cox C, et al. The role of copper suppression as an antiangiogenic strategy in head and neck squamous cell carcinoma. Laryngoscope. 2001;111(4 Pt 1):696–701. doi: 10.1097/00005537-200104000-00024. [DOI] [PubMed] [Google Scholar]
- 7.Yoshii J, et al. The copper-chelating agent, trientine, suppresses tumor development and angiogenesis in the murine hepatocellular carcinoma cells. Int J Cancer. 2001;94(6):768–773. doi: 10.1002/ijc.1537. [DOI] [PubMed] [Google Scholar]
- 8.Pan Q, et al. Copper deficiency induced by tetrathiomolybdate suppresses tumor growth and angiogenesis. Cancer Res. 2002;62(17):4854–4859. [PubMed] [Google Scholar]
- 9.Ishida S, McCormick F, Smith-McCune K, Hanahan D. Enhancing tumor-specific uptake of the anticancer drug cisplatin with a copper chelator. Cancer Cell. 2010;17(6):574–583. doi: 10.1016/j.ccr.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hanahan D. Heritable formation of pancreatic β-cell tumours in transgenic mice expressing recombinant insulin/simian virus 40 oncogenes. Nature. 1985;315(6015):115–122. doi: 10.1038/315115a0. [DOI] [PubMed] [Google Scholar]
- 11.Folkman J, Watson K, Ingber D, Hanahan D. Induction of angiogenesis during the transition from hyperplasia to neoplasia. Nature. 1989;339(6219):58–61. doi: 10.1038/339058a0. [DOI] [PubMed] [Google Scholar]
- 12.Brewer GJ, et al. Initial therapy of patients with Wilson’s disease with tetrathiomolybdate. Arch Neurol. 1991;48(1):42–47. doi: 10.1001/archneur.1991.00530130050019. [DOI] [PubMed] [Google Scholar]
- 13.Turnlund JR. Human whole-body copper metabolism. Am J Clin Nutr. 1998;67(5) Suppl:960S–964S. doi: 10.1093/ajcn/67.5.960S. [DOI] [PubMed] [Google Scholar]
- 14.Efrat S, et al. β-cell lines derived from transgenic mice expressing a hybrid insulin gene-oncogene. Proc Natl Acad Sci USA. 1988;85(23):9037–9041. doi: 10.1073/pnas.85.23.9037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miniowitz-Shemtov S, Teichner A, Sitry-Shevah D, Hershko A. ATP is required for the release of the anaphase-promoting complex/cyclosome from inhibition by the mitotic checkpoint. Proc Natl Acad Sci USA. 2010;107(12):5351–5356. doi: 10.1073/pnas.1001875107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hudson DF, Marshall KM, Earnshaw WC. Condensin: Architect of mitotic chromosomes. Chromosome Res. 2009;17(2):131–144. doi: 10.1007/s10577-008-9009-7. [DOI] [PubMed] [Google Scholar]
- 17.Nasmyth K, Haering CH. Cohesin: Its roles and mechanisms. Annu Rev Genet. 2009;43:525–558. doi: 10.1146/annurev-genet-102108-134233. [DOI] [PubMed] [Google Scholar]
- 18.Sweet S, Singh G. Accumulation of human promyelocytic leukemic (HL-60) cells at two energetic cell cycle checkpoints. Cancer Res. 1995;55(22):5164–5167. [PubMed] [Google Scholar]
- 19.Nair PM, Mason HS. Reconstitution of cytochrome c oxidase from an apo-enzyme and Cu (I) Biochem Biophys Res Commun. 1966;23(1):12–17. doi: 10.1016/0006-291x(66)90261-0. [DOI] [PubMed] [Google Scholar]
- 20.Paynter DI, Moir RJ, Underwood EJ. Changes in activity of the Cu-Zn superoxide dismutase enzyme in tissues of the rat with changes in dietary copper. J Nutr. 1979;109(9):1570–1576. doi: 10.1093/jn/109.9.1570. [DOI] [PubMed] [Google Scholar]
- 21.Hardie DG, Ross FA, Hawley SA. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13(4):251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Basu S. Personalized versus evidence-based medicine with PET-based imaging. Nat Rev Clin Oncol. 2010;7(11):665–668. doi: 10.1038/nrclinonc.2010.121. [DOI] [PubMed] [Google Scholar]
- 24.Brem S, Tsanaclis AM, Zagzag D. Anticopper treatment inhibits pseudopodial protrusion and the invasive spread of 9L gliosarcoma cells in the rat brain. Neurosurgery. 1990;26(3):391–396. doi: 10.1097/00006123-199003000-00003. [DOI] [PubMed] [Google Scholar]
- 25.Pan Q, Bao LW, Merajver SD. Tetrathiomolybdate inhibits angiogenesis and metastasis through suppression of the NFkappaB signaling cascade. Mol Cancer Res. 2003;1(10):701–706. [PubMed] [Google Scholar]
