

Significance
Site-2 proteases are ubiquitously distributed, and yet their regulation and substrate recognition remain poorly understood. Here, we describe a model for site-2 protease regulation in which regulated intramembrane protease 1 (Rip1), a site-2 protease crucial for virulence in Mycobacterium tuberculosis, is tethered to one of its substrates by an adapter protein, PDZ-interacting protease regulator (Ppr) 1.
Keywords: intramembrane proteolysis, signal transduction, Tuberculosis, Sigma factor
Abstract
Site-2 proteases (S2Ps) are intramembrane metalloproteases that cleave transmembrane substrates in all domains of life. Many S2Ps, including human S2P and Mycobacterium tuberculosis Rip1, have multiple substrates in vivo, which are often transcriptional regulators. However, S2Ps will also cleave transmembrane sequences of nonsubstrate proteins, suggesting additional specificity determinants. Many S2Ps also contain a PDZ domain, the function of which is poorly understood. Here, we identify an M. tuberculosis protein, PDZ-interacting protease regulator 1 (Ppr1), which bridges between the Rip1 PDZ domain and anti-sigma factor M (Anti-SigM), a Rip1 substrate, but not Anti-SigK or Anti-SigL, also Rip1 substrates. In vivo analyses of Ppr1 function indicate that it prevents nonspecific activation of the Rip1 pathway while coupling Rip1 cleavage of Anti-SigM, but not Anti-SigL, to site-1 proteolysis. Our results support a model of S2P substrate specificity in which a substrate-specific adapter protein tethers the S2P to its substrate while holding the protease inactive through its PDZ domain.
Regulated intramembrane proteolysis (RIP) is a widely distributed mechanism of signal transduction across membranes in which specialized intramembrane cleaving proteases (iCLIPs) cleave the transmembrane segments of substrate proteins (1, 2). The site-2 (S2) proteases (S2Ps) are one class of iCLIP that is widely distributed from bacteria to human cells and participate in such diverse pathways as lipid biosynthesis (1, 3, 4), sporulation (5), membrane stress (6, 7), alginate production (8), and polar morphogenesis (9). In bacteria, PDZ domain containing S2Ps often control the release of extracytoplasmic function sigma factors from the membrane through proteolysis of their cognate membrane-bound anti-sigma factors (10, 11). S2Ps are so named because the intramembrane cleavage event follows cleavage by a site-1 (S1) protease (S1P). The S1P first cleaves the periplasmic domain of the anti-sigma factor in response to an extracellular signal. Only after S1 cleavage does the S2P cleave near the cytoplasmic side of the transmembrane domain of the anti-sigma factor, liberating the anti-sigma/sigma factor complex from the membrane into the cytoplasm where the sigma factor can associate with RNA polymerase. A specific extracytoplasmic stimulus can thus be coupled to a transcriptional response through a coordinated proteolytic cascade.
One of the major unanswered questions in S2P-mediated RIP is how these proteases achieve specificity in signaling. In many cases, S2Ps have multiple substrates, including human S2P, which cleaves SREBPs, ATF6, and CREB-H (12, 13); Bacillus subtilis RasP, which cleaves both RsiW as well as FtsL (14, 15); and Mycobacterium tuberculosis regulated intramembrane protease 1 (Rip1), which cleaves three anti-sigma factors, Anti-SigK (RskA), Anti-SigL (RslA), and Anti-SigM (RsmA) (3), as well as PBP3 (16). However, there is substantial evidence that S2Ps will cleave transmembrane segments nonspecifically both in vitro and in vivo. RseP will cleave the LacY transmembrane domain both in vitro and in vivo (17), and Methanocaldococcus jannaschii S2P will cleave the Caenorhabditis elegans transmembrane protein Ced-9 (18). These results strongly suggest that the substrate specificity of S2Ps is not determined by the primary sequence of the cleaved transmembrane segment and that additional determinants of protease specificity exist in vivo (19).
Another poorly understood feature of S2Ps is the mechanism by which S1P and S2P cleavage events are coupled. Many S2Ps, including E. coli RseP, Salmonella enterica RseP, Human S2P, and M. tuberculosis Rip1, contain PDZ domains. Substantial evidence indicates that these PDZ domains integrate S1 and site-2 cleavage by preventing the S2P from cleaving before the S1P. In E. coli, RseP lacking its PDZ domain can complement the essential function of RseP but decouples RseP activity from S1 proteolysis (20–23). However, the mechanism by which the PDZ domain modulates S2P activity is not known. Despite the usual function of PDZ domains in protein–protein interactions, and particularly in nucleating signaling complexes (24), no S2P PDZ protein interactors have been identified.
In M. tuberculosis, we have previously identified the Rip1 S2P as a major virulence determinant with three anti-sigma factor substrates (3, 4). Here, we identify a Rip1-PDZ interacting protein, PDZ-interacting protease regulator (Ppr)1, which holds Rip1 inactive while tethering Rip1 to one of its substrates, RsmA. After S1 cleavage, Ppr1 specifically facilitates Rip1 cleavage of RsmA but not other Rip1 substrates. We propose a model of S2P substrate specificity and coupling in which the S2P PDZ domain nucleates distinct signaling complexes through substrate-specific adapter proteins that both hold the S2P inactive and tether it to its substrate.
Results
Membrane Topology of Rip1 and Its Three Anti-Sigma Factor Substrates.
Many S2Ps, including E. coli RseP, are predicted to have four transmembrane (TM) domains with a PDZ domain between the second and third TMs (Fig. 1A and ref. 25), although the reported structure of M. jannaschii S2P has six TM segments (18). However, the topology of a PDZ-containing S2P has not been directly examined experimentally. We investigated the topology of M. tuberculosis Rip1 in the mycobacterial membrane by constructing enzymatic fusions to alkaline phosphatase (encoded by phoA) and β-galactosidase (encoded by lacZ). Alkaline phosphatase is active in the periplasm and produces a blue colony in the presence of BCIP, whereas β-galactosidase is active in the cytoplasm and produces a blue colony in the presence of X-gal (26). Mycobacterium smegmatis expressing fusions to phoA or lacZ between TM1 and TM2 or TM3 and TM4 (fusions 1 and 4, respectively, in Fig. 1A) demonstrated β-gal activity, but not PhoA activity, consistent with localization to the cytoplasm (Fig. 1B). Conversely, two fusions within the PDZ domain (amino acids 124–230) at residues 145 and 218 exhibited PhoA activity, but not β-galactosidase activity, indicating localization to the periplasm (fusions 2 and 3, respectively; Fig. 1B). Thus, expression of these fusions in M. smegmatis indicated that the PDZ domain of Rip1 is extracytoplasmic.
