

Abstract
Aims
The assessment of heart rate-corrected QT (QTc) interval prolongation relies on the evidence of drug effects in healthy subjects. This study demonstrates the relevance of pharmacokinetic–pharmacodynamic (PKPD) relationships to characterize drug-induced QTc interval prolongation and explore the discrepancies between clinical trials and real-life conditions.
Methods
d,l-Sotalol data from healthy subjects and from the Rotterdam Study cohort were used to assess treatment response in a phase I setting and in a real-life conditions, respectively. Using modelling and simulation, drug effects at therapeutic doses were predicted in both populations.
Results
Inclusion criteria were shown to restrict the representativeness of the trial population in comparison to real-life conditions. A significant part of the typical patient population was excluded from trials due to weight and baseline QTc interval criteria. Relative risk was significantly different between sotalol users with and without heart failure, hypertension, diabetes and myocardial infarction (P < 0.01). Although drug effects do cause an increase in the relative risk of QTc interval prolongation, the presence of diabetes represented an increase from 4.0 [95% confidence interval (CI) 2.7–5.8] to 6.5 (95% CI 1.6–27.1), whilst for myocardial infarction it increased from 3.4 (95% CI 2.3–5.13) to 15.5 (95% CI 4.9–49.3).
Conclusions
Our findings show that drug effects on QTc interval do not explain the observed QTc values in the population. The prevalence of high QTc values in the real-life population can be assigned to co-morbidities and concomitant medications. These findings substantiate the need to account for these factors when evaluating the cardiovascular risk of medicinal products.
Keywords: model-based drug development, risk management; observational cohorts; pharmacokinetic–pharmacodynamic modelling; QTc interval prolongation; sotalol
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Safety signals regarding drug effects on cardiac conductivity have been found after the approval of medicines, despite evidence suggesting that they could be deemed safe during development.
Such a discrepancy may be caused by the known differences between real-life conditions and clinical trial protocols, which represent a subset of the patient population, as defined by the many inclusion and exclusion criteria. No formal quantitative method is available to assess the clinical implications of such differences.
Modelling and simulation have been successfully applied as a tool for evidence synthesis in drug development. In conjunction with Bayesian statistics, modelling and simulation allow inferences to be made about the response to treatment, taking into account drug- and disease-specific properties as well as population characteristics.
WHAT THIS STUDY ADDS
The concept of not-in-trial simulations is introduced as a tool for risk management, integrating pharmacokinetic–pharmacodynamic relationships as the basis for discriminating drug-specific properties from other relevant factors in noncontrolled settings.
Using d,l-sotalol as an example compound, we show that a model-based approach can be used to evaluate heart rate-corrected QT (QTc) interval prolongation in clinical trials and predict drug-induced effects in real-life conditions.
In addition, our results clearly indicate that the effect of concomitant medications and co-morbidities needs to be considered in parallel when assessing the propensity for the prolongation of QTc interval.
Introduction
In the last two decades, there have been increased concerns around the rising number of pro-arrhythmia cases due to non-antiarrhythmic drug-induced QTc prolongation [1]. Physiologically, the duration of the QT interval on the surface electrocardiogram (ECG) represents the ventricular action potential duration. As such, prolonged heart rate-corrected QT (QTc) interval may result in early after-depolarizations, which may induce re-entry and provoke Torsade de Pointes [2, 3]. Even though epidemiological evidence regarding the correlation between drug-induced QTc interval prolongation and the risk of fatal arrhythmias remains under scrutiny [4, 5], QTc prolongation has become the second most common cause for postmarket drug withdrawal [6, 7].
Given that in most cases the pro-arrhythmic effects were unclear or unobserved during clinical development, regulatory bodies started to monitor closely the liability for QTc interval prolongation in the submissions of new chemical entities. Guidelines have also emerged outlining the prerequisites for the evaluation of drug-induced effects on heart conductivity and QTc interval during development [8, 9]. More recently, a thorough QT (TQT) study has been defined as a requirement for the clinical evaluation of the pro-arrhythmic potential of non-antiarrhythmic drugs [10].
From a methodological perspective, the TQT protocol design relies on the use of a positive control as well as supratherapeutic doses of the investigational drug to assess QT/QTc interval prolongation. Whilst the sensitivity of the proposed data analysis methods has been the subject of debate in numerous publications, less attention has been paid to the implications of overlooking the underlying concentration–QTc effect relationship and consequently to the availability of supporting evidence for extrapolation of the findings to real-life conditions [11, 12].
Taking into account PKPD relationships, we explore the implications of the known differences between clinical trial populations and real-life conditions, with special focus on the potential impact of protocol inclusion and exclusion criteria. Although it may be understandable and desirable to characterize the safety profile of a drug in healthy subjects, we show how inclusion/exclusion criteria lead to a somewhat biased estimation of the drug effects in the patient population who will receive the treatment after approval. In particular, the exclusion of severely ill subgroups or those with higher risk factors for Torsade de Pointes is usually not factored in a quantitative manner when evaluating clinical trial results. These subgroups include females, elderly subjects, those with predisposed cardiac or noncardiac diseases associated with diminished repolarization reserve, those with pharmacogenetic defects of drug-metabolising enzymes or pharmacological targets such as the potassium channels, those susceptible to bradycardia or electrolyte imbalance, or those receiving drugs with a potential for pharmacokinetic or pharmacodynamic interactions [13, 14]. In addition, little importance has been ascribed to the role of other factors contributing to QTc interval prolongation in the patient population, in comparison to the observed drug effect in healthy subjects [12, 15]. Consequently, no formal procedures exist to mitigate the impact of such differences or support the management of cardiovascular risk in the target population.
