

Significance
The cell cytoplasm contains a complex array of macromolecules at concentrations exceeding 300 g/L. The natural, most relevant state of a biological macromolecule is thus a “crowded” one. Moving quantitative protein chemistry from dilute solution to the inside of living cells represents a major frontier that will affect not only our fundamental biological knowledge, but also efforts to produce and stabilize protein-based pharmaceuticals. We show that the bacterial cytosol actually destabilizes our test protein, contradicting most theoretical predictions, but in agreement with a novel Escherichia coli model.
Abstract
Protein stability is usually studied in simple buffered solutions, but most proteins function inside cells, where the heterogeneous and crowded environment presents a complex, nonideal system. Proteins are expected to behave differently under cellular crowding owing to two types of contacts: hard-core repulsions and weak, chemical interactions. The effect of hard-core repulsions is purely entropic, resulting in volume exclusion owing to the mere presence of the crowders. The weak interactions can be repulsive or attractive, thus enhancing or diminishing the excluded volume, respectively. We used a reductionist approach to assess the effects of intracellular crowding. Escherichia coli cytoplasm was dialyzed, lyophilized, and resuspended at two concentrations. NMR-detected amide proton exchange was then used to quantify the stability of the globular protein chymotrypsin inhibitor 2 (CI2) in these crowded solutions. The cytosol destabilizes CI2, and the destabilization increases with increasing cytosol concentration. This observation shows that the cytoplasm interacts favorably, but nonspecifically, with CI2, and these interactions overcome the stabilizing hard-core repulsions. The effects of the cytosol are even stronger than those of homogeneous protein crowders, reinforcing the biological significance of weak, nonspecific interactions.
Macromolecules in Escherichia coli reach concentrations of 300–400 g/L and occupy up to 40% of the cellular volume (1), but proteins are normally studied in buffer alone. The effects of crowding arise from two phenomena, hard-core repulsions and nonspecific chemical (soft) interactions (2–9). Hard-core repulsions limit the volume available to biological macromolecules for the simple reason that two molecules cannot be in the same place at the same time. This press for space favors compact states over expanded states. The second phenomenon arises because crowders not only exclude volume, but also participate in chemical interactions. Even though individually weak, the high concentration of macromolecules can lead to a large net effect. Repulsive nonspecific interactions reinforce the hard-core repulsions, whereas attractive nonspecific interactions oppose them. We use the term “nonspecific attractive interactions” to distinguish these from specific chemical interactions, such as ligand binding.
Our aim is to understand how the crowded and heterogeneous, intracellular environment affects the equilibrium thermodynamic stability of globular proteins. Globular proteins are marginally stable in buffer at room temperature (10), with Gibbs free energy differences of 5–15 kcal/mol between the efficiently packed native (N) state and the ensemble of higher-energy denatured (D) states 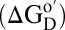 (11). Crowding effects arise from entropic and enthalpic contributions; hard-core repulsions are entropic, whereas the consequent nonspecific chemical interactions are also enthalpic. Hard-core repulsions always increase
(11). Crowding effects arise from entropic and enthalpic contributions; hard-core repulsions are entropic, whereas the consequent nonspecific chemical interactions are also enthalpic. Hard-core repulsions always increase 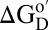 for globular proteins because D occupies more space than N (12–14). However, nonspecific interactions can favor N or D, depending on their nature (2). Nonspecific repulsions increase
for globular proteins because D occupies more space than N (12–14). However, nonspecific interactions can favor N or D, depending on their nature (2). Nonspecific repulsions increase 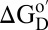 because they make the crowder seem even larger. Nonspecific attractions decrease
because they make the crowder seem even larger. Nonspecific attractions decrease  because more favorable interactions are able to form as the protein unfolds (2–9).
because more favorable interactions are able to form as the protein unfolds (2–9).
Most efforts to understand macromolecular crowding use uncharged synthetic polymers as crowding agents (2, 14, 15). Such polymers tend to emphasize hard-core repulsions, and therefore stabilize globular proteins, but they are not accurate physiological mimics because most synthetic polymers are nonglobular and relatively inert (15, 16). Protein crowders are better mimics because cells contain large amounts of protein. Protein crowders can be destabilizing because they interact with proteins via attractive, weak, nonspecific contacts with the backbone (8, 16, 17). Nevertheless, cells are not crowded with one particular protein. For instance, an E. coli cell possesses 2.4 million protein molecules representing ∼4,000 different proteins (18, 19), resulting in a diverse population of charges, sizes, and amino acid compositions.
The intracellular environment is optimal for understanding the physicochemical effects of the cytoplasm, but measuring stability in living cells is challenging because application of reductionist-based methods involving alteration of the cytosol is inconsistent with viability (20). Studies of in-cell stability by urea denaturation suggest that the cytoplasm does not affect stability (21, 22), whereas results from McGuffee and Elcock in their pioneering molecular dynamics simulation of the E. coli cytoplasm (23) point to destabilization.
A nonperturbing method is clearly advantageous for quantifying the effect of the cellular interior. NMR-detected backbone amide proton exchange (24) is one such method and, unlike others, reports stability at the residue level. As discussed below, under certain specific conditions (25) the technique yields the free energy required to expose amide protons to solvent  . For a particular protein, the largest values of this parameter occur upon global unfolding and are equivalent to the denaturation free energy,
. For a particular protein, the largest values of this parameter occur upon global unfolding and are equivalent to the denaturation free energy, 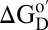 (25).
(25).
Detecting resonances from the backbone of globular proteins by NMR in E. coli is difficult because nonspecific interactions limit rotational motion, broadening 15N-1H cross-peaks from heteronuclear single quantum coherence (HSQC) spectra into the background (26–29). Here, we use lyophilized E. coli cytosol (16, 30–32) to mimic the intracellular environment and quantify its effects on the stability of the test protein chymotrypsin inhibitor 2 (CI2), which is amenable to NMR-detected amide proton exchange (33, 34). The cell extracts not only allow us to more directly assess the results of the molecular dynamics simulation (23), but also allow the application of a reductionist approach not possible using live cells in that the cytosol concentration can be varied.
