

Abstract
Aims
This study investigated the tolerability, safety, pharmacokinetics and pharmacodynamics of ponesimod, a novel oral selective sphingosine-1-phosphate (S1P1) receptor modulator in development for the treatment of auto-immune diseases.
Methods
This was a double-blind, placebo-controlled, ascending, single-dose study. Healthy male subjects received doses of 1–75 mg or placebo control.
Results
Ponesimod was well tolerated. Starting with a dose of 8 mg, transient asymptomatic reductions in heart rate were observed. Ponesimod pharmacokinetics were dose proportional. The median time to maximal concentration ranged from 2.0 to 4.0 h, and ponesimod was eliminated with a mean half-life varying between 21.7 and 33.4 h. Food had a minimal effect on ponesimod pharmacokinetics. Doses of ≥8 mg reduced total lymphocyte count in a dose-dependent manner. Lymphocyte counts returned to normal ranges within 96 h. A pharmacokinetic/pharmacodynamic model was developed that adequately described the observed effects of ponesimod on total lymphocyte counts.
Conclusions
Single doses of ponesimod up to and including 75 mg were well tolerated. The results of this ascending single-dose study indicate an immunomodulator potential for ponesimod and a pharmacokinetic/pharmacodynamic profile consistent with once-a-day dosing.
Keywords: pharmacodynamics, pharmacokinetics, ponesimod, S1P1 receptor modulator, safety and tolerability, total lymphocyte count
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Fingolimod, a nonselective sphingosine-1-phosphate (S1P) immunomodulator, reduces circulating lymphocytes via the S1P1 receptor and decreases heart rate, which may be associated with its effects on the S1P3 receptor.
Ponesimod is selective for S1P1 receptors. It has the potential to reduce lymphocytes without cardiac effects.
WHAT THIS STUDY ADDS
This study provides information on the tolerability, safety, pharmacokinetics and pharmacodynamics of ponesimod, a selective S1P1 modulator, a potential new treatment for auto-immune diseases.
Ponesimod induces a clear dose-dependent effect on circulating lymphocytes in accordance with animal data.
Ponesimod has a shorter half-life than other S1P modulators, allowing a faster reversibility of its effects.
Although preclinical data indicated no cardiac effects with ponesimod, human data showed a clear effect on heart rate. This suggests that cardiac effects of S1P modulators in humans are not only mediated by the S1P3 receptor, but also by the S1P1 receptor.
Introduction
The adaptive immune system relies on constant circulation of lymphocytes between lymphoid organs and other tissues of the body. After maturation, lymphocytes leave the bone marrow or thymus, enter the circulation, and travel via the blood and the lymphatic system, surveying for cognate antigen [1]. In the secondary lymphoid organs, which include lymph nodes, Peyer's patches and the spleen, naïve lymphocytes encounter antigen-presenting cells and may be activated. Once activated, T cells must egress from lymphoid organs to reach target tissues, whereas antibody-producing B cells are produced in bone marrow [1, 2]. Circulation of lymphocytes between blood, lymphatic system and nonlymphoid tissues is tightly regulated, and the lysophospholipid sphingosine-1-phosphate (S1P) has been shown to play a central role in lymphocyte trafficking [3–5].
Sphingosine-1-phosphate is synthesized and secreted by many cell types and elicits a variety of physiological responses [3, 6] mediated via five G-protein-coupled S1P receptors, S1P1–S1P5 [7–9]. This receptor nomenclature conforms to a published guide [10]. Sphingosine-1-phosphate receptor agonists block the egress of lymphocytes from lymphoid organs and reduce the availability of circulating effector T cells that can invade target organs. This effect of S1P is mediated via the S1P1 receptor [11]. Hence, S1P1 receptor agonism may be a promising therapeutic principle for a variety of auto-immune disorders, such as multiple sclerosis, rheumatoid arthritis, psoriasis, type 1 diabetes and Crohn's disease, which involve an aberrant attack of T cells against certain tissues [12]. Efficacy of the nonselective S1P receptor agonist fingolimod has been reported in models of auto-immune diseases [13, 14] and in clinical trials for multiple sclerosis [15, 16]. The most common adverse events of fingolimod in multiple sclerosis patients were nasopharyngitis, dyspnoea, headache, diarrhoea and nausea [16], whereas in healthy subjects and renal-transplant patients bradycardia and lymphopenia were commonly reported [16–18]. Heart rate reduction [19, 20], vaso- and bronchoconstriction and pulmonary epithelial leakage [21] have been related to activation of the S1P3 pathway. Therefore, while retaining efficacy, i.e. inhibition of lymphocyte egress without affecting their function (e.g. cytokine production), selective S1P1 receptor agonists may be safer than nonselective S1P receptor agonists.
Ponesimod (ACT-128800, (Z,Z)-5-[3-chloro-4-((2R)-2,3-dihydroxy-propoxy)-benzylidene]-2-propylimino-3-o-tolylthiazolidin-4-one), is a selective, reversible, orally active S1P receptor modulator specifically targeting the S1P1 receptor [22]. Preclinical experiments have shown that ponesimod reduced blood lymphocyte counts and was efficacious in experimental models of lymphocyte-mediated tissue inflammation [23]. In this study, we investigated the safety, tolerability, pharmacokinetics, food effect and effects on blood lymphocyte counts of ascending single oral doses of ponesimod in healthy male subjects.
Methods
Subjects
Forty-eight healthy male subjects were enrolled. The subjects were in good health, as assessed by medical history, physical examination, 12-lead electrocardiogram (ECG) and clinical laboratory tests (including haematology, clinical chemistry, urinalysis, virus serology and drug screening). Supine blood pressure and heart rate had to be within the normal range, defined as 100–140 mmHg for systolic blood pressure, 50–90 mmHg for diastolic blood pressure and 45–90 beats min−1 for heart rate. Low lymphocyte counts (<1100 lymphocytes μl−1) and abnormal C-reactive protein levels (>10 mg l−1) were among the exclusion criteria. Written informed consent was obtained from all subjects before any study assessment was conducted. The Ethics Committee of the ‘Ärztekammer’ Schleswig Holstein, Bad Segeberg, Germany and the German Health Authorities approved the protocol. The study was in accordance with the principles of the Declaration of Helsinki and conducted according to good clinical practice.