In fact, from a clinical standpoint one should raise questions about the reliability of the inferences made from pre-approval clinical trials. Given the patient population enrolled, the background noise and the relatively low frequency of clinically relevant arrhythmogenic effects, the signal arising from clinical trials may or may not accurately detect the rate and extent of QTc interval prolongation [1]. Therefore, sponsors, regulatory agencies and other stakeholders need to consider how best to predict the pro-arrhythmic liability of a medicinal product, taking into account the contribution of other factors in the target population in real-life conditions.
Using d,l-sotalol as an example compound, the primary objective of this investigation is to characterize the differences in QTc interval following administration of therapeutic doses of this compound to healthy subjects in clinical trials and to patients in real life. Based on imputation procedures, we subsequently use the predicted concentration–effect relationship to identify the effect of other influential covariates contributing to QTc interval prolongation in patients. Our approach relies on hierarchical modelling and simulation principles as a tool for evidence synthesis in the evaluation of drug safety [16, 17]. In contrast to previous applications of modelling and simulation [18–20], we show how simulations can be used to explore the role of design factors that have been omitted or excluded from a randomized trial. Thus, we anticipate that our approach will represent a natural extension of ongoing efforts within the pharmaceutical industry to improve safety signal detection [21–25].
Methods
Real-life study population
The real-life population setting was derived from the Rotterdam Study. The study is a prospective population-based cohort study, which started with a baseline visit between 1990 and 1993. All inhabitants of a suburb in Rotterdam, Ommoord, aged 55 years and over were invited to participate. Of the 10 275 subjects invited, 7983 (78%) gave their written informed consent and took part in the baseline examination. The study was approved by the Medical Ethics Committee of the Erasmus Medical Center, Rotterdam, The Netherlands. Objectives and methods of investigation have been described in detail in other publications [26]. All participants were visited at home for a standardized questionnaire, and 7151 were subsequently examined at the research centre. A second, third and fourth follow-up visit took place, respectively, during 1993–1995, 1997–1999 and 2002–2004. In addition to follow-up examinations, the cohort is continuously monitored for major morbidity and mortality through linkage with general practitioner and municipality records. Drug prescriptions dispensed to participants by automated pharmacies have been routinely stored in the database since 1 January 1991. For the present study, we included all participants in the Rotterdam Study with at least two consecutive ECG assessments.
All subjects who started treatment with d,l-sotalol during follow-up were included in the present study, with the exception of prevalent users and subjects with left ventricular hypertrophy, right bundle-branch block and left bundle-branch block, who were excluded. The reason for the exclusion of these patients is that left ventricular hypertrophy, right bundle-branch block and left bundle-branch block can cause secondary repolarization changes and atrial fibrillation can cause difficulties in measuring QT intervals. From the remaining population, ‘new’ sotalol users were identified as those receiving their first prescription after enrolment into the study. Our analysis included all QTc measurements after the start of sotalol treatment for all new users.
During the research centre visit, nonfasting blood samples were obtained [27]. Body mass index was computed as weight divided by height squared. Co-morbidities were screened and identified according to standard clinical criteria. Hypertension was defined as systolic blood pressure >160 mmHg or diastolic blood pressure >100 mmHg and/or use of antihypertensive medications, encompassing grade 2 and grade 3 hypertension compounds, according to the World Health Organization criteria [28]. Diabetes mellitus was defined as the use of blood glucose-lowering medication and/or a nonfasting or postload serum glucose level of 11.1 nmol l−1 or higher, according to the World Health Organization [29]. A history of myocardial infarction was assessed by self-report checked with records from the general practitioner or cardiologist and/or electrocardiographic evidence. All reported myocardial infarctions were verified against the medical records as described in detail previously [30]. Assessment of heart failure at baseline and during follow-up was assessed by reviewing all medical records for the occurrence of at least two signs and symptoms suggestive of the disease or the use of medication for heart failure and hospital discharge letters. Cases of incident heart failure were obtained by continuously monitoring the participants [31, 32]. The ankle brachial pressure index was used as potential predictor of cardiovascular diseases and mortality [33].
Healthy subject population
A study on healthy subjects using d,l-sotalol was selected from GlaxoSmithKline's clinical data repository (study number EXP20001). The primary objective of this study was to assess the role of intrinsic and extrinsic factors on the variability of QTc interval and was powered to detect a 10 ms increase in QTc. The study had a placebo-controlled, three-way crossover design, with two placebo periods and one active treatment period. Blood concentrations were taken at −30, 5, 15, 30 and 45 min relative to dosing time, as well as 1, 2, 4, 8, 10, 18 and 24 h after the dose. The study population comprised 30 subjects (12 females and 18 males) between the ages of 19 and 47 years.
