

Significance
Amyloid formation is a hallmark of a range of human diseases. The polypeptide hormone amylin forms amyloid deposits in type 2 diabetes, and amyloid formation is thought to be a contributor to the decline in β-cell mass in the disease, however the basis of amylin-induced cytotoxicity is not fully understood. Amylin-induced membrane disruption has attracted considerable interest, and the interactions of amylin with model membranes have been characterized, but the relationship between studies with model membranes and toxicity is not understood. In this article the relationship between cell toxicity and the ability to disrupt model membranes is examined. There is no direct correlation between the two processes.
Abstract
Islet amyloid polypeptide (IAPP) is responsible for amyloid formation in type 2 diabetes and contributes to the failure of islet cell transplants, however the mechanisms of IAPP-induced cytotoxicity are not known. Interactions with model anionic membranes are known to catalyze IAPP amyloid formation in vitro. Human IAPP damages anionic membranes, promoting vesicle leakage, but the features that control IAPP–membrane interactions and the connection with cellular toxicity are not clear. Kinetic studies with wild-type IAPP and IAPP mutants demonstrate that membrane leakage is induced by prefibrillar IAPP species and continues over the course of amyloid formation, correlating additional membrane disruption with fibril growth. Analyses of a set of designed mutants reveal that membrane leakage does not require the formation of β-sheet or α-helical structures. A His-18 to Arg substitution enhances leakage, whereas replacement of all of the aromatic residues via a triple leucine mutant has no effect. Biophysical measurements in conjunction with cytotoxicity studies show that nonamyloidogenic rat IAPP is as effective as human IAPP at disrupting standard anionic model membranes under conditions where rat IAPP does not induce cellular toxicity. Similar results are obtained with more complex model membranes, including ternary systems that contain cholesterol and are capable of forming lipid rafts. A designed point mutant, I26P-IAPP; a designed double mutant, G24P, I26P-IAPP; a double N-methylated variant; and pramlintide, a US Food and Drug Administration–approved IAPP variant all induce membrane leakage, but are not cytotoxic, showing that there is no one-to-one relationship between disruption of model membranes and induction of cellular toxicity.
Amyloid formation is a key feature of a range of diseases including Alzheimer’s disease, Parkinson’s disease, systemic amyloidosis, and type 2 diabetes (T2D) (1, 2). Islet amyloid polypeptide (IAPP, amylin) forms amyloid deposits in the pancreas in T2D. Islet amyloid, or species produced during islet amyloid formation, is toxic to cultured β cells in vitro (3–6). Increasing evidence highlights a role for β-cell loss in T2D, making islet amyloid deposition clinically relevant as it is an important contributor to the decline in β-cell mass (7). IAPP amyloid formation is also a factor in the failure of islet cell transplants, and prevention of amyloid formation enhances graft survival (8, 9).
IAPP is stored with insulin in the β-cell secretory granules, and is cosecreted in response to the same stimuli that lead to insulin release (10, 11). The polypeptide plays a role in the control of adiposity, gastric emptying, glucose homeostasis, and other metabolic activities (12, 13). IAPP is 37 residues in length, has a disulfide bond between Cys-2 and Cys-7, and has an amidated C terminus. The hormone is relatively hydrophobic, but is cationic at physiological pH due to its free N terminus, Lys-1, Arg-11, and His-18 (Fig. 1).
Fig. 1.

The primary sequence of the IAPP variants. Residues that differ from hIAPP are underlined and in italics. Each peptide has an amidated C terminus and a disulfide bridge between Cys-2 and Cys-7. Meth, N-methylated.
The basis of IAPP-induced cytotoxicity is not fully understood, despite its clinical importance, and a wide range of mechanisms have been proposed. These include receptor-mediated mechanisms, ER stress, defects in autophagy, increased production of proinflammatory cytokines, and permeabilization of the plasma and mitochondria membranes (6, 13). Membrane disruption has attracted considerable interest, and a range of studies have examined interactions of IAPP with model membranes. Vesicles containing a significant fraction of anionic lipids accelerate amyloid formation by IAPP in vitro, and human IAPP (hIAPP) induces membrane leakage in these systems (14–17). This has led to the hypothesis that membrane leakage may be a critical factor in islet amyloidosis cytotoxicity, but the relationship between studies with model membranes and in vivo or in vitro toxicity are not clear, especially as the β-cell membrane differs significantly from standard model membranes. This is an important issue, as the physiological basis of hIAPP cytotoxicity in T2D is not defined. The features that control IAPP–model membrane interactions are also not fully understood.
IAPP–membrane interactions are sensitive to the fraction of anionic lipids, and the majority of studies use membranes consisting of a pure anionic lipid such as phosphatidylglycerol or phosphatidylserine (PS) or mixtures of an anionic lipid with zwitterionic lipids, such as phosphatidylcholine (PC). However, these systems are notably different from the β-cell membrane, and their physiological relevance is not clear. The content of anionic lipid in model membranes typically ranges from 20 to 50 mol %, which is significantly higher than the 2.5–13.2 mol % reported for pancreatic β cells (14, 15, 18). Furthermore, the phospholipid composition of β-cell membranes is different from model membranes. The plasma membrane is also asymmetric, and anionic lipids are preferentially located in the inner leaflet. The inner leaflet is enriched in PS, whereas the outer leaflet is enriched in sphingolipids and PC, although it does contain gangliosides (19). The β-cell membrane contains cholesterol, and cholesterol has been proposed to be important for IAPP–membrane interactions (20). These differences naturally lead to the question of how interactions with model membranes correlate with cellular IAPP toxicity.