Study design
This was a double-blind, placebo-controlled, ascending, single-dose study. Each dose level was investigated in a separate group of eight subjects (six on active drug and two on placebo). Ponesimod doses were 1, 3, 8, 20, 50 and 75 mg, and effects were compared with placebo. The starting dose of 1 mg was based on the pharmacokinetic and pharmacodynamic properties in rats and dogs and allometric scaling. Based on this, a single oral dose of 1 mg of ponesimod should not deplete or should minimally deplete the number of circulating lymphocytes in man, whereas a single dose of 10–15 mg of ponesimod was predicted to reduce the lymphocyte count in man by >50% for about 12 h. The study treatments were administered to subjects in the fasted state except for subjects receiving the 20 mg dose, who were administered ponesimod twice. After they received ponesimod in the fasted state and following a washout of 1 week, these subjects again received 20 mg ponesimod after intake of a high-calorie breakfast. Dosing was done to subjects in the sitting position and, except for the recording of vital signs and 12-lead ECG, subjects remained in the sitting position for at least 4 h after dosing.
Safety and tolerability were evaluated by monitoring adverse events, clinical laboratory variables, vital signs (blood pressure, heart rate and body temperature), 12-lead ECG and physical examination. Blood pressure and heart rate were recorded from subjects in the supine position after having rested for a 5 min period and subsequently after standing for exactly 1 min. A decrease in total lymphocyte count was an expected effect of ponesimod and, therefore, not considered adverse. Venous blood samples were taken at regular time points up to 48 h after treatment for measuring ponesimod levels and total lymphocyte counts. Subjects were in the clinic until 48 h after dosing. Plasma was separated and stored at −20°C pending analysis. After a per-protocol interim analysis of the plasma concentration–time profiles, the observation phase in the clinic was extended from 48 h to 7 days for the 75 mg dose level. In addition, after review of the 50 mg data, three-lead telemetry ECG monitoring for the first 8 h postdose was implemented, and a specific inclusion criterion for heart rate was added (65–90 beats min–1), as well as lung function tests (forced expiratory volume in 1 s and forced vital capacity) and neurological examinations, including Romberg's test for sensory ataxia [24]. The decision to proceed with the next higher dose level was taken after review of the safety, tolerability and pharmacodynamic data of the previous dose group.
Bioanalytical analysis
A validated liquid chromatography coupled to tandem mass spectrometry method was used to measure ponesimod concentrations in plasma. Ponesimod and the internal standard, a deuterated analogue of ponesimod, were extracted by protein precipitation using a mixture of acetonitrile and ethanol (1:1, vol/vol). After protein precipitation, plasma samples were filtered through a protein precipitation plate, and the filtrate was diluted with an equal volume of water containing 0.1% formic acid prior to injection onto the HPLC column. The chromatographic system consisted of a pump and an analytical column (Atlantis dC18; Waters, Milford, MA, USA; 3 μm particle size, 2.1 mm × 20 mm). Mobile phases to elute the column consisted of water containing 0.1% formic acid, acetonitrile containing 0.1% formic acid, and acetonitrile/water (4:1, vol/vol) containing 0.5% formic acid. Mass spectrometric detection was performed with a triple quadrupole mass spectrometer (TSQ Quantum; ThermoFinnigan, San Jose, CA, USA) operating in electron spray-ionization positive-ion mode. Samples were quantified using peak area ratios.
The method was linear in the concentration range 1.0−2000 ng ml−1. The limit of quantification was 1.0 ng ml−1. The performance of the method was monitored using quality control samples. Precision (CV%) was ≤8.1, whereas inaccuracy ranged from −12.0 to −3.5%.
Pharmacokinetic analysis
Pharmacokinetic parameters were determined by model-independent methods (WinNonlin Professional Version 4.0.1; Pharsight, Mountain View, CA, USA) whereby the area under the concentration–time curve (AUC) was calculated using the linear trapezoidal method [25]. At doses ≤50 mg, the AUC from time zero to the last time point with a measurable ponesimod concentration represented less than 80% of AUC0–∞.
Pharmacodynamic assessments and analysis
Total lymphocyte counts were measured by haemocytometry as part of the standard haematological parameters. The pharmacodynamic variable, time to nadir, was read directly from the effect–time profiles. The phenotype of peripheral blood lymphocytes and other leukocyte lineages was determined using fluorochrome-conjugated monoclonal antibodies and flow cytometric techniques [26]. By flow cytometry, changes in the amount of specific phenotypically defined lymphocyte subsets could indicate a response to ponesimod treatment.
Pharmacokinetic/pharmacodynamic modelling
Pharmacokinetic and pharmacokinetic/pharmacodynamic modelling was performed using the nonlinear mixed-effects modelling technique using NONMEM® V [27]. No covariate analysis was conducted.
Adequate description of the ponesimod plasma concentration–time profiles was obtained with a two-compartment model with a first-order absorption process. Total lymphocyte counts were decreased with ponesimod doses ≥8 mg and, therefore, pharmacodynamic modelling was done with these doses and with placebo. The change of total lymphocyte counts showed a circadian pattern. Based on the known effects of corticosteroids on total lymphocyte count and that cortisol plasma concentrations follow a circadian rhythm, a pharmacokinetic/pharmacodynamic model incorporating the cortisol circadian rhythm was developed. In this model, the cortisol circadian rhythm was the driving force for the total lymphocyte trafficking described by an indirect response model. A previously published pharmacokinetic/pharmacodynamic model with a linear release rate was used to describe the endogenous cortisol circadian rhythm, and an indirect response model was used to characterize the ponesimod-induced transient reduction in total lymphocyte count [28, 29]. Briefly, the change of the total cortisol level in plasma at baseline is determined by endogenous cortisol release and elimination. Cortisol release rate is described by two linear functions to account for the circadian rhythm. The transient depletion of the percentage of lymphocytes in the blood (L) induced by the concentration of ponesimod (C) is described by:
where kin and kout are the rate constants for lymphocyte influx into and efflux from blood, Imax is the maximal inhibition effect and IC50 is the ponesimod plasma concentration to give half of the maximal effect [28, 29].
