

Significance
Understanding the effect of near-surface environments on protein conformation is critical in many fields, including biosensing, cell culture, tissue engineering, biocatalysis, and pharmaceutical formulation. However, methods to elucidate both protein structure and interfacial dynamics in heterogeneous near-surface environments are virtually nonexistent. This article describes an approach to characterize changes in protein structure on surfaces using dynamic single-molecule microscopy. Specifically, this approach exploits single-molecule Förster resonance energy transfer tracking to elucidate changes in protein structure at the single-molecule level. Using this approach, structural changes in the protein organophosphorus hydrolase were monitored upon adsorption to fused silica in the presence of BSA on a molecule-by-molecule basis. This method, which is widely applicable to virtually any protein, provides the framework for developing surfaces and surface modifications with improved biocompatibility.
Keywords: protein adhesion, single-molecule fluorescence, total internal reflection fluorescence microscopy
Abstract
A method was developed to monitor dynamic changes in protein structure and interfacial behavior on surfaces by single-molecule Förster resonance energy transfer. This method entails the incorporation of unnatural amino acids to site-specifically label proteins with single-molecule Förster resonance energy transfer probes for high-throughput dynamic fluorescence tracking microscopy on surfaces. Structural changes in the enzyme organophosphorus hydrolase (OPH) were monitored upon adsorption to fused silica (FS) surfaces in the presence of BSA on a molecule-by-molecule basis. Analysis of >30,000 individual trajectories enabled the observation of heterogeneities in the kinetics of surface-induced OPH unfolding with unprecedented resolution. In particular, two distinct pathways were observed: a majority population (∼ 85%) unfolded with a characteristic time scale of 0.10 s, and the remainder unfolded more slowly with a time scale of 0.7 s. Importantly, even after unfolding, OPH readily desorbed from FS surfaces, challenging the common notion that surface-induced unfolding leads to irreversible protein binding. This suggests that protein fouling of surfaces is a highly dynamic process because of subtle differences in the adsorption/desorption rates of folded and unfolded species. Moreover, such observations imply that surfaces may act as a source of unfolded (i.e., aggregation-prone) protein back into solution. Continuing study of other proteins and surfaces will examine whether these conclusions are general or specific to OPH in contact with FS. Ultimately, this method, which is widely applicable to virtually any protein, provides the framework to develop surfaces and surface modifications with improved biocompatibility.
Understanding the effect of near-surface environments on protein conformation is critical in many bioengineering and biomedical applications, including biosensing, cell culture, tissue engineering, biocatalysis, and pharmaceutical formulation. Importantly, surface interactions that perturb protein structure can inactivate proteins, as has widely been observed in the case of surface-immobilized enzymes (1–6). Such interactions, by inducing unfolding and subsequent accumulation of freely absorbing proteins on biomaterial surfaces, may trigger unfavorable cellular responses (7–10). However, experimental methods to elucidate both protein structure and interfacial dynamics (e.g., adsorption, diffusion, desorption), particularly in heterogeneous near-surface environments, are virtually nonexistent.
Conventional methods to determine surface effects on protein structure are largely ensemble-averaging techniques that provide limited mechanistic insight. Such limitations can lead to misinterpretations of apparent interfacial phenomena, which are used to explain the biocompatibility of biomaterials. For example, conventional biophysical methods often find that the average surface protein conformation relaxes from native-like to non-native-like upon surface adsorption, which is interpreted as evidence for unfolding (11–14). However, the time scales over which these conformational changes reportedly occur are orders of magnitude longer than typical surface residence times of isolated proteins (15), suggesting that the picture of surface-induced spreading may be oversimplified. In particular, this interpretation overlooks alternative hypotheses, including that the surface may “collect” nonnative protein molecules or that the surface may indirectly influence protein–protein interactions, causing aggregation. In part, ambiguity stems from the fact that molecules can continuously absorb and desorb, complicating the analysis of surface-induced unfolding kinetics. In addition, such methods are unable to capture heterogeneous behavior, which can arise from statistical variation in protein structure, surface aggregation, or irregularities in the underlying surface.
Recent advances in dynamic single-molecule (SM) fluorescence microscopy techniques present considerable opportunities for structure determination of adsorbed and immobilized proteins. Importantly, by measuring the entire distribution of protein structure (as opposed to only the ensemble average), such techniques can better capture the effects of protein heterogeneity. For example, using SM total internal reflection fluorescence microscopy (TIRFM), Kastantin and colleagues (15) uncovered the dynamic behavior of fibrinogen on biomaterial surfaces, showing that fibrinogen–surface interactions were highly transient and, moreover, that fibrinogen oligomers diffused more slowly on surfaces and had longer residence times than monomers. Furthermore, when intermolecular interactions were maximized by formation of an organized protein layer, residence times increased (16). These works suggested that protein aggregation, which may be mediated by surface chemistry, plays a prominent role in the formation of stable protein layers.
