

Abstract
Aims
To characterize the pharmacokinetics (PK) of recombinant human C1 inhibitor (rhC1INH) in healthy volunteers and hereditary angioedema (HAE) patients.
Methods
Plasma levels of C1INH following 294 administrations of rhC1INH in 133 subjects were fitted using nonlinear mixed-effects modelling. The model was used to simulate maximal C1INH levels for the proposed dosing scheme.
Results
A one-compartment model with Michaelis–Menten elimination kinetics described the data. Baseline C1INH levels were 0.901 [95% confidence interval (CI): 0.839–0.968] and 0.176 U ml−1 (95% CI: 0.154–0.200) in healthy volunteers and HAE patients, respectively. The volume of distribution of rhC1INH was 2.86 l (95% CI: 2.68–3.03). The maximal rate of elimination and the concentration corresponding to half this maximal rate were 1.63 U ml−1 h−1 (95% CI: 1.41–1.88) and 1.60 U ml−1 (95% CI: 1.14–2.24), respectively, for healthy volunteers and symptomatic HAE patients. The maximal elimination rate was 36% lower in asymptomatic HAE patients. Peak C1INH levels did not change upon repeated administration of rhC1INH. Bodyweight was found to be an important predictor of the volume of distribution. Simulations of the proposed dosing scheme predicted peak C1INH concentrations above the lower level of the normal range (0.7 U ml−1) for at least 94% of all patients.
Conclusions
The population PK model for C1INH supports a dosing scheme on a 50 U kg−1 basis up to 84 kg, with a fixed dose of 4200 U above 84 kg. The PK of rhC1INH following repeat administration are consistent with the PK following the first administration.
Keywords: hereditary angioedema, NONMEM, pharmacokinetics, recombinant human C1 inhibitor
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Hereditary angioedema (HAE) results from a heterozygous deficiency of the plasma protein C1 inhibitor (C1INH) and is characterized by the occurrence of acute attacks of angioedema that can be life threatening or disabling.
Recombinant human C1INH (rhC1INH) is approved for the treatment of acute angioedema in patients with HAE.
A previous publication has presented the pharmacokinetics (PK) of rhC1INH in a limited number of asymptomatic HAE patients.
The population PK of plasma-derived C1INH (pdC1INH) in HAE patients has been previously described.
WHAT THIS STUDY ADDS
A population PK analysis of rhC1INH incorporating all available PK data from six studies using mixed-effects modelling was conducted.
The population PK modelling demonstrates different PK for rhC1INH in comparison to pdC1INH.
The PK of rhC1INH was similar following both single and multiple dosing.
Introduction
The C1 esterase inhibitor (C1INH) is an endogenous human protein and regulates several inflammatory pathways by inhibiting proteases that are part of the human immune and inflammatory systems. Hereditary angioedema (HAE) is a rare disease, with an estimated prevalence of 1:50 000, caused by a heterozygous mutation in the gene for C1INH [1]. This results in low levels of C1INH activity, leading to excessive activation of the complement and contact systems and subsequent release of vasoactive peptides, causing acute attacks of angioedema [2, 3]. Although patients have chronically low C1INH levels, attacks occur episodically, usually without an apparent trigger. These attacks are characterized by acute and often painful swellings of submucosal or subcutaneous soft tissues, which can be life threatening when located in the upper airway. Attacks develop over the course of hours and take several days to subside if not treated [1].
A rational treatment of HAE attacks is the administration of C1INH protein, because this can normalize the levels of functional C1INH and stop the biochemical activation processes underlying the angioedema attack. Typically, one administration of C1INH is sufficient to ameliorate clinical symptoms quickly, indicating that only short exposure to C1INH is required to treat an HAE attack. Plasma-derived C1INH (pdC1INH) products have been used for decades for this indication, but their use has been limited by both supply issues and the risk of transmission of blood-borne pathogens. Ruconest® (Pharming Technologies BV, Leiden, The Netherlands), a recombinant human C1INH (rhC1INH), is manufactured from the milk of transgenic rabbits, allowing for the commercial production of large quantities of high-grade product of consistent quality, which is devoid of the potential risk for transmission of blood-borne pathogens.
Although C1INH products have been used for decades to treat HAE patients with an acute angioedema attack, appropriate dosing studies with individual products have not been performed, presumably because of the low prevalence of the disease. Comparison of data from controlled trials with different C1INH products indicates that for optimal clinical efficacy a dose of 50 U kg−1 should be given [4].
During the clinical development program of rhC1INH, blood samples were taken before and after administration of rhC1INH. These samples were assessed for levels of C1INH activity to evaluate the pharmacokinetics (PK) of rhC1INH. Studies were performed in healthy volunteers, in asymptomatic patients with HAE and in HAE patients with acute attacks. The present population PK analysis of rhC1INH incorporates all available PK data from six clinical studies using mixed-effects modelling.
Methods
Study design
Table 1 summarizes the design of each of the six studies included in the population PK analysis.
Table 1.