The estimated parameters for kin, kout, Imax and IC50 are listed in Table 3. Fitting results were plotted with observed data as total lymphocyte percentage (circles) in Figure 4, and the continuous lines are model-predicted profiles based on the mean percentage change from baseline for total lymphocytes.
Table 3.
Population pharmacokinetic and pharmacokinetic/pharmacodynamic variables of ponesimod and its effects on total lymphocyte count
| Variables | Estimate | Interindividual variability (%) | CV (%) |
|---|---|---|---|
| Pharmacokinetic variables | |||
| ka (h−1) | 0.67 | 55.0 | 12.1 |
| Vcentral/F (l) | 304 | 14.3 | 3.4 |
| Vperipheral/F (l) | 979 | 119 | 5.6 |
| CL/F (l h−1) | 6.7 | 14.3 | 10.6 |
| CLD/F (l h−1) | 3.6 | 0.002 | 21.8 |
| Pharmacokinetic/pharmacodynamic variables | |||
| kin (%) | 45.8 | – | 10.5 |
| kout (h−1) | 0.36 | – | 9.86 |
| IC50 (ng ml−1) | 65.1 | – | 25.4 |
| Imax (%) | 94 | – | 9.85 |
Abbreviations are as follows: CL/F, apparent clearance; CLD/F, distribution clearance; CV, coefficient of variation; IC50, half-maximal inhibitory concentration; Imax, maximal inhibitory effect; ka, first-order absorption rate constant; kin, zero-order lymphocyte influx rate constant; kout, first-order lymphocyte efflux rate constant; Vcentral/F, apparent volume of central compartment; and Vperipheral/F, apparent volume of peripheral compartment.
Figure 4.
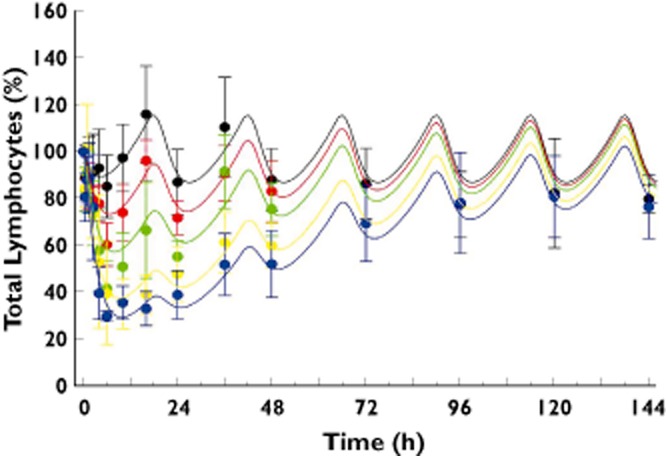
Pharmacokinetic/pharmacodynamic model including the circadian rhythm of cortisol: observed time course (symbols) and predicted time course (lines) of the percentage change from baseline in total lymphocyte count after administration of 8, 20, 50 or 75 mg of ponesimod. The observed values represent the mean (and SD) of data from n = 12 subjects on placebo and n = 6 subjects on the ponesimod doses. The continuous lines are model-predicted population profiles based on mean total lymphocyte percentage data.  , placebo;
, placebo;  , 8 mg;
, 8 mg;  , 20 mg;
, 20 mg;  , 50 mg;
, 50 mg;  , 75 mg
, 75 mg
Statistical analysis
Safety and tolerability data were analysed descriptively by treatment group, and data from placebo subjects were pooled. A power model [30] was used to explore the dose proportionality of ponesimod pharmacokinetics.
Results
Forty-eight healthy male subjects with a mean age of 33.3 years (range, 21–47 years) and a mean bodyweight of 81.7 kg (range, 64–98 kg) participated in this study. The different dose groups were well balanced for all demographic variables, and no subject withdrew prematurely from the study.
Safety and tolerability
No serious and severe adverse events occurred, and all adverse events resolved without sequelae. The adverse event profile was similar to that of placebo for ponesimod doses ≤8 mg. The most frequently reported adverse events were vertigo, headache, fatigue, bradycardia and dyspnoea (Table 1). The severity and/or incidence of these adverse events increased with increasing ponesimod dose. At the highest dose level of 75 mg, the incidence was 83.3% for fatigue, bradycardia and dyspnoea, 66.7% for vertigo and 50.0% for headache. Except for one case of headache and one of fatigue, these adverse events were not reported by placebo-treated subjects.
Table 1.
Overview of reported adverse events by treatment
| Treatment | Placebo* | Ponesimod (mg) | |||||
|---|---|---|---|---|---|---|---|
| 1 | 3 | 8 | 20* | 50 | 75 | ||
| Number of subjects | 12 | 6 | 6 | 6 | 6 | 6 | 6 |
| Number of subjects reporting at least one adverse event | 2 | 1 | 1 | 1 | 4 | 5 | 6 |
| Adverse event | |||||||
| Headache | 1 | 1 | – | – | 1 | 4 | 3 |
| Vertigo | – | – | – | 1 | 4 | 2 | 4 |
| Fatigue | 1 | – | – | – | – | 5 | 5 |
| Bradycardia | – | – | – | – | – | 2 | 5 |
| Dyspnoea | – | – | – | – | – | 2 | 5 |
| Nausea | 1 | – | – | – | 1 | 1 | – |
| Asthenia | – | – | – | – | – | – | 2 |
| Alanine aminotransferase increased | – | – | – | – | – | – | 1 |
| C-reactive protein increased | – | – | 1 | – | – | – | – |
| Dizziness | – | – | – | – | – | – | – |
| Euphoric mood | – | – | – | – | – | – | 1 |
| Forced expiratory volume decreased | – | – | – | – | – | – | 1 |
| Herpes simplex | – | – | – | – | – | – | 1 |
| Nasopharyngitis | – | – | 1 | – | – | – | – |
| Pyrexia | – | – | – | – | – | – | 1 |
| Vomiting | 1 | – | – | – | – | – | – |
Adverse events reported by subjects in the fed state are not shown.