This article describes a unique approach to characterizing dynamic changes in protein structure in near-surface environments using dynamic SM microscopy. Specifically, this approach exploits SM-Förster resonance energy transfer (FRET) tracking to elucidate changes in protein structure at the SM level. To enable SM resolution, this approach was used in conjunction with protein engineering, using unnatural amino acids as a means to site-specifically introduce donor and acceptor fluorophores. Importantly, by incorporating donor and acceptor fluorophores at different sites via protein engineering, interfacial effects on local protein structure may be precisely probed. Structural changes in different regions of a protein also may be combined with dynamic interfacial, as well as functional, measurements, allowing direct conclusions to be drawn with respect to ways in which protein conformation, dynamics, and activity are connected. Although site-specific labeling methods have been used with FRET to monitor conformational changes in single protein molecules in bulk solution (17–19) or covalently tethered to surfaces (20–22), such methods have not been applied to the analysis of proteins undergoing dynamic adsorption, desorption, and diffusion while interacting directly with material interfaces. SM-FRET tracking, with temporal resolution of 100 ms, has previously been used to monitor dynamic changes in conformation of mobile DNA at the solution–surface interface (23, 24), using high-throughput tracking algorithms that permit the observation of 104–106 molecules.
The utility of SM-FRET tracking microscopy, when combined with protein engineering, for surface characterization was demonstrated by studying the interaction of the enzyme organophosphorus hydrolase (OPH) with FS. OPH, which catalyzes the hydrolysis of organophosphate compounds, is structurally and functionally well-characterized and is also of interest because of the relevance of OPH–surface interactions in the development of biosensors and self-decontaminating surfaces for sensing and destroying toxic nerve agents and pesticides (25, 26). The unnatural amino acid p-azido-l-phenylalanine (AzF) was incorporated in OPH so that donor and acceptor fluorophores could be site-specifically attached. Analysis of FRET as a function of time after adsorption enabled characterization of surface-induced unfolding kinetics of OPH and the fate of the unfolded OPH. SM-FRET tracking revealed that the mean unfolding time of individual molecules is 0.19 s and that 99.99% of molecules resided on the surface for less than 30 s. In contrast, ensemble unfolding analysis found that formation of an unfolded layer of OPH on FS required longer than 9 h. This profound discrepancy suggests that the mechanisms behind apparent surface-induced protein unfolding and layer formation should be reconsidered.
Results
Genetic Encoding of AzF in OPH.
To enable SM-FRET TIRFM to probe dynamic structural changes in protein molecules at interfaces, site-specific FRET labeling based on AzF incorporation was used. This strategy entailed replacement of lysines 175 in monomers A and B of OPH, which is a homodimer, with AzF (Fig. 1). AzF-containing OPH was produced by amber codon suppression in Escherichia coli, using orthogonal tyrosyl-tRNA synthetase and tyrosyl-tRNACUA from Methanococcus jannaschii that incorporate AzF in response to the TAG codon, which were evolved previously (27, 28). Lysine 175 (Lys175), which protrudes from the enzyme, is highly solvent-exposed and does not interact with other residues. In addition, the intermonomer distance between Lys175, according to the enzyme’s crystal structure, is 3.1 nm. Labeling of Lys175 thus enables monitoring of FRET between monomers to ultimately provide insight into the unfolding of OPH, which unfolds via a dimeric intermediate (29).
Fig. 1.

Structure of OPH showing the position of site-specific donor and acceptor labeling. OPH is a homodimer (C2 symmetry) that consists of two (α/β)8 monomers. The position K175, which was replaced with AzF in monomers A and B of OPH, is highlighted (yellow).
Production of OPH AzF175 resulted in soluble enzyme of the correct molecular weight, as characterized by SDS-PAGE, which showed a single prominent band (SI Appendix, Fig. S1A). Incorporation of AzF was further confirmed by electrospray ionization (ESI) mass spectrometry (SI Appendix, Fig. S1B). In the ESI spectra, the mass of the major peak (37,417 Da) matched that of the predicted mass for OPH AzF175 monomer. The minor peak at 37,389 Da presumably corresponded to a fraction of OPH in which tyrosine (Tyr) was misincorporated (the theoretical mass of OPH Tyr175 is 37,391 Da). Tyrosine misincorporation may occur as a result of nonspecific recognition of tyrosine by the mutant tRNA synthetase encoded by pDule2 pCNF RS (30). Alternatively, the minor peak may also correspond to OPH AzF175, in which the azide group of AzF is reduced to an amine. The predicted mass of reduced OPH AzF175 is identical to that of OPH Tyr175, making it impossible to differentiate between these species by mass spectrometry. Additional differences in the catalytic efficiency (kcat/Km) of wild-type OPH and OPH AzF175 were minimal, suggesting that replacement of lysine with AzF at position 175 did not drastically affect the activity, and thus the folding state, of OPH (SI Appendix, Fig. S4).
Labeling of Engineered AzF Sites with FRET Probes.
Engineered OPH AzF175 was labeled with FRET probes via modification with an approximate 2:1 molar ratio of dibenzocyclooctyne-activated Alexa Fluor (AF) 55 (donor) and AF647 (acceptor) to enzyme. Results of in-gel fluorescent image analysis of OPH AzF175 labeling with AF555 and AF647 in separate reactions found that near-complete labeling was obtained after 18 h (SI Appendix, Fig. S2). Imaging of dual-labeled OPH AzF175 confirmed the attachment of both fluorophores (SI Appendix, Fig. S3). As a FRET pair, AF555 and AF647 have an appropriate Förster radius (5.1 nm) for measuring changes in the separation of labeled AzF sites in OPH AzF175. The kcat/Km of OPH AzF175 after double-labeling with AF555 and AF647 was similar to that of the unlabeled enzyme, indicating that the effect of labeling on OPH conformation was negligible.
SM-FRET Analysis of Interfacial OPH Conformation.