Relevant eligibility criteria for inclusion into [items (i)–(iii)] and exclusion from [items (iv)–(viii)] the study for the purposes of our investigation were as follows: (i) age between 18 and 55 years, inclusive; (ii) nonsmoker (subject must have been a nonsmoker for at least 3 months prior to screening); (iii) body mass index between 19 and 30 kg m−2, with a weight of 50–95 kg, inclusive; (iv) at screening, ECG recording showed abnormal QRST complex morphology, sinus bradycardia (heart rate <45 beats min−1, PR interval >210 ms) or QTc interval values above 420 or 440 ms for males and females, respectively; (v) any medical history which was contra-indicated in the SOTACOR™ product label; (vi) history of hypertension, asthma, bronchial hyperactivity or peripheral vascular disease, including diabetes; (vii) the subject had a history of alcohol or drug abuse; and (viii) the subject was positive for HIV, hepatitis C or hepatitis B surface antigen.
Drug-induced QTc interval prolongation
The simulation of QTc intervals was performed according to a two-step approach. First, drug concentrations in the target population (i.e. sotalol users) were derived using a pharmacokinetic model. The QTc intervals were subsequently obtained using final population pharmacokinetic–pharmacodynamic (PKPD) parameter estimates.
The pharmacokinetics of d,l-sotalol was described by a two-compartment, first-order absorption pharmacokinetic model, with weight as a covariate on clearance, using nonlinear mixed-effects modelling, as implemented in NONMEM (v5.1; ICON Development Solutions, Ellicott City, MD, USA) [34]. Heart rate-corrected QT intervals were described by a PKPD model, in which physiological (QT and RR) and drug-specific (concentration) parameters are parameterized in an independent manner (equation 1) [35]. A detailed description of the model development was described in a previous publication [11].
| (equation 1) |
where QT0 (in milliseconds) is the intercept of the QT–RR relationship (sex was included as a covariate for this parameter whenever applicable, RR (in milliseconds) is the interval between successive R waves, α is the individual heart rate correction factor, A (in milliseconds) is the amplitude of the circadian rhythm, t is the clock time, φ is the phase, slope (in milliseconds per nanogram per millilitre) is the linear pharmacodynamic relationship, and C is the predicted concentration of drug at the time of QT interval measurements. It should be noted that we have chosen to express the QTc interval in milliseconds, as accepted in clinical practice. However, dimensional analysis of the formula, taking into account the heart rate variation, shows that the proper dimensions of QTc should be (milliseconds)1/2, not milliseconds. Ambiguities involving formulae in terms of heart rate are eliminated by expressing them in terms of RR. When viewed from the physical rather than the statistical perspective, the formulae for QT as a function of RR are equivalent to the duration of the systole as a function of heart rate [36].
The effect of drug-induced QTc prolongation in the Rotterdam Study population was then simulated using the PKPD model, taking into account the demographic and clinical characteristics of the patients in this cohort. Population mean estimates were used for the simulations (slope = 0.02 ms ng−1 ml−1 and intercept (QT0) = 380 ms), with the exception of the drug concentrations, which were simulated using the individual body weight of the patients. Full compliance to treatment was assumed for the purposes of our analysis. Finally, QTc values in the Rotterdam Study population were compared nonparametrically with the observed data.
Electrocardiographic measurements
For the Rotterdam Study cohort, a 10 s 12-lead ECG, resulting on average in 8–10 heart beats, was recorded with an ACTA electrocardiograph (ESAOTE, Florence, Italy) at a sampling frequency of 500 Hz and stored digitally. All ECGs were processed by the Modular ECG Analysis System (MEANS) to obtain the ECG parameters of interest. The MEANS program has been evaluated extensively [37, 38]. MEANS determines common onset and offset for all 12 leads together for one representative averaged beat, with the use of template matching techniques, until the end of the T wave. To adjust for heart rate, Bazett's formula (QTc = QT/RR1/2) was used [39]. A total of 1387 digitally stored ECG records were used for this study.
In contrast, ECG measurements in healthy volunteers were recorded in triplicate and read manually from lead II by a blinded cardiographer. The average value from the triplicates was used for data analysis and development of the PKPD model.
Comparison between controlled trials and real-life conditions
After applying the target population selection criteria to the Rotterdam Study, 608 subjects were classified as ‘new’ sotalol users without left ventricular hypertrophy, right bundle-branch block or left bundle-branch block. The total accumulated follow-up was 4864 years (i.e. a mean ± SD of 8 ± 1.5 years per subject), during which they all had at least one ECG measurement. At the end, there were a total of 1375 available ECGs, with an average of two per person. Inclusion and exclusion criteria from the phase I sotalol trial were then applied to this subset to allow further characterization of the differences between the real-life conditions and the clinical trial protocol.
Statistical analysis
Observed and expected QTc intervals were nonparametrically compared visually and statistically, by determining the significance of the differences between the two distributions. Specifically, the Wilcoxon signed-rank test was used to evaluate whether the distributions differed significantly from each other, with a P value < 0.05.