Summary of rhC1INH studies included in population pharmacokinetic analysis
| Study | Population | rhC1INH dose | Number of subjects* | Number of rhC1INH administrations† | PK samples | Number of PK samples above LLOQ | Number of PK samples below LLOQ |
|---|---|---|---|---|---|---|---|
| C1 1101-01 | Asymptomatic HAE | 6.25–100 U kg−1 | 12 | 24 | Screening, baseline, 0.25, 0.5, 1, 2, 4, 8, 12, 16, 24, 48, 96 and 504 h postdose | 359 | 7 |
| C1 1106-02 | Healthy volunteers | 100 U kg−1 | 14 | 36 | Screening, baseline, 0.267, 0.5, 1, 2, 4, 8, 12 and 168 h postdose | 355 | 0 |
| C1 1202-01 | Symptomatic HAE | 100 U kg−1 | 4 | 6 | Screening, baseline, 0.267, 0.5, 1, 2, 4, 8, 12, 24, 168 and 528 h postdose | 41 | 10 |
| C1 1203-01 | Symptomatic HAE | 100 U kg−1 | 10 | 15 | Screening, baseline, 0.267, 0.5, 1, 2, 4, 8, 12, 24, 168 and 528 h postdose | 129 | 28 |
| C1 1205-01 | Symptomatic HAE | 50 and 100 U kg−1 | 52 | 111 | Screening, baseline, 0.25, 0.5, 1, 2, 4, 8, 12, 16, 24, 144 and 504 h postdose | 209 | 254 |
| C1 1304-01 | Symptomatic HAE | Up to three vials of 2100 U | 41 | 102 | Screening, baseline, 0.25, 0.5, 1, 2, 4, 8, 12, 24 and 504 h postdose | 269 | 357 |
| Total | 133 | 294 | 1362 | 656 |
Abbreviations are as follows: HAE, hereditary angioedema; LLOQ, lower limit of quantification; PK, pharmacokinetics; rhC1INH, recombinant human C1 inhibitor.
Refers to the number of patients for whom there were associated PK samples.
Refers to the number of administrations for which there were associated PK samples. For Study C1 1205-01, seven of these administrations were second doses given within 4 h of the initial treatment of an attack. For Study C1 1304-01, 26 of these administrations were second doses given within 4 h of the initial treatment of an attack. Therefore, PK data from the treatment of 201 attacks in symptomatic HAE patients were included in the data set.
Study C1 1101-01
Twelve HAE patients, who were asymptomatic at the time of drug administration, were infused on two separate occasions, with an interval of at least 5 weeks, with escalating doses (6.25–100 U kg−1) of rhC1INH. Serial PK sampling was performed on each of the two occasions.
Study C1 1106-02
Fourteen healthy volunteers were enrolled to receive rhC1INH at 100 U kg−1 on five occasions, with intervals of approximately 3 weeks between doses. Eleven subjects completed the study. Serial PK sampling was performed following the first, third and fifth occasion when rhC1INH was administered. Serial PK samples for at least one administration were available from all subjects.
Study C1 1202-01
This was an open-label exploratory study in four patients with symptomatic HAE to assess the safety, tolerability and efficacy as well as PK and pharmacodynamics (PD) of rhC1INH dosed at 100 U kg−1. Two patients received a repeat treatment for a subsequent angioedema attack.
Study C1 1203-01
This was an open-label exploratory study in 10 patients with symptomatic HAE to assess the safety, tolerability and efficacy as well as PK and PD of rhC1INH dosed at 100 U kg−1. Five patients received a repeat treatment for a subsequent angioedema attack.
Study C1 1205-01
This was a randomized, placebo-controlled, double-blind study of the safety and efficacy of rhC1INH for the treatment of acute attacks in patients with HAE, evaluating doses of 50 and 100 U kg−1. In the randomized, controlled portion of the study, 38 patients were treated and three dose groups were evaluated, namely 50 U kg−1 (12 patients), 100 U kg−1 (13 patients) and saline (13 patients). In the open-label extension part of this study, 39 patients, including 11 who had received rhC1INH in the randomized phase, received a dose of 50 U kg−1, with an option of an additional 50 U kg−1 within 4 h of the first dose. This second dose was given at the discretion of the investigator depending on the patient's clinical condition. Each 50 U kg−1 dose was delivered as a fixed volume of 90 ml. The study also allowed for repeat treatments of subsequent angioedema attacks.
Study C1 1304-01
This was a randomized, placebo-controlled, double-blind study of the safety and efficacy of rhC1INH for the treatment of acute attacks in patients with HAE. Thirty-two patients were randomized to receive rhC1INH at 100 U kg−1 (16 patients) or saline (16 patients). In the open-label extension phase, 41 symptomatic patients, including four who had received rhC1INH in the randomized phase, were treated with a single vial of 2100 U of rhC1INH. At the discretion of the investigator and depending on the patient's condition, in the open-label extension phase, an additional dose of one or two vials (2100 or 4200 U) could be administered within 4 h from the initial dose of 2100 U. Therefore, the maximal total amount of rhC1INH that could be given to a patient during a single attack in the open-label phase was 6300 U. The study also allowed for repeat treatments of subsequent angioedema attacks.
All together, the PK data set contained data from the treatment of 201 attacks in symptomatic HAE patients. In 33 of these attacks, a second dose of rhC1INH was administered within 4 h of the initial treatment. The rhC1INH was administered intravenously over 2–30 min.
Subjects
All subjects participating in the clinical studies, for whom a sufficient number of pre- and postexposure C1INH activity values were available, were included in this population PK analysis (Table 1). Data from 294 administrations from 133 subjects were included, as follows: 14 healthy volunteers and 12 asymptomatic and 107 symptomatic HAE patients. Sixty-eight and 33 subjects had two and three or more administrations, respectively, with 431 of the 1362 quantifiable concentrations resulting from the second administration, and 202 quantifiable concentrations resulting from three administrations or more. The age of the subjects included ranged from 13 to 66 years (median of 33 years), while their weight ranged from 45 to 128 kg (median of 72 kg). Of the 133 subjects, 47 were male and 86 were female.
Bioanalysis
A chromogenic assay was used to measure levels of functional C1INH as described [3]. This assay is based on the property of active complement component 1, s subcomponent (C1s) to convert a chromogenic substrate. The chromogenic assay measures the inhibitory effect of C1INH on cleavage of the chromogenic substrate by a fixed amount of C1s in the sample to be tested. The test sample is incubated with an excess of C1s for 15 min to allow C1s to interact with C1INH. The remaining C1s activity is subsequently measured by addition of a chromogenic substrate. The difference between the amount of C1s added and the C1s activity measured reflects the amount of C1INH present in plasma. Results are expressed as units per millilitre, with 1 U ml−1 representing the amount of functional C1INH in pooled normal human plasma.