Fig. 1.

Membrane topology of Rip1 and its three anti-sigma factor substrates. (A) Schematic representation of M. tuberculosis Rip1 illustrating the four predicted transmembrane domains (TM1 through -4, in gray) and the PDZ domain between TM2 and 3 (in green). The vertical lines marked with numbers 1–5 mark the fusion points to either β-galactosidase or alkaline phosphatase as follows: 1, Y89; 2, V145; 3, W218; 4, G361; and 5, L383. (B) Membrane topology of Rip1. lacZ (Upper) and phoA (Lower) fusions to Rip1 at the positions indicated in A were expressed in M. smegmatis along with a vector control (vect) or a positive control (+) for lacZ (consisting of unfused lacZ) or phoA [an antigen85-phoA fusion (26)]. The upper image shows cells bearing each β-galactosidase fusion cultured on media containing X-gal and the bottom panel shows each PhoA fusion cultured on media containing BCIP. (C) Membrane topology of Rip1 substrates. β-Gal or PhoA was fused to the C terminus of Anti-SigK (RskA), Anti-SigL (RslA), or Anti-SigM (RsmA), and the plasmids encoding these fusions were transformed into M. smegmatis and cultured on media containing X-gal (Upper) or BCIP (Lower). The positive controls for X-gal and BCIP are the same as used in B. (D) Experimentally determined topology of Rip1 and its substrates.
We next determined the membrane topology of the three identified Rip1 substrates, Anti-SigK, Anti-SigL, and Anti-SigM (3). M. smegmatis expressing phoA or lacZ fusions of the C termini of these anti-sigma factors were blue on agar containing BCIP, but not X-gal, indicating localization of the C termini to the periplasm (Fig. 1C). Taken together, these data indicate that the Rip1-PDZ domain and the C termini of Rip1 substrates colocalize in the extracytoplasmic compartment (Fig. 1D).
Protein Interactors of the Rip1-PDZ Domain.
The PDZ domain is a widely distributed protein–protein interaction motif that often organizes signaling complexes through binding C-terminal peptides (24). Deletion of the PDZ domain of E. coli RseP causes hyperactivation of the SigE pathway, DegS (S1P), independent cleavage of RseA, and insensitivity of SigE activation to RseB inhibition (20, 21, 23, 27). However, no proteins have been identified that interact with the RseP-PDZ specifically, or S2P-PDZs more broadly, that may mediate these effects. We screened for M. tuberculosis proteins that interact directly with the Rip1-PDZ domain by performing a yeast two-hybrid screen of the Rip1-PDZ domain against a genome-wide M. tuberculosis protein library (28). Candidate Rip1-PDZ interactors were counterscreened against Ku, a cytoplasmic protein involved in the nonhomologous end-joining pathway of DNA repair (29, 30), in addition to the PDZ domains from PepA, PepD, and HtrA, three PDZ-containing proteases in the M. tuberculosis proteome (31). Of 7 × 104 candidate fusions screened, we identified four specific Rip1 PDZ interactors after counter screening. Two of these activation domain (AD) fusions contained fusions to the uncharacterized proteins encoded by rv3333c and rv3439c, which we named Ppr1 and Ppr2, respectively. Both Ppr1 and Ppr2 interact with the Rip1 PDZ, but not with Ku or the PDZ domains from PepA, PepD, or HtrA (Fig. 2A). Ppr1 and Ppr2 are both proline- and alanine-rich proteins that share significant sequence similarity in their C-terminal regions (Fig. S1B). The M. tuberculosis yeast two-hybrid library was constructed from partial digests of genomic DNA and therefore the AD-Ppr1 fusions identified in the screen encoded truncated versions of the Ppr1 protein, all of which included the C terminus of Ppr1 beginning at amino acid 131. The Ppr1 protein contains a predicted transmembrane domain at its N terminus (amino acids 6–25) but has no identifiable enzymatic function or conserved domains. BLAST searching of the M. tuberculosis Ppr1 protein sequence identified Ppr1 in pathogenic slow-growing mycobacteria, including Mycobacterium bovis, Mycobacterium africanum, and Mycobacterium marinum, which contains three Ppr1 paralogs (Fig. S1A). We did not identify Ppr1 orthologs in M. smegmatis or in any other bacterial taxa outside of pathogenic mycobacteria. M. smegmatis expressing a PhoA fusion to the C terminus of Ppr1 had PhoA activity, indicating localization of the C terminus of Ppr1 after amino acid 70 to the extracytoplasmic space (Fig. S1C).
Fig. 2.

Ppr1 is a specific interactor of the Rip1-PDZ domain. (A) EGY48 yeast cells containing plasmids encoding AD fusions to Ppr1 (encoded by M. tuberculosis Rv3333c) or Ppr2 (encoded by M. tuberculosis Rv3439c) and a LexA-LacZ reporter plasmid were tested with LexA-DNA BD fusions to Rip1-PDZ, the NHEJ protein Ku, PepA-PDZ, PepD-PDZ, or HtrA-PDZ. The Ppr1 and Ppr2 AD fusions are under the control of a galactose-inducible promoter, which is repressed by glucose. (B) Specificity of the Ppr1-Rip1-PDZ interaction. Ppr1-Myc was coexpressed in E. coli with either Rip1-PDZ-His, PepA-PDZ-His, or HtrA-PDZ-His. Soluble lysates from the indicated combinations of expressed proteins or vector controls were subjected to metal-affinity chromatography to purify histidine tagged proteins. Supernatants (sn) and eluted proteins (e) were analyzed by immunoblotting with anti-Myc or anti-His antibodies. The lanes labeled “-” are no-isopropyl β-D-1-thiogalactopyranoside (IPTG) control samples, and the lanes labeled “x” are blank lanes. (C) Ppr1 and Rip1-PDZ interact in vitro. Ppr1 lacking its transmembrane domain (Ppr1-no TM; amino acids 32–281) and either His-SUMO or His-SUMO-Rip1-PDZ were mixed and purified by metal-affinity chromatography. Bound proteins were eluted with imidazole, and the input protein (I), wash (W), and elution (E) were separated by SDS/PAGE and proteins visualized by staining with Coomassie blue.