We extracted the cytoplasm from saturated cultures and removed the low-molecular-weight components to observe only the macromolecular effects. After exchanging the labile protons with deuterons, we lyophilized the sample to obtain powdered cytosol. We used NMR-detected amide proton exchange (25) to obtain residue level stabilities of 15N-enriched CI2 in solutions crowded with 100.0 g dry weight/L and 130.0 g dry weight/L reconstituted cytosol.
Results
Suitability of Reconstituted Cytosol.
McGuffee and Elcock (23) used an inventory of E. coli proteins compiled by Link et al. (35) to design a model cytosol. We made lyophilized cytosol and used mass spectrometry and proteomics to identify the proteins. Our list of 233 proteins (SI Appendix, Table S1) contains 33 of the 45 proteins from the model. The difference probably reflects the fact that McGuffee and Elcock used the list from growth on minimal media and we used cytosol from cells grown in rich media. We also assessed the ratio of the protein mass in the lyophilized cytosol to its dry weight. Triplicate Lowry analyses of the 100.0 and 130.0 g dry weight/L reconstituted samples showed they contained 52 ± 4 g/L and 87 ± 3 g/L protein, respectively. Thus, half to two-thirds of the mass of our lyophilized cytosol comprises proteins, the remainder being mostly nucleic acids, which is reasonable (18). These observations indicate that the results from its use are appropriate for comparisons to the results from McGuffee and Elcock’s study (23).
Weak Interactions.
For stable, globular proteins such as CI2, NMR chemical shifts reflect the highly populated N state. Significant chemical shift changes are expected if a protein interacts strongly with surrounding molecules (36). We studied the weighted chemical-shift difference from 15N–1H HSQC spectra (8, 37–39) of the I29A;I37H variant in 130.0 g dry weight/L sample and in buffer alone. Out of the 45 observable residues, only 5 showed significant shift changes (Gly-10, Glu-26, Leu-32, Met-40, and Asn-56; SI Appendix, Fig. S1). Apart from Leu-32, these residues are in unstructured regions. A plausible explanation is that the cytosol induces small structural changes in flexible regions. The fact that only small chemical shift changes are observed throughout the rest of the protein indicates that CI2–crowder interactions are nonspecific, weak, and transient, in agreement with previous studies (16). This result also suggests that the N state of CI2 is largely unperturbed in reconstituted cytosol.
Measuring  .
.
Amide proton exchange experiments provide equilibrium thermodynamic data about protein stability if four assumptions are valid. First, the test protein must be stable 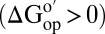 . Second, it must remain intact during the experiment. Third, the intrinsic rate of exchange, kint, must be rate determining. Fourth, kint values, which are calculated in buffer (40), must be applicable in the presence of cosolutes. These assumptions are true in buffer (41), in several synthetic crowders (38, 39), and in protein solutions (8). Here, we assessed these assumptions for reconstituted cytosol.
. Second, it must remain intact during the experiment. Third, the intrinsic rate of exchange, kint, must be rate determining. Fourth, kint values, which are calculated in buffer (40), must be applicable in the presence of cosolutes. These assumptions are true in buffer (41), in several synthetic crowders (38, 39), and in protein solutions (8). Here, we assessed these assumptions for reconstituted cytosol.
The cytosol does not induce unfolding or misfolding, because changes in CI2 backbone chemical shift are small (SI Appendix, Fig. S1). Nevertheless, the cytosol could cause CI2 to aggregate, and cytosolic proteases could degrade it. The signal decay from aggregation or proteolysis would compete with the signal decay from exchange, complicating interpretation. To test for these problems we performed a pseudo amide proton exchange experiment using nonexchanged cytosol in H2O at our highest concentration of reconstituted cytosol (130.0 g dry weight/L). There was no significant change in CI2 cross-peak intensities over 24 h (SI Appendix, Table S2). These observations indicate that the first two assumptions are valid; CI2 remained folded, intact, and unaggregated in the reconstituted cytosol for the duration of the amide proton exchange experiments.
The third assumption is that kint is rate determining. Unfortunately, the methods commonly used to verify this assumption (29)—changing the pH and NOESY-detected amide proton exchange (25, 41, 42)—do not work in cytosol. The pH change method fails because CI2 stability depends on pH (8) and the reconstituted cytosol aggregates upon altering the pH. Nor did we observe cross-peaks in a 30 min, time-resolved NOESY-detected amide proton exchange spectrum. Increasing the time gave more signal, but the diagonal cross-peaks were lost owing to the exchange. Instead, we turned to a thermodynamic cycle (43) to ensure we are measuring 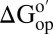 (Fig. 1).
(Fig. 1).
Fig. 1.

Thermodynamic cycle (cyt, cytosol; dil, dilute).
The cycle comprises two CI2 variants, I29A;I37H and I57A;I37H, and two solvent conditions, dilute and reconstituted cytosol at 100.0 g dry weight/L. These variants were chosen because they allow us to observe complete amide proton exchange within 24 h. We calculated 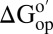 assuming kint is rate-determining. If the assumption is correct, the effect of crowding on the variants (horizontal sides) should be equal, as should the effect of the mutation on the solvent conditions (vertical sides). These conditions are met (Figs. 2 and 3), suggesting that we are measuring
assuming kint is rate-determining. If the assumption is correct, the effect of crowding on the variants (horizontal sides) should be equal, as should the effect of the mutation on the solvent conditions (vertical sides). These conditions are met (Figs. 2 and 3), suggesting that we are measuring 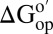 . This result could be fortuitous, but two observations support its veracity. First, we know that kint is rate-determining for the I29A;I37H variant in buffer alone, in 100.0 g/L polyvinylpyrrolidone (PVP), BSA, and lysozyme at pH 6.5, 20 °C (8, 38, 44). Second, the folding rates of the I29A and I57A variants are 11 s−1 and 31 s−1 (45), respectively, much larger than the intrinsic rates (∼1 s−1).