Plasma levels of functional C1INH in Study 1101-01 were measured in a specialized laboratory, whereas levels in the other studies were measured in a central contract laboratory using the same chromogenic assay. The lower limit of quantification (LLOQ) of the assay in the specialized laboratory was 0.07 U ml−1, while that in the central contract laboratory was 0.28 U ml−1.
Population pharmacokinetic analysis
The population PK analysis was conducted using the nonlinear mixed effects modelling program, NONMEM (version 7.2.0) [5] to estimate the population mean parameters, interindividual (η) and residual (ε) random effects. The interindividual variability (IIV) of each of the structural parameters of the basic model was modelled using the following exponential model:
where Pi is the value of the parameter in the ith individual, and ηi is the random variable for the difference between Pi and PTV, which is the value of the parameter in a typical individual. It was assumed that the values of ηi are normally distributed, with a mean of zero and a variance of ω2. The residual variability was evaluated to describe the intrasubject variability. The combined additive and proportional error model based on the following equation best described the residual variability:
where Cij is the jth observed value in the ith subject, Cpred,ij is the jth predicted value in the ith subject, and εpro,ij and εadd,ij are the residual intrasubject variability, with means of zero and variances of σpro2 and σadd2, respectively.
A composite of methods [iterative two-stage (ITS) followed by stochastic approximation expectation maximization (SAEM) followed by IMP (EONLY = 1)] was used throughout the model development process by using multiple $EST statements so that final estimates from one method served as initial estimates for the next. ‘Mu Referencing’ [5] was used to improve the efficiency of computations.
The base model, which included key parameters, was selected based on goodness-of-fit plots, precision of the estimates and the likelihood ratio test within NONMEM.
For linear models, endogenous C1INH levels were assumed to be constant, and it was also assumed that the observed functional C1INH levels were the sum of the endogenous baseline levels and exogenous drug administered. Baseline concentration was estimated, and it was assumed that the rate of production was constant with respect to the baseline clearance. For nonlinear models, baseline concentration was estimated, and the rate of production was a function of the nonlinear clearance and baseline concentration.
Baseline samples were included in the NONMEM data set to allow these samples to provide information on the endogenous baseline functional C1INH levels.
Concentrations below LLOQ were included in the NONMEM data set, and the likelihood was maximized for all the data, treating concentrations below LLOQ as censored, with the likelihood for data above and below LLOQ conditioned on the observations being greater than zero (M4) [6]. Overall, concentrations below LLOQ accounted for 33% of the total data.
Population (healthy volunteers, symptomatic patients and asymptomatic patients), weight, age, gender, dose and study were evaluated as covariates. Covariates were added to the base model if a relationship was evident in the individual random effects against covariates plots. All significant covariate–parameter relationships [greater than 10.8 point decrease in objective function value (OFV)] were retained in the model.
Model evaluation
The accuracy and robustness of the final population model were evaluated by performing a visual predictive check as follows. The predicted concentration–time profiles for 100 data sets at each time point after each administration of rhC1INH were generated from the parameters and associated variances of the final model. The estimated median, fifth and 95th percentiles of the predicted concentrations from the simulations were plotted against time, with the observed concentrations stratified by dose (50 and 100 U kg−1) and population.
Simulations
The clinical studies have revealed optimal efficacy as a treatment for an acute angioedema attack with a dose of rhC1INH of 50 U kg−1 [7]. Simulations were used to investigate the percentage of patients who achieved a maximal plasma concentration (Cmax) of ≥0.7 U kg−1 upon administration of the proposed dosing regimen of 50 U kg−1 dose for bodyweights up to 84 kg and a fixed dose of 4200 U for bodyweights >84 kg. Functional C1INH levels were simulated using the parameter estimates from the final PK model for a range of bodyweights (40–180 kg) following a single administration of rhC1INH in accordance with the proposed dosing regimen. For each bodyweight, 1000 profiles were simulated within NONMEM.
Results
Population pharmacokinetic analysis
A one-compartment model with Michaelis–Menten elimination kinetics has been previously reported to describe the PK of rhC1INH in asymptomatic HAE patients and served as a starting point for the present analysis [8]. A one-compartment linear model had higher objective function (∼91 points) and poorer predictions. However, attempts to include a parallel linear elimination pathway in the model provided no further improvement in the predicted C1INH levels. Separate proportional residual errors were explored for each study; the proportional errror was comparable for studies 1101-01, 1106-02, 1202-01 and 1203-01, and a single proportional residual error was estimated for these studies without a significant change in the OFV. Exploration of a two-compartment model, although resulting in a reduction in the OFV by approximately 32 points, showed no improvement in the overall population predicted C1INH levels compared with the one-compartment linear model. Therefore, a one-compartment model with Michaelis–Menten elimination kinetics was selected as the base model.
A separate IIV for the baseline levels in healthy volunteers and all HAE patients was estimated with a 23-point decrease in the OFV.
The estimation of a separate volume of distribution for studies 1202-01 and 1203-01 resulted in a 22-point drop in the OFV, and volume was 32% lower in these two studies compared with all other studies.
The inclusion of bodyweight as a covariate on the volume term resulted in an additional drop in the OFV of 20 points and a reduction in the IIV from 27.0 to 21.8%, indicating a statistically significant effect. Volume increased with weight, with a power coefficient of 0.612 [95% confidence interval (CI): 0.351, 0.873; i.e. as (bodyweight/70)0.612]. For subjects with the lowest and highest bodyweights in the analysis (45 and 128 kg), volume would be 24% lower and 45% higher compared with a 70 kg subject.
The estimation of a separate maximal rate (Vmax) for asymptomatic HAE patients resulted in a 17-point drop in OFV, and Vmax was 36% lower in this population compared with the healthy volunteers and the symptomatic HAE patient population.