There are a number of other outstanding issues in IAPP–membrane interactions, and model membranes can be useful tools to address these questions. The mechanisms of model membrane disruption are not fully defined, and the nature of the membrane active species is not fully elucidated. Pore formation has been proposed to be a key for membrane disruption (21–23), but a carpeting, detergent-like mechanism has also been advocated (24, 25). There is experimental evidence that intermediates permeabilize membranes (26–28), whereas other studies implicate fibril growth at the membrane surface (29, 30). The structural requirements for membrane activity are also not completely clear. Membrane binding is linked to α-helix formation under certain conditions, and membrane-bound helical structures can associate and convert to β-sheet–rich amyloid fibrils (14, 15, 31). However, variants of IAPP that do not convert from the α-helical membrane-bound state to amyloid fibrils disrupt model membranes, showing that membrane permeabilization does not require fibril formation. It is not known if less structured conformers of IAPP disrupt membranes, and the features of the IAPP sequence that control the peptide’s ability to interact with membranes are not completely defined.
We analyze human and rat IAPP (rIAPP) and a set of IAPP variants (Fig. 1) to probe the factors that control IAPP-mediated membrane leakage and to explore the potential connection between leakage of standard model membranes and cytotoxicity. We use membranes composed of 25% or 100% anionic lipids as well as two more physiologically relevant model systems. Kinetic studies demonstrate that membrane leakage is induced by prefibrillar IAPP species and continues over the course of amyloid formation, correlating additional membrane disruption with fibril growth. Analysis of a set of mutants reveals that neither β-sheet nor α-helix structure is required for membrane disruption. A H18R substitution is shown to enhance membrane disruption, but replacement of the aromatic residues with Leu has no effect. We demonstrate that there is no one-to-one relationship between standard model membrane leakage and cytotoxicity. Similar results are obtained with the more complicated membranes. The data dissociate model membrane leakage from primary pathogenic mechanisms underlying IAPP-mediated cellular toxicity, highlighting the difficulty of extrapolating from biophysical studies to the situation in vivo.
Results and Discussion
The sequences of the polypeptides studied here are listed in Fig. 1. rIAPP, which is nontoxic and does not form amyloid in vivo or in vitro, differs from hIAPP at six sites, including three proline residues at positions 25, 28, and 29 and the replacement of H18 by R. These substitutions significantly influence the aggregation behavior of the polypeptide, whereas the others are more conservative: F23 in the human peptide is replaced by L, and I26 by V. We used the following series of mutations to test the role of specific residues in membrane interactions: First, pramlintide (PM) is an analog of hIAPP, which contains the three proline substitutions found in rIAPP. The molecule does not form amyloid in dilute solution, is not toxic, and has been approved as a complement to insulin therapy for the treatment of diabetes (32). Comparison of hIAPP and PM allows us to probe the role of the three prolines. Second, replacement of H18 by R in PM leads to the H18R-PM mutant; comparison of PM and H18R-PM tests the role of H18. Third, the I26P point mutant (I26P-IAPP) does not form amyloid in solution and inhibits amyloid formation by hIAPP (33). Fourth, the G24P, I26P double mutant (DM-IAPP), is also nonamyloidogenic (33). I26P-IAPP and DM-IAPP provide additional tests of the membrane activity of nonamyloidogenic variants. N-methyl–IAPP corresponds to hIAPP with a N-methylated glycine substitution at G24 and a N-methylated isoleucine substitution at I26. This peptide is not toxic and is a potent inhibitor of amyloid formation and cytotoxicity by wild-type hIAPP (34). Fifth, the 3XL-IAPP peptide is a triple mutant of hIAPP in which the three aromatic residues, F15, F23, and Y37, have been replaced by L (35). It forms amyloid approximately ninefold more slowly than wild-type hIAPP and allows us to test the role of the aromatic residues in membrane disruption.
Toxic and Nontoxic Variants of IAPP Induce Model Membrane Leakage.
PM, N-methyl–IAPP, and rIAPP have been reported to be nontoxic (32, 34, 36). We tested the cytotoxicity of I26P-IAPP, DM-IAPP, rIAPP, and N-methyl–IAPP using rat insulinoma-1 (INS-1) β cells. INS-1 β cells are a standard pancreatic cell line that is widely used in β-cell biology and studies of IAPP cytotoxicity. None of the polypeptides, except hIAPP, decreased cell viability as judged by AlamarBlue assays (Fig. 2A). rIAPP was examined over a concentration range of 15–60 µM and was nontoxic even at the highest concentration (Fig. 2B).
Fig. 2.