To confirm the specificity of Ppr1 for the Rip1-PDZ domain, we performed copurification experiments with Ppr1 (30-281)-Myc and His-tagged PDZ domains from M. tuberculosis expressed in E. coli. Ppr1(30-281)-Myc copurified with Rip1-PDZ-His, but not with PepA-PDZ-His nor HtrA-PDZ-His (Fig. 2B). Taken together, these experiments confirm that Ppr1 interacts specifically with the PDZ domain of Rip1 but not the PDZ domains from other membrane-embedded proteases from M. tuberculosis. To confirm the interaction of full-length Ppr1 with the Rip1-PDZ domain in vitro, we purified the Rip1-PDZ as a His-SUMO fusion protein and Ppr1 lacking its N-terminal transmembrane domain (residues 30–281). Nickel agarose affinity chromatography of mixtures of His-SUMO-Rip1 PDZ and Ppr1 (30-281) copurified Ppr1, whereas a control His-SUMO protein did not (Fig. 2C). These experiments confirm that Ppr1 interacts directly with the PDZ domain of Rip1.
Ppr1 Suppresses the Rip1 Pathway but Does Not Affect Virulence.
To explore the function of ppr1 in the Rip1 pathway, we constructed a deletion mutant of ppr1 in M. tuberculosis. Through Southern blot analysis, we confirmed deletion of ppr1 and thus demonstrated that ppr1 is not essential (Fig. S2). Because rip1 is critical for M. tuberculosis virulence (4) and Ppr1 interacts with the Rip1-PDZ domain, we assessed the role of ppr1 on virulence using the mouse model of aerosol infection. M. tuberculosis Δppr1 displayed similar growth kinetics to wild type (WT), as measured by lung colony-forming units in the first three weeks of infection (Fig. 3A). There was also no significant difference between titers of WT M. tuberculosis and Δppr1 during the chronic phase of infection (Fig. 3A). Because the Δrip1 strain is attenuated in both acute and chronic infection (4), the lack of substantial virulence phenotype of the Δppr1 strain indicates that ppr1 is not an essential cofactor for all Rip1 functions.
Fig. 3.

Loss of Ppr1 hyperactivates the Rip1 pathway. (A) C57Bl6 mice were infected by aerosol with either WT M. tuberculosis Erdman (WT, inverted triangle), or M. tuberculosis Δppr1::hyg (circle). Bacterial titers in the lung were determined over time with 3–5 mice per group at each time point. (B) Role of Ppr1 and the Rip1-PDZ in Rip1 pathway activation. Quantitative RT-PCR was used to measure the abundance of the mRNA encoding RpfC in the indicated M. tuberculosis strains. Error bars represent the SEM. Strains are WT, WT carrying a plasmid directing the expression of Rip1 lacking the PDZ domain (WT+ Rip1ΔPDZ, MGM575), Δrip1(MGM350), Δrip1+ a WT copy of rip1 (MGM350), Δppr1 (JSS001), Δppr1 complemented strain (Δppr1+ppr1, JSS002), Δrip1 with ppr1 (Δrip1+ppr1, JSS004), and WT with ppr1 (WT+ppr1, JSS003).
We next explored direct effects of ppr1 on the Rip1 pathway in vivo through analysis of the mRNA for rpfC, a gene positively regulated by the Rip1 pathway (3, 4). As previously reported, rpfC mRNA is underexpressed in the Δrip1 strain and restored in the Δrip1 strain complemented by a WT copy of rip1 (ref. 3 and Fig. 3B). WT M. tuberculosis expressing Rip1 lacking its PDZ domain (Rip1-ΔPDZ) overexpressed rpfC, consistent with a hyperactive Rip1 pathway conferred by loss of the Rip1 PDZ domain (Fig. 3B). Loss of ppr1 resulted in increased rpfC mRNA levels similar to the overexpression observed in M. tuberculosis expressing Rip1ΔPDZ (Fig. 3B), a phenotype that was reversed by complementation of the Δppr1 strain with a WT copy of ppr1 (Fig. 3B and Fig. S3D). Conversely, overexpression of Ppr1 repressed rpfC levels in WT M. tuberculosis but had no effect on rpfC levels in the Δrip1 strain (Fig. 3B). Thus, loss of Ppr1 phenocopies loss of the Rip1-PDZ and overexpression of Ppr1 phenocopies loss of Rip1, consistent with Ppr1 repressing the Rip1 pathway through the Rip1-PDZ.
Ppr1 Scaffolds Rip1 to an Anti-Sigma Factor Substrate via the PDZ Domain.
The data above indicate that Ppr1 interacts directly with Rip1-PDZ in the periplasmic space and that the C termini of the three known anti-sigma factor substrates also localize to this compartment. In E. coli, the RseB protein interacts directly with the C terminus of RseA and negatively regulates the SigE pathway (32, 33). To understand the protein interactors of the C termini of the three Rip1 anti-sigma factor substrates, we performed yeast two-hybrid screens using the C termini of these anti-sigma factors as baits. Surprisingly, this screen identified Ppr1 as a specific interactor with the extracytoplasmic domain of RsmA (RsmA-C), but not RskA (Anti-SigK) (Fig. 4A). The RsmA-C interacting Ppr1 fragments isolated from the two-hybrid screen included Ppr1 amino acids 59–281 (Fig. 4A), suggesting that RsmA may interact with a distinct region of Ppr1 from that of Rip1-PDZ. To define the interacting regions of Ppr1 with Rip-PDZ and RsmA-C, we constructed serial truncations of Ppr1 and tested them for interaction with Rip1-PDZ, RsmA, and RskA in the yeast two-hybrid system. Ppr1 (59-281) and Ppr1 (73-281) interacted with both RsmA and Rip1-PDZ, but not RskA (Fig. 4A). Ppr1 (87-281) failed to interact with RsmA, but retained interaction with Rip1-PDZ. Deletion of the C-terminal 33 amino acids of Ppr1 did not affect interaction with Rip1-PDZ, indicating that, in contrast to many ligand-PDZ interactions, the Ppr1-Rip1PDZ interaction does not require the C terminus of Ppr1. Truncation of Ppr1 beyond amino acid 248 from the C terminus abolished interaction with Rip1-PDZ (Fig. 4A).
Fig. 4.