After both placebo and ponesimod administration, a transient decrease in heart rate was observed with a maximal effect generally 2.5 h postdose and a return to baseline values 6–10 h after study drug intake (Figure 1). At ponesimod doses ≥8 mg, decreases in heart rate were greater and dose dependent when compared with placebo. These heart rate reductions were more pronounced in the standing than in the supine position. A temporal correlation between the decrease in heart rate and the reporting of the adverse events of vertigo, fatigue and dyspnoea was observed. There was no apparent effect of ponesimod on supine and standing systolic blood pressure and supine diastolic blood pressure. A trend for a decrease in standing diastolic blood pressure was observed at doses ≥20 mg (maximal mean decrease of 17 mmHg at 6 h postdose with 50 mg and 12 mmHg at 2.5 h postdose with 75 mg compared with 8 mmHg at 8 h postplacebo), which followed a similar time course to that observed for heart rate (data not shown).
Figure 1.
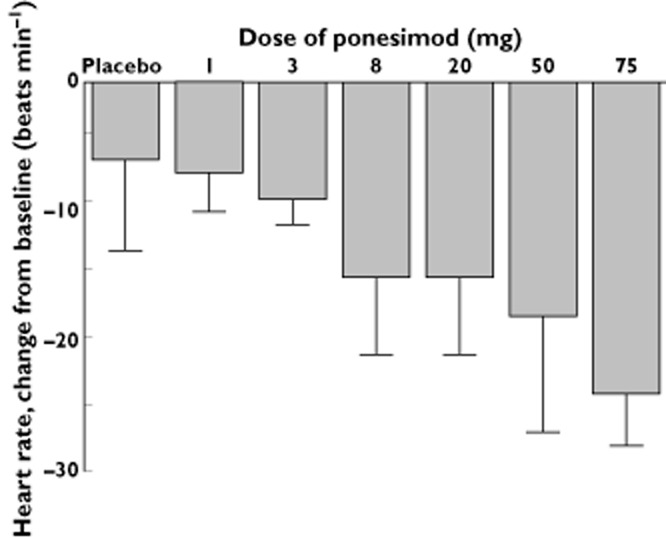
Arithmetic mean maximal change from baseline (and SD) for heart rate as recorded by 12-lead electrocardiogram in the supine position after single-dose administration of placebo (n = 12) or ponesimod at doses of 1, 3, 8, 20, 50 or 75 mg (n = 6 per dose group)
There were no apparent drug-related effects on 12-lead ECG, body temperature, clinical chemistry variables, coagulation variables or physical examination. Exceptions were heart rate reductions and a transient increase in C-reactive protein in the 50 and 75 mg dose groups 24 h after study drug intake. Treatment with ponesimod decreased total lymphocyte count (see ‘Pharmacodynamics’ section below), transiently increased monocytes and neutrophils at doses ≥20 mg, but had no effect on haemoglobin and platelets.
At the 75 mg dose level, ponesimod had no relevant effects on pulmonary function tests that could be related to the observed dyspnoea. Neurological assessments were normal except for three observations of nystagmus reported in subjects on active treatment.
Pharmacokinetics
Ponesimod concentration–time courses across the studied doses are plotted in Figure 2 (means ± SD). Pharmacokinetic parameters from the noncompartmental analysis are summarized in Table 2. In fasting conditions, ponesimod was rapidly absorbed (Figure 2), with the median time to maximal concentration (tmax) ranging from 2.0 to 4.0 h at the various ponesimod doses. The elimination phase was characterized by an apparent terminal elimination half-life (t1/2) of about 30 h, which appeared independent of dose (Table 2). Exposure to ponesimod increased proportionally with dose. Values (95% confidence interval) for the dose-proportionality coefficient, β, were 0.98 (0.93–1.02) and 1.02 (0.96–1.07) for the peak plasma concentration (Cmax) and the AUC0–∞, respectively. The intersubject variability (CV%) did not exceed 36%. In the presence of food, absorption of ponesimod was delayed, as indicated by a value for tmax of 5.0 h when compared with 2.5 h in the fasted state. Furthermore, plasma concentrations of ponesimod were slightly higher in the fed state, resulting in greater values for Cmax and AUC0–∞ (Table 2). The ratio (fed/fasted) of geometric mean values (90% confidence interval) were 1.1 (1.0–1.3) and 1.2 (1.1–1.3) for Cmax and AUC0–∞, respectively.
Figure 2.
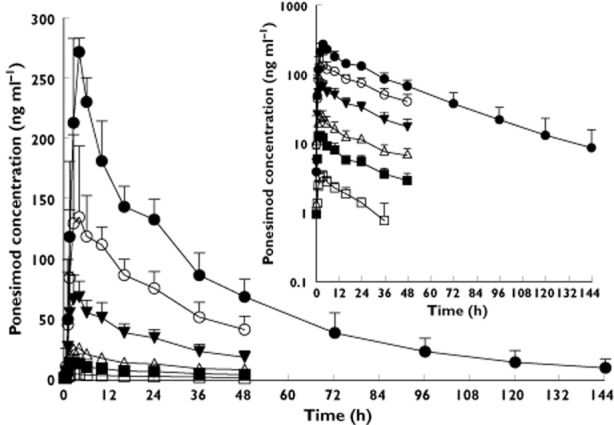
Arithmetic mean (and SD) plasma concentration–time profiles of ponesimod in healthy subjects (n = 6 per dose group) after administration of single doses of 1, 3, 8, 20, 50 or 75 mg of ponesimod (linear scale). The inset shows the profiles on a semilogarithmic scale. For clarity, SD is displayed only for ponesimod doses ≥20 mg for the profiles on the linear scale.  , 1 mg;
, 1 mg;  , 3 mg;
, 3 mg;  , 8 mg;
, 8 mg;  , 20 mg;
, 20 mg;  , 50 mg;
, 50 mg;  , 75 mg
, 75 mg
Table 2.