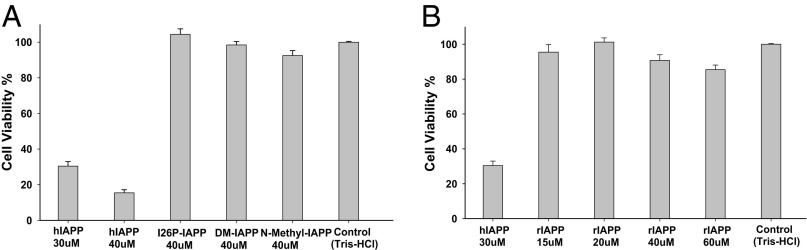
hIAPP is toxic to rat INS-1 β cells, but the other peptides are not. (A) Comparison of hIAPP and IAPP variants. (B) rIAPP was examined over a concentration range of 15–60 µM. The results of AlamarBlue assays are plotted. Percent cell viability is normalized to buffer control.
We examined the effect of the polypeptides on the integrity of standard model vesicles. We first tested the ability of a subset of IAPP variants to induce leakage of 100-nm-diameter model membrane vesicles comprising just the negatively charged lipid, 1,2-dioleoyl-sn-glycero-3-phospho-(1'-rac-glycerol) (DOPG). The fluorescent dye carboxyfluorescein was used to measure vesicle leakage. The fluorescence of the dye is self-quenched when it is encapsulated in vesicles (37). Loss of membrane integrity allows the dye to escape, relieving self-quenching and leading to enhanced fluorescence. The percent leakage was measured after 10 min incubation with peptide; this is a standard protocol in the field, and most studies are conducted with incubation times of 100–1,000 s. The peptides were tested over a concentration range of 2–40 µM, leading to lipid/peptide ratios (10:1–200:1) that are within the range used in other studies. All of the variants are effective at inducing leakage of 400 µM anionic membrane vesicles (Fig. 3A and Fig. S1A), and all are more disruptive above 20 µM, where lipid/peptide ratios are lower. hIAPP is the most effective, but all of the other polypeptides induced over 60% leakage at 40 µM, even though they are nontoxic to β cells. rIAPP, which is nontoxic, induced 86% leakage at 40 µM, identical to that induced by 20 µM hIAPP (89%), which is toxic.
Fig. 3.

Membrane leakage induced by hIAPP and IAPP variants after incubating with 400 µM anionic model membranes (100% DOPG) for (A) 10 min or (B) 48 h. Experiments were performed at 25 °C in 20 mM Tris⋅HCl, 100 mM NaCl, pH 7.4.
We next examined the effect of incubating the peptide with the model membranes for 48 h. This is much longer than the time required for hIAPP to form amyloid under these conditions; this experiment helps to test whether fibril growth plays a role in membrane leakage. The percent leakage is higher after 48 h incubation; hIAPP at 1 µM induces 14% leakage after 10 min incubation and 33% leakage after 48 h of incubation. Little additional enhancement upon longer incubation is observed for the higher concentrations; 40 µM hIAPP induces 93% leakage after 10 min and 98% after 48 h. hIAPP is somewhat more effective than the variants at inducing leakage under these conditions, but the differences are very modest. A total of 40 µM hIAPP induces 98% leakage after 48 h incubation, whereas 40 µM rIAPP induces 88%. rIAPP, I26P-IAPP, and DM-IAPP are all still highly effective at inducing membrane leakage under conditions where they are not toxic; thus, there is no direct one-to-one correlation between cytotoxicity and the ability to induce leakage of DOPG vesicles (Fig. 3B and Fig. S1B).
We next tested the ability of the full set of polypeptides to induce leakage of 100 nm diameter, 400 µM vesicles comprising 75% zwitterionic phospholipid, 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), and 25% DOPG. The percentage of anionic lipids is comparable to or lower than that used in most studies, but is still higher than that found in the β-cell plasma membrane in vivo. The interaction of IAPP with vesicles also depends on the lipid/peptide ratio (14); thus, we tested lipid/peptide ratios from 200:1–6.7:1, a set of values that span the range used in other studies (15). We first compared the concentration dependence of polypeptide-induced leakage after 10 min incubation, a time which is shorter than required for hIAPP to form amyloid (Fig. 4A and Fig. S2A). The human peptide readily induces leakage, but 60 µM rIAPP is as effective as 30 µM hIAPP, even though 60 µM rIAPP is not toxic to β cells, and 30 µM hIAPP is (Fig. 2). This result shows that there is no one-to-one correlation between cellular toxicity and leakage in this system. The single and double mutants, N-methyl–IAPP, and PM all induce leakage, but are less effective than rIAPP. DM-IAPP is the least effective, inducing around 20% leakage at 60 µM, whereas I26P-IAPP, N-methyl–IAPP, and PM induced comparable levels of leakage (25%) (Fig. 4A).
Fig. 4.

Membrane leakage induced by hIAPP and IAPP variants after incubating with 400 µM 25% anionic model membranes (25% DOPG + 75% DOPC) for (A) 10 min or (B) 48 h. The dashed line in A compares 30 µM hIAPP with 60 µM rIAPP. Experiments were performed at 25 °C in 20 mM Tris⋅HCl, 100 mM NaCl, pH 7.4.