SM-FRET TIRFM was used to perform wide-field imaging and object tracking of dual-labeled OPH AzF175 on adsorption to FS surfaces in the presence of BSA to stabilize OPH. Protein conformation was monitored by the dynamic relative fluorophore-to-fluorophore distance, d = (FD/FA)1/6, which is a dimensionless measure of the physical separation between donor and acceptor (see SI Appendix, section 1.5 for further explanation of the calculation of d). The d-value is related to the apparent energy transfer efficiency, E = FA/(FA + FD), by the relationship E = (1 + d6)-1. After exciting the labeled enzyme at 532 nm, fluorescence intensity of the donor (FD) and acceptor (FA) fluorophores were imaged in spectrally distinct channels for individual molecules as they diffused on the FS surface. To eliminate mislabeled molecules (i.e., molecules without one donor and one acceptor), only molecules that exhibited signatures of both folded and unfolded conformations during their trajectories were considered in the analysis of unfolding. Trajectories were collected for >30,000 molecules that underwent an initial folded-to-unfolded transition. Sample movies and raw trajectories of FD and FA for these molecules are shown in SI Appendix, Fig. S6A.
SM-FRET TIRFM trajectories of dual-labeled OPH AzF175 showed two distinct structural states. The distribution of d at the midpoint of each trajectory (Fig. 2) has peaks near d = 0.77 (E = 0.83) and d = 1.42 (E = 0.11). Notably, the center of the folded peak at low d corresponds to an absolute distance of ∼3.9 nm, which is similar to the intermonomer distance between positions 175 in the crystal structure of OPH. Accordingly, it can be reasonably assumed that the low d state represents the natively folded state of the enzyme. The second peak, at larger d, presumably represents molecules that have a nonnative (unfolded) conformation. As such, a criterion for identifying folded OPH (d ≤ 0.9 or E = 0.65) was assigned while noting that the distribution of d for the nonnative conformation was approximately normal, with a SD of 0.14. Because of this distribution, molecules with d ≤ 0.9 could be differentiated from the unfolded population with 99.9% confidence. Notably, the center of the apparent unfolded peak corresponds to an absolute distance of ∼7.2 nm, which is indicative of significant interfacial spreading, and thus loss of tertiary structure of the OPH dimer relative to the native state. A more complete picture of the structure of the unfolded state could be obtained by labeling OPH in different positions, although this was not explored in the context of this work.
Fig. 2.

Distribution of the relative fluorophore-to-fluorophore distance (d) of OPH on FS surfaces immediately after adsorption (initial), at the midpoint of each trajectory (midpoint), and immediately before desorption (final). The “box-like” ends of each distribution represent the fraction of molecules that presented near-zero intensity in one channel or the other, leading to apparent d-values either near zero or that tended to infinity. Corresponding values of apparent FRET efficiency (E) are provided on the secondary axis.
After adsorption to FS, dynamic changes in OPH structure were observed. Specifically, for most molecules that initially absorbed in a presumably folded state, a significant shift toward larger values of d was detected before desorption. The apparent shift toward increasing d demonstrated the ability to distinguish between different conformational states while identifying surface-induced conformational bias. The precise d-value of conformations with extreme d-values below 0.5 (E = 0.98) or above 1.8 (E = 0.03) could not be accurately determined because of sensitivity limits of fluorescence intensities in the donor and acceptor channels. These observations were represented by d = 0 or d = 2 in Fig. 3A and with bins at extreme d-values in Fig. 2 and Fig. 4. Finally, anomalous effects such as channel cross-talk, direct acceptor excitation, photobleaching, and photoblinking were found to be insignificant (SI Appendix, section 4).
Fig. 3.

(A) Trajectories of d as a function of time for selected OPH molecules after adsorption to FS surfaces. At values of d below 0.9 (blue area), molecules are presumed to be in a folded state. At values of d ≥ 1.1 (green area), molecules are presumed to be in an unfolded state. The folded-state residence time, indicated by the red line in each trajectory, is the time between adsorption and the first observation of d ≥ 1.1 and is independent of the surface residence time. (B) Observations of folded-state residence times, similar to those shown in A, were used to create the integrated folded-state residence time distribution. This distribution was modeled with Eq. 1 to extract first-order unfolding time constants for two subpopulations, and the resulting fit is shown as a solid line. Note that the distribution starts at (0.1,1) rather than (0,1) because a folded-state residence time of 0 s cannot be measured.
Fig. 4.

Distribution of the probability of d as a function of time constructed from individual trajectories. Time = 0 represents the first observation of the unfolded state for each trajectory, and extreme d-values (d ≤ 0.5 and d ≥ 1.8) were placed into a single bin. The probability distribution in the d-direction integrates to 1 at any given time.
SM-FRET Analysis of OPH Unfolding Kinetics on FS.
Trajectories of individual molecules were further analyzed to characterize the unfolding kinetics of OPH on FS. Surface-induced OPH unfolding was characterized by measuring the folded-state residence time of molecules after adsorption. For this analysis, criteria based on the observed d-value were used to identify states as folded (d ≤ 0.9 or E ≥ 0.65) or unfolded (d ≥ 1.1 or E ≤ 0.36). Analysis of sample trajectories using this criterion is shown in Fig. 3A, in which regions that correspond to the presumed folded (blue) and unfolded (green) states are shaded. The folded-state residence time is the time between adsorption and the first observation of unfolding (i.e., d ≥ 1.1).