. This result could be fortuitous, but two observations support its veracity. First, we know that kint is rate-determining for the I29A;I37H variant in buffer alone, in 100.0 g/L polyvinylpyrrolidone (PVP), BSA, and lysozyme at pH 6.5, 20 °C (8, 38, 44). Second, the folding rates of the I29A and I57A variants are 11 s−1 and 31 s−1 (45), respectively, much larger than the intrinsic rates (∼1 s−1).
Fig. 2.

Differences in 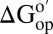 corresponding to the vertical sides of Fig. 1. Black bars,
corresponding to the vertical sides of Fig. 1. Black bars, 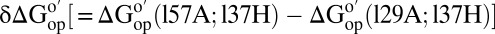 in reconstituted cytosol (100.0 g dry weight/L); red bars,
in reconstituted cytosol (100.0 g dry weight/L); red bars, 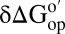 values in dilute solution. Data are shown for residues observed in both proteins whose
values in dilute solution. Data are shown for residues observed in both proteins whose 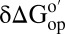 values are statistically different from zero.
values are statistically different from zero.
Fig. 3.

Differences in  corresponding to the horizontal sides of Fig. 1. Green bars,
corresponding to the horizontal sides of Fig. 1. Green bars,  for I57A;I37H variant; blue bars,
for I57A;I37H variant; blue bars, 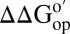 values for I29A;I37H variant. Data are shown for residues observed in both proteins under both conditions whose
values for I29A;I37H variant. Data are shown for residues observed in both proteins under both conditions whose 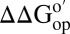 values are statistically different from zero.
values are statistically different from zero.
The final assumption is that the cytosol does not change kint. Even though the disordered loop (residues 35–45) is not observed in our exchange experiments, owing to rapid solvent exchange, it serves as an internal control for assessing the effect of the cytosol. Previously, we used the loop to show that a synthetic polymer and protein crowders do not change kint (8, 38, 44). This assumption was tested for the cytosol by applying the 15NH/D SOLEXSY experiment (46). We found that the intrinsic exchange rates remain unchanged in reconstituted cytosol (30).
Discussion
NMR-detected amide proton exchange (24, 25) allows quantification of protein stability at the level of individual residues. We measured the stability, compared with buffer alone, at two cytosol concentrations at pH 6.5 and 20 °C. The 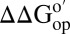 values (cytosol minus buffer) for the I29A;I37H variant are superimposed on the backbone of CI2 in Fig. 4. Increasing the cytosol concentration from 100.0 g dry weight/L to 130.0 g dry weight/L decreases the stability further, especially in α-helix and β-sheet regions. Even though reconstituted cytosol is destabilizing, a bar chart of
values (cytosol minus buffer) for the I29A;I37H variant are superimposed on the backbone of CI2 in Fig. 4. Increasing the cytosol concentration from 100.0 g dry weight/L to 130.0 g dry weight/L decreases the stability further, especially in α-helix and β-sheet regions. Even though reconstituted cytosol is destabilizing, a bar chart of 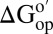 versus residue number (SI Appendix, Fig. S2) shows similar trends in reconstituted cytosol and buffer alone, suggesting that the folding mechanism is unchanged. We were unable to quantify
versus residue number (SI Appendix, Fig. S2) shows similar trends in reconstituted cytosol and buffer alone, suggesting that the folding mechanism is unchanged. We were unable to quantify 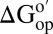 in certain residues for two reasons. First, some are in unstructured regions (e.g., the loop, residues 35–45). These amide protons exchange too fast to assess rates. Second, the cytosol broadens some resonances beyond detection.
in certain residues for two reasons. First, some are in unstructured regions (e.g., the loop, residues 35–45). These amide protons exchange too fast to assess rates. Second, the cytosol broadens some resonances beyond detection.
Fig. 4.

Impact of cytosol on stability. The backbone of CI2 (I29A;I37H) is color-coded by  in 100.0 g dry weight/L (A) and 130.0 g dry weight/L (B) of reconstituted cytosol [blue,
in 100.0 g dry weight/L (A) and 130.0 g dry weight/L (B) of reconstituted cytosol [blue, 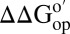 (kcal/mol) > 0.3; cyan,
(kcal/mol) > 0.3; cyan, 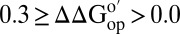 ; orange,
; orange, 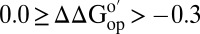 ; red,
; red, 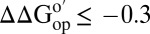 ]. White areas denote prolines and amide protons that either exchanged completely before the second time point or were too broad to observe under one or more conditions. Changes in the average global stability of CI2 under crowded conditions (C). The SDs of the mean of the
]. White areas denote prolines and amide protons that either exchanged completely before the second time point or were too broad to observe under one or more conditions. Changes in the average global stability of CI2 under crowded conditions (C). The SDs of the mean of the  values for protons that exchange upon global unfolding (41) are indicated by the size of the circle. Cytosol-100 and Cytosol-130 indicate 100.0 g dry weight/L and 130.0 g dry weight/L of the reconstituted cytosol, respectively at pH 6.5, 20 °C. Ficoll (100 g/L, pH 5.4, 37 °C) data are from ref. 39. PVP (100 g/L, pH 5.4, 37 °C) data are from ref. 38. BSA (100 g/L, pH 6.5, 20 °C) and lysozyme (100 g/L, pH 6.5, 20 °C) data are from ref. 8. The lysate, BSA, and lysozyme data were acquired at 20 °C. The stabilizing effects Ficoll and PVP are probably slightly exaggerated because these data were acquired at 37 °C, and crowding-induced stability is predicted to increase with increasing temperature (57).