We also measured the leakage induced by the peptides after 48 h incubation with the 25% anionic model membranes. hIAPP induces higher leakage after 48 h relative to 10 min of incubation, further suggesting that amyloid formation or fibril growth contributes to membrane disruption in vitro (Fig. 4B and Fig. S2B). A total of 10 µM hIAPP induces over 40% leakage after 48 h incubation, comparable to the leakage induced by 60 µM rIAPP (35%). hIAPP has been reported to be toxic at 10 µM (38), whereas 60 µM rIAPP is not.
Fibril Growth Promotes Additional Leakage.
Fibril formation at the membrane surface has been proposed to promote leakage (29, 30). Comparison of the 10 min vs. 48 h incubation data strongly supports this conclusion. We monitored the full time course of leakage of the 25% anionic vesicles induced by human and rIAPP in parallel with kinetic studies of amyloid formation to directly address this issue (Fig. 5). hIAPP displays a typical amyloid formation profile with a lag phase of about 2 h under these conditions. In contrast, rIAPP does not form amyloid even after 48 h of incubation. A multistep process for hIAPP-induced leakage is detected, but only a single step is observed for rIAPP (Fig. 5C). Closer examination of the human data shows that the percent leakage plateaued after 80 min and then increased again after 2 h (Fig. 5D). The first phase, observed for both hIAPP and rIAPP, is likely due to the initial interactions of the peptide and membrane. The second phase is observed only for hIAPP, correlating it strongly with the growth of amyloid. The data suggest that the differences in leakage observed between hIAPP and the nonamyloidogenic rat peptide after 48 h are due to additional damage caused by amyloid fibril growth (30). This helps to rationalize the lower leakage induced by the nonamyloidogenic mutants relative to hIAPP for long incubation times.
Fig. 5.

Additional leakage correlates with the time course of amyloid formation. (A) Comparison of the kinetics of amyloid formation. hIAPP, black circles; rIAPP, open squares. (B) An expansion of the first 7 h of A. (C) The kinetics of membrane leakage. hIAPP, black circles; rIAPP, open squares. (D) An expansion of the first 7 h of C. Experiments were performed at 25 °C in 20 mM Tris⋅HCl, 100 mM NaCl, pH 7.4, with 20 µM peptide.
Aromatic Residues Are Not Required for Membrane Damage, but Membrane Damage Is Sensitive to Substitutions at Position-18.
The triple leucine mutant, 3XL-IAPP, was originally designed to test the role of the aromatic residues in IAPP amyloid formation (35). 3XL-IAPP forms amyloid fibrils with a similar morphology to those derived from wild-type hIAPP, but does so more slowly. The ability of 3XL-IAPP to induce leakage was tested over the concentration range of 2–60 µM using the 25% anionic lipid membrane model. The mutant induced leakage as effectively as hIAPP, for both the 10 min and 48 h incubation times (Fig. S3), showing that aromatic residues are not required to induce membrane leakage.
Comparison of rIAPP with PM provides insight into the relative importance of the nonproline substitutions in rIAPP, as the two peptides share the same proline residues, but PM contains the human residues at other locations. The major difference between rIAPP and PM is the R18H substitution. rIAPP is more effective at inducing membrane leakage than PM, suggesting that R18 in rIAPP plays an important role. We further tested the importance of position-18 by comparing PM with the designed H18R-PM variant. H18R-PM is as effective at inducing leakage as rIAPP and is more effective than PM, confirming that R18 is important for IAPP-mediated membrane damage.
Neither β-Sheet Formation nor Significant α-Helical Structure Is Required for Membrane Leakage.
We used CD and transmission electron microscopy (TEM) to examine the conformation of the peptides after 10 min and 48 h of incubation with model membranes. Neither rIAPP, I26P-IAPP, DM-IAPP, N-methyl–IAPP, PM, nor H18R-PM formed signficant amounts of β-sheet structure in the presence of 25% DOPG/75% DOPC vesicles at a 6.7:1 lipid/peptide ratio as judged by CD (Fig. 6 and Fig. S4). None of the variants formed amyloid fibrils, as judged by TEM, even after 48 h of incubation (Fig. S5). The data show that neither the formation of β-sheet structure nor the formation of amyloid fibrils is required for membrane disruption under these condtions.
Fig. 6.

Membrane disruption does not require formation of detectable α-helix or β-sheet structure. CD spectra of IAPP and IAPP mutants after incubating with 400 µM 25% anionic model membranes (25% DOPG + 75% DOPC) for 10 min. Note the lack of α-helical or β-sheet signal from all of the IAPP mutants except 3XL-IAPP. The peptide concentration was 60 µM, and experiments were performed at 25 °C in 20 mM Tris⋅HCl, 100 mM NaCl, pH 7.4.
The mutants did not form significant α-helical structure under this condition, arguging that it is also not strictly required for membrane leakage. Other studies that made use of vesicles with higher anionic lipid content have shown that helical structure is compatible with membrane disruption. Thus, the data do not imply that model membranes are not able to promote helical structure and should not be intreperted to mean that helical formation does not play a role in membrane disruption in other cases; rather, it indicates that membrane leakage can be induced by IAPP variants even in the absence of significant, detectable α-helical or β-sheet structure.