The effect of influential covariates was assessed by the changes in relative risk. The relative risk together with the 95% confidence interval of the association between ‘prolonged’ QTc, defined as >450 ms for males and >470 ms for females, and concomitant use of medications and presence of co-morbidities while on sotalol prescription were estimated using a Cox's proportional hazards analysis [40]. At each ECG measurement, all information concerning medications and co-morbidities was updated. Repeated measurements in a given subject were treated independently, because within-subject variability could not be considered in this analysis.
Results
The use of inclusion and exclusion criteria from the phase I trial for the subset of patients selected from the Rotterdam Study revealed important differences between clinical trial protocols and real-life conditions. Figure 1 shows that 10.8% of the male population and 3.4% of the female population from the Rotterdam Study would have been excluded based on their body weight. Likewise, 21.9% of male and 14.9% of female subjects would not be included due to their baseline QT interval measurements (Figure 2).
Figure 1.
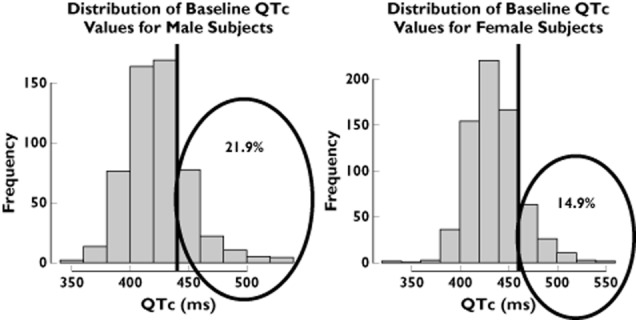
Baseline QTc interval distribution of males and females in the Rotterdam Study. The vertical lines represent the inclusion and exclusion cut-off values for healthy subjects in a thorough QT study. Numbers in the encircled area indicate the proportion of patients in the tails of the distribution
Figure 2.
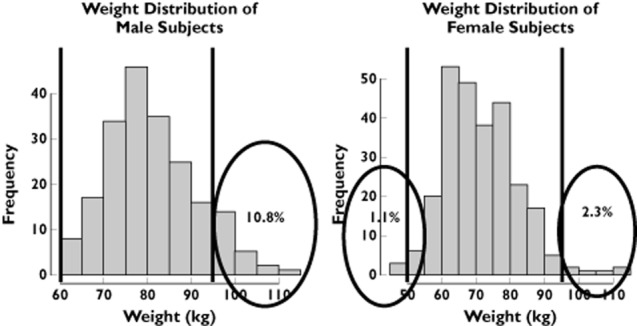
Weight distribution of males and females in the Rotterdam Study. The vertical lines represent the inclusion and exclusion cut-off values for healthy subjects in a thorough QT study. Numbers in the encircled area indicate the proportion of patients in the tails of the distribution
Pharmacokinetic and PKPD model parameters were used to test whether drug effects alone could explain the observed QTc values in the observational real-life cohort. The simulated QTc intervals (i.e. the expected drug-induced QTc interval prolongation) were found to differ from the observed values (Figure 3), especially in the male population. Using the Wilcoxon signed-rank test, our analysis shows that the two distributions were significantly different from each other in both genders (P < 0.05). The distribution of observed QTc values in sotalol users in the elderly population revealed some dangerously high measurements. These observations contrast with model-based predictions, suggesting that drug-induced effects alone cannot describe the observed QTc interval.
Figure 3.
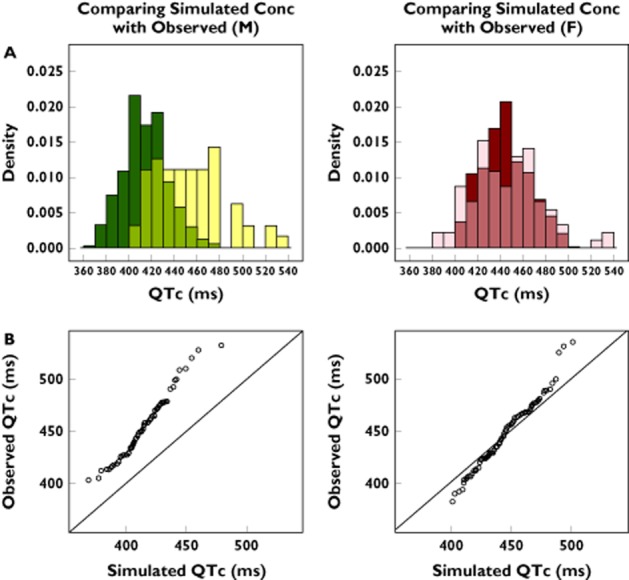
(A) Not-in-trial simulation results show overlapping distributions and discrepancies between observed and predicted QTc interval in the male (M) (left panel) and female (F) population (right panel). The darker colours represent the predicted drug-induced QTc values and the light colours represent the observed overall QTc intervals. The medium shades denote the overlapping areas. (B) QQ plots comparing the distributions of the QTc values for the male (left panel) and female population (right panel). The deviation from the line of identity reflects the residual difference between the observed QTc intervals and model-predicted sotalol effects under the assumption of comparable pharmacokinetic–pharmacodynamic relationship, as determined in phase I clinical trials
Further evaluation of the differences between the two populations was performed by binary logistic regression, which revealed the contribution of concomitant medications and co-morbidities as QTc-prolonging factors (Table 1). As expected, a 4.0-fold increase in relative risk for QTc interval prolongation was found in sotalol users, in comparison to nonsotalol users. In contrast, it was also clear that in the absence of any drug treatment, co-morbidities caused a significant increase in the relative risk for QTc interval prolongation (Table 1), which ranged in ascending order from 3.4 to 7.4 times for myocardial infarction, diabetes, heart failure and hypertension. The prescription of sotalol modifies the underlying relative risks. A reduction in the impact caused by the co-morbidities was observed for heart failure and hypertension, whilst a further increase was seen for diabetes (1.62 times higher) and myocardial infarction (3.87 times higher). The magnitude of the interaction between treatment and co-morbidities cannot be explained by differences in the pharmacokinetics or pharmacodynamics of sotalol.