values for protons that exchange upon global unfolding (41) are indicated by the size of the circle. Cytosol-100 and Cytosol-130 indicate 100.0 g dry weight/L and 130.0 g dry weight/L of the reconstituted cytosol, respectively at pH 6.5, 20 °C. Ficoll (100 g/L, pH 5.4, 37 °C) data are from ref. 39. PVP (100 g/L, pH 5.4, 37 °C) data are from ref. 38. BSA (100 g/L, pH 6.5, 20 °C) and lysozyme (100 g/L, pH 6.5, 20 °C) data are from ref. 8. The lysate, BSA, and lysozyme data were acquired at 20 °C. The stabilizing effects Ficoll and PVP are probably slightly exaggerated because these data were acquired at 37 °C, and crowding-induced stability is predicted to increase with increasing temperature (57).
It is tempting to correlate structural features of CI2 with the observed destabilization, but interpreting amide proton exchange data in such detail remains controversial. For instance, it has been argued that caution should be used in attempts to correlate rates with solvent-accessible area (47–50). In addition, CI2 possesses fast-exchanging amide protons in buried regions and slow-exchanging protons in nonhydrogen-bonded regions (34). In particular, its α-helix contains both partially (Ala-16 and Lys-24) and completely (Ile-20 and Leu-21) buried residues (34). These regions are marginally stabilized in 100.0 g dry weight/L reconstituted cytosol (<0.3 kcal/mol) but destabilized at the higher concentration. Maximum destabilization was observed in the β-strand comprising Asp-45 to Asp-52, and most of these residues are buried in that part of the core that exchanges only upon global unfolding (34). Among the residues we observe, only Val-34 is significantly stabilized (>0.3 kcal/mol) at both concentrations. Interestingly, this residue is immediately before the extended loop whose structure, as we described above, may be affected in cytosol. We did not observe a change in  for Val-34 in our previous studies using PVP, BSA, or lysozyme (8) as crowding agents. One explanation could be that components of the reconstituted cytosol interact specifically with this region. We looked for correlations between changes in stability and solvent-accessible surface area and hydrogen-bond lengths but have found none (SI Appendix, Figs. S3 and S4). Taken together, these observations support both the idea that the interactions between components of the reconstituted cytosol and the protein backbone are mostly nonspecific and, as discussed next, we focus on positions that exchange only by global unfolding.
for Val-34 in our previous studies using PVP, BSA, or lysozyme (8) as crowding agents. One explanation could be that components of the reconstituted cytosol interact specifically with this region. We looked for correlations between changes in stability and solvent-accessible surface area and hydrogen-bond lengths but have found none (SI Appendix, Figs. S3 and S4). Taken together, these observations support both the idea that the interactions between components of the reconstituted cytosol and the protein backbone are mostly nonspecific and, as discussed next, we focus on positions that exchange only by global unfolding.
We averaged the 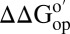 for CI2 residues that exchange on global unfolding (34) and compared (Fig. 4) the effects of the reconstituted cytosol to the effects of inert synthetic polymers and biologically relevant protein crowders (8, 39). Ficoll shows maximum stabilization, followed by PVP. The more biologically relevant crowders BSA, lysozyme, and cytosol destabilize CI2. We also know that Ficoll and PVP interact only weakly with the protein, whereas the biologically relevant crowders have stronger interactions (16, 51). We conclude that transient, nonspecific interactions between the test protein and other proteins cause destabilization.
for CI2 residues that exchange on global unfolding (34) and compared (Fig. 4) the effects of the reconstituted cytosol to the effects of inert synthetic polymers and biologically relevant protein crowders (8, 39). Ficoll shows maximum stabilization, followed by PVP. The more biologically relevant crowders BSA, lysozyme, and cytosol destabilize CI2. We also know that Ficoll and PVP interact only weakly with the protein, whereas the biologically relevant crowders have stronger interactions (16, 51). We conclude that transient, nonspecific interactions between the test protein and other proteins cause destabilization.
Although our reconstituted cytosol is a reasonable model for testing McGuffee and Elcock’s simulation (23) it is not a perfect mimic of the intracellular environment. First, the DNA is sheared during preparation and the mRNA has been degraded. Second, E. coli contains a large number of small molecules (52–54), but our reconstituted cytosol was dialyzed against water, leaving only the ions required for charge neutralization. Many of the absent small molecules, especially osmolytes (55), affect protein stability, and we have shown that salt mitigates the destabilizing effect of crowding by BSA (8). Thus, a unified theory of intracellular crowding cannot be deduced from our reconstituted cytosol. Perhaps the absence of small molecules explains the difference between our observation of destabilization and findings from in-cell studies in which no change in stability was observed (21, 22). We note, however, that small changes in the conditions used in those studies lead to the conclusion that the cytoplasm is slightly destabilizing (56).
Despite these shortcomings, reconstituted cytosol is probably a better mimic than homogenous crowders such as Ficoll, PVP, BSA, and so on, and a similar preparation has been successfully used to study changes in side-chain dynamics of calmodulin (31, 32). Reconstituted cytosol provides an intermediate step between dilute solution and in-cell experiments. Moreover, direct residue-level NMR studies of globular proteins are not possible in E. coli because, with a few exceptions (27), it is difficult to observe high-quality HSQC spectra from proteins in E. coli cells (26–29).
Our residue-level analysis validates the cytoplasmic model (23), and our results are generally consistent with investigations of global protein stability in cells (21, 22, 56). All these studies indicate that the intracellular environment offers enough nonspecific interactions to overcome the stabilizing effects of hard-core repulsion. Our results highlight the need to incorporate nonspecific, weak interactions in the modeling of macromolecular crowding.