Folded, Monomeric Proteins Are Less Effective at Inducing Model Membrane Leakage.
A range of IAPP mutants are effective at inducing leakage, and it is natural to inquire if any similarly sized cationic polypeptide will do so. We tested the ability of NTL9, a 56-residue α–β structure, and the villin headpiece helical subdomain (HP36), a 36-residue helical protein, to induce leakage of the 25% anionic vesicles (Fig. S6). The estimated net charge of NTL9 is +6 at neutral pH and that of HP36 is +2. No detectable leakage was observed for NTL9 after 10 min incubation, and only very low levels were detected after 48 h, even though NTL9 has a higher net charge than IAPP. HP36 was more effective than NTL9 at inducing leakage, but the effects were still modest and noticablely less than what were detected for IAPP. The results demonstrate that the effects observed with IAPP are not due simply to the net charge of the monomer.
Nontoxic Variants of IAPP Disrupt More Complex Model Membranes.
We examined two more complicated model systems: total brain extract lipids (TBE-lipids) and a ternary system consisting of DOPC, 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) and cholesterol at a 1:2:1 ratio. TBE-lipids have been widely used as models of the plasma membrane and have been used in studies of IAPP–membrane interactions (28, 39–41). They are considerably more complex than standard one- or two-component model membrane systems. We examined the effect of TBE-lipids on amyloid formation by rIAPP, I26P-IAPP, and hIAPP, and tested the ability of the peptides to permeabilize large unilamellar vesicles (LUVs) prepared from TBE-lipids (Fig. S7). TBE-lipid vesicles catalyze amyloid formation by hIAPP, reducing the lag time by a factor of 4, but are less effective than vesicles composed of 25% DOPG/75% DOPC, which reduced the lag time by a factor of 11. TBE-lipid vesicles do not induce amyloid formation by rIAPP or I26P-IAPP, even after 48 h of incubation.
hIAPP is less effective at inducing leakage of TBE-lipid vesicles than simpler model vesicles. A total of 20 µM hIAPP led to 17% leakage of the TBE-vesicles after 10 min of incubation versus 27% leakage of the 25% anionic vesicles and 89% of 100% anionic vesicles. A total of 60 µM hIAPP led to 30% leakage of the TBE-vesicles and 40% leakage of the 25% anionic lipid vesicles after 10 min of incubation. Similar trends were observed after 48 h of incubation. rIAPP and the I26P point mutant are also more effective at disrupting the two-component model vesicles. However, rIAPP, under conditions where it is not toxic, induces similar leakage of the TBE-vesicles as hIAPP does under conditions where it is toxic: 60 µM rat is as effective as 20 µM hIAPP at inducing leakage after 10 min of incubation, even though 60 µM rIAPP is not cytotoxic, and 20 µM hIAPP is. Similar effects are observed after 48 h of incubation. In this case, 60 µM rIAPP is as effective at inducing leakage as 10 µM hIAPP, even though the former is not toxic whereas the latter has been reported to be (38).
No significant α-helical or β-sheet structure was detected for any of the peptides after 10 min of incubation with the TBE-vesicles as judged by CD (Fig. S8A), and no detectable α-helical or β-sheet structure was observed for I26P-IAPP or rIAPP after 48 h of incubation, confirming that neither α-helical nor β structure is required to induce membrane leakage under these conditions (Fig. S8B).
The DOPC, DPPC, cholesterol system can form lipid rafts and has been used in other studies of IAPP–model membrane interactions (28, 39, 40). Amyloid formation by wild-type hIAPP is accelerated by this membrane. rIAPP and I26P-IAPP were as effective at inducing leakage as hIAPP after both 10 min and 48 h of incubation. These results further demonstrate that there is no 1:1 correspondence between cytotoxicity and model membrane leakage. CD indicates that I26P-IAPP and rIAPP fail to form significant amouts of helical or β structure under the conditions of these studies, confirming that the formation of detectable amounts of α-helical or β-sheet structure is not required for leakage (Fig. S9).
Conclusions
The data demonstrate that there is no one-to-one relationship between IAPP cytotoxicity and the ability to disrupt standard model membranes. We do not wish to imply that loss of membrane integrity is unimportant in vivo; it may be one mechanism contributing to cytotoxicity along with a range of other cellular events. IAPP amyloid has been shown to cluster at or near membranes, and exogenously added IAPP can perturb cell membranes (21, 26, 42, 43), but it can be difficult to extrapolate from in vitro studies with model membranes to the situation in vivo. The fact that differences in the ability to induce leakage are observed for the 48 h incubation in the presence of the 25% anionic vesicle might suggest that there is some correlation with in vitro leakage and toxicity. However, it is important to reiterate that 60 µM rIAPP (nontoxic) induces leakage at the same level as 10 µM hIAPP, which has been reported to be toxic. In addition, comparable levels of leakage are observed for hIAPP and rIAPP in the ternary system.