Table 1.
Results of the relative risk calculations for prolonged QTc interval, as defined in epidemiological cohorts
| Co-morbidity and corresponding co-medications | Relative risk (95% confidence interval) in sotalol nonusers | P value | Relative risk (95% confidence interval) in sotalol users | P value |
|---|---|---|---|---|
| Using sotalol alone | – | <0.01 | 4.0 (2.7–5.8) | <0.01 |
| Diabetes | 4.0 (2.7–5.8) | <0.01 | 6.5 (1.6–27.1) | <0.01 |
| Heart failure | 4.4 (3.0–6.6) | <0.01 | 0.8 (0.2–3.4) | <0.01 |
| Hypertension | 7.4 (4.3–12.7) | <0.01 | 2.4 (1.3–4.2) | <0.01 |
| Myocardial infarction | 3.4 (2.3–5.127) | <0.01 | 15.5 (4.9–49.3) | <0.01 |
The effect of sotalol alone, i.e. drug-specific effect, is shown on the first row. Significance level was set at P < 0.05.
Discussion
Our results show that the population selection and inclusion/exclusion criteria applied to clinical protocols in early drug development may lead to significant differences between drug-induced and overall treatment effect size in the target population. Thus far, the evidence of pro-arrhythmic effects in real-life patients has been primarily assigned to the pharmacological treatment, i.e. to drug effects. The present investigation strongly suggests that the signal detected during a TQT study does not accurately predict the interaction between drug effects and other disease-related factors, which together may lead to further increase in the cardiovascular risk associated with the arrhythmogenic effects of a drug.
The use of a model-based approach enables us to make inferences about drug exposure in patients and to evaluate, in an integrated manner, how different covariates and other sources of variability affect the observed QTc values in real-life patients. Our results indicate that other causal factors are present, which significantly affect the observed QTc values in uncontrolled, real-life conditions. In fact, our findings are in agreement with previous publications, which show that heart failure, hypertension, diabetes and myocardial infarction increase the risk of QTc prolongation [41–44]. However, the absolute changes in relative risk associated with co-morbidities varied considerably when considered together with the use of sotalol. A clear reduction in the relative risk was observed for hypertension and heart failure, with further increases for diabetes and myocardial infarction. Given that patients were also being prescribed treatment for these co-morbidities, a distinction between the effect of disease and other drugs is not possible. Furthermore, in this specific case, no evidence exists to support that the prescribed co-medications could lead to potential pharmacokinetic and/or pharmacodynamic interactions with sotalol.
The concepts outlined in the present investigation also show how clinical trial and epidemiological data can be used in an integrated manner for the purpose of signal detection and improved risk management. Our findings appear to confirm the points highlighted by Black on ‘the false conflict between those who advocate randomized trials in all situations and those who believe observational data provide sufficient evidence needs to be replaced with mutual recognition of the complementary roles of the two approaches’ [45]. Others have also advocated the synergistic potential for using both kinds of data to aid decision making [46–49]. In the design of this study, we applied a parametric approach, in which simulations played a central role. As a matter of fact, this is the first time that simulations based on nonlinear hierarchical models have been used to characterize the implications of exclusion criteria on the overall safety profile.
By applying PKPD modelling concepts to epidemiological data, we have also shown that the PKPD relationships derived from healthy volunteers do not necessarily describe or predict what will happen in real-life conditions. New approaches are required to assess the impact of QTc prolongation, taking into account not only the drug effects. This prerequisite clearly challenges the validity of the TQT study as the state-of-the-art approach for determining the risk of pro-arrhythmia or Torsade de Pointes. In this context, an important advantage of the use of PKPD relationships is that even if pharmacokinetics has been evaluated at supratherapeutic levels, drug exposure can be extrapolated to reflect drug concentrations observed after therapeutic doses, so that accurate inferences and recommendations can be made about the potential drug effects on QTc interval.
Indirectly, our results highlight the importance of assessing the clinical implications of adverse drug effects in vulnerable or high-risk population subgroups (e.g. drug–drug interactions) in a more quantitative manner. We therefore anticipate the use of our approach to assess treatment effects in these subgroups by introducing such patients into simulation scenarios.