Traditionally, investigations of macromolecular crowding effects focused on hard-core repulsions, which predict only stabilization. Our result contradicts these predictions. The reason may be that most studies used synthetic polymers, such as Ficoll and Dextran. These polymers have only weak interactions, whereas proteins and cytosol have stronger nonspecific interactions with CI2 (16, 51). Synthetic, homogenous crowding systems have provided fundamental insight into macromolecular crowding, but, as stated by Elcock (15), they may not resemble the intracellular environment because they cannot mimic the interactions in the cytosol. Only recently has the importance of both nonspecific attractive interactions and steric repulsions been recognized (2–9). Furthermore, a new idea about the temperature dependence of crowding effects suggests that there is a cross-over temperature at which the stabilizing and destabilizing effects cancel (57). Clearly, combining experimental and modeling efforts will increase our understanding of how the intracellular environment affects protein stability.
Nonspecific interactions arise from several sources. For example, we know that coulombic interactions (8), hydrogen bonds, and the hydrophobic effect are key players (27, 28). These interactions are probably of functional importance in cells. For instance, it has been hypothesized that weak protein–protein interactions give rise to cellular organization (58), as observed by centrifugation of whole cells (59) and in protein clusters (60). Such clustering leads to the formation of ‟supercrowded” regions surrounded by zones of dilute cytoplasm, which help direct cellular metabolism (61). In summary, our results provide strong evidence for the importance of nonspecific attractive interactions in determining protein stability in cells and highlight the significance of using reconstituted cytosol to mimic cellular crowding.
Materials and Methods
The CI2 gene was mutated and the variants expressed and purified as described (8, 44). 1H and 15N chemical shift assignments for the 15N- and 13C-enriched I57A;I37H variant were obtained in dilute solution (50 mM sodium phosphate, pH 6.5, 20 °C) by using the HNCACB (62) and CBCA(CO)NH (63) experiments on a 600-MHz Varian Inova spectrometer equipped with a triple resonance probe. The 1H, 13C, and 15N spectral widths were 11,990 Hz, 12,064 Hz, and 2,500 Hz, respectively. The HNCACB spectrum was acquired with 60 and 32 complex increments and the CBCA(CO)NH spectrum was acquired with 48 and 24 complex increments in the 13C and 15N dimensions, respectively. The number of complex points in the 1H dimension was 1,024. Data were processed with NMRpipe (64) and NMRView (65). Assignments for the I57A;I37H variant are given in SI Appendix, Table S6. Assignments for I29A;I37H variant are from ref. 37.
The preparation of reconstituted cytosol has been described (16, 30), but in this instance a 28-L culture was used. The lyophilized and deuterated cytosol (1.20 g) was suspended in enough deuterated 50 mM sodium phosphate buffer to give 12.0 mL of solution. The pHread [ i.e., uncorrected for the isotope effect (66)] was adjusted to 6.5. The solution was centrifuged at 14,000 × g for 40 min at 4 °C. The supernatant, which contained 100.0 g dry weight/L of reconstituted cytosol, was used for six trials: three trials each on the I29A;I37H and I57A;I37H variants. A small portion of the reconstituted cytosol was retained for analysis (discussed below).
Amide proton exchange experiments were performed as described (8, 29). Before starting an experiment, the shims were optimized with 1 mL of 1 mM CI2 in 50 mM sodium phosphate, pH 6.5, containing 10% (vol/vol) D2O (25). The spectra were acquired with 1,024 complex points in the 1H dimension and 48 complex increments in the 15N dimension. Sufficient 15N-enriched CI2 was added to 1 mL of the deuterated, reconstituted cytosol to give a final concentration of 1 mM CI2. The solution was immediately transferred to a 5-mm NMR tube (Norell) and 22 serial HSQC datasets were acquired at 20 °C using the 600-MHz spectrometer. Exchange experiments were performed thrice to obtain the mean and its SD. The intrinsic rates of amide exchange (kint, s−1) were obtained from the online platform SPHERE (40) at pHread 6.5, 20 °C in 50 mM sodium phosphate buffer and 100% D2O, because the cytosol does not change intrinsic rates (30).
The I29A;I37H variant was also studied at a higher cytosol concentration, 130.0 g dry weight/L. These experiments were performed on a 500-MHz Varian Inova spectrometer with a triple resonance HCN cold probe with 1H and 15N spectral widths of 8,401.6 Hz and 2,200 Hz, respectively.
A modified Lowry assay (Thermo Scientific) was performed after 100-fold dilution of the two reconstituted cytosol samples with NMR buffer (50 mM sodium phosphate, pH 6.5). The total protein concentration was obtained from the absorbance at 750 nm by using a standard curve made from 0.25, 0.50, 1.0, and 1.5 mg/mL solutions of BSA. The assay was performed thrice to obtain a mean and its SD. The samples contained 52 ± 4 g/L and 87 ± 3 g/L proteins, respectively.