Fig. 3B shows the integrated folded-state residence time distribution (f) for OPH on FS, which indicates the fraction of molecules with a folded-state residence time greater than or equal to the time shown on the abscissa. In analyzing the folded-state residence time distribution, it was assumed that OPH unfolding on the surface at low OPH coverage was a first-order process with respect to OPH coverage and that molecules may unfold via multiple pathways. On the basis of these assumptions, the probability distribution in Fig. 3B can be described by Eq. 1:
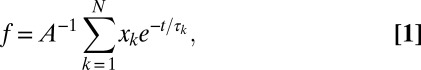 |
where xk is the fraction of molecules that follow unfolding pathway k, with time constant τk, and there are N independent pathways. Fitting the distribution to Eq. 1 found two subpopulations with different characteristic unfolding time constants. A large fraction of molecules (x1 = 85 ± 5%) underwent rapid unfolding with a time constant of 0.10 ± 0.03 s (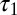 ); the remaining fraction unfolded significantly more slowly (
); the remaining fraction unfolded significantly more slowly (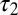 = 0.7 ± 0.1 s). Reported values represent the mean and SD of 12 independent analyses, each performed on a different location with an approximate area of 2,000 μm2. In direct contrast, the mean unfolding time constant for OPH in contact with FS measured by ensemble TIRF was 3.30 (± 0.01) × 104 s or ∼9.2 h (SI Appendix, Fig. S7).
= 0.7 ± 0.1 s). Reported values represent the mean and SD of 12 independent analyses, each performed on a different location with an approximate area of 2,000 μm2. In direct contrast, the mean unfolding time constant for OPH in contact with FS measured by ensemble TIRF was 3.30 (± 0.01) × 104 s or ∼9.2 h (SI Appendix, Fig. S7).
Analysis of SM unfolding kinetics may be influenced by fluctuations in the observed d-value that are independent of protein structural change. In principle, such fluctuations can be caused by statistical variations in quantifying the fluorescence intensity of a single fluorophore or by photophysical phenomena such as photoblinking. The presence of such phenomena was analyzed by aligning trajectories at their apparent unfolding point and observing the d-distribution before and after unfolding. Fig. 4 indicates that the kinetic analysis was not affected by fluctuations or photophysical anomalies. In particular, we see that all molecules exhibit d ≥ 1.1 for times long after their perceived unfolding transition. If the perceived transition were actually caused by photophysical artifacts, we would expect to see large fluctuations of d at random times after the transition. In general, this phenomenon was not observed, although a small fraction of molecules did exhibit temporary fluctuations to a low d state at short times after unfolding only. These fluctuations were likely a result of interconversion between conformations with low and high d before reaching a permanently unfolded state.
Fig. 4 also provides evidence that OPH remains as a homodimer in the denatured state. Specifically, d fluctuates about a finite value of d ∼ 1.4 after unfolding, indicating measurable fluorescence in the acceptor channel. If apparent OPH unfolding were caused by dimer dissociation, d would be expected to irreversibly increase to d > 1.8, as this value is reserved for observations in which FA ∼ 0. A similar trend can also be seen in the individual trajectories shown in Fig. 3A and SI Appendix, Fig. S4A, in which nonzero fluorescence, leading to d < 1.8, is typically observed in the acceptor channel in many frames after unfolding. Importantly, it should be noted that the apparent unfolding kinetics occur in the presence of adsorbed BSA, which was added in micromolar amounts. Although BSA is often added to passivate surfaces, our results clearly show that the addition of BSA does not entirely prevent OPH unfolding on FS. Thus, rather than rendering the surface inert, passivation with BSA, in this case, likely decreased the direct contact of OPH with the FS surface. Native and unfolded monomer on the surface may also participate in unfolding pathways, although this effect was likely negligible given the low absolute surface density of OPH in these experiments (<<1 molecule μm−2). Analysis of the total intensity in both the acceptor and donor channels also confirmed that high-order OPH aggregates (i.e., tetramer, etc.) were not observed (SI Appendix, section 5).
Characterization of Conformation-Specific Differences in OPH Surface Residence Time.
In addition to monitoring OPH unfolding after surface adsorption, we analyzed the relationship between OPH conformation and surface residence time. Specifically, surface residence times were compared between OPH populations that were always folded, always unfolded, initially folded but later unfolded (i.e., molecules that were analyzed in the previous section), and initially unfolded but underwent apparent refolding. For this analysis, different filters were applied to the SM-FRET data than the filter used in the kinetic analysis of unfolding to permit observation of each population. Notably, to exclude mislabeled molecules in the analysis of always-folded or always-unfolded populations, we ignored trajectories without measurable fluorescence intensity for at least two consecutive frames at any time in the donor or acceptor channel, respectively. Integrated surface residence time distributions, plotting the fraction of trajectories that exhibited a given residence time or longer, are shown for each population in Fig. 5, where desorption of both native and denatured OPH occurs over time scales on the order of 1 s or less. Importantly, this indicated that surface-induced conformational changes did not lead to irreversible protein binding, as often assumed. These time scales were significantly smaller than the measured time constant for photobleaching (170 ± 10 s; SI Appendix, section 4.3), indicating that the disappearance of objects was not significantly affected by photobleaching. Rigorous analysis of the observed trajectories further indicated that the large majority of desorption events were not artifacts of photoblinking (SI Appendix, section 4.1).
Fig. 5.

Integrated surface residence time distributions for molecules that were always folded, always unfolded, initially folded but later unfolded, and initially unfolded but subsequently in an apparent folded state.