Given that a substantial number of subjects received rhC1INH on multiple occasions, the inclusion of interoccasion variability (IOV) was evaluated. Sixty-eight subjects had PK data for two occasions or more, 33 subjects had three occasions or more, and only nine subjects had PK data for four or more occasions. The IOV was tested on both the volume of distribution (V) and Vmax parameters. The IOV in Vmax was not significant once the IOV on V was incorporated into the model. The IIV in V decreased from 21.8 to 16.2% following inclusion of IOV on V.
No other relationships were evident between the individual random effects and covariates.
The population parameter values and inter- and intraindividual variability estimated by the final model are shown in Table 2. The PK parameters were generally well estimated, with the relative standard error (%RSE) for estimation ranging from 3 to 37%. The typical baseline functional C1INH levels in healthy volunteers and HAE patients were estimated to be 0.901 and 0.176 U ml−1, respectively. The associated IIV for the baseline levels was 12.7 and 54.4% for the healthy volunteers and HAE patients, respectively. The individual predicted baseline levels ranged from 0.09 to 0.64 U ml−1 for symptomatic HAE patients, consistent with the observed data. The central volume was estimated to be 2.86 l, while the Michaelis–Menten parameters, Vmax and Km, were estimated to be 1.63 U ml−1 h−1 and 1.60 U ml−1, respectively, for healthy volunteers and symptomatic HAE patients. The maximal elimination rate was 36% lower in asymptomatic HAE patients. All the parameters were estimated with good precision, with the highest %RSE of 36.6% being associated with Km. The IIV was estimated to be 16.2% for the volume parameter and 29.2% for Vmax. The proportional residual error was estimated to be 23.6 and 53.6% for Studies C1 1205-01 and C1 1304-01, respectively, and 10.5% for the remaining studies. The additive residual error (standard deviation) was estimated to be 0.0556 U ml−1.
Table 2.
Population pharmacokinetic parameters for C1INH following administration of rhC1INH
| Parameter | Final model estimate | 95% Confidence interval |
|---|---|---|
| Baseline levels in HAE patients (U ml−1) | 0.176 | 0.154–0.200 |
| Baseline levels in healthy volunteers (U ml−1) | 0.901 | 0.839–0.968 |
| Volume of distribution (l) | 2.86 | 2.68–3.03 |
| Fractional change in volume for studies C1 1202-01 and C1 1203-01 | 0.681 | 0.579–0.801 |
| Effect of bodyweight on volume | 0.612 | 0.351–0.873 |
| Vmax (U ml−1 h−1) in healthy volunteers and symptomatic HAE patients | 1.63 | 1.41–1.88 |
| Fractional change in Vmax (U ml−1 h−1) for asymptomatic HAE patients | 0.644 | 0.517–0.803 |
| Km (U ml−1) | 1.60 | 1.14–2.24 |
| Inter- and intrasubject variability | %RSE | |
|---|---|---|
| Interindividual variability in HAE patient baseline levels (%CV)† | 54.4 | 21.0% |
| Interindividual variability in healthy volunteer baseline levels (%CV)† | 12.7 | 43.1% |
| Interindividual variability in volume (%CV)* | 16.2 | 37.8% |
| Interoccasion variability in volume (%CV)* | 18.3 | 25.8% |
| Interindividual variability in Vmax (%CV)* | 29.2 | 27.7% |
| Proportional residual variability for Study C1 1205-01 (%CV)* | 23.6 | 8.09% |
| Proportional residual variability for Study C1 1304-01 (%CV)* | 53.6 | 7.31% |
| Proportional residual variability for all other studies (%CV)* | 10.5 | 4.30% |
| Additive residual variability | 0.0556 | 6.21% |
The %CV for both intersubject and residual variability is an approximation taken as the square root of the variance for the proportional error term × 100.
The SD was calculated as the square root of the variance for the additive error term. Abbreviations are as follows: %CV, coefficient of variation; HAE, hereditary angioedema; Km, concentration at which rate is half of Vmax; Vmax, maximal rate.
The basic goodness-of-fit plots are shown for both the overall data and for the 50 and 100 U kg−1 dosing regimens in Figure 1. The diagnostic plots for the final population PK model revealed no systematic bias, with good agreement between the individual predicted and observed values. The individual weighted residuals were generally distributed around zero and were relatively symmetric. The population and individual post hoc predictions were distributed around the line of identity.
Figure 1.