Pharmacokinetic parameters of ponesimod in healthy subjects after administration of single doses of 1, 3, 8, 20 (fasted and fed), 50 and 75 mg ponesimod (n = 6 per dose group)
| Dose (mg) | Cmax (ng ml−1) | tmax (h) | AUC0–∞ (ng h ml−1) | t1/2 (h) |
|---|---|---|---|---|
| 1 | 3.4 (2.8–4.2) | 3.3 (1.5–4.0) | 96.0 (75.9–121) | 21.7 (18.1–26.0) |
| 3 | 13.7 (11.9–15.7) | 2.0 (1.5–2.5) | 405 (323–509) | 30.1 (21.6–41.9) |
| 8 | 27.2 (23.7–31.2) | 3.3 (1.5–6.0) | 913 (751–1110) | 33.4 (28.1–39.7) |
| 20 (fasted) | 71.0 (56.0–90.1) | 2.5 (1.5–4.0) | 2344 (1796–3058) | 27.7 (19.2–39.8) |
| 20 (fed) | 81.2 (65.6–100) | 5.0 (4.0–6.0) | 2837 (2264–3356) | 29.7 (21.5–41.1) |
| 50 | 163 (130–205) | 4.0 (2.5–10.0) | 5266 (3942–7035) | 28.8 (20.3–40.8) |
| 75 | 274 (256–292) | 4.0 (2.5–6.0) | 9153 (7456–11 238) | 31.4 (24.4–40.5) |
Data are expressed as geometric means (and 95% confidence intervals) or, for tmax, the median (and range). Abbreviations are as follows: AUC0–∞, area under the plasma concentration–time curve; Cmax, peak plasma concentration; t1/2, terminal elimination half-life; and tmax, time to maximal concentration.
Pharmacodynamics
Figure 3 shows the effect–time profiles for the change from baseline of ponesimod and placebo on total lymphocyte count. Mean baseline total lymphocyte count for the different treatment groups varied between 2100 and 2630 cells μl−1. The time to nadir for doses ≥8 mg varied from 6.0 to 12.0 h. The total lymphocyte count for placebo-treated subjects and in subjects treated with ponesimod doses ≤20 mg showed a circadian rhythm. Higher ponesimod doses appeared to suppress this circadian rhythm (Figure 3). Treatment with ponesimod resulted in a dose-dependent reduction in total lymphocyte count. This reduction of total lymphocyte count was most pronounced during the first 6 h after administration, with peak effects typically occurring 6 h after administration (Figure 3). A maximal mean percentage (±SD) reduction from baseline of 70.3 ± 2.3% was observed in the 75 mg dose group. At this dose level, 96 h after study drug treatment, total lymphocyte count was similar to that observed in placebo subjects (data not shown).
Figure 3.
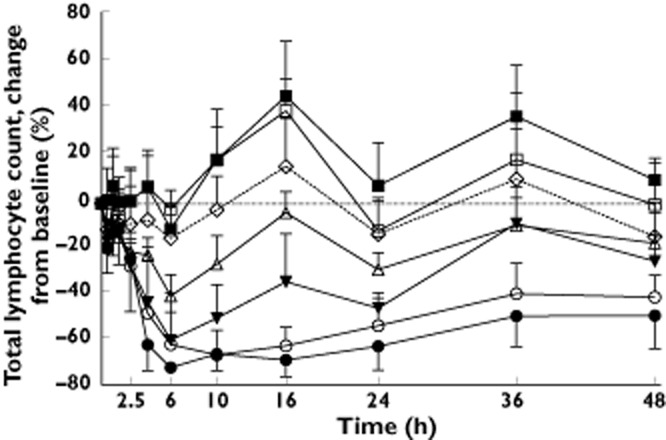
Effect–time profiles of the mean (and SD) change from baseline (%) for total lymphocyte count after single-dose administration of placebo (n = 12) or ponesimod at doses of 1, 3, 8, 20, 50 or 75 mg (n = 6 per dose group).  , 1 mg;
, 1 mg;  , 3 mg;
, 3 mg;  , 8 mg;
, 8 mg;  , 20 mg;
, 20 mg;  , 50 mg;
, 50 mg;  , 75 mg;
, 75 mg;  , placebo
, placebo
Ponesimod elicited a clear reduction in almost all circulating lymphocyte subsets at doses of 20 mg or higher. The extent of reduction of the lymphocyte subsets was dose related. A dose of 8 mg elicited a transient depletion of lymphocyte subsets 6 h after study drug administration. The time course of the lymphocyte subset reduction was comparable for most of the different subsets and was comparable to that of total lymphocytes, with some nuances, as follows. T cells (including T-helper, T-cytotoxic and regulatory T cells) were strongly affected, with the active T cells being more affected than the memory T cells. B cells were also affected, but slightly less than T cells. Some cell types, such as the NK cells (CD3–16+) and the CD8+CD25+ regulatory T cells were not affected by ponesimod.
Pharmacokinetic/pharmacodynamic modelling
A pharmacokinetic and pharmacodynamic model was developed to describe the ponesimod concentration–time course and its impact on the total lymphocyte trafficking adequately. The final variables and estimates are shown in Table 3. Large volumes of distribution were estimated for the central and peripheral compartments, but a small apparent clearance. The observed and modelled effects of ponesimod on total lymphocyte count are shown in Figure 4. The observed pharmacodynamic effect of ponesimod could be well described by the pharmacokinetic/pharmacodynamic model including the circadian rhythm of cortisol. A maximal inhibition of total lymphocyte count of 94% is predicted by this model (Table 3), which was not attained in this study.