Ppr1 interacts with both Rip1-PDZ and Anti-SigM. (A) Interaction of Ppr1 with RsmA and Rip1-PDZ. Yeast strains carrying a LexA-LacZ reporter plasmid and plasmids encoding AD fusions to Ppr1 with the amino acid boundaries listed above each column and LexA-DNA BD fusion to Rip1-PDZ, RsmA-C, or RskA-C. The strains are shown on X-gal–containing media with either glucose (Upper) or galactose (Lower), which induces expression of the AD fusion proteins. (B) Mixtures of His-SUMO-RslA-C/Ppr1 (Left) or His-SUMO-RsmA-C/Ppr1 (Right) were purified by metal-affinity chromatography. Bound proteins were eluted with imidazole, and the input protein (I), wash (W), and elution (E) were separated by SDS/PAGE and proteins visualized by staining with Coomassie blue. (C) Mixtures of Ppr1/His-SUMO/RsmA-C or Ppr1/His-SUMO-Rip1-PDZ-/RsmA-C were purified by metal-affinity chromatography. Bound proteins were eluted with imidazole, and the input protein (I), wash (W), and elution (E) were separated by SDS/PAGE and proteins visualized by staining with Coomassie blue. (D) Specificity of the Ppr1-RsmA interaction. Ppr1-Myc was coexpressed in E. coli with the C-terminal fragments of RsmA-His, RskA-His, or RslA-His. Soluble lysates from the indicated combinations of expressed proteins or vector controls were subjected to metal-affinity chromatography to purify histidine-tagged proteins. Supernatants (sn) and eluted proteins (e) were analyzed by immunoblotting with anti-Myc or anti-His antibodies. The lanes labeled “-” are no-IPTG control samples, and the lanes labeled “x” are blank lanes.
To further confirm the direct interaction between Rip1-PDZ, Ppr1, and RsmA, we purified Rip-PDZ, Ppr1, and the C-terminal fragment of RsmA (RsmA-C). Affinity purification of His-SUMO-RsmA coprecipitated Ppr1, whereas His-SUMO-RslA did not (Fig. 4B). When all three proteins were mixed, affinity purification of His-SUMO-Rip1-PDZ copurified both Ppr1 and RsmA-C, whereas a control His-SUMO protein did not purify either RsmA-C or Ppr1 (Fig. 4C). We further confirmed the specificity of the Ppr1-RsmA interaction by coexpression in E. coli. When coexpressed with anti-sigma factor C termini in E. coli, Ppr1 only copurified with RsmA-C but not RslA-C or RskA-C (Fig. 4D). These experiments indicate that the Rip1-PDZ and RsmA both interact with Ppr1, RsmA at the Ppr1 N terminus and Rip1-PDZ through the Ppr1 C terminus. These interactions are exclusive to RsmA and do not extend to the two other known anti-sigma factor substrates of Rip1, RskA and RslA, indicating that Ppr1 is a substrate-specific adapter protein that bridges between Rip1 and one of its substrates.
Ppr1 Modulates the Sigma Factor M Branch of the Rip1 Pathway.
Based on the interaction of Ppr1 with both Rip1-PDZ and RsmA, we hypothesized that Ppr1 may affect activation of the SigM branch of the Rip1 pathway. SigM positively auto-regulates its own transcription (34), and therefore we measured the abundance of the mRNA encoding SigM by two methods, quantitative RT-PCR and NanoString (35), in the Δppr1 strain. By both methods, we observed that the mRNA encoding SigM was overexpressed in Δppr1 cells compared with either WT or Δppr1 complemented cells (Fig. S3 A–C). We were unable to detect an effect of ppr1 deletion on the SigK or SigL pathways, but these experiments were hampered by the low expression levels of these regulons and the unknown inducing signals of these pathways.
Ppr1 Accelerates Rip1 Cleavage of S1P-Cleaved RsmA but Not RslA.
The data above suggest that Ppr1 may specifically control the interaction of Rip1 with RsmA. One model is that Ppr1 tethers Rip1 to RsmA while simultaneously inhibiting Rip1 cleavage through the Rip1 PDZ domain, thereby inhibiting RsmA cleavage. With SigM pathway activation, Ppr1 might facilitate Rip1 mediated proteolysis of S1P-cleaved RsmA by tethering Rip1 to RsmA. Loss of Ppr1 leads to broad activation of the Rip1 pathway that includes both SigM-dependent (Fig. S3) and SigM-independent genes (Fig. 3B and Fig. S3), indicating that one effect of Ppr1 is to generally prevent Rip1 activation. To test the effect of Ppr1 on RsmA cleavage, we sought to quantify the half-lives of the cleavage products of RsmA in vivo. In vitro reconstitution of S1P/S2P systems has been difficult and our prior efforts to identify the S1Ps in the Rip1 pathway have been unsuccessful (3), precluding in vitro analysis. In WT E. coli (6), B. subtilis (36), and M. tuberculosis (3), the S1P-cleaved anti-sigma factor intermediate is very short-lived and not detectable in WT cells. In cells lacking the S2P, this intermediate accumulates. To enhance our ability to quantify the S1-cleaved anti-sigma factor intermediates in WT cells, we adapted the O6-alkylguanine-DNA-alkyltransferases (hAGT, SNAP tag) protein fusion system (37, 38) for mycobacteria, which allows covalent labeling of fusion proteins in vivo. We constructed N-terminal SNAP-RsmA and SNAP-RslA fusions. Preliminary experiments confirmed that the SNAP fusions were expressed in M. smegmatis, were labeled efficiently by SNAP-505 (a green fluorescent O6 benzylguanine derivative), and labeling was quenched by unlabeled O6 benzylguanine (O6-BG). We expressed SNAP-RsmA or SNAP-RslA in M. smegmatis carrying a tetracycline inducible HA-Ppr1, allowing us to temporally control Ppr1 expression.