Surface residence time distributions were nearly indistinguishable between the populations that were always unfolded, initially folded but subsequently unfolded, and initially unfolded but subsequently in an apparent folded state. In each of these populations, OPH was at one point in contact with the surface in an unfolded state. The similarity between the residence time distributions of the always-unfolded and initially unfolded populations is not surprising, given that the trajectories for these states (SI Appendix, Figs. S5B and S6B) were very similar. Although the appearance of a low d-value at a later time suggests refolding in the initially unfolded population, OPH may instead assume a partially folded state (i.e., a state in which the donor and acceptor are in relatively close proximity) that interacts with FS similarly to unfolded OPH, suggesting that surface-induced unfolding was largely irreversible. Compared with the surface residence time distribution of these populations, molecules that were always folded had much shorter residence times, indicating that the attraction between the surface and unfolded OPH is stronger than with folded OPH. Increased interaction between the surface and unfolded state of OPH likely contributes to the rapid unfolding of OPH.
In constructing the surface residence time distributions of the different populations, we found that 46% and 16% of the total molecules were always unfolded or initially unfolded, respectively. It is important to note that although these molecules may have indeed adsorbed in an unfolded state, they also may have unfolded very quickly after adsorption, before the initial measurement of conformation. On the basis of the apparent unfolding kinetic parameters, we predicted that 56% of the protein molecules were likely to unfold within the first 0.1 s on adsorption. This value is consistent with the apparent fraction of the enzyme that unfolded in the first frame (62%), suggesting that only a small fraction of OPH truly adsorbs in an unfolded state. In addition, OPH molecules that were always folded made up only 22% of the total observed trajectories, confirming that most of the molecules unfolded before desorption.
Discussion
Because of the lack of available experimental methods, interfacial effects on protein conformation and function are, at best, poorly understood. Widely used methods for probing such effects entail measuring changes in infrared absorbance, circular dichroism, or intrinsic fluorescence after exposure of the interface to protein solution (2, 31–36). These methods can detect shifts in the average secondary structure of the adsorbed protein layer over time without a priori knowledge of protein structure. However, such methods, which are protein-intensive, may not capture the process of protein unfolding at interfaces because of indiscernible concurrent adsorption and desorption events that may mask unfolding.
The use of SM techniques to isolate adsorption and desorption events provides unique perspective into the interaction of proteins with surfaces, which was previously inaccessible. Specifically, SM analysis of the interaction of OPH with FS indicated that desorption of denatured protein from the surface was highly dynamic. This observation contradicts the ensemble view of protein relaxation as resulting in irreversible adsorption for most proteins, which paints a static picture of the exchange of protein molecules (37, 38). Thus, ensemble measurements that have traditionally been interpreted as showing slow unfolding of adsorbed proteins may actually reflect an average phenomenon resulting from the highly dynamic net replacement of native by denatured proteins and, therefore, differences in adsorption and desorption rates of various species. To this point, the mean apparent unfolding time constant for OPH on FS was several orders of magnitude larger than the surface residence time of virtually all OPH molecules. The marked difference between the ensemble and SM unfolding time constants highlights the potential to incorrectly interpret surface effects on conformation, using ensemble methods. In addition, our results suggest that desorption of denatured OPH may result in an increase in the concentration of denatured protein in solution, which may induce aggregation in solution. Desorption from a denatured state has been proposed to explain the observation of denatured proteins accumulating in solution after surface contact (35, 39). However, to our knowledge, these results are the first to confirm the occurrence of this phenomenon at the molecular level. Characterization of other proteins and surfaces is required to determine whether these conclusions can be generalized or whether they are specific to OPH in contact with FS.
Use of protein engineering for site-specific labeling represents a major advance in the application of SM fluorescence techniques for investigating protein–surface interactions. Site-specific labeling enables the characterization of structural information at multiple levels (i.e., secondary, tertiary, quaternary) by SM-FRET. Such levels of structural information may be inferred either directly from a known structure or by labeling multiple positions. Site-specific labeling strategies may also be paired with fluorescence correlation spectroscopy to obtain SM data that are complementary to widefield TIRFM (40). Although fluorescence correlation spectroscopy has superior temporal resolution, interpretation of these data is model-dependent, making it difficult to distinguish adsorption and desorption from surface diffusion. In addition, because of the size of the diffraction limited field, the frequency that molecules both absorb and unfold in the field of view is typically low, limiting the quantification of unfolding pathways.
An important strength of SM tracking over ensemble methods is the ability to identify and eliminate mislabeled proteins by eliminating trajectories without specific spectral signatures. This strength was demonstrated while analyzing the unfolding kinetics of OPH in which molecules that did not exhibit signatures of both folded and unfolded states were excluded. Excluding such molecules, including those mislabeled with either two AF555 or AF647 molecules, eliminated artifacts that skew the results of the kinetic analysis. The effect of mislabeled species was apparent when comparing SM data with ensemble-averaged measurements of OPH conformation in solution, which we also measured (SI Appendix, Fig. S8). The average d-value in solution was calculated at varying concentrations of guanidine hydrochloride, using the ratio of OPH fluorescence at 568 nm (FD) to that at 670 nm (FA). Although significant unfolding was expected when the denaturant concentration was increased from 0 to 3 M, the average d-value varied only slightly over this range. In contrast, the SM approach was able to resolve a relatively large dynamic range of relative fluorophore-to-fluorophore distances (Fig. 2). A smaller dynamic range of d-values in ensemble measurements is likely a result of molecules that are mislabeled with two donors, which cannot be eliminated. The ability to measure a large range of d-values at the SM level could enable identification of distinct intermediates within protein folding pathways.