Discussion
This study provides the first safety, tolerability, pharmacokinetic and pharmacodynamic data for ponesimod, a selective oral S1P1 receptor modulator, in healthy males. Single doses of up to 75 mg were well tolerated. The adverse events reported by subjects receiving ponesimod were similar to those reported for fingolimod in previous studies [15–17]. Based on the fact that ponesimod showed an ∼20-fold higher affinity for S1P1 compared with S1P3 receptors [23] (i.e. 600-fold more selective than the natural agonist S1P for the S1P1 vs. the S1P3 receptor) and that published data in rodents [19, 20] suggested a key role for the S1P3 receptor in the heart rate reduction induced by S1P, the effects on heart rate observed in this study were unexpected. These findings could be explained by species differences, but other explanations cannot be ruled out, such as, for example, an in vivo selectivity of ponesimod for S1P1 receptors in man which is smaller than expected based on in vitro data. No pharmacokinetic/pharmacodynamic modelling was performed for the effects of ponesimod on heart rate but, given the dose-dependent nature of this effect and the fact that the times to maximal changes in heart rate and maximal plasma concentrations are similar, a relationship between these two might very well exist.
The mechanism via which fingolimod, and most probably ponesimod, decreases heart rate is by agonism of S1P1 receptors, resulting in a slowing of the sinoatrial node due to activation of inwardly rectifying G-protein-activated potassium channels 1 and 4 in atrial myocytes [31]. In the present study, the decreases in heart rate were not associated with relevant changes in blood pressure. In contrast, high but not low, single-dose fingolimod reduced blood pressure in healthy subjects [32], whereas this compound tended to increase blood pressure in patients during prolonged treatment [16].
The adverse events of vertigo, fatigue and dyspnoea were temporally associated with the reductions in heart rate, suggesting that there could be a causal relationship. However, direct effects of ponesimod on the central nervous system causing fatigue and vertigo cannot be excluded. The cases of dyspnoea following administration of ponesimod at doses of ≥50 mg are suggestive of S1P3 activation [22], although, similar to the effect on heart rate, species differences cannot be excluded. Effects on lung function (i.e. dyspnoea and reduction in forced expiratory volume in 1 s) have been previously reported with fingolimod in patients with multiple sclerosis [15]. In the present study, no clear effects on lung function could be discerned after intake of a dose of 75 mg.
The pharmacokinetics of ponesimod were dose proportional, showed moderate intersubject variability and are consistent with a once-a-day dosing regimen. The observed t1/2 of ponesimod (∼30 h) was longer than expected from allometric scaling of pharmacokinetic results obtained in animals (2–3 h in rats and 9–11 h in dogs; data on file; Actelion Pharmaceuticals Ltd, Allschwil, Switzerland), which necessitated a protocol amendment to extend the blood sampling scheme during the study. Allometric scaling has been used successfully in drug development to predict human pharmacokinetics of new compounds based on animal data [33, 34]. However, more recent methods, such as physiologically based pharmacokinetic modelling, have provided superior results in most cases but require far more experimental input [35]. The effect of food on the pharmacokinetics of ponesimod was minimal. Thus, no special instructions regarding the timing of ponesimod intake in relation to the consumption of food appear necessary.
Agonists of S1P1 receptors prevent the egress of lymphocytes from lymph nodes and other secondary lymphoid organs. This results in a decrease in circulating lymphocytes, as observed preclinically in rats dosed with ponesimod [23] or clinically with other S1P agonists, such as fingolimod [15–18]. Consistent with preclinical findings [22, 23], single-dose administration of ponesimod resulted in a dose-dependent decrease in circulating total lymphocyte count in this study. The time to nadir was longer than the tmax (6.0 vs. 2.0–4.0 h) indicating an indirect response to ponesimod. Consistent with the observed t1/2 (∼30 h), total lymphocyte count was still decreased 24 h after administration, and at the highest dose the count was comparable to placebo at 96 h postdose. One month after stopping treatment with fingolimod, total lymphocyte counts had recovered to only within 75 and 50% of baseline in the low- and high-dose groups, respectively. This slower recovery rate following fingolimod when compared with ponesimod is in line with the longer t1/2 of ∼1 week of fingolimod [36]. A circadian rhythm in total lymphocyte count was observed in placebo-treated subjects. Such circadian variations in lymphocyte subpopulations have been described previously [37]. Doses of 8 and 20 mg ponesimod reduced total lymphocyte count while preserving the circadian rhythm, whereas higher doses suppressed this circadian rhythm. The importance of circadian variations in lymphocyte subpopulations for normal functioning of the immune system is unknown. At the recommended dose of 0.5 mg once a day for the treatment of multiple sclerosis, fingolimod decreased total lymphocyte count by 73% [16], i.e. an effect equivalent to that seen at the highest dose tested in this study.
The effects of ponesimod on total lymphocyte count were well described by the pharmacokinetic/pharmacodynamic model including the cortisol circadian rhythm. The effects of ponesimod on cortisol levels were not investigated in this study, and any possible role of this hormone in the mechanism of action of ponesimod will need to be explored. Furthermore, the model does not account for the suppression of the circadian rhythm of total lymphocyte levels in blood observed at ponesimod doses ≥50 mg. Refinement of the model will be necessary once more information becomes available.
In conclusion, single doses of ponesimod up to and including 75 mg were well tolerated. The tolerability and safety profile of ponesimod and fingolimod appears to be similar. The results of this ascending single-dose study indicate an immunomodulator potential for ponesimod and a pharmacokinetic/pharmacodynamic profile consistent with once-a-day dosing. However, future studies in patients will need to be conducted to investigate whether specific S1P1 receptor agonism by ponesimod will provide a superior approach to nonspecific agonism for treating auto-immune diseases such as multiple sclerosis.