The experimental scheme is depicted in Fig. 5A. We induced Ppr1 expression with anhydrotetracycline (ATC) 45 min before labeling SNAP-RsmA with SNAP-505 for 30 min. Labeling was terminated by addition of unlabeled O6-BG and samples collected over a 180-min time course for analysis. Cells carrying the ATC-inducible vector served as controls for any effects of ATC treatment on RsmA stability. The intensities of RsmA-SNAP-505 fluorescent species were normalized to total protein abundance by immunoblotting for CarD (39). We detected three fluorescent protein species of SNAP-RsmA, corresponding to full-length (full), S1-cleaved (S1), and final site-2–cleaved (Fig. S2 and Fig. 5B). The S1 product was observed as two closely migrating species, consistent with prior data showing anti-sigma factor trimming by multiple proteases (40). Over the 180-min time course, the half-life of S1-cleaved RsmA was unaffected by ATC treatment in vector bearing control cells (Fig. 5 B and C). In contrast, expression of Ppr1 modestly accelerated the clearance of RsmA-S1 (P = 0.01 for comparison between Ppr1 and Ppr1 plus ATC at the 180-min time point; Fig. 5C). The half-life of RsmA-S1 was significantly shorter in the presence of Ppr1 (117 min with Ppr1 compared with 181 min in control cells; P = 0.025 for comparison of half-lives). We did not observe a consistent or significant difference in the rate of clearance of full-length RsmA-SNAP with Ppr1 expression, indicating that Ppr1 specifically affects clearance of the S1-cleaved RsmA.
Fig. 5.

Ppr1 accelerates Rip1 cleavage of S1-cleaved RsmA. (A) Experimental Schematic. ATC induces expression of HA-Ppr1. A control strain carries an ATC-inducible vector without Ppr1 sequences. At time −30 min, cells are labeled with SNAP-505, which labels SNAP-tagged proteins covalently, for 30 min. SNAP-505 labeling is terminated by addition of O6 benzylguanine (O6-BG). Aliquots are taken at time 0, 60 min, and 180 min, and protein lysates are prepared. The right side of A depicts the full-length RsmA-SNAP, full-length RsmA-SNAP 505, and S1P-cleaved RsmA-SNAP 505, which is the substrate for Rip1. (B) Kinetics of RsmA degradation with and without Ppr1. Protein lysates from WT M. smegmatis expressing RsmA-SNAP and either vector (JSS182) (left blots) or ATC-inducible Ppr1 (JSS183, right blots) were prepared at the indicated time points and separated by SDS/PAGE. Fluorescently labeled RsmA-SNAP was detected by gel fluorometric scanning (upper blots). The positions of full-length RsmA-SNAP, S1P-cleaved RsmA-SNAP (S1) and the S2P-cleaved product (S2P) are indicated at the side of the gel image. Ppr1 protein was detected by immunoblotting with anti-HA antibodies and protein loading was normalized using antibodies to CarD. (C) The normalized fraction of RsmA-S1 over time in vector –ATC (red), vector +ATC (black), Ppr1 –ATC (green), and Ppr1 +ATC (blue). Error bars are SEM, and each point is the mean of four independent experiments. The bar graph depicts data from the 180-min time point. (D) The accelerated degradation of RsmA-S1 by Ppr1 is dependent on Rip1. The experimental design is identical to B, but the parent strain is M. smegmatis Δrip1.
To confirm that Rip1 mediates the acceleration of S1 clearance with Ppr1 expression, we quantitated the clearance of the S1 intermediate in Δrip1 cells with or without Ppr1. As previously reported (3), the RsmA-S1 intermediate accumulates in Δrip1 cells (Fig. 5D). In the absence of Rip1, Ppr1 did not accelerate the clearance of RsmA-S1 in comparison with control cells (Fig. 5D). To determine whether the effect of Ppr1 on RsmA-S1 clearance was specific for RsmA, we measured the clearance of SNAP-RslA with and without Ppr1 using the same experimental design (with an additional longer induction of Ppr1 of 6 h; Fig. S4). We did not observe any effect of Ppr1 on clearance of SNAP-RslA-S1 (Fig. S4), indicating that the effect of Ppr1 on S1 clearance is specific for RsmA. These data indicate that Ppr1 specifically accelerates Rip1 cleavage of the S1-cleaved RsmA, but not other Rip1 substrates.
Discussion
Despite the wide phylogenetic distribution of S2Ps and diverse biologic functions, their regulation remains poorly understood. In most cases, S2P-mediated signaling cascades follow the same general paradigm: (i) a S1P first cleaves the extracytoplasmic domain of a transmembrane substrate in response to a specific inducing signal [e.g., unfolded OMPs in the case of the S1P (DegS) in the E. coli SigE pathway] upon which (ii) S2P cleavage within the transmembrane segment of the substrate rapidly ensues, thereby liberating a cytosolic fragment of the substrate. This S1P/S2P coupling necessitates that the S2P remain inactive until the S1P cleavage has occurred, a coupling event that is not well understood. Observations in prokaryotic systems suggest that the PDZ domains of S2Ps participate in this S1P-S2P coupling because this domain is necessary to hold the protease inactive until site one proteolysis occurs (20, 21, 23, 27, 41). Recent structural data suggested that one function of the RseP PDZ domain is to bind the cleaved C terminus of the RseA anti-sigma factor generated by DegS (42), potentially providing a mechanism of coupling. However, recent in vivo examination of this model did not support this mechanism (27). Other recent data indicate that acid can activate RseA cleavage by RseP independent of DegS in S. typhimurium (41) and that LPS biosynthetic intermediates are a second signal for RseA degradation through binding RseB (43). We propose that Ppr1 in M. tuberculosis provides an alternative model to understand the role of the S2P PDZ domain in coupling and S2P self-inhibition. By binding the PDZ at its C terminus and RsmA at its N terminus, Ppr1 is able to simultaneously tether Rip1 to its substrate, while also holding Rip1 inactive (Fig. 6). When the S1P cleaves RsmA, the presence of Ppr1 accelerates Rip1 cleavage, consistent with a role in coupling the two proteolytic events.
Fig. 6.
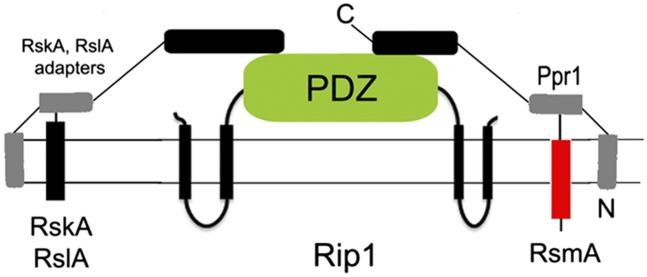
Adapter protein model of Rip1 substrate specificity. Ppr1 binds to both the Rip1 PDZ domain and the C terminus of the RsmA Rip1 substrate. Rip1 is tethered to RsmA by Ppr1 through the Rip1-PDZ, thereby holding Rip1 inactive. S1 cleavage of RsmA, which is not inhibited by Ppr1, releases the inhibitory effect of Ppr1 on the PDZ domain, allowing Rip1 cleavage. The model predicts additional unidentified substrate-specific adapters that tether Rip1 to Anti-SigK and Anti-SigL.