Correlation studies of model membrane damage and cytotoxicity have interesting implications for biological studies of IAPP pathogenesis in T2D, however more complicated and physiologically relevant model membranes will be needed to provide the most translational insight. Commonly used model systems are very different from β-cell membranes. Along these lines, it is interesting to note that IAPP is much more effective at inducing leakage of 100% anionic vesicles than vesicles containing 25% anionic lipids, and both are more susceptible to the effects of IAPP than are the TBE-lipid vesicles.
Model membranes studies have been presented for other amyloidogenic peptides including α-synuclein and the Aβ-peptide, but the type of analysis described here has not been reported. It may be of interest to do so, especially as α-synuclein has been shown to adopt different structures on different model membranes (44).
Our results also provide information about the factors that control the ability of IAPP to induce membrane leakage. The data confirm that prefibrillar species induce leakage, but also show that progression of leakage correlates with fibril formation in agreement with earlier work (30). The data strongly support a recently proposed two-stage mechanism where the first stage involves interaction of prefibrillar species with the membrane and the second stage correlates with fibril growth on the membrane (29). Neither significant detectable α-helical or β-sheet structure is required to induce leakage in the model systems examined here. Aromatic residues are not obligatory, but the identity of residue-18 is important. The importance of postion-18 is consistent with NMR studies of IAPP fragments in the presence of model membranes (45). hIAPP1–19 and rIAPP1–19, which differ only in the identity of residue-18, bind membranes in different orientations, and these differences are believed to correlate with the difference in the potential for membrane disruption (45, 46).
In summary, our data dissociate IAPP-induced leakage of standard model membranes from direct cellular toxicity, thereby indicating that further studies to identify the precise mechanism(s) of IAPP cellular toxicity are essential for the optimal development of therapeutic strategies to prevent T2D and islet graft failure.
Materials and Methods
A detailed description of sample preparation, amyloid assays, membrane leakage assays, and cell viability assays are provided in the SI Materials and Methods along with a description of the spectroscopic methods. Analytical HPLC was used to check the purity of the peptides before each experiment. This is important because deamidation can be a complicating factor in studies of IAPP amyloid formation (47). Thioflavin-T binding assays were used to follow the time course of amyloid formation. Each experiment was repeated three times using different IAPP stock solutions. Membrane leakage was monitored using carboxyfluorescein-filled LUVs. Cell viability was measured by AlamarBlue reduction assays. Values were calculated relative to those of control cells treated with buffer only. All values represent means ± SEM (n = 3).
Supplementary Material
Acknowledgments
We thank Professor Erwin London for advice on experimental design and for numerous helpful discussions. We also thank Ivan Peran for supplying NTL9 and Bowu Laun for supplying HP-36. This work was supported by National Institutes of Health Grants GM078114 (to D.P.R.) and F32 DK089734-02 (to A.A.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1305517110/-/DCSupplemental.
References
- 1.Selkoe DJ. Cell biology of protein misfolding: The examples of Alzheimer’s and Parkinson’s diseases. Nat Cell Biol. 2004;6(11):1054–1061. doi: 10.1038/ncb1104-1054. [DOI] [PubMed] [Google Scholar]
- 2.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 3.Clark A, et al. Islet amyloid, increased alpha-cells, reduced beta-cells and exocrine fibrosis: Quantitative changes in the pancreas in type-2 diabetes. Diabetes Res Clin Ex. 1988;9(4):151–159. [PubMed] [Google Scholar]
- 4.Lorenzo A, Razzaboni B, Weir GC, Yankner BA. Pancreatic islet cell toxicity of amylin associated with type-2 diabetes mellitus. Nature. 1994;368(6473):756–760. doi: 10.1038/368756a0. [DOI] [PubMed] [Google Scholar]
- 5.Konarkowska B, Aitken JF, Kistler J, Zhang S, Cooper GJ. The aggregation potential of human amylin determines its cytotoxicity towards islet beta-cells. FEBS J. 2006;273(15):3614–3624. doi: 10.1111/j.1742-4658.2006.05367.x. [DOI] [PubMed] [Google Scholar]