Acknowledgments
The authors thank Paul van Giersbergen (Van Giersbergen Consulting) for editorial assistance, which was funded by Actelion Pharmaceuticals Ltd, Marzia Cavallaro, Actelion Pharmaceuticals Ltd, for the statistical work, and Christoph Siethoff from Swiss Bioanalytics, Reinach, Switzerland for the bioanalytical work.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: P.B., H.D., H.M., A.H., and J.D. had support from Actelion Pharmaceuticals Ltd for the submitted work; P.B. and J.D. are full-time employees of Actelion Pharmaceuticals Ltd, A.H. and H.M. are full-time employees of Clinical Research Services Kiel GmbH, H.D. is a full-time employee of the University of Florida, J.X. was a student and received no financial compensation; no financial relationships with any organisations that might have an interest in the submitted work in the previous 3 years; no other relationships or activities that could appear to have influenced the submitted work.
Author contributions
P.B. and J.D. contributed to the study design and protocol. A.H. and H.M. conducted the study. P.B. directed the study at Actelion. H.D. and J.X. performed the pharmacokinetic/pharmacodynamic modelling. All authors reviewed, interpreted the data and agreed on the content. P.B. was the lead author, wrote the first draft and directed the manuscript content of each draft, supervising the medical writer.
All authors approved the final version for submission.
References
- 1.Tanaka T, Ebisuno Y, Kanemitsu N, Umemoto E, Yang BG, Jang MH, Miyasaka M. Molecular determinants controlling homeostatic recirculation and tissue-specific trafficking of lymphocytes. Int Arch Allergy Immunol. 2004;134:120–134. doi: 10.1159/000078497. [DOI] [PubMed] [Google Scholar]
- 2.Cyster JG. Homing of antibody secreting cells. Immunol Rev. 2003;194:48–60. doi: 10.1034/j.1600-065x.2003.00041.x. [DOI] [PubMed] [Google Scholar]
- 3.Cyster JG. Chemokines, sphingosine-1-phosphate, and cell migration in secondary lymphoid organs. Annu Rev Immunol. 2005;23:127–159. doi: 10.1146/annurev.immunol.23.021704.115628. [DOI] [PubMed] [Google Scholar]
- 4.Brinkmann V. Sphingosine 1-phosphate receptors in health and disease: mechanistic insights from gene deletion studies and reverse pharmacology. Pharmacol Ther. 2007;115:84–105. doi: 10.1016/j.pharmthera.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 5.Schwab SR, Pereira JP, Matloubian M, Xu Y, Huang Y, Cyster JG. Lymphocyte sequestration through S1P lyase inhibition and disruption of S1P gradients. Science. 2005;309:1735–1739. doi: 10.1126/science.1113640. [DOI] [PubMed] [Google Scholar]
- 6.Alvarez SE, Milstien S, Spiegel S. Autocrine and paracrine roles of sphingosine-1-phosphate. Trends Endocrinol Metab. 2007;18:300–307. doi: 10.1016/j.tem.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 7.Chun J. International Union of Pharmacology. XXXIV. Lysophospholipid receptor nomenclature. Pharmacol Rev. 2002;54:265–269. doi: 10.1124/pr.54.2.265. [DOI] [PubMed] [Google Scholar]
- 8.Ishii I, Fukushima N, Ye X, Chun J. Lysophospholipid receptors: signaling and biology. Annu Rev Biochem. 2004;73:321–354. doi: 10.1146/annurev.biochem.73.011303.073731. [DOI] [PubMed] [Google Scholar]
- 9.Meyer zu Heringdorf D, Jakobs KH. Lysophospholipid receptors: signalling, pharmacology and regulation by lysophospholipid metabolism. Biochim Biophys Acta. 2007;1768:923–940. doi: 10.1016/j.bbamem.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 10.Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V, Allende ML, Proia RL, Cyster JG. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427:355–360. doi: 10.1038/nature02284. [DOI] [PubMed] [Google Scholar]
- 12.Rose NR, MacKay IR. The Autoimmune Diseases. 4th edn. San Diego, CA: Elsevier Academic Press; 2006. [Google Scholar]
- 13.Kataoka H, Sugahara K, Shimano K, Teshima K, Koyama M, Fukunari A, Chiba K. FTY720, sphingosine 1-phosphate receptor modulator, ameliorates experimental autoimmune encephalomyelitis by inhibition of T cell infiltration. Cell Mol Immunol. 2005;2:439–448. [PubMed] [Google Scholar]
- 14.Brinkmann V. FTY720 (fingolimod) in Multiple Sclerosis: therapeutic effects in the immune and the central nervous system. Br J Pharmacol. 2009;158:1173–1182. doi: 10.1111/j.1476-5381.2009.00451.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kappos L, Antel J, Comi G, Montalban X, O'Connor P, Polman CH, Haas T, Korn AA, Karlsson G, Radue EW. Oral fingolimod (FTY720) for relapsing multiple sclerosis. N Engl J Med. 2006;355:1124–1140. doi: 10.1056/NEJMoa052643. [DOI] [PubMed] [Google Scholar]
- 16.Cohen JA, Barkhof F, Comi G, Hartung HP, Khatri BO, Montalban X, Pelletier J, Capra R, Gallo P, Izquierdo G, Tiel-Wilck K, de Vera A, Jin J, Stites T, Wu S, Aradhye S, Kappos L. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362:402–415. doi: 10.1056/NEJMoa0907839. [DOI] [PubMed] [Google Scholar]
- 17.Budde K, Schmouder RL, Brunkhorst R, Nashan B, Lucker PW, Mayer T, Choudhury S, Skerjanec A, Kraus G, Neumayer HH. First human trial of FTY720, a novel immunomodulator, in stable renal transplant patients. J Am Soc Nephrol. 2002;13:1073–1083. doi: 10.1681/ASN.V1341073. [DOI] [PubMed] [Google Scholar]