We also propose that Ppr1 provides a model for understanding the substrate selection of S2Ps, which is poorly understood. Although amino acid residues have been identified in cleaved TM segments that affect iCLIP cleavage, these residues affect cleavage of both physiologic and nonphysiologic substrates (44, 45) and a conserved S2P cleavage motif has not been identified. Multiple studies have demonstrated that S2Ps will cleave transmembrane segments of proteins that are not natural substrates (17, 18), strongly suggesting that additional determinants must limit S2P cleavage to physiologic substrates while excluding other transmembrane proteins. We propose that Ppr1 is a trans-acting factor that provides S2P specificity by tethering Rip1 to one substrate (among three that have been identified), thereby facilitating RsmA cleavage while inhibiting nonspecific cleavage. This model implies that there may be additional adapter proteins which function analogously to Ppr1 in tethering Rip1 to its other anti-sigma factor substrates, and leading candidates are the additional Ppr proteins that we have identified as binding the Rip1-PDZ. We also speculate that the adapter protein model of S2P-substrate bridging could apply to other PDZ containing S2Ps outside of mycobacteria for which PDZ-interacting proteins have yet to be identified.
Materials and Methods
Bacterial Strains and Growth Conditions.
All bacterial strains for this study are listed in Table S1. M. tuberculosis strains (all based on the WT strain Erdman EG1, which is animal-passaged) were grown at 37 °C in 7H9 (broth) or 7H10 (agar) (Difco) media with oleic acid, albumin, dextrose, catalase (OADC) enrichment, 0.5% glycerol, and 0.05% Tween 80 (broth media only). M. smegmatis strains were cultured at 37 °C on Luria–Bertani (LB) medium containing 0.5% dextrose, 0.5% glycerol, and 0.05% Tween 80 (broth). When appropriate, the following were added into growth medium for E. coli/Mycobacteria, respectively: chloramphenicol (Sigma) at 25 μg⋅mL−1, hygromycin B (Boehringer Mannheim) at 150/50 μg⋅mL−1, kanamycin (Sigma) at 40/20 μg⋅mL−1, streptomycin (MP Biomedicals) at 40/20 μg⋅mL−1, ATC (Sigma) at 50 ng⋅mL−1, 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal) (Fisher Scientific) at 50 μg⋅mL−1, 5-bromo-4-chloro-3-indolyl phosphate (BCIP) (Sigma) at 60 μg⋅mL−1.
Yeast Strains and Growth Conditions.
All yeast strains for this study are listed in Table S2. See SI Materials and Methods for yeast growth conditions. All yeast and bacterial plasmids are listed in Table S3.
Yeast Two-Hybrid Screen for M. tuberculosis Rip1-PDZ–Binding Proteins.
The bait plasmid comprised a fusion of the LexA DNA-binding domain (BD) encoded in pEG202 to the N terminus of the Rip1 PDZ domain. Characterization of the bait fusion protein and the interaction screen against an M. tuberculosis activation domain (AD) fusion library were performed as previously described (28). DNA-BD fusions to the N terminus of Ku, HtrA-PDZ, PepA-PDZ, and PepD-PDZ were similarly constructed and characterized for use in the yeast two-hybrid system (see strain plasmid table for specific amino acid boundaries). Expression of full-length LexA-DNA BD bait fusions and AD–prey fusions was confirmed by immunoblotting with anti-LexA and anti-HA antibodies, respectively.
Protein Purification, Coexpression, Copurification, and in Vitro Protein Interactions.
See SI Materials and Methods for details.
Deletion of ppr1 (rv3333c) from M. tuberculosis.
M. tuberculosis Δppr1 was constructed via specialized transduction using the temperature-sensitive phage phAE87 as previously described (46). ppr1 was replaced with a hygromycin resistance cassette by deleting all but the start and stop codons of ppr1. The Δppr1 allele was verified by Southern blotting using the 3′ flanking region of ppr1 as a probe of chromosomal DNA fragmented with NcoI. The predicted size for WT is 884 bp, and the Δppr1 allele is 3,431 bp.
Quantitative RT-PCR.
mRNA levels were measured with quantitative RT-PCR as previously described (3). See SI Materials and Methods for additional details. Oligonucleotides are listed in Table S4.
SNAP Pulse-Chase Analysis.
Cultures were grown in LB medium with appropriate antibiotics to an OD600 of ∼0.6, induced with ATC for the time indicated, and then pulsed with 1 μM SNAP-Cell 505 reagent (NEB) for 30 min. Cells were then collected by centrifugation and resuspended in fresh LB with 1 mM O6-benzylguanine (Sigma) in DMSO (a concentration determined in preliminary experiments to block further labeling); 1-mL aliquots from the time points indicated were snap-frozen. Cells were lysed at 37 °C in Tris⋅EDTA with 10 mg⋅mL−1 lysozyme for 45 min, incubated for 10 min at 100 °C in SDS/PAGE loading buffer. Proteins were resolved on NuPAGE 4–12% Bis-Tris polyacrylamide gels (Invitrogen). Fluorescently labeled SNAP proteins were detected on a Typhoon Trio (GE Healthcare) using the 526-nm Fluorescein emission filter and Green (532-nm) laser, with the photomultiplier set to 450, and visualized with ImageQuant (GE Healthcare). The same gels used for SNAP detection were transferred to nitrocellulose membranes and probed with anti-HA antibody (HA.11 Clone 16B12; Covance) to detect HA-Ppr1, and monoclonal anti-CarD as a loading control. Quantification of Western blots and fluorescently labeled SNAP-tagged proteins was performed with ImageJ by calculating the area under the curves measuring band intensity. The fluorescence intensity of SNAP-tagged proteins was adjusted to the intensity of the loading control CarD for the same protein sample. For analysis of the S1-cleaved intermediates of RsmA and RslA, we quantitated the sum of both intermediates and each intermediate independently, with similar results. Data shown are for the sum of the two intermediates.
Murine Infection.