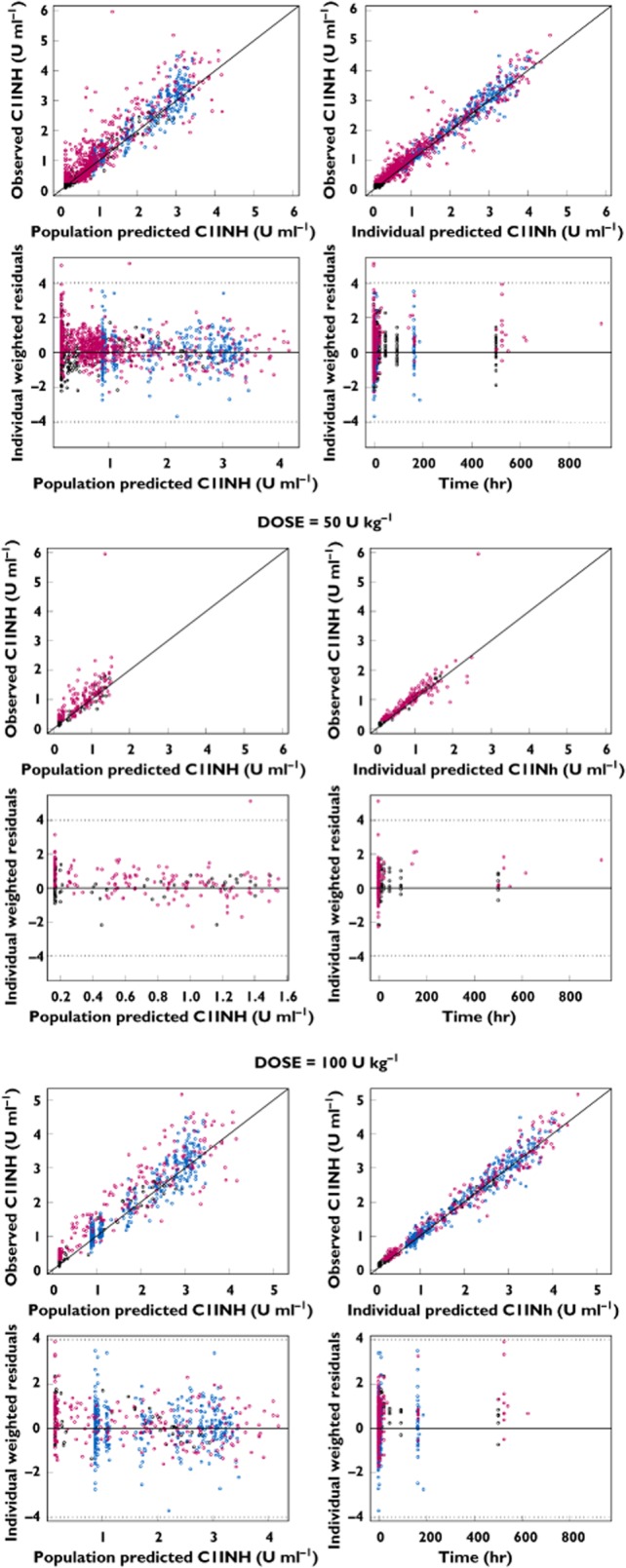
Goodness-of fit-plots for the final model. Top panel, overall population; middle panel, 50 U kg−1 dosing regimen; and bottom panel, 100 U kg−1 dosing regimen.  , asymptomatic patients;
, asymptomatic patients;  , healthy volunteers;
, healthy volunteers;  , symptomatic patients
, symptomatic patients
The diagnostic plots for the final model indicated that there was no systematic change in functional C1INH levels upon repeat administration of rhC1INH. This was investigated further by tabulating the observed peak functional C1INH levels for HAE patients on each occasion when rhC1INH was administered (Figure 2). There was no evidence for changes in the functional C1INH levels upon repeat administration.
Figure 2.
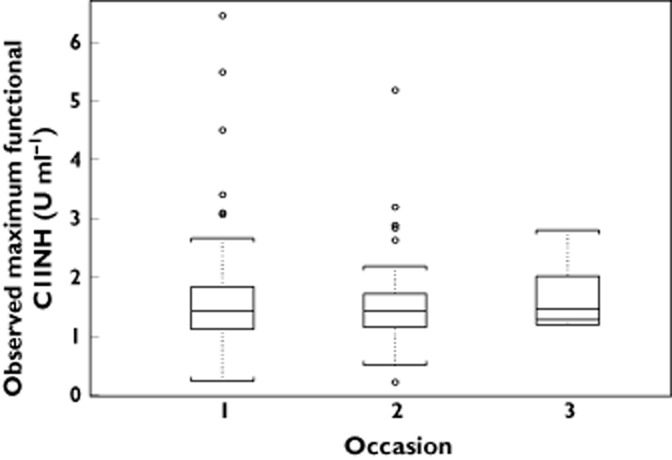
Observed peak functional C1INH levels upon repeated administration of rhC1INH in hereditary angioedema patients (normalized to a dose of 3550 U rhC1INH, i.e. 50 U kg−1 for a median weight of 72 kg). The box represents the 25th and 75th percentile with the median value. The ends of the whiskers represent 1.5 times the interquartile range from the 25th and 75th percentiles, and any observations beyond 1.5 times the interquartile range are presented as open circles
Model evaluation
The predicted concentrations for 100 simulated data sets by the final PK model for functional C1INH following administration of rhC1INH are depicted in Figure 3 by population for the 50 and 100 U kg−1 dosing regimens. The majority of the observed C1INH levels were within the range of the lower (5%) and upper (95%) percentile of the simulated concentrations.
Figure 3.

Visual predictive check of the final pharmacokinetic model. A total of 100 data sets were simulated using the final pharmacokinetic parameter estimates. Abbreviations are as follows: Asymp, asymptomatic patients; HV, healthy volunteers; Symp, symptomatic patients. Depicted are the observations (open circles), median (dashed line), 25th and 75th percentiles (dotted lines) and 5th and 95th percentiles (continuous lines) of the predictions
Simulations
The predicted maximal functional C1INH concentrations in symptomatic HAE patients for the proposed dosing regimen of 50 U kg−1 up to and including 84 kg and thereafter a fixed dose of 4200 U are presented in Figure 4. These simulations indicate that the maximal concentrations of functional C1INH will increase based on the proposed dosing regimen up to a bodyweight of 84 kg and that the fixed dose of 4200 U would result in maximal concentrations that would only decrease slightly with bodyweight. However, for each weight between 40 and 180 kg, at least 94% of patients were predicted to have a maximal C1INH level of at least 0.7 U ml−1, based on the proposed dosing scheme. The predicted median peak levels (Cmax) increased from 1.18 to 1.45 U ml−1 for the 50 U kg−1 (40 to ≤84 kg) dosing and decreased from 1.50 to 1.01 U ml−1 across the proposed fixed dose of 4200 U for the weight range from >84 to 180 kg. The highest predicted peak level was 3.23 U ml−1 for an 80 kg patient.
Figure 4.