- 18.Kovarik JM, Schmouder R, Barilla D, Wang Y, Kraus G. Single-dose FTY720 pharmacokinetics, food effect, and pharmacological responses in healthy subjects. Br J Clin Pharmacol. 2004;57:586–591. doi: 10.1111/j.1365-2125.2003.02065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Forrest M, Sun SY, Hajdu R, Bergstrom J, Card D, Doherty G, Hale J, Keohane C, Meyers C, Milligan J, Mills S, Nomura N, Rosen H, Rosenbach M, Shei GJ, Singer II, Tian M, West S, White V, Xie J, Proia RL, Mandala S. Immune cell regulation and cardiovascular effects of sphingosine 1-phosphate receptor agonists in rodents are mediated via distinct receptor subtypes. J Pharmacol Exp Ther. 2004;309:758–768. doi: 10.1124/jpet.103.062828. [DOI] [PubMed] [Google Scholar]
- 20.Sanna MG, Liao J, Jo E, Alfonso C, Ahn MY, Peterson MS, Webb B, Lefebvre S, Chun J, Gray N, Rosen H. Sphingosine 1-phosphate (S1P) receptor subtypes S1P1 and S1P3, respectively, regulate lymphocyte recirculation and heart rate. J Biol Chem. 2004;279:13839–13848. doi: 10.1074/jbc.M311743200. [DOI] [PubMed] [Google Scholar]
- 21.Gon Y, Wood MR, Kiosses WB, Jo E, Sanna MG, Chun J, Rosen H. S1P3 receptor-induced reorganization of epithelial tight junctions compromises lung barrier integrity and is potentiated by TNF. Proc Natl Acad Sci U S A. 2005;102:9270–9275. doi: 10.1073/pnas.0501997102. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Bolli MH, Abele S, Binkert C, Bravo R, Buchmann S, Bur D, Gatfield J, Hess P, Kohl C, Mangold C, Mathys B, Menyhart K, Muller C, Nayler O, Scherz M, Schmidt G, Sippel V, Steiner B, Strasser D, Treiber A, Weller T. 2-imino-thiazolidin-4-one derivatives as potent, orally active S1P1 receptor agonists. J Med Chem. 2010;53:4198–4211. doi: 10.1021/jm100181s. [DOI] [PubMed] [Google Scholar]
- 23.Piali L, Froidevaux S, Hess P, Nayler O, Bolli MH, Schlosser E, Kohl C, Steiner B, Clozel M. The selective sphingosine 1-phosphate receptor 1 agonist ponesimod protects against lymphocyte-mediated tissue inflammation. J Pharmacol Exp Ther. 2011;337:547–556. doi: 10.1124/jpet.110.176487. [DOI] [PubMed] [Google Scholar]
- 24.Khasnis A, Gokula RM. Romberg's test. J Postgrad Med. 2003;49:169–172. [PubMed] [Google Scholar]
- 25.Gibaldi M, Perrier D. Pharmacokinetics. New York: Dekker; 1982. [Google Scholar]
- 26.De Rosa SC. Multicolor immunophenotyping: human mature immune system. Methods Cell Biol. 2004;75:577–594. doi: 10.1016/s0091-679x(04)75024-4. [DOI] [PubMed] [Google Scholar]
- 27.Beal SL, Sheiner LB, Boeckmann AJ. NONMEM Users Guides. San Francisco, CA: NONMEM Project Group, University of California at San Francisco; 1998. [Google Scholar]
- 28.Mager DE, Lin SX, Blum RA, Lates CD, Jusko WJ. Dose equivalency evaluation of major corticosteroids: pharmacokinetics and cell trafficking and cortisol dynamics. J Clin Pharmacol. 2003;43:1216–1227. doi: 10.1177/0091270003258651. [DOI] [PubMed] [Google Scholar]
- 29.Stark JG, Werner S, Homrighausen S, Tang Y, Krieg M, Derendorf H, Moellmann H, Hochhaus G. Pharmacokinetic/pharmacodynamic modeling of total lymphocytes and selected subtypes after oral budesonide. J Pharmacokinet Pharmacodyn. 2006;33:441–459. doi: 10.1007/s10928-006-9013-5. [DOI] [PubMed] [Google Scholar]
- 30.Gough K, Hutchison M, Keene O, Byrom B, Ellis S, Lacey L, McKellar J. Assessment of dose proportionality: report from the statisticians in the pharmaceutical industry/ pharmacokinetics UK joint working group. Drug Inf J. 1995;29:1039–1048. [Google Scholar]
- 31.Mazurais D, Robert P, Gout B, Berrebi-Bertrand I, Laville MP, Calmels T. Cell type-specific localization of human cardiac S1P receptors. J Histochem Cytochem. 2002;50:661–670. doi: 10.1177/002215540205000507. [DOI] [PubMed] [Google Scholar]
- 32.Schmouder R, Serra D, Wang Y, Kovarik JM, Dimarco J, Hunt TL, Bastien MC. FTY720: placebo-controlled study of the effect on cardiac rate and rhythm in healthy subjects. J Clin Pharmacol. 2006;46:895–904. doi: 10.1177/0091270006289853. [DOI] [PubMed] [Google Scholar]
- 33.Boxenbaum H. Interspecies scaling, allometry, physiological time, and the ground plan of pharmacokinetics. J Pharmacokinet Biopharm. 1982;10:201–227. doi: 10.1007/BF01062336. [DOI] [PubMed] [Google Scholar]
- 34.Mahmood I. Application of allometric principles for the prediction of pharmacokinetics in human and veterinary drug development. Adv Drug Deliv Rev. 2007;59:1177–1192. doi: 10.1016/j.addr.2007.05.015. [DOI] [PubMed] [Google Scholar]
- 35.Jones HM, Parrott N, Jorga K, Lave T. A novel strategy for physiologically based predictions of human pharmacokinetics. Clin Pharmacokinet. 2006;45:511–542. doi: 10.2165/00003088-200645050-00006. [DOI] [PubMed] [Google Scholar]
- 36.Kovarik JM, Schmouder R, Barilla D, Riviere GJ, Wang Y, Hunt T. Multiple-dose FTY720: tolerability, pharmacokinetics, and lymphocyte responses in healthy subjects. J Clin Pharmacol. 2004;44:532–537. doi: 10.1177/0091270004264165. [DOI] [PubMed] [Google Scholar]
- 37.Carandente F, Angeli A, De Vecchi A, Dammacco F, Halberg F. Multifrequency rhythms of immunological functions. Chronobiologia. 1988;15:7–23. [PubMed] [Google Scholar]