Mass spectrometric analysis was performed at the UNC Michael Hooker Proteomics Center. The lyophilized cytosol was resuspended in a solution containing 8 M urea and 100 mM NH4HCO3 and reduced for 30 min with 10 mM DTT. The proteins were then acetylated with 40 mM iodoacetamide in the dark for 30 min. The reaction was quenched by adjusting the reducant concentration to 40 mM. The urea concentration was reduced to 0.5 M by dilution with 100 mM NH4HCO3, and the sample was digested with trypsin (1:10 enzyme:protein by weight) overnight at 37 °C. The peptides were purified by using C-18 Spin Columns (Pierce Biotechnology) and lyophilized. The peptides were suspended in 5% (vol/vol) acetic acid and loaded onto a 2-cm × 360-µm o.d. × 100-µm i.d. microcapillary fused silica precolumn packed with Magic 5 µm C18AQ resin (Michrom Biosciences). The loaded precolumn was washed with 95% (vol/vol) Solvent A (0.1% vol/vol aqueous formic acid)/5% Solvent B (0.1% vol/vol formic acid in acetonitrile) for 20 min at a flow rate of 2 µL/min and then connected to a 360-µm o.d. × 75-µm i.d. analytical column packed with 14 cm of 5 µm C18 resin constructed with an integrated electrospray emitter tip. The peptides were eluted at a flow rate of 250 nL/min by increasing solvent B to 40% (vol/vol) with a Nano-Acquity HPLC solvent delivery system (Waters). The system was directly connected through an electrospray ionization source interfaced to a LTQ Orbitrap XL ion trap mass spectrometer (Thermo Fisher Scientific). The mass spectrometer was controlled with Xcalibur software and operated in the data-dependent mode, in which the initial scan recorded the mass-to-charge ratios of ions from 400 to 2,000. The 10 most abundant ions were automatically selected for subsequent collision-activated dissociation. Raw files were searched by using MASCOT software (version 2.3.02; Matrix Science) via Proteome Discoverer (version 1.3.0.339; Thermo) against the UniProt E. coli reference database (67). Search parameters included a peptide mass tolerance of 10 ppm, fragment ion tolerance of 0.8 Da, and variable modifications for methionine oxidation and carbamidomethylation of cysteine. Identification of two or more peptides for a given protein was considered sufficient evidence to state the protein was present. Proteins with more than two identified peptides are listed in SI Appendix, Table S1.
Supplementary Material
Acknowledgments
We thank Greg Young and Marc ter Horst for help with the NMR, David Smalley for help with proteomics, and Elizabeth Pielak and Linda Spremulli for helpful comments. This work is supported by National Science Foundation Grant MCB 1051819.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1312678110/-/DCSupplemental.
References
- 1.Zimmerman SB, Trach SO. Estimation of macromolecule concentrations and excluded volume effects for the cytoplasm of Escherichia coli. J Mol Biol. 1991;222(3):599–620. doi: 10.1016/0022-2836(91)90499-v. [DOI] [PubMed] [Google Scholar]
- 2.Sarkar M, Li C, Pielak GJ. Soft interactions and crowding. Biophys Rev. 2013;5(2):187–194. doi: 10.1007/s12551-013-0104-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Minton AP. Quantitative assessment of the relative contributions of steric repulsion and chemical interactions to macromolecular crowding. Biopolymers. 2013;99(4):239–244. doi: 10.1002/bip.22163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Knowles DB, LaCroix AS, Deines NF, Shkel I, Record MT., Jr Separation of preferential interaction and excluded volume effects on DNA duplex and hairpin stability. Proc Natl Acad Sci USA. 2011;108(31):12699–12704. doi: 10.1073/pnas.1103382108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fodeke AA, Minton AP. Quantitative characterization of temperature-independent and temperature-dependent protein-protein interactions in highly nonideal solutions. J Phys Chem B. 2011;115(38):11261–11268. doi: 10.1021/jp2049266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou HX. Influence of crowded cellular environments on protein folding, binding, and oligomerization: biological consequences and potentials of atomistic modeling. FEBS Lett. 2013;587(8):1053–1061. doi: 10.1016/j.febslet.2013.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Phillip Y, Schreiber G. Formation of protein complexes in crowded environments—from in vitro to in vivo. FEBS Lett. 2013;587(8):1046–1052. doi: 10.1016/j.febslet.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miklos AC, Sarkar M, Wang Y, Pielak GJ. Protein crowding tunes protein stability. J Am Chem Soc. 2011;133(18):7116–7120. doi: 10.1021/ja200067p. [DOI] [PubMed] [Google Scholar]
- 9.Wang Y, Sarkar M, Smith AE, Krois AS, Pielak GJ. Macromolecular crowding and protein stability. J Am Chem Soc. 2012;134(40):16614–16618. doi: 10.1021/ja305300m. [DOI] [PubMed] [Google Scholar]
- 10.Anfinsen CB. Principles that govern the folding of protein chains. Science. 1973;181(4096):223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 11. Creighton TE (2010) The Biophysical Chemistry of Nucleic Acids and Proteins (Helvetian), p 774.