Murine experiments were performed as previously described (39) and in accordance with National Institutes of Health guidelines and for housing and care of laboratory animals, and were approved by the Memorial Sloan-Kettering Institutional Animal Care and Use Committee (IACUC).
Supplementary Material
Acknowledgments
We thank Miriam Braunstein for providing phoA fusion vectors and Allison Fay for insights into use of the SNAP tag. This work was supported by National Institutes of Health (NIH) Grant AI53417 (to M.S.G.). J.S.S. is supported by NIH Grant T32 AI007621 (Weill Cornell Graduate School of Medical Sciences) and the Dorris J. Hutchison Fellowship (Sloan-Kettering Institute). H.W.E. is supported by the Gerstner Sloan Kettering Summer Undergraduate Research Program.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. M.S.W. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1305934110/-/DCSupplemental.
References
- 1.Brown MS, Ye J, Rawson RB, Goldstein JL. Regulated intramembrane proteolysis: A control mechanism conserved from bacteria to humans. Cell. 2000;100(4):391–398. doi: 10.1016/s0092-8674(00)80675-3. [DOI] [PubMed] [Google Scholar]
- 2.Lemberg MK. Intramembrane proteolysis in regulated protein trafficking. Traffic. 2011;12(9):1109–1118. doi: 10.1111/j.1600-0854.2011.01219.x. [DOI] [PubMed] [Google Scholar]
- 3.Sklar JG, Makinoshima H, Schneider JS, Glickman MS. M. tuberculosis intramembrane protease Rip1 controls transcription through three anti-sigma factor substrates. Mol Microbiol. 2010;77(3):605–617. doi: 10.1111/j.1365-2958.2010.07232.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Makinoshima H, Glickman MS. Regulation of Mycobacterium tuberculosis cell envelope composition and virulence by intramembrane proteolysis. Nature. 2005;436(7049):406–409. doi: 10.1038/nature03713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rudner DZ, Fawcett P, Losick R. A family of membrane-embedded metalloproteases involved in regulated proteolysis of membrane-associated transcription factors. Proc Natl Acad Sci USA. 1999;96(26):14765–14770. doi: 10.1073/pnas.96.26.14765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kanehara K, Ito K, Akiyama Y. YaeL (EcfE) activates the sigma(E) pathway of stress response through a site-2 cleavage of anti-sigma(E), RseA. Genes Dev. 2002;16(16):2147–2155. doi: 10.1101/gad.1002302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ades SE, Connolly LE, Alba BM, Gross CA. The Escherichia coli sigma(E)-dependent extracytoplasmic stress response is controlled by the regulated proteolysis of an anti-sigma factor. Genes Dev. 1999;13(18):2449–2461. doi: 10.1101/gad.13.18.2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Damron FH, Goldberg JB. Proteolytic regulation of alginate overproduction in Pseudomonas aeruginosa. Mol Microbiol. 2012;84(4):595–607. doi: 10.1111/j.1365-2958.2012.08049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen JC, Viollier PH, Shapiro L. A membrane metalloprotease participates in the sequential degradation of a Caulobacter polarity determinant. Mol Microbiol. 2005;55(4):1085–1103. doi: 10.1111/j.1365-2958.2004.04443.x. [DOI] [PubMed] [Google Scholar]
- 10.Heinrich J, Wiegert T. Regulated intramembrane proteolysis in the control of extracytoplasmic function sigma factors. Res Microbiol. 2009;160(9):696–703. doi: 10.1016/j.resmic.2009.08.019. [DOI] [PubMed] [Google Scholar]
- 11.Urban S. Making the cut: Central roles of intramembrane proteolysis in pathogenic microorganisms. Nat Rev Microbiol. 2009;7(6):411–423. doi: 10.1038/nrmicro2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ye J, et al. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell. 2000;6(6):1355–1364. doi: 10.1016/s1097-2765(00)00133-7. [DOI] [PubMed] [Google Scholar]
- 13.Zhang K, et al. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell. 2006;124(3):587–599. doi: 10.1016/j.cell.2005.11.040. [DOI] [PubMed] [Google Scholar]
- 14.Bramkamp M, Weston L, Daniel RA, Errington J. Regulated intramembrane proteolysis of FtsL protein and the control of cell division in Bacillus subtilis. Mol Microbiol. 2006;62(2):580–591. doi: 10.1111/j.1365-2958.2006.05402.x. [DOI] [PubMed] [Google Scholar]
- 15.Schöbel S, Zellmeier S, Schumann W, Wiegert T. The Bacillus subtilis sigmaW anti-sigma factor RsiW is degraded by intramembrane proteolysis through YluC. Mol Microbiol. 2004;52(4):1091–1105. doi: 10.1111/j.1365-2958.2004.04031.x. [DOI] [PubMed] [Google Scholar]
- 16.Mukherjee P, et al. Novel role of Wag31 in protection of mycobacteria under oxidative stress. Mol Microbiol. 2009;73(1):103–119. doi: 10.1111/j.1365-2958.2009.06750.x. [DOI] [PubMed] [Google Scholar]
- 17.Akiyama Y, Kanehara K, Ito K. RseP (YaeL), an Escherichia coli RIP protease, cleaves transmembrane sequences. EMBO J. 2004;23(22):4434–4442. doi: 10.1038/sj.emboj.7600449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng L, et al. Structure of a site-2 protease family intramembrane metalloprotease. Science. 2007;318(5856):1608–1612. doi: 10.1126/science.1150755. [DOI] [PubMed] [Google Scholar]
- 19.Chen G, Zhang X. New insights into S2P signaling cascades: Regulation, variation, and conservation. Protein Sci. 2010;19(11):2015–2030. doi: 10.1002/pro.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Inaba K, et al. A pair of circularly permutated PDZ domains control RseP, the S2P family intramembrane protease of Escherichia coli. J Biol Chem. 2008;283(50):35042–35052. doi: 10.1074/jbc.M806603200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grigorova IL, et al. Fine-tuning of the Escherichia coli sigmaE envelope stress response relies on multiple mechanisms to inhibit signal-independent proteolysis of the transmembrane anti-sigma factor, RseA. Genes Dev. 2004;18(21):2686–2697. doi: 10.1101/gad.1238604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bohn C, Collier J, Bouloc P. Dispensable PDZ domain of Escherichia coli YaeL essential protease. Mol Microbiol. 2004;52(2):427–435. doi: 10.1111/j.1365-2958.2004.03985.x. [DOI] [PubMed] [Google Scholar]