- 6.Cao P, et al. Islet amyloid: From fundamental biophysics to mechanisms of cytotoxicity. FEBS Lett. 2013;587(8):1106–1118. doi: 10.1016/j.febslet.2013.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ashcroft FM, Rorsman P. Diabetes mellitus and the β cell: The last ten years. Cell. 2012;148(6):1160–1171. doi: 10.1016/j.cell.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Potter KJ, et al. Islet amyloid deposition limits the viability of human islet grafts but not porcine islet grafts. Proc Natl Acad Sci USA. 2010;107(9):4305–4310. doi: 10.1073/pnas.0909024107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Westermark GT, Westermark P, Berne C, Korsgren O, Transpla NNCI. Nordic Network for Clinical Islet Transplantation Widespread amyloid deposition in transplanted human pancreatic islets. N Engl J Med. 2008;359(9):977–979. doi: 10.1056/NEJMc0802893. [DOI] [PubMed] [Google Scholar]
- 10.Lukinius A, Wilander E, Westermark GT, Engström U, Westermark P. Co-localization of islet amyloid polypeptide and insulin in the B cell secretory granules of the human pancreatic islets. Diabetologia. 1989;32(4):240–244. doi: 10.1007/BF00285291. [DOI] [PubMed] [Google Scholar]
- 11.Kahn SE, et al. Evidence of cosecretion of islet amyloid polypeptide and insulin by beta-cells. Diabetes. 1990;39(5):634–638. doi: 10.2337/diab.39.5.634. [DOI] [PubMed] [Google Scholar]
- 12.Lutz TA. Control of energy homeostasis by amylin. Cell Mol Life Sci. 2012;69(12):1947–1965. doi: 10.1007/s00018-011-0905-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Westermark P, Andersson A, Westermark GT. Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol Rev. 2011;91(3):795–826. doi: 10.1152/physrev.00042.2009. [DOI] [PubMed] [Google Scholar]
- 14.Knight JD, Hebda JA, Miranker AD. Conserved and cooperative assembly of membrane-bound alpha-helical states of islet amyloid polypeptide. Biochemistry. 2006;45(31):9496–9508. doi: 10.1021/bi060579z. [DOI] [PubMed] [Google Scholar]
- 15.Hebda JA, Miranker AD. The interplay of catalysis and toxicity by amyloid intermediates on lipid bilayers: Insights from type II diabetes. Annu Rev Biophys. 2009;38:125–152. doi: 10.1146/annurev.biophys.050708.133622. [DOI] [PubMed] [Google Scholar]
- 16.Jayasinghe SA, Langen R. Membrane interaction of islet amyloid polypeptide. Biochim Biophys Acta. 2007;1768(8):2002–2009. doi: 10.1016/j.bbamem.2007.01.022. [DOI] [PubMed] [Google Scholar]
- 17.Engel MFM. Membrane permeabilization by Islet Amyloid Polypeptide. Chem Phys Lipids. 2009;160(1):1–10. doi: 10.1016/j.chemphyslip.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 18.Seeliger J, Weise K, Opitz N, Winter R. The effect of Aβ on IAPP aggregation in the presence of an isolated β-cell membrane. J Mol Biol. 2012;421(2-3):348–363. doi: 10.1016/j.jmb.2012.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng HT, Megha, London E. Preparation and properties of asymmetric vesicles that mimic cell membranes: Effect upon lipid raft formation and transmembrane helix orientation. J Biol Chem. 2009;284(10):6079–6092. doi: 10.1074/jbc.M806077200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trikha S, Jeremic AM. Clustering and internalization of toxic amylin oligomers in pancreatic cells require plasma membrane cholesterol. J Biol Chem. 2011;286(41):36086–36097. doi: 10.1074/jbc.M111.240762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mirzabekov TA, Lin MC, Kagan BL. Pore formation by the cytotoxic islet amyloid peptide amylin. J Biol Chem. 1996;271(4):1988–1992. doi: 10.1074/jbc.271.4.1988. [DOI] [PubMed] [Google Scholar]
- 22.Anguiano M, Nowak RJ, Lansbury PT., Jr Protofibrillar islet amyloid polypeptide permeabilizes synthetic vesicles by a pore-like mechanism that may be relevant to type II diabetes. Biochemistry. 2002;41(38):11338–11343. doi: 10.1021/bi020314u. [DOI] [PubMed] [Google Scholar]
- 23.Last NB, Rhoades E, Miranker AD. Islet amyloid polypeptide demonstrates a persistent capacity to disrupt membrane integrity. Proc Natl Acad Sci USA. 2011;108(23):9460–9465. doi: 10.1073/pnas.1102356108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Green JD, et al. Atomic force microscopy reveals defects within mica supported lipid bilayers induced by the amyloidogenic human amylin peptide. J Mol Biol. 2004;342(3):877–887. doi: 10.1016/j.jmb.2004.07.052. [DOI] [PubMed] [Google Scholar]
- 25.Sparr E, et al. Islet amyloid polypeptide-induced membrane leakage involves uptake of lipids by forming amyloid fibers. FEBS Lett. 2004;577(1-2):117–120. doi: 10.1016/j.febslet.2004.09.075. [DOI] [PubMed] [Google Scholar]
- 26.Janson J, Ashley RH, Harrison D, McIntyre S, Butler PC. The mechanism of islet amyloid polypeptide toxicity is membrane disruption by intermediate-sized toxic amyloid particles. Diabetes. 1999;48(3):491–498. doi: 10.2337/diabetes.48.3.491. [DOI] [PubMed] [Google Scholar]
- 27.Porat Y, Kolusheva S, Jelinek R, Gazit E. The human islet amyloid polypeptide forms transient membrane-active prefibrillar assemblies. Biochemistry. 2003;42(37):10971–10977. doi: 10.1021/bi034889i. [DOI] [PubMed] [Google Scholar]
- 28.Weise K, Radovan D, Gohlke A, Opitz N, Winter R. Interaction of hIAPP with model raft membranes and pancreatic beta-cells: Cytotoxicity of hIAPP oligomers. ChemBioChem. 2010;11(9):1280–1290. doi: 10.1002/cbic.201000039. [DOI] [PubMed] [Google Scholar]