- 12.Wilf J, Minton AP. Evidence for protein self-association induced by excluded volume. Myoglobin in the presence of globular proteins. Biochim Biophys Acta. 1981;670(3):316–322. doi: 10.1016/0005-2795(81)90103-3. [DOI] [PubMed] [Google Scholar]
- 13.Minton AP, Wilf J. Effect of macromolecular crowding upon the structure and function of an enzyme: Glyceraldehyde-3-phosphate dehydrogenase. Biochemistry. 1981;20(17):4821–4826. doi: 10.1021/bi00520a003. [DOI] [PubMed] [Google Scholar]
- 14.Zhou HX, Rivas G, Minton AP. Macromolecular crowding and confinement: Biochemical, biophysical, and potential physiological consequences. Annu Rev Biophys. 2008;37:375–397. doi: 10.1146/annurev.biophys.37.032807.125817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elcock AH. Models of macromolecular crowding effects and the need for quantitative comparisons with experiment. Curr Opin Struct Biol. 2010;20(2):196–206. doi: 10.1016/j.sbi.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y, Li C, Pielak GJ. Effects of proteins on protein diffusion. J Am Chem Soc. 2010;132(27):9392–9397. doi: 10.1021/ja102296k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feig M, Sugita Y. Variable interactions between protein crowders and biomolecular solutes are important in understanding cellular crowding. J Phys Chem B. 2012;116(1):599–605. doi: 10.1021/jp209302e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Neidhardt FC. Chemical Composition of Escherichia coli. Escherichia coli and Salmonella typhimurium. Vol 1. Washington, DC: Am Soc Microbiol; 1987. [Google Scholar]
- 19.Blattner FR, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277(5331):1453–1462. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 20.Gierasch LM, Gershenson A. Post-reductionist protein science, or putting Humpty Dumpty back together again. Nat Chem Biol. 2009;5(11):774–777. doi: 10.1038/nchembio.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghaemmaghami S, Oas TG. Quantitative protein stability measurement in vivo. Nat Struct Biol. 2001;8(10):879–882. doi: 10.1038/nsb1001-879. [DOI] [PubMed] [Google Scholar]
- 22.Ignatova Z, Gierasch LM. Monitoring protein stability and aggregation in vivo by real-time fluorescent labeling. Proc Natl Acad Sci USA. 2004;101(2):523–528. doi: 10.1073/pnas.0304533101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McGuffee SR, Elcock AH. Diffusion, crowding & protein stability in a dynamic molecular model of the bacterial cytoplasm. PLOS Comput Biol. 2010;6(3):e1000694. doi: 10.1371/journal.pcbi.1000694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Englander SW, Kallenbach NR. Hydrogen exchange and structural dynamics of proteins and nucleic acids. Q Rev Biophys. 1983;16(4):521–655. doi: 10.1017/s0033583500005217. [DOI] [PubMed] [Google Scholar]
- 25.Miklos AC, Li C, Pielak GJ. Using NMR-detected backbone amide 1H exchange to assess macromolecular crowding effects on globular-protein stability. Methods Enzymol. 2009;466:1–18. doi: 10.1016/S0076-6879(09)66001-8. [DOI] [PubMed] [Google Scholar]
- 26.Barnes CO, Monteith WB, Pielak GJ. Internal and global protein motion assessed with a fusion construct and in-cell NMR spectroscopy. ChemBioChem. 2011;12(3):390–391. doi: 10.1002/cbic.201000610. [DOI] [PubMed] [Google Scholar]
- 27.Crowley PB, Chow E, Papkovskaia T. Protein interactions in the Escherichia coli cytosol: An impediment to in-cell NMR spectroscopy. ChemBioChem. 2011;12(7):1043–1048. doi: 10.1002/cbic.201100063. [DOI] [PubMed] [Google Scholar]
- 28.Wang Q, Zhuravleva A, Gierasch LM. Exploring weak, transient protein—protein interactions in crowded in vivo environments by in-cell nuclear magnetic resonance spectroscopy. Biochemistry. 2011;50(43):9225–9236. doi: 10.1021/bi201287e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pielak GJ, et al. Protein nuclear magnetic resonance under physiological conditions. Biochemistry. 2009;48(2):226–234. doi: 10.1021/bi8018948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith AE, Sarkar M, Young GB, Pielak GJ. Amide proton exchange of a dynamic loop in cell extracts. Protein Sci. 2013;22(10):1313–1319. doi: 10.1002/pro.2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Latham MP, Kay LE. Probing non-specific interactions of Ca²⁺-calmodulin in E. coli lysate. J Biomol NMR. 2013;55(3):239–247. doi: 10.1007/s10858-013-9705-2. [DOI] [PubMed] [Google Scholar]
- 32.Latham MP, Kay LE. Is buffer a good proxy for a crowded cell-like environment? A comparative NMR study of calmodulin side-chain dynamics in buffer and E. coli lysate. PLoS ONE. 2012;7(10):e48226. doi: 10.1371/journal.pone.0048226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Itzhaki LS, Neira JL, Fersht AR. Hydrogen exchange in chymotrypsin inhibitor 2 probed by denaturants and temperature. J Mol Biol. 1997;270(1):89–98. doi: 10.1006/jmbi.1997.1049. [DOI] [PubMed] [Google Scholar]
- 34.Neira JL, Itzhaki LS, Otzen DE, Davis B, Fersht AR. Hydrogen exchange in chymotrypsin inhibitor 2 probed by mutagenesis. J Mol Biol. 1997;270(1):99–110. doi: 10.1006/jmbi.1997.1088. [DOI] [PubMed] [Google Scholar]
- 35.Link AJ, Robison K, Church GM. Comparing the predicted and observed properties of proteins encoded in the genome of Escherichia coli K-12. Electrophoresis. 1997;18(8):1259–1313. doi: 10.1002/elps.1150180807. [DOI] [PubMed] [Google Scholar]
- 36.Lian L-Y. NMR studies of weak protein-protein interactions. Prog Nucl Magn Reson Spectrosc. 2013;71:59–72. doi: 10.1016/j.pnmrs.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 37. Charlton LM (2008) Protein behavior in crowded environments. PhD dissertation (Univ of North Carolina at Chapel Hill, Chapel Hill, NC)
- 38.Miklos AC, Li C, Sharaf NG, Pielak GJ. Volume exclusion and soft interaction effects on protein stability under crowded conditions. Biochemistry. 2010;49(33):6984–6991. doi: 10.1021/bi100727y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Benton LA, Smith AE, Young GB, Pielak GJ. Unexpected effects of macromolecular crowding on protein stability. Biochemistry. 2012;51(49):9773–9775. doi: 10.1021/bi300909q. [DOI] [PubMed] [Google Scholar]