Using SM-FRET, kinetic analysis of OPH unfolding on FS highlighted the heterogeneous nature of protein structure at biomaterial interfaces. Heterogeneity in OPH unfolding transitions was modeled as a combination of two distinct subpopulations with first-order time constants of different magnitude. Notably, the time constant of the small population would have little effect on the mean stability of OPH measured using ensemble methods. Values of these time constants may ultimately be useful in identifying mechanistic information about the unfolding pathways they represent. For example, one could observe how each unfolding time constant varies with temperature or ionic strength to understand the nature of the interactions. In addition to quantifying unfolding kinetics, SM-FRET was used to elucidate differences in the surface residence times of subpopulations as a function of conformation, shedding light on differences between the attraction of folded and unfolded OPH with FS.
In conclusion, SM-FRET tracking, when combined with site-specific labeling methods, can provide insight into the interaction of proteins with biomaterials with unprecedented resolution. Insight that can be obtained using this approach, which is not accessible using conventional methods, will enable a more complete understanding of interfacial biophysical phenomena. Ultimately, the method described here is widely applicable to the characterization of biomaterials, permitting a unique level of understanding of the ways in which surface chemistry influences molecular conformation and, in turn, function.
Materials and Methods
Cloning, Expression, and Purification of Wild-Type and AzF-Containing OPH.
The gene encoding wild-type OPH from Brevundimonas diminuta (amino acids 30–365) was amplified from pUC19 opd (provided by Tony Reeves, Southwest Research Institute, Texas) and cloned into pET-21b (Novagen) with a C-terminal 6×his tag. Introduction of the TAG stop codon at amino acid position 175 was enabled via QuikChange mutagenesis (Agilent). Wild-type OPH and OPH AzF175 were expressed in BL21 (DE3) E. coli in the presence of pDule2 pCNF RS (provided by Ryan Mehl, Oregon State University, Corvallis, Oregon) (41, 42) and purified via affinity chromatography.
Labeling of OPH AzF175 with FRET Probes.
Dibenzocyclooctyne-activated AF555 and AF 647 (Invitrogen) were added to OPH AzF175 at a dye-to-protein molar ratio of 2:1. The reaction was incubated in the dark at room temperature for up to 18 h and periodically quenched with azidoethanol (0.5 M) and urea (4 M). The time-course of labeling was monitored by in-gel fluorescent imaging. For ensemble-averaged and SM-FRET, the enzyme was labeled for 18 h, after which excess fluorophore was removed using a desalting column.
Folded-State Residence Time Distribution Filtering and Analysis.
Details of TIRFM experiment and analysis are given in the SI Appendix, sections 1.3 and 1.4. Briefly, molecular trajectories were created from dual-channel images using previously described methods (43, 44). Trajectories lasting less than 0.5 s were neglected, minimizing noise. For assessing unfolding kinetics, trajectories that did not indicate unfolding (i.e., without d ≥ 1.1 for one frame) were also eliminated. This criterion eliminated mislabeled proteins as well as observations in which the folded-state residence time was uncertain.
The folded-state residence time distribution of OPH was determined for initially folded molecules with d ≤ 0.9. The d-value of each molecule was tracked from adsorption until unfolding was observed, and the length of time that each molecule persisted in the folded state was recorded as the folded-state residence time (ti). From these raw folded-state residence times, an integrated probability distribution of folded-state residence times was constructed (24). Counting the fraction of trajectories that exhibit specific folded-state residence times yields the probability distribution of folded-state residence times [p(ti)] after correcting for the finite residence times of proteins at the interface. The integrated folded-state residence time distribution, f(tj) is subsequently given by Eq. 2:
 |
Finally, regarding the fit of Eq. 1 to the experimental probability distribution, the normalization constant, A, is not left as a free parameter but is given by 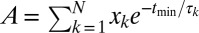 , such that f(t) = 1 at t = tmin, where tmin is the minimum observable folded-state residence time (0.1 s). A value of A ≥ 1 accounts for the fact that finite time resolution of 0.1 s inherently neglects unfolding events that occur on faster time scales (45).
, such that f(t) = 1 at t = tmin, where tmin is the minimum observable folded-state residence time (0.1 s). A value of A ≥ 1 accounts for the fact that finite time resolution of 0.1 s inherently neglects unfolding events that occur on faster time scales (45).
Supplementary Material
Acknowledgments
The authors are grateful to Ryan Mehl for technical help with pDule2 pCNF RS. This work was supported by a US Army Research Office Young Investigator Award (W911NF-12-1-0115; to J.L.K.) and the National Institutes of Health (1R21EB015532-01).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1311761110/-/DCSupplemental.