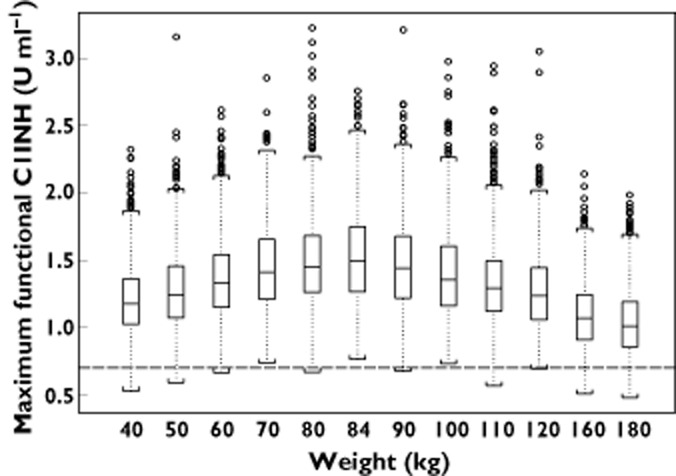
Simulation of maximal functional C1INH concentration (Cmax) for levels the proposed dosing regimen of 50 U kg−1 up to 84 kg and a fixed dose of 4200 U for a bodyweight >84 kg. The boxes represent the 25th and 75th percentile with the median value. The ends of the whiskers represent 1.5 times the interquartile range from the 25th and 75th quartiles, and any observations beyond 1.5 times the interquartile range are represented as open circles. The dashed line represents the lower limit of normal C1INH activity (0.7 U ml−1)
Discussion
The present study provides a population PK analysis of functional C1INH levels following administration of rhC1INH in healthy volunteers and patients with HAE. The data set included functional C1INH levels from 133 subjects following 294 administrations of rhC1INH doses ranging from 6.25 to 121 U kg−1. There were only six profiles associated with doses of 12.5 U kg−1 or less, with the majority of profiles being associated with doses of either 50 or 100 U kg−1. Within the data set, there were 33 occasions when an attack in a symptomatic HAE patient was treated with a second administration of rhC1INH within 4 h of the initial administration.
The population PK of C1INH was well described by a one-compartment model with Michaelis–Menten elimination kinetics. The final PK model estimated the typical baseline functional C1INH levels in HAE patients to be 0.176 U ml−1, with an associated 95% CI of 0.154–0.200 U ml−1. Many of these predicted baseline values were lower than the 0.28 U ml−1 LLOQ of the assay used in these studies. These low baseline levels of C1INH are typical and diagnostic for HAE patients [98]. The final estimate of the volume of distribution of rhC1INH was 2.86 l, consistent with the reported volume of plasma [10], suggesting that there is negligible distribution of rhC1INH. These results are consistent with those presented in a previous PK analysis in asymptomatic HAE patients only [8], and are also comparable to the distribution volumes reported for pdC1INH [98, 11]. Moreover, endogenous C1INH levels were assumed to be constant and not influenced by the administration of rhC1INH, because the latter is mainly cleared via receptors for mannose and galactose exposed on the glycans, whereas endogenous C1INH does not interact with these receptors [8]. Indeed, baseline levels of C1INH in HAE patients are not influenced by administration of rhC1INH at doses up to 100 U kg−1 [8].
The half-life of rhC1INH, estimated to be approximately 3 h after administration of 100 U kg−1 rhC1INH, is shorter than that of pdC1INH, which has been reported to be approximately 30 h [11, 12]. This difference in half-life between rhC1INH and pdC1INH is most likely to be related to the differential glycosylation [13].
It has been suggested that a shorter half-life of treatments could lead to increased risk of relapses of HAE attacks [14, 15]. Relapses, however, have not been observed with rhC1INH. Both plasma-derived and recombinant human C1INH will bind irreversibly to various proteases and restore both the complement and contact systems to their normal state, irrespective of their differences in half-life. Hence, the mechanism of action and pharmacodynamic effect, rather than half-life, may explain the sustained duration of action of rhC1INH and why therapies directed only at kallikrein or bradykinin have been associated with clinical relapse. Target proteases of C1INH include other mediators of the complement pathways, besides activated C1s. Also, several mediators of the haemostatic and fibrinolytic pathways are target proteases of C1INH, specifically Factor XIIa, kallikrein and Factor XIa and, to a lesser extent, tissue plasminogen activator, thrombin and plasmin [16]– [20].
Bodyweight was identified as a predictor of C1INH PK. The allometric scalor estimated from the available data was 0.612 (95% CI: 0.351–0.873), indicating that the volume of distribution for C1INH (in effect, plasma volume) does not increase in a directly proportional manner with bodyweight, with the relationship becoming more divergent for heavier subjects. The estimate of the allometric scalor for C1INH is consistent with previous reports that prediction of plasma volume measurements based solely on bodyweight is inappropriate, particularly in obese individuals because adipose tissue is relatively avascular [10]. In addition, there is a greater divergence between plasma volume and total bodyweight at higher bodyweights [21]. With increasing bodyweight, C1INH concentrations will increase when C1INH is dosed at similar units per kilogram, as the volume of distribution remains relatively constant.
The IIV associated with the PK parameters ranged from 12.7% for the baseline C1INH levels in healthy volunteers to 54.4% for the baseline C1INH levels in HAE patients; the precision of the estimates of the interindividual variability ranged from 21.0 to 43.1%. The goodness-of-fit plots indicate that the PK is well described by the final model, both on an overall basis and also for each of the three populations (healthy volunteers and asymptomatic and symptomatic HAE patients). The data at rhC1INH doses at 12.5 U kg−1 or lower are not as well described by the final model; however, these data represent a very small proportion of the overall functional C1INH data available for the present analysis. The model provides a very good description of data resulting from doses of 50 U kg−1, which is the proposed dosing regimen for rhC1INH for bodyweights up to 84 kg.