- 40.Bai Y, Milne JS, Mayne L, Englander SW. Primary structure effects on peptide group hydrogen exchange. Proteins. 1993;17(1):75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nölting B, et al. The folding pathway of a protein at high resolution from microseconds to seconds. Proc Natl Acad Sci USA. 1997;94(3):826–830. doi: 10.1073/pnas.94.3.826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wagner G. A novel application of nuclear Overhauser enhancement (NOE) in proteins: Analysis of correlated events in the exchange of internal labile protons. Biochem Biophys Res Commun. 1980;97(2):614–620. doi: 10.1016/0006-291x(80)90308-3. [DOI] [PubMed] [Google Scholar]
- 43.Fersht A. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding. New York: Freeman; 1999. [Google Scholar]
- 44.Charlton LM, et al. Residue-level interrogation of macromolecular crowding effects on protein stability. J Am Chem Soc. 2008;130(21):6826–6830. doi: 10.1021/ja8005995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Itzhaki LS, Otzen DE, Fersht AR. The structure of the transition state for folding of chymotrypsin inhibitor 2 analysed by protein engineering methods: Evidence for a nucleation-condensation mechanism for protein folding. J Mol Biol. 1995;254(2):260–288. doi: 10.1006/jmbi.1995.0616. [DOI] [PubMed] [Google Scholar]
- 46.Chevelkov V, Xue Y, Rao DK, Forman-Kay JD, Skrynnikov NR. 15N H/D-SOLEXSY experiment for accurate measurement of amide solvent exchange rates: Application to denatured drkN SH3. J Biomol NMR. 2010;46(3):227–244. doi: 10.1007/s10858-010-9398-8. [DOI] [PubMed] [Google Scholar]
- 47.Hernández G, LeMaster DM. NMR analysis of native-state protein conformational flexibility by hydrogen exchange. Methods Mol Biol. 2009;490:285–310. doi: 10.1007/978-1-59745-367-7_12. [DOI] [PubMed] [Google Scholar]
- 48.Skinner JJ, Lim WK, Bédard S, Black BE, Englander SW. Protein hydrogen exchange: Testing current models. Protein Sci. 2012;21(7):987–995. doi: 10.1002/pro.2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wagner G, Wüthrich K. Amide protein exchange and surface conformation of the basic pancreatic trypsin inhibitor in solution. Studies with two-dimensional nuclear magnetic resonance. J Mol Biol. 1982;160(2):343–361. doi: 10.1016/0022-2836(82)90180-2. [DOI] [PubMed] [Google Scholar]
- 50.Anderson JS, Hernández G, Lemaster DM. A billion-fold range in acidity for the solvent-exposed amides of Pyrococcus furiosus rubredoxin. Biochemistry. 2008;47(23):6178–6188. doi: 10.1021/bi800284y. [DOI] [PubMed] [Google Scholar]
- 51.Li C, Pielak GJ. Using NMR to distinguish viscosity effects from nonspecific protein binding under crowded conditions. J Am Chem Soc. 2009;131(4):1368–1369. doi: 10.1021/ja808428d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bennett BD, Yuan J, Kimball EH, Rabinowitz JD. Absolute quantitation of intracellular metabolite concentrations by an isotope ratio-based approach. Nat Protoc. 2008;3(8):1299–1311. doi: 10.1038/nprot.2008.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bennett BD, et al. Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat Chem Biol. 2009;5(8):593–599. doi: 10.1038/nchembio.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cayley S, Lewis BA, Guttman HJ, Record MT., Jr Characterization of the cytoplasm of Escherichia coli K-12 as a function of external osmolarity. Implications for protein-DNA interactions in vivo. J Mol Biol. 1991;222(2):281–300. doi: 10.1016/0022-2836(91)90212-o. [DOI] [PubMed] [Google Scholar]
- 55.Burg MB, Ferraris JD. Intracellular organic osmolytes: Function and regulation. J Biol Chem. 2008;283(12):7309–7313. doi: 10.1074/jbc.R700042200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ignatova Z, et al. From the test tube to the cell: Exploring the folding and aggregation of a beta-clam protein. Biopolymers. 2007;88(2):157–163. doi: 10.1002/bip.20665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou HX. Polymer crowders and protein crowders act similarly on protein folding stability. FEBS Lett. 2013;587(5):394–397. doi: 10.1016/j.febslet.2013.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McConkey EH. Molecular evolution, intracellular organization, and the quinary structure of proteins. Proc Natl Acad Sci USA. 1982;79(10):3236–3240. doi: 10.1073/pnas.79.10.3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Srere PA. Macromolecular interactions: Tracing the roots. Trends Biochem Sci. 2000;25(3):150–153. doi: 10.1016/s0968-0004(00)01550-4. [DOI] [PubMed] [Google Scholar]
- 60.Greenfield D, et al. Self-organization of the Escherichia coli chemotaxis network imaged with super-resolution light microscopy. PLoS Biol. 2009;7(6):e1000137. doi: 10.1371/journal.pbio.1000137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Spitzer J, Poolman B. How crowded is the prokaryotic cytoplasm? FEBS Lett. 2013;587(14):2094–2098. doi: 10.1016/j.febslet.2013.05.051. [DOI] [PubMed] [Google Scholar]
- 62.Wittekind M, Mueller L. HNCACB, a high-sensitivity 3D NMR experiment to correlate amide-proton and nitrogen resonances with the alpha-carbon and beta-carbon resonances in proteins. J Magn Reson B. 1993;101(2):201–205. [Google Scholar]
- 63.Grzesiek S, Bax A. Correlating backbone amide and side chain resonances in larger proteins by multiple relayed triple resonance NMR. J Am Chem Soc. 1992;114:6291–6293. [Google Scholar]
- 64.Delaglio F, et al. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6(3):277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 65.Johnson BA, Blevins RA. NMR View: A computer program for the visualization and analysis of NMR data. J Biomol NMR. 1994;4(5):603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 66.Schowen KB, Schowen RL. Solvent isotope effects of enzyme systems. Methods Enzymol. 1982;87:551–606. [PubMed] [Google Scholar]
- 67. The UniProt Consortium (2012) Reorganizing the protein space at the Universal Protein Resource (UniProt). Nucleic Acids Res 40 (Database issue):D71–D75. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.