Despite the potential impact of a tool for not-in-trial simulations, the proposed approach in this investigation has some limitations. First, it should be noted that population estimated parameters rather than the individual estimates were used in the simulations. This reduces the overall interindividual variability that exists within a population. In addition, an assumption was made that all the subjects in the Rotterdam Study were fully compliant with medication consumption so that the same plasma concentration range was reached as in the subjects in the clinical trial. Differences in the procedures, timing and equipment used for ECG monitoring in the Rotterdam Study and in the clinical trial were assumed to contribute to increased random variation without introducing significant bias. The implications of the aforementioned assumptions cannot be fully evaluated with the existing data. An external validation approach is desirable and required to dismiss potential biases and confounders. We have therefore initiated a follow-up research protocol, in which the same approach is applied to another cohort, including different study drugs.
In summary, the concept of not-in-trial simulations has been introduced as a tool for the evaluation of QTc interval prolongation in real-life conditions, integrating PKPD relationships as the basis for differentiating drug-specific properties from other relevant factors in noncontrolled settings. Moreover, we have shown that co-morbidities can have a significant contribution to the observed QT prolongation in the overall population. The impact of such factors cannot be derived empirically from a TQT study in healthy subjects.
Acknowledgments
We would like to thank Dr Jan Kors from the Department of Medical Informatics and Dr Jacqueline Witteman from the Department of Epidemiology of the Erasmus University Medical Center for their invaluable contribution towards the data used for the work presented here.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: A.S.Y.C. had financial support from the Top Institute Pharma (a tripartite consortium sponsored by The Netherlands Ministry of Education, Culture and Science) for the submitted work; no financial relationships with any organizations that might have an interest in the submitted work in the previous 3 years; no other relationships or activities that could appear to have influenced the submitted work.
References
- 1.Shah RR. Drug-induced QT interval prolongation: regulatory perspectives and drug development. Ann Med. 2004;36(Suppl. 1):47–52. doi: 10.1080/17431380410032445. [DOI] [PubMed] [Google Scholar]
- 2.Yap YG, Camm AJ. Drug induced QT prolongation and torsades de pointes. Heart. 2003;89:1363–1372. doi: 10.1136/heart.89.11.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dessertenne F. [Ventricular tachycardia with 2 variable opposing foci] Arch Mal Coeur Vaiss. 1966;59:263–272. [PubMed] [Google Scholar]
- 4.Straus SM, Kors JA, De Bruin ML, van der Hooft CS, Hofman A, Heeringa J, Deckers JW, Kingma JH, Sturkenboom MC, Stricker BH, Witteman JC. Prolonged QTc interval and risk of sudden cardiac death in a population of older adults. J Am Coll Cardiol. 2006;47:362–367. doi: 10.1016/j.jacc.2005.08.067. [DOI] [PubMed] [Google Scholar]
- 5.Straus SM, Sturkenboom MC, Bleumink GS, Dieleman JP, van der Lei J, de Graeff PA, Kingma JH, Stricker BH. Non-cardiac QTc-prolonging drugs and the risk of sudden cardiac death. Eur Heart J. 2005;26:2007–2012. doi: 10.1093/eurheartj/ehi312. [DOI] [PubMed] [Google Scholar]
- 6.Darpo B, Nebout T, Sager PT. Clinical evaluation of QT/QTc prolongation and proarrhythmic potential for nonantiarrhythmic drugs: the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use. E14 guideline. J Clin Pharmacol. 2006;46:498–507. doi: 10.1177/0091270006286436. [DOI] [PubMed] [Google Scholar]
- 7.Shah RR. Drug-induced prolongation of the QT interval: regulatory dilemmas and implications for approval and labelling of a new chemical entity. Fundam Clin Pharmacol. 2002;16:147–156. doi: 10.1046/j.1472-8206.2002.00083.x. [DOI] [PubMed] [Google Scholar]
- 8.Anonymous Points to Consider: the assessmet of the potential for QT interval prolongation by non-cardiovascular medicinal products. In: edProducts CfPM, London, 1997.
- 9.Anonymous Note for Guidance on the Investigation of Drug Interactions. In: edProducts CfPM, London, 1997.
- 10.USA Food and Drug Administration. 2005. Guidance for Industry: E14 Clinical Evaluation of QT/QTc Interval Prolongation and Proarrythmic Potential for Non-Antiarrythmic Drugs.