Previously, Späth and coworkers postulated a circulating level of 38% of normal (0.38 U ml−1) as the minimal C1INH activity level that provides protection against the occurrence of angioedema attacks [22]. However, a target level for C1INH treatment in acute HAE attacks has never been determined. Data obtained from the clinical trials performed with the various C1INH preparations suggest that maximal efficacy of rhC1INH is achieved when plasma levels of C1INH activity are raised above the lower level of the normal range, which is 0.7 U ml−1 [4]. In a phase I study [8], a dose of 50 U kg−1 was shown to restore C1INH levels into the normal range (mean Cmax ∼0.7 U ml−1). In addition, C1INH levels below 0.7 U ml−1 were shown to be associated with increasing complement activation [8], indicating that 0.7 U ml−1 is also the target level to restore homeostatic control of plasma cascade systems by C1INH. Hence, simulations with doses of 50 U kg−1 for bodyweights up to 84 kg and a fixed dose of 4200 U (corresponding to two vials of Ruconest®) for bodyweights 84 kg or greater were undertaken using the final PK model to address what proportion of patients would achieve restoration of normal C1INH levels with an rhC1INH dose of 50 U kg−1. These simulations indicate that the maximal concentrations of functional C1 inhibitor will remain within an acceptable range for adult patients up to 84 kg, increasing somewhat with increasing bodyweights. For patients with bodyweights 84 kg or greater, maximal concentrations are predicted to decrease across the weight range. Nevertheless, the proposed 50 U kg−1 dose for bodyweights up to 84 kg and the fixed dosing of 4200 U for bodyweights above 84 kg ensures that the vast majority (≥94%) of patients will achieve a peak concentration above 0.7 U ml−1, regardless of bodyweight.
Nonclinical studies of rhC1INH have revealed no safety concerns regarding the treatment of acute angioedema attacks with the proposed dosing regimen. This conclusion is based on the absence of toxicological effects in rat and dog toxicity studies and a no-observed-adverse-effect level of 2000 U kg−1 day−1 in a 14 day repeated dose toxicity study in cynomolgus monkeys. Plasma peak levels of rhC1INH at this high dose surpassed 22 U ml−1, more than six times the predicted plasma peak levels of rhC1INH in human subjects [23].
In the clinical studies of rhC1INH, doses up to 121 U kg−1 were evaluated. In the exploratory efficacy open label studies, HAE patients were administered doses of 100 U kg−1. Median peak plasma levels obtained at this dose were 4.3 U ml−1, with a highest peak level of 4.6 U ml−1, which are beyond the highest individual peak levels of 3.5 U ml−1 predicted by the simulations of the proposed dosing regimen. There was no evidence of an increase in the rate of adverse events, or in seriousness or severity of adverse events at this higher rhC1INH dose, with treatment of higher numbers of attacks, or with treatment of an attack with an additional dose. Taken together, the nonclinical and clinical data do not suggest a safety risk at the peak plasma levels predicted by the simulations.
The observed plasma levels of C1INH following repeat administration showed no systematic deviation from the corresponding levels following single dose administration, and were consistent across repeat treatments with rhC1INH. These findings suggest a lack of time-dependent changes in the PK. These data further support observations in the clinical studies that neutralizing antibodies against C1INH have not been found upon repeated treatment with rhC1INH, and corroborate the immunosafety of rhC1INH [24].
In conclusion, the PK of C1INH following administration of rhC1INH is well described by a one-compartment model, in which the relationship between the volume of distribution and bodyweight supports a proposed dosing scheme of 50 U kg−1 up to a bodyweight of 84 kg, with a fixed dose of 4200 U for those patients 84 kg or greater.
Competing Interests
AR, ESvA and RP are/were employees of Pharming Technologies. CF, SH and CEH provided consulting services to Pharming Technologies.
References
- 1.Zuraw BL. Clinical practice. Hereditary angioedema. N Engl J Med. 2008;359:1027–1036. doi: 10.1056/NEJMcp0803977. [DOI] [PubMed] [Google Scholar]
- 2.Cicardi M, Agostini A. Hereditary angioedema. N Engl J Med. 1996;334:1666–1667. doi: 10.1056/NEJM199606203342510. [DOI] [PubMed] [Google Scholar]
- 3.Landerman NS, Webster ME, Becker EL, Ratcliffe HE. Hereditary angioneurotic edema. II. Deficiency of inhibitor for serum globulin permeability factor and/or plasma kallikrein. J Allergy. 1962;33:330–341. doi: 10.1016/0021-8707(62)90032-1. [DOI] [PubMed] [Google Scholar]
- 4.Hack CE, Relan A, van Amersfoort ES, Cicardi M. Target levels of functional C1-inhibitor in hereditary angioedema. Allergy. 2012;67:123–130. doi: 10.1111/j.1398-9995.2011.02716.x. [DOI] [PubMed] [Google Scholar]
- 5.Beal S, Sheiner LB, Boeckmann A, Bauer RJ. NONMEM User's Guides. (1989–2011) Ellicott City, MD: Icon Development Solutions; 2011. 7.2.0 ed. [Google Scholar]
- 6.Ahn JE, Karlsson MO, Dunne A, Ludden TM. Likelihood based approaches to handling data below the quantification limit using NONMEM VI. J Pharmacokinet Pharmacodyn. 2008;35:401–421. doi: 10.1007/s10928-008-9094-4. [DOI] [PubMed] [Google Scholar]