- 26.Huang TT, et al. Superoxide-mediated cytotoxicity in superoxide dismutase-deficient fetal fibroblasts. Arch Biochem Biophys. 1997;344(2):424–432. doi: 10.1006/abbi.1997.0237. [DOI] [PubMed] [Google Scholar]
- 27.Chandel NS, et al. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci USA. 1998;95(20):11715–11720. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Semenza GL. Oxygen homeostasis. Wiley Interdiscip Rev Syst Biol Med. 2010;2(3):336–361. doi: 10.1002/wsbm.69. [DOI] [PubMed] [Google Scholar]
- 29.Kowaltowski AJ, de Souza-Pinto NC, Castilho RF, Vercesi AE. Mitochondria and reactive oxygen species. Free Radic Biol Med. 2009;47(4):333–343. doi: 10.1016/j.freeradbiomed.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 30.Warburg O, Posener K, Negelein E. Über den stoffwechsel der carcinomzelle. Biochem Z. 1924;152:309–344. [Google Scholar]
- 31.Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11(2):85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 32.Weinhouse S. On respiratory impairment in cancer cells. Science. 1956;124(3215):267–269. doi: 10.1126/science.124.3215.267. [DOI] [PubMed] [Google Scholar]
- 33.Zu XL, Guppy M. Cancer metabolism: Facts, fantasy, and fiction. Biochem Biophys Res Commun. 2004;313(3):459–465. doi: 10.1016/j.bbrc.2003.11.136. [DOI] [PubMed] [Google Scholar]
- 34.Moreno-Sánchez R, Rodríguez-Enríquez S, Marín-Hernández A, Saavedra E. Energy metabolism in tumor cells. FEBS J. 2007;274(6):1393–1418. doi: 10.1111/j.1742-4658.2007.05686.x. [DOI] [PubMed] [Google Scholar]
- 35.Funes JM, et al. Transformation of human mesenchymal stem cells increases their dependency on oxidative phosphorylation for energy production. Proc Natl Acad Sci USA. 2007;104(15):6223–6228. doi: 10.1073/pnas.0700690104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fogal V, et al. Mitochondrial p32 protein is a critical regulator of tumor metabolism via maintenance of oxidative phosphorylation. Mol Cell Biol. 2010;30(6):1303–1318. doi: 10.1128/MCB.01101-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.El-Mir MY, et al. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. 2000;275(1):223–228. doi: 10.1074/jbc.275.1.223. [DOI] [PubMed] [Google Scholar]
- 38.Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. 2000;348(Pt 3):607–614. [PMC free article] [PubMed] [Google Scholar]
- 39.Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330(7503):1304–1305. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kourelis TV, Siegel RD. Metformin and cancer: New applications for an old drug. Med Oncol. 2012;29(2):1314–1327. doi: 10.1007/s12032-011-9846-7. [DOI] [PubMed] [Google Scholar]
- 41.Hirst J, King MS, Pryde KR. The production of reactive oxygen species by complex I. Biochem Soc Trans. 2008;36(Pt 5):976–980. doi: 10.1042/BST0360976. [DOI] [PubMed] [Google Scholar]
- 42.Kushnareva Y, Murphy AN, Andreyev A. Complex I-mediated reactive oxygen species generation: modulation by cytochrome c and NAD(P)+ oxidation-reduction state. Biochem J. 2002;368(Pt 2):545–553. doi: 10.1042/BJ20021121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shackelford DB, et al. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell. 2013;23(2):143–158. doi: 10.1016/j.ccr.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arnold S. The power of life—cytochrome c oxidase takes center stage in metabolic control, cell signalling and survival. Mitochondrion. 2012;12(1):46–56. doi: 10.1016/j.mito.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 45.Gerlach W. Über den kupfergehalt menschlicher tumoren in beziehung zum kupfergehalt der leber. Z Krebsforsch. 1935;42:290–294. [Google Scholar]
- 46.Farquharson MJ, et al. The distribution of trace elements Ca, Fe, Cu and Zn and the determination of copper oxidation state in breast tumour tissue using muSRXRF and muXANES. Phys Med Biol. 2008;53(11):3023–3037. doi: 10.1088/0031-9155/53/11/018. [DOI] [PubMed] [Google Scholar]
- 47.Ishida S, Lee J, Thiele DJ, Herskowitz I. Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc Natl Acad Sci USA. 2002;99(22):14298–14302. doi: 10.1073/pnas.162491399. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.