- 11.Chain AS, Krudys KM, Danhof M, Della Pasqua O. Assessing the Probability of Drug-induced QTc-interval prolongation during clinical drug development. Clin Pharmacol Ther. 2011;90:867–875. doi: 10.1038/clpt.2011.202. [DOI] [PubMed] [Google Scholar]
- 12.Garnett CE, Beasley N, Bhattaram VA, Jadhav PR, Madabushi R, Stockbridge N, Tornoe CW, Wang Y, Zhu H, Gobburu JV. Concentration-QT relationships play a key role in the evaluation of proarrhythmic risk during regulatory review. J Clin Pharmacol. 2008;48:13–18. doi: 10.1177/0091270007307881. [DOI] [PubMed] [Google Scholar]
- 13.Letsas KP, Efremidis M, Kounas SP, Pappas LK, Gavrielatos G, Alexanian IP, Dimopoulos NP, Filippatos GS, Sideris A, Kardaras F. Clinical characteristics of patients with drug-induced QT interval prolongation and torsade de pointes: identification of risk factors. Clin Res Cardiol. 2009;98:208–212. doi: 10.1007/s00392-008-0741-y. [DOI] [PubMed] [Google Scholar]
- 14.Wolbrette DL. Drugs that cause Torsades de pointes and increase the risk of sudden cardiac death. Curr Cardiol Rep. 2004;6:379–384. doi: 10.1007/s11886-004-0041-8. [DOI] [PubMed] [Google Scholar]
- 15.Bonate PL, Russell T. Assessment of QTc prolongation for non-cardiac-related drugs from a drug development perspective. J Clin Pharmacol. 1999;39:349–358. doi: 10.1177/00912709922007912. [DOI] [PubMed] [Google Scholar]
- 16.USA Food and Drug Administration. Innovation or stagnation? Challenge and opportunity on the critical path to new medical products. In: edServices DoHaH, 2004.
- 17.Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov. 2004;3:711–715. doi: 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- 18.Chan PL, Holford NH. Drug treatment effects on disease progression. Annu Rev Pharmacol Toxicol. 2001;41:625–659. doi: 10.1146/annurev.pharmtox.41.1.625. [DOI] [PubMed] [Google Scholar]
- 19.Gobburu JV, Marroum PJ. Utilisation of pharmacokinetic-pharmacodynamic modelling and simulation in regulatory decision-making. Clin Pharmacokinet. 2001;40:883–892. doi: 10.2165/00003088-200140120-00001. [DOI] [PubMed] [Google Scholar]
- 20.Kimko HHC, Peck CC. Clinical Trial Simulations: Applications and Trends. New York: Springer; 2011. [Google Scholar]
- 21.DiMasi JA, Feldman L, Seckler A, Wilson A. Trends in risks associated with new drug development: success rates for investigational drugs. Clin Pharmacol Ther. 2010;87:272–277. doi: 10.1038/clpt.2009.295. [DOI] [PubMed] [Google Scholar]
- 22.Lalonde RL, Kowalski KG, Hutmacher MM, Ewy W, Nichols DJ, Milligan PA, Corrigan BW, Lockwood PA, Marshall SA, Benincosa LJ, Tensfeldt TG, Parivar K, Amantea M, Glue P, Koide H, Miller R. Model-based drug development. Clin Pharmacol Ther. 2007;82:21–32. doi: 10.1038/sj.clpt.6100235. [DOI] [PubMed] [Google Scholar]
- 23.Laverty H, Benson C, Cartwright E, Cross M, Garland C, Hammond T, Holloway C, McMahon N, Milligan J, Park B, Pirmohamed M, Pollard C, Radford J, Roome N, Sager P, Singh S, Suter T, Suter W, Trafford A, Volders P, Wallis R, Weaver R, York M, Valentin J. How can we improve our understanding of cardiovascular safety liabilities to develop safer medicines? Br J Pharmacol. 2011;163:675–693. doi: 10.1111/j.1476-5381.2011.01255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pater C. Methodological considerations in the design of trials for safety assessment of new drugs and chemical entities. Curr Control Trials Cardiovasc Med. 2005;6:1. doi: 10.1186/1468-6708-6-1. DOI: 10.1186/1468-6708-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pollard CE, Valentin JP, Hammond TG. Strategies to reduce the risk of drug-induced QT interval prolongation: a pharmaceutical company perspective. Br J Pharmacol. 2008;154:1538–1543. doi: 10.1038/bjp.2008.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hofman A, Grobbee DE, de Jong PT, van den Ouweland FA. Determinants of disease and disability in the elderly: the Rotterdam Elderly Study. Eur J Epidemiol. 1991;7:403–422. doi: 10.1007/BF00145007. [DOI] [PubMed] [Google Scholar]
- 27.van Gent CM, van der Voort HA, de Bruyn AM, Klein F. Cholesterol determinations. A comparative study of methods with special reference to enzymatic procedures. Clin Chim Acta. 1977;75:243–251. doi: 10.1016/0009-8981(77)90195-4. [DOI] [PubMed] [Google Scholar]
- 28.Guidelines Subcommittee. 1999 World Health Organization-International Society of Hypertension Guidelines for the Management of Hypertension. J Hypertens. 1999;17:151–183. [PubMed] [Google Scholar]
- 29.Diabetes Mellitus. Technical Reports Series 894. Geneva, 1985.
- 30.Bots ML, Hoes AW, Koudstaal PJ, Hofman A, Grobbee DE. Common carotid intima-media thickness and risk of stroke and myocardial infarction: the Rotterdam Study. Circulation. 1997;96:1432–1437. doi: 10.1161/01.cir.96.5.1432. [DOI] [PubMed] [Google Scholar]
- 31.Bleumink GS, Knetsch AM, Sturkenboom MC, Straus SM, Hofman A, Deckers JW, Witteman JC, Stricker BH. Quantifying the heart failure epidemic: prevalence, incidence rate, lifetime risk and prognosis of heart failure The Rotterdam Study. Eur Heart J. 2004;25:1614–1619. doi: 10.1016/j.ehj.2004.06.038. [DOI] [PubMed] [Google Scholar]
- 32.Mosterd A, Hoes AW, de Bruyne MC, Deckers JW, Linker DT, Hofman A, Grobbee DE. Prevalence of heart failure and left ventricular dysfunction in the general population; The Rotterdam Study. Eur Heart J. 1999;20:447–455. [PubMed] [Google Scholar]
- 33.World Health Organization. International Classification of Diseases and Health Related Problems. Geneva: World Health Organization; 1992. [Google Scholar]
- 34.Beal S, Sheiner L. NONMEM user's guide In: NONMEM project group, San Francisco, 1999.