- 29.Brender JR, et al. Biphasic effects of insulin on islet amyloid polypeptide membrane disruption. Biophys J. 2011;100(3):685–692. doi: 10.1016/j.bpj.2010.09.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Engel MFM, et al. Membrane damage by human islet amyloid polypeptide through fibril growth at the membrane. Proc Natl Acad Sci USA. 2008;105(16):6033–6038. doi: 10.1073/pnas.0708354105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jayasinghe SA, Langen R. Lipid membranes modulate the structure of islet amyloid polypeptide. Biochemistry. 2005;44(36):12113–12119. doi: 10.1021/bi050840w. [DOI] [PubMed] [Google Scholar]
- 32.Ratner RE, et al. Amylin replacement with pramlintide as an adjunct to insulin therapy improves long-term glycaemic and weight control in Type 1 diabetes mellitus: A 1-year, randomized controlled trial. Diabet Med. 2004;21(11):1204–1212. doi: 10.1111/j.1464-5491.2004.01319.x. [DOI] [PubMed] [Google Scholar]
- 33.Meng FL, Raleigh DP, Abedini A. Combination of kinetically selected inhibitors in trans leads to highly effective inhibition of amyloid formation. J Am Chem Soc. 2010;132(41):14340–14342. doi: 10.1021/ja1046186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yan LM, Tatarek-Nossol M, Velkova A, Kazantzis A, Kapurniotu A. Design of a mimic of nonamyloidogenic and bioactive human islet amyloid polypeptide (IAPP) as nanomolar affinity inhibitor of IAPP cytotoxic fibrillogenesis. Proc Natl Acad Sci USA. 2006;103(7):2046–2051. doi: 10.1073/pnas.0507471103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marek P, et al. Aromatic interactions are not required for amyloid fibril formation by islet amyloid polypeptide but do influence the rate of fibril formation and fibril morphology. Biochemistry. 2007;46(11):3255–3261. doi: 10.1021/bi0621967. [DOI] [PubMed] [Google Scholar]
- 36.Cao P, et al. Sensitivity of amyloid formation by human islet amyloid polypeptide to mutations at residue 20. J Mol Biol. 2012;421(2-3):282–295. doi: 10.1016/j.jmb.2011.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen RF, Knutson JR. Mechanism of fluorescence concentration quenching of carboxyfluorescein in liposomes: Energy transfer to nonfluorescent dimers. Anal Biochem. 1988;172(1):61–77. doi: 10.1016/0003-2697(88)90412-5. [DOI] [PubMed] [Google Scholar]
- 38.Lee TY, et al. A Co(III) complex cleaving soluble oligomers of h-IAPP in the presence of polymeric aggregates of h-IAPP. Bioorg Med Chem Lett. 2012;22(17):5689–5693. doi: 10.1016/j.bmcl.2012.06.089. [DOI] [PubMed] [Google Scholar]
- 39.Radovan D, Opitz N, Winter R. Fluorescence microscopy studies on islet amyloid polypeptide fibrillation at heterogeneous and cellular membrane interfaces and its inhibition by resveratrol. FEBS Lett. 2009;583(9):1439–1445. doi: 10.1016/j.febslet.2009.03.059. [DOI] [PubMed] [Google Scholar]
- 40.Jha S, Sellin D, Seidel R, Winter R. Amyloidogenic propensities and conformational properties of ProIAPP and IAPP in the presence of lipid bilayer membranes. J Mol Biol. 2009;389(5):907–920. doi: 10.1016/j.jmb.2009.04.077. [DOI] [PubMed] [Google Scholar]
- 41.Díaz RS, Monreal J, Regueiro P, Lucas M. Preparation of a protein-free total brain white matter lipid fraction: Characterization of liposomes. J Neurosci Res. 1992;31(1):136–145. doi: 10.1002/jnr.490310119. [DOI] [PubMed] [Google Scholar]
- 42.Demuro A, et al. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J Biol Chem. 2005;280(17):17294–17300. doi: 10.1074/jbc.M500997200. [DOI] [PubMed] [Google Scholar]
- 43.Khemtémourian L, Killian JA, Höppener JWM, Engel MFM. Recent insights in islet amyloid polypeptide-induced membrane disruption and its role in beta-cell death in type 2 diabetes mellitus. Exp Diabetes Res. 2008;2008:421287. doi: 10.1155/2008/421287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nath A, Rhoades E. A flash in the pan: Dissecting dynamic amyloid intermediates using fluorescence. FEBS Lett. 2013;587(8):1096–1105. doi: 10.1016/j.febslet.2013.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nanga RP, Brender JR, Xu J, Veglia G, Ramamoorthy A. Structures of rat and human islet amyloid polypeptide IAPP(1-19) in micelles by NMR spectroscopy. Biochemistry. 2008;47(48):12689–12697. doi: 10.1021/bi8014357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brender JR, et al. Amyloid fiber formation and membrane disruption are separate processes localized in two distinct regions of IAPP, the type-2-diabetes-related peptide. J Am Chem Soc. 2008;130(20):6424–6429. doi: 10.1021/ja710484d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nilsson MR, Driscoll M, Raleigh DP. Low levels of asparagine deamidation can have a dramatic effect on aggregation of amyloidogenic peptides: Implications for the study of amyloid formation. Protein Sci. 2002;11(2):342–349. doi: 10.1110/ps.48702. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.