References
- 1.Maste MCL, Norde W, Visser AJWG. Adsorption-induced conformational changes in the serine proteinase savinase: A tryptophan fluorescence and circular dichroism study. J Colloid Interface Sci. 1997;196(2):224–230. doi: 10.1006/jcis.1997.5205. [DOI] [PubMed] [Google Scholar]
- 2.Vertegel AA, Siegel RW, Dordick JS. Silica nanoparticle size influences the structure and enzymatic activity of adsorbed lysozyme. Langmuir. 2004;20(16):6800–6807. doi: 10.1021/la0497200. [DOI] [PubMed] [Google Scholar]
- 3.Norde W. My voyage of discovery to proteins in flatland ...and beyond. Colloids Surf B Biointerfaces. 2008;61(1):1–9. doi: 10.1016/j.colsurfb.2007.09.029. [DOI] [PubMed] [Google Scholar]
- 4.Fears KP, Latour RA. Assessing the influence of adsorbed-state conformation on the bioactivity of adsorbed enzyme layers. Langmuir. 2009;25(24):13926–13933. doi: 10.1021/la900799m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Di Risio S, Yan N. Adsorption and inactivation behavior of horseradish peroxidase on cellulosic fiber surfaces. J Colloid Interface Sci. 2009;338(2):410–419. doi: 10.1016/j.jcis.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 6.Di Risio S, Yan N. Adsorption and inactivation behavior of horseradish peroxidase on various substrates. Colloids Surf B Biointerfaces. 2010;79(2):397–402. doi: 10.1016/j.colsurfb.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 7.Tsai WB, Grunkemeier JM, Horbett TA. Human plasma fibrinogen adsorption and platelet adhesion to polystyrene. J Biomed Mater Res. 1999;44(2):130–139. doi: 10.1002/(sici)1097-4636(199902)44:2<130::aid-jbm2>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 8.Hylton DM, Shalaby SW, Latour RA., Jr Direct correlation between adsorption-induced changes in protein structure and platelet adhesion. J Biomed Mater Res A. 2005;73(3):349–358. doi: 10.1002/jbm.a.30295. [DOI] [PubMed] [Google Scholar]
- 9.Sperling C, Fischer M, Maitz MF, Werner C. Blood coagulation on biomaterials requires the combination of distinct activation processes. Biomaterials. 2009;30(27):4447–4456. doi: 10.1016/j.biomaterials.2009.05.044. [DOI] [PubMed] [Google Scholar]
- 10.Sivaraman B, Latour RA. The relationship between platelet adhesion on surfaces and the structure versus the amount of adsorbed fibrinogen. Biomaterials. 2010;31(5):832–839. doi: 10.1016/j.biomaterials.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wertz CF, Santore MM. Adsorption and relaxation kinetics of albumin and fibrinogen on hydrophobic surfaces: Single-species and competitive Behavior. Langmuir. 1999;15(26):8884–8894. [Google Scholar]
- 12.Wertz CF, Santore MM. Effect of surface hydrophobicity on adsorption and relaxation kinetics of albumin and fibrinogen: Single-species and competitive behavior. Langmuir. 2001;17(10):3006–3016. [Google Scholar]
- 13.Wertz CF, Santore MM. Fibrinogen adsorption on hydrophilic and hydrophobic surfaces: Geometrical and energetic aspects of interfacial relaxations. Langmuir. 2002;18(3):706–715. [Google Scholar]
- 14.Yano YF. Kinetics of protein unfolding at interfaces. J Phys Condens Matter. 2012;24(50):503101. doi: 10.1088/0953-8984/24/50/503101. [DOI] [PubMed] [Google Scholar]
- 15.Kastantin M, Langdon BB, Chang EL, Schwartz DK. Single-molecule resolution of interfacial fibrinogen behavior: Effects of oligomer populations and surface chemistry. J Am Chem Soc. 2011;133(13):4975–4983. doi: 10.1021/ja110663u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kastantin M, Keller TF, Jandt KD, Schwartz DK. Single-molecule tracking of fibrinogen dynamics on nanostructured poly(ethylene) films. Adv Funct Mater. 2012;22(12):2617–2623. [Google Scholar]
- 17.Brustad EM, Lemke EA, Schultz PG, Deniz AA. A general and efficient method for the site-specific dual-labeling of proteins for single molecule fluorescence resonance energy transfer. J Am Chem Soc. 2008;130(52):17664–17665. doi: 10.1021/ja807430h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chakraborty A, et al. Opening and closing of the bacterial RNA polymerase clamp. Science. 2012;337(6094):591–595. doi: 10.1126/science.1218716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Milles S, et al. Click strategies for single-molecule protein fluorescence. J Am Chem Soc. 2012;134(11):5187–5195. doi: 10.1021/ja210587q. [DOI] [PubMed] [Google Scholar]
- 20.Seo MH, et al. Efficient single-molecule fluorescence resonance energy transfer analysis by site-specific dual-labeling of protein using an unnatural amino acid. Anal Chem. 2011;83(23):8849–8854. doi: 10.1021/ac202096t. [DOI] [PubMed] [Google Scholar]
- 21.Kim SY, Miller EJ, Frydman J, Moerner WE. Action of the chaperonin GroEL/ES on a non-native substrate observed with single-molecule FRET. J Mol Biol. 2010;401(4):553–563. doi: 10.1016/j.jmb.2010.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim J, et al. Simple and efficient strategy for site-specific dual labeling of proteins for single-molecule fluorescence resonance energy transfer analysis. Anal Chem. 2013;85(3):1468–1474. doi: 10.1021/ac303089v. [DOI] [PubMed] [Google Scholar]