- 35.Lunn DJ, Thomas A, Best N, Spiegelhalter D. WinBUGS – a Bayesian modelling framework: concepts, structure, and extensibility. Stat Comput. 2000;10:325–337. [Google Scholar]
- 36.Kovács SJ., Jr The duration of the QT interval as a function of heart rate: a derivation based on physical principles and a comparison to measured values. Am Heart J. 1985;110:872–878. doi: 10.1016/0002-8703(85)90472-7. [DOI] [PubMed] [Google Scholar]
- 37.de Bruyne MC, Kors JA, Hoes AW, Kruijssen DA, Deckers JW, Grosfeld M, van Herpen G, Grobbee DE, van Bemmel JH. Diagnostic interpretation of electrocardiograms in population-based research: computer program research physicians, or cardiologists? J Clin Epidemiol. 1997;50:947–952. doi: 10.1016/s0895-4356(97)00100-5. [DOI] [PubMed] [Google Scholar]
- 38.Willems JL, Abreu-Lima C, Arnaud P, van Bemmel JH, Brohet C, Degani R, Denis B, Gehring J, Graham I, van Herpen G, Machado H, Macfarlane PW, Michaelis, Moulopoulos SD, Rubel P, Zywietz C. The diagnostic performance of computer programs for the interpretation of electrocardiograms. N Engl J Med. 1991;325:1767–1773. doi: 10.1056/NEJM199112193252503. [DOI] [PubMed] [Google Scholar]
- 39.Bazett HC. The time relations of the blood-pressure changes after excision of the adrenal glands, with some observations on blood volume changes. J Physiol. 1920;53:320–339. doi: 10.1113/jphysiol.1920.sp001881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cox DR. Ression models and life-tables. J R Stat Soc Series B Stat Methodol. 1972;34:187–220. [Google Scholar]
- 41.Choy AM, Darbar D, Dell'Orto S, Roden DM. Exaggerated QT prolongation after cardioversion of atrial fibrillation. J Am Coll Cardiol. 1999;34:396–401. doi: 10.1016/s0735-1097(99)00226-0. [DOI] [PubMed] [Google Scholar]
- 42.Makkar RR, Fromm BS, Steinman RT, Meissner MD, Lehmann MH. Female gender as a risk factor for torsades de pointes associated with cardiovascular drugs. JAMA. 1993;270:2590–2597. doi: 10.1001/jama.270.21.2590. [DOI] [PubMed] [Google Scholar]
- 43.Nowinski K, Gadler F, Jensen-Urstad M, Bergfeldt L. Transient proarrhythmic state following atrioventricular junction radiofrequency ablation: pathophysiologic mechanisms and recommendations for management. Am J Med. 2002;113:596–602. doi: 10.1016/s0002-9343(02)01274-3. [DOI] [PubMed] [Google Scholar]
- 44.Torp-Pedersen C, Moller M, Bloch-Thomsen PE, Kober L, Sandoe E, Egstrup K, Agner E, Carlsen J, Videbaek J, Marchant B, Camm AJ. Dofetilide in patients with congestive heart failure and left ventricular dysfunction. Danish Investigations of Arrhythmia and Mortality on Dofetilide Study Group. N Engl J Med. 1999;341:857–865. doi: 10.1056/NEJM199909163411201. [DOI] [PubMed] [Google Scholar]
- 45.Black N. Why we need observational studies to evaluate the effectiveness of health care. BMJ. 1996;312:1215–1218. doi: 10.1136/bmj.312.7040.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Atkins D. Creating and synthesizing evidence with decision makers in mind: integrating evidence from clinical trials and other study designs. Med Care. 2007;45:S16–22. doi: 10.1097/MLR.0b013e3180616c3f. [DOI] [PubMed] [Google Scholar]
- 47.Hannan EL. Randomized clinical trials and observational studies: guidelines for assessing respective strengths and limitations. JACC Cardiovasc Interv. 2008;1:211–217. doi: 10.1016/j.jcin.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 48.Landewe R, van der Heijde D. Primer: challenges in randomized and observational studies. Nat Clin Pract Rheumatol. 2007;3:661–666. doi: 10.1038/ncprheum0626. [DOI] [PubMed] [Google Scholar]
- 49.Yang W, Zilov A, Soewondo P, Bech OM, Sekkal F, Home PD. Observational studies: going beyond the boundaries of randomized controlled trials. Diabetes Res Clin Pract. 2010;88(Suppl. 1):S3–9. doi: 10.1016/S0168-8227(10)70002-4. [DOI] [PubMed] [Google Scholar]