- 7.Zuraw B, Cicardi M, Levy RJ, Nuijens JH, Relan A, Visscher S, Haase G, Kaufman L, Hack CE. Recombinant human C1-inhibitor for the treatment of acute angioedema attacks in patients with hereditary angioedema. J Allergy Clin Immunol. 2010;126:821–827. doi: 10.1016/j.jaci.2010.07.021. [DOI] [PubMed] [Google Scholar]
- 8.van Doorn MBA, Burggraaf J, van Dam T, Eerenberg A, Levi M, Hack CE, Schoemaker RC, Cohen AF, Nuijens J. A phase I study of recombinant human C1 inhibitor in asymptomatic patients with hereditary angioedema. J Allergy Clin Immunol. 2005;116:876–883. doi: 10.1016/j.jaci.2005.05.019. [DOI] [PubMed] [Google Scholar]
- 98.Cicardi M, Zingale L. Clinical manifestations of hereditary angioedema. J Allergy Clin Immunol. 2004;114:S55–58. doi: 10.1016/j.jaci.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 10.Pearson TC, Guthrie DL, Simpson J, Chinn S, Barosi G, Ferrant A, Lewis SM, Najean Y. Interpretation of measured red cell mass and plasma volume in adults: expert panel on radionuclides of the international council for standardization in haematology. Br J Haematol. 1995;89:748–756. doi: 10.1111/j.1365-2141.1995.tb08411.x. [DOI] [PubMed] [Google Scholar]
- 11.Martinez-Sauger I, Rusicke E, Aygören-Pürsün E, von Hentig N, Klingebiel T, Kruez W. Pharmacokinetic analysis of human plasma-derived pasteurized C1-inhibitor concentrate in adults and children with hereditary angioedema: a prospective study. Transfusion. 2010;50:354–360. doi: 10.1111/j.1537-2995.2009.02394.x. [DOI] [PubMed] [Google Scholar]
- 12.Bernstein JA, Ritchie B, Levy RJ, Wasserman RL, Bewtra AK, Hurewitz DS, Obtulowicz K, Reshef A, Moldovan D, Shirov T, Grivcheva-Panov Ska V, Kiessling PC, Schindel F, Craig TJ. Population pharmacokinetics of plasma-derived C1 esterase inhibitor concentrate used to treat hereditary angioedema attacks. Ann Allergy Asthma Immunol. 2010;105:149–154. doi: 10.1016/j.anai.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 13.Koles K, Van Berkel PH, Pieper FR, Nuijens JH, Mannesse ML, Vliegenthart JF. N- and O-glycans of recombinant human C1 inhibitor expressed in the milk of transgenic rabbits. Glycobiology. 2004;14:51–64. doi: 10.1093/glycob/cwh010. [DOI] [PubMed] [Google Scholar]
- 14.Cicardi M, Banerji A, Bracho F, Malbrán A, Rosenkranz B, Riedl M, Bork K, Lumry W, Aberer W, Bier H, Bas M, Greve J, Hoffmann TK, Farkas H, Reshef A, Ritchie B, Yang W, Grabbe J, Kivity S, Kreuz W, Levy RJ, Luger T, Obtulowicz K, Schmid-Grendelmeier P, Bull C, Sitkauskiene B, Smith WB, Toubi E, Werner S, Anné S, Björkander J, Bouillet L, Cillari E, Hurewitz D, Jacobson KW, Katelaris CH, Maurer M, Merk H, Bernstein HA, Feighery C, Floccard B, Gleich G, Hébert J, Kaatz M, Keith P, Kirkpatrick CH, Langton D, Martin L, Pichler C, Resnick D, Wombolt D, Fernández Romero DS, Zanichelli A, Arcoleo F, Knolle J, Kravec I, Dong L, Zimmermann J, Rosen K, Fan W-T. Icatibant, a new bradykinin-receptor antagonist, in hereditary angioedema. N Engl J Med. 2010;363:532–541. doi: 10.1056/NEJMoa0906393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.FDA Advisory Committee Briefing Document Ecallantide. 2009. Available at http://www.fda.gov/downloads/advisorycommittees/committeesmeetingmaterials/drugs/pulmonary-allergydrugsadvisorycommittee/ucm170334.pdf (last accessed 11 July 2013)
- 16.Bos IG, Hack CE, Abrahams JP. Structural and functional aspects of C1-inhibitor. Immunobiology. 2002;205:518–533. doi: 10.1078/0171-2985-00151. [DOI] [PubMed] [Google Scholar]
- 17.Wuillemin WA, Eldering E, Citarella F, de Ruig CP, ten Cate H, Hack CE. Modulation of contact system proteases by glycosaminoglycans. Selective enhancement of the inhibition of factor XIa. J Biol Chem. 1996;271:12913–12918. doi: 10.1074/jbc.271.22.12913. [DOI] [PubMed] [Google Scholar]
- 18.Cugno M, Bos I, Lubbers Y, Hack CE, Agostoni A. In vitro interaction of C1-inhibitor with thrombin. Blood Coagul Fibrinolysis. 2001;12:253–256. doi: 10.1097/00001721-200106000-00005. [DOI] [PubMed] [Google Scholar]
- 19.Eldering E, Huijbregts CC, Lubbers YT, Longstaff C, Hack CE. Characterization of recombinant C1 inhibitor P1 variants. J Biol Chem. 1992;267:7013–7020. [PubMed] [Google Scholar]
- 20.Sulikowski T, Patston PA. The inhibition of TNK-t-PA by C1-inhibitor. Blood Coagul Fibrinolysis. 2001;12:75–77. doi: 10.1097/00001721-200101000-00011. [DOI] [PubMed] [Google Scholar]
- 21.Morgan DJ, Bray KM. Lean body mass as a predictor of drug dosage – implications for drug therapy. Clin Pharmacokinet. 1994;26:292–307. doi: 10.2165/00003088-199426040-00005. [DOI] [PubMed] [Google Scholar]
- 22.Späth PJ, Wüthrich B, Bütler R. Quantification of C1-inhibitor functional activities by immunodiffusion assay in plasma of patients with hereditary angioedema – evidence of a functionally critical level of C1-inhibitor concentration. Complement. 1984;1:147–159. doi: 10.1159/000467830. [DOI] [PubMed] [Google Scholar]
- 23.European Medicines Agency. CHMP Assessment Report: Ruconest. 24 June 2010. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/001223/WC500098546.pdf (last assessed 11 July 2013)
- 24.Baboeram A, Relan CE, Hack M, Mannesse G, Haase B, Oortwijn S, Visscher R. Pijpstra. 29th European Academy of Allergy and Clinical Immunology Congress. 5–9 June 2010, London. Immunogenicity assessment of recombinant human C1 Inhibitor (rhC1INH)