- 23.Kastantin M, Schwartz DK. Connecting rare DNA conformations and surface dynamics using single-molecule resonance energy transfer. ACS Nano. 2011;5(12):9861–9869. doi: 10.1021/nn2035389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kastantin M, Schwartz DK. DNA hairpin stabilization on a hydrophobic surface. Small. 2013;9(6):933–941. doi: 10.1002/smll.201202335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mulchandani A, Chen W, Mulchandani P, Wang J, Rogers KR. Biosensors for direct determination of organophosphate pesticides. Biosens Bioelectron. 2001;16(4-5):225–230. doi: 10.1016/s0956-5663(01)00126-9. [DOI] [PubMed] [Google Scholar]
- 26.Singh BK, Walker A. Microbial degradation of organophosphorus compounds. FEMS Microbiol Rev. 2006;30(3):428–471. doi: 10.1111/j.1574-6976.2006.00018.x. [DOI] [PubMed] [Google Scholar]
- 27.Schultz KC, et al. A genetically encoded infrared probe. J Am Chem Soc. 2006;128(43):13984–13985. doi: 10.1021/ja0636690. [DOI] [PubMed] [Google Scholar]
- 28.Young DD, et al. An evolved aminoacyl-tRNA synthetase with atypical polysubstrate specificity. Biochemistry. 2011;50(11):1894–1900. doi: 10.1021/bi101929e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grimsley JK, Scholtz JM, Pace CN, Wild JR. Organophosphorus hydrolase is a remarkably stable enzyme that unfolds through a homodimeric intermediate. Biochemistry. 1997;36(47):14366–14374. doi: 10.1021/bi971596e. [DOI] [PubMed] [Google Scholar]
- 30.Young TS, Ahmad I, Yin JA, Schultz PG. An enhanced system for unnatural amino acid mutagenesis in E. coli. J Mol Biol. 2010;395(2):361–374. doi: 10.1016/j.jmb.2009.10.030. [DOI] [PubMed] [Google Scholar]
- 31.Giacomelli CE, Norde W. Influence of hydrophobic Teflon particles on the structure of amyloid beta-peptide. Biomacromolecules. 2003;4(6):1719–1726. doi: 10.1021/bm034151g. [DOI] [PubMed] [Google Scholar]
- 32.Giacomelli CE, Norde W. Conformational changes of the amyloid beta-peptide (1-40) adsorbed on solid surfaces. Macromol Biosci. 2005;5(5):401–407. doi: 10.1002/mabi.200400189. [DOI] [PubMed] [Google Scholar]
- 33.Brandes N, Welzel PB, Werner C, Kroh LW. Adsorption-induced conformational changes of proteins onto ceramic particles: Differential scanning calorimetry and FTIR analysis. J Colloid Interface Sci. 2006;299(1):56–69. doi: 10.1016/j.jcis.2006.01.065. [DOI] [PubMed] [Google Scholar]
- 34.Ganesan A, et al. Optical spectroscopic methods for probing the conformational stability of immobilised enzymes. ChemPhysChem. 2009;10(9-10):1492–1499. doi: 10.1002/cphc.200800759. [DOI] [PubMed] [Google Scholar]
- 35.Felsovalyi F, Mangiagalli P, Bureau C, Kumar SK, Banta S. Reversibility of the adsorption of lysozyme on silica. Langmuir. 2011;27(19):11873–11882. doi: 10.1021/la202585r. [DOI] [PubMed] [Google Scholar]
- 36.Pan H, Qin M, Meng W, Cao Y, Wang W. How do proteins unfold upon adsorption on nanoparticle surfaces? Langmuir. 2012;28(35):12779–12787. doi: 10.1021/la302258k. [DOI] [PubMed] [Google Scholar]
- 37.Li JJ, et al. Mechanistic understanding of protein-silicone oil interactions. Pharm Res. 2012;29(6):1689–1697. doi: 10.1007/s11095-012-0696-6. [DOI] [PubMed] [Google Scholar]
- 38.Karlsson M, Ekeroth J, Elwing H, Carlsson U. Reduction of irreversible protein adsorption on solid surfaces by protein engineering for increased stability. J Biol Chem. 2005;280(27):25558–25564. doi: 10.1074/jbc.M503665200. [DOI] [PubMed] [Google Scholar]
- 39.Felsovalyi F, Patel T, Mangiagalli P, Kumar SK, Banta S. Effect of thermal stability on protein adsorption to silica using homologous aldo-keto reductases. Protein Sci. 2012;21(8):1113–1125. doi: 10.1002/pro.2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wallace MI, Ying L, Balasubramanian S, Klenerman D. Non-Arrhenius kinetics for the loop closure of a DNA hairpin. Proc Natl Acad Sci USA. 2001;98(10):5584–5589. doi: 10.1073/pnas.101523498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hammill JT, Miyake-Stoner S, Hazen JL, Jackson JC, Mehl RA. Preparation of site-specifically labeled fluorinated proteins for 19F-NMR structural characterization. Nat Protoc. 2007;2(10):2601–2607. doi: 10.1038/nprot.2007.379. [DOI] [PubMed] [Google Scholar]
- 42.Peeler JC, Mehl RA. Site-specific incorporation of unnatural amino acids as probes for protein conformational changes. Methods Mol Biol. 2012;794:125–134. doi: 10.1007/978-1-61779-331-8_8. [DOI] [PubMed] [Google Scholar]
- 43.Walder R, Kastantin M, Schwartz DK. High throughput single molecule tracking for analysis of rare populations and events. Analyst (Lond) 2012;137(13):2987–2996. doi: 10.1039/c2an16219a. [DOI] [PubMed] [Google Scholar]
- 44.Kastantin M, Walder R, Schwartz DK. Identifying mechanisms of interfacial dynamics using single-molecule tracking. Langmuir. 2012;28(34):12443–12456. doi: 10.1021/la3017134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kastantin M, Schwartz DK. Identifying multiple populations from single-molecule lifetime distributions. ChemPhysChem. 2013;14(2):374–380. doi: 10.1002/cphc.201200838. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.