- 23.Kanehara K, Ito K, Akiyama Y. YaeL proteolysis of RseA is controlled by the PDZ domain of YaeL and a Gln-rich region of RseA. EMBO J. 2003;22(23):6389–6398. doi: 10.1093/emboj/cdg602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harris BZ, Lim WA. Mechanism and role of PDZ domains in signaling complex assembly. J Cell Sci. 2001;114(Pt 18):3219–3231. doi: 10.1242/jcs.114.18.3219. [DOI] [PubMed] [Google Scholar]
- 25.Kinch LN, Ginalski K, Grishin NV. Site-2 protease regulated intramembrane proteolysis: Sequence homologs suggest an ancient signaling cascade. Protein Sci. 2006;15(1):84–93. doi: 10.1110/ps.051766506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Braunstein M, et al. Identification of genes encoding exported Mycobacterium tuberculosis proteins using a Tn552’phoA in vitro transposition system. J Bacteriol. 2000;182(10):2732–2740. doi: 10.1128/jb.182.10.2732-2740.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hizukuri Y, Akiyama Y. PDZ domains of RseP are not essential for sequential cleavage of RseA or stress-induced σ(E) activation in vivo. Mol Microbiol. 2012;86(5):1232–1245. doi: 10.1111/mmi.12053. [DOI] [PubMed] [Google Scholar]
- 28.Sinha KM, Stephanou NC, Gao F, Glickman MS, Shuman S. Mycobacterial UvrD1 is a Ku-dependent DNA helicase that plays a role in multiple DNA repair events, including double-strand break repair. J Biol Chem. 2007;282(20):15114–15125. doi: 10.1074/jbc.M701167200. [DOI] [PubMed] [Google Scholar]
- 29.Stephanou NC, et al. Mycobacterial nonhomologous end joining mediates mutagenic repair of chromosomal double-strand DNA breaks. J Bacteriol. 2007;189(14):5237–5246. doi: 10.1128/JB.00332-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gong C, et al. Mechanism of nonhomologous end-joining in mycobacteria: A low-fidelity repair system driven by Ku, ligase D and ligase C. Nat Struct Mol Biol. 2005;12(4):304–312. doi: 10.1038/nsmb915. [DOI] [PubMed] [Google Scholar]
- 31.Mohamedmohaideen NN, et al. Structure and function of the virulence-associated high-temperature requirement A of Mycobacterium tuberculosis. Biochemistry. 2008;47(23):6092–6102. doi: 10.1021/bi701929m. [DOI] [PubMed] [Google Scholar]
- 32.Cezairliyan BO, Sauer RT. Inhibition of regulated proteolysis by RseB. Proc Natl Acad Sci USA. 2007;104(10):3771–3776. doi: 10.1073/pnas.0611567104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De Las Peñas A, Connolly L, Gross CA. The sigmaE-mediated response to extracytoplasmic stress in Escherichia coli is transduced by RseA and RseB, two negative regulators of sigmaE. Mol Microbiol. 1997;24(2):373–385. doi: 10.1046/j.1365-2958.1997.3611718.x. [DOI] [PubMed] [Google Scholar]
- 34.Raman S, et al. Mycobacterium tuberculosis SigM positively regulates Esx secreted protein and nonribosomal peptide synthetase genes and down regulates virulence-associated surface lipid synthesis. J Bacteriol. 2006;188(24):8460–8468. doi: 10.1128/JB.01212-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barczak AK, et al. RNA signatures allow rapid identification of pathogens and antibiotic susceptibilities. Proc Natl Acad Sci USA. 2012;109(16):6217–6222. doi: 10.1073/pnas.1119540109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ellermeier CD, Losick R. Evidence for a novel protease governing regulated intramembrane proteolysis and resistance to antimicrobial peptides in Bacillus subtilis. Genes Dev. 2006;20(14):1911–1922. doi: 10.1101/gad.1440606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Keppler A, Pick H, Arrivoli C, Vogel H, Johnsson K. Labeling of fusion proteins with synthetic fluorophores in live cells. Proc Natl Acad Sci USA. 2004;101(27):9955–9959. doi: 10.1073/pnas.0401923101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Keppler A, et al. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat Biotechnol. 2003;21(1):86–89. doi: 10.1038/nbt765. [DOI] [PubMed] [Google Scholar]
- 39.Stallings CL, et al. CarD is an essential regulator of rRNA transcription required for Mycobacterium tuberculosis persistence. Cell. 2009;138(1):146–159. doi: 10.1016/j.cell.2009.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heinrich J, Hein K, Wiegert T. Two proteolytic modules are involved in regulated intramembrane proteolysis of Bacillus subtilis RsiW. Mol Microbiol. 2009;74(6):1412–1426. doi: 10.1111/j.1365-2958.2009.06940.x. [DOI] [PubMed] [Google Scholar]
- 41.Muller C, et al. Acid stress activation of the sigma(E) stress response in Salmonella enterica serovar Typhimurium. Mol Microbiol. 2009;71(5):1228–1238. doi: 10.1111/j.1365-2958.2009.06597.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li X, et al. Cleavage of RseA by RseP requires a carboxyl-terminal hydrophobic amino acid following DegS cleavage. Proc Natl Acad Sci USA. 2009;106(35):14837–14842. doi: 10.1073/pnas.0903289106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lima S, Guo MS, Chaba R, Gross CA, Sauer RT. Dual molecular signals mediate the bacterial response to outer-membrane stress. Science. 2013;340(6134):837–841. doi: 10.1126/science.1235358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koide K, Ito K, Akiyama Y. Substrate recognition and binding by RseP, an Escherichia coli intramembrane protease. J Biol Chem. 2008;283(15):9562–9570. doi: 10.1074/jbc.M709984200. [DOI] [PubMed] [Google Scholar]
- 45.Strisovsky K, Sharpe HJ, Freeman M. Sequence-specific intramembrane proteolysis: Identification of a recognition motif in rhomboid substrates. Mol Cell. 2009;36(6):1048–1059. doi: 10.1016/j.molcel.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barkan D, Liu Z, Sacchettini JC, Glickman MS. Mycolic acid cyclopropanation is essential for viability, drug resistance, and cell wall integrity of Mycobacterium tuberculosis. Chem Biol. 2009;16(5):499–509. doi: 10.1016/j.chembiol.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.