

Abstract
Deregulation of the phosphatidylinositide 3-kinase (PI3K) and mammalian target of rapamycin (mTOR) signaling pathway occurs frequently in a wide range of human cancers and is a major driving force in tumorigenesis. Thus, small molecules targeting this pathway are under active development as anticancer therapeutics. Although small-molecule inhibitors of the PI3K-mTOR pathway have shown promising clinical efficacy against human cancers, the emergence of drug resistance may limit their success in the clinic. To date, several resistance mechanisms, including both PI3K-dependent and -independent mechanisms, have been described. Here, we summarize the current understanding of resistance mechanisms to PI3K-mTOR inhibitors and discuss potential strategies for overcoming resistance for potential clinical application.
Keywords: PI3K-mTOR pathway, drug resistance, PDK1, MYC, targeted therapy
The phosphatidylinositide 3-kinase and mammalian target of rapamycin (PI3K-mTOR) pathway regulates cell growth, proliferation, and metabolism and is a deregulated signaling pathway in human cancer[1],[2]. Genetic aberrations in this pathway, such as activating mutations of phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha (PIK3CA) or inactivating mutations of phosphatase and tensin homolog (PTEN), occur in most epithelial tumors[3],[4], leading to constitutive hyperactivation of downsteam effector kinases such as AKT (protein kinase B) and mTOR[5],[6]. In addition to the alterations of upstream activators, genetic amplification of downstream effectors, such as eukaryotic initiation factors 4E (eIF4E), p70 ribosomal protein S6 kinase1 (S6K1), and eukaryotic translation initiation factor 4E binding protein 1 (4E-BP1), also result in enhanced mTOR activation, which contributes to its hyperactivity in human cancers[7],[8]. Because enhanced activity of the mTOR pathway is frequently observed in cancer cells, inhibition of mTOR signaling has become a viable and attractive option for molecular targeted therapy in human cancer.
Mechanisms of Resistance to PI3K-mTOR Inhibitors
PI3K-dependent feedback mechanism of resistance to mTOR inhibitors
Several small-molecule inhibitors of the PI3K-mTOR pathway have entered preclinical and clinical development as anticancer therapeutics. These inhibitors, including rapamycin and its analogs, have shown promising rationale for their use in cancer therapy[9]. However, clinical responses to these drugs in the absence of patient stratification are generally unpredictable, and patients often develop drug resistance, limiting clinical efficacy. The underlying molecular basis of resistance, either intrinsic or acquired, remains largely unknown and has emerged as a challenging question that needs to be addressed. To date, multiple mechanisms of resistance to kinase inhibitors have been described, including secondary target mutations, activation of parallel pro-survival signaling pathways, and amplification of downstream lesions in the same pathway. The well-documented mechanisms of rapamycin resistance are often linked to its context-dependent negative feedback loops. In one loop, mTORC1 inhibition leads to up-regulation of receptor tyrosine kinases (RTKs or substrates) such as platelet-derived growth factor receptors (PDGFRs) and insulin receptor substrate 1 (IRS-1), resulting in increased PI3K-dependent AKT phosphorylation at Ser473. In another loop, mTORC1 inhibition leads to PI3K-Ras activation, which leads to increased mitogen-activated protein kinase (MAPK) signaling[10]–[12]. The clinical relevance of these feedback mechanisms are also confirmed by their existence in cancer patients[13]. These findings provide a strong rationale for dual targeting of mTORC1 and PI3K activity to escape the feedback resistance. Consistent with this idea, several dual inhibitors of PI3K and mTOR activity have been developed, including PI-103 and NVP-BEZ235[14],[15]. These agents have shown greater anticancer efficacy compared with analogs of rapamycin (rapalogs). However, the clinical therapeutic efficacy and tolerability of dual PI3K-mTOR inhibitors remain to be examined. Second-generation ATP-competitive mTOR inhibitors, such as PP242 and Torin1, suppress the catalytic activities of both mTORC1 and mTORC2 by binding to the kinase domain[16]. Because mTORC2 leads to an increase in AKT activation[17], second-generation mTOR inhibitors, which can block the feedback activation of AKT and phosphorylation of 4E-BP1, may be superior to rapalogs. In breast cancer cells, rapalogs such as RAD001 can induce MAPK activation instead of AKT activation. In these cells, pharmacologic inhibition of the MAPK pathway has been shown to enhance the sensitivity of rapamycin both in vitro and in vivo[12]. As expected, pharmacologic or genetic inhibition of these pathways may overcome resistance to mTOR-targeted therapy.
Sensitivity to mTOR inhibitors is thought to be related to deregulation of critical upstream components of the PI3K-mTOR pathway. Results from preclinical models suggest that genetic alterations and aberrant gene expression drive pathway activation, which associates with sensitivity to mTOR inhibition. For example, HER2 amplification and PIK3CA activating mutations lead to elevated levels of phosphorylated AKT in many human cancers and have been identified in preclinical models as putative predictors of rapalog response[18]–[20]. However, the use of HER2 and PIK3CA alterations and AKT phosphorylation for predicting rapalog sensitivity has not been fully validated. This outcome may be due to the complexity of the PI3K-AKT-mTOR signaling network, which includes feedback loops and points of crosstalk with oncogenic signaling pathways that drive malignant transformation and determine the sensitivity of mTOR inhibitors. Thus, it is critical to understand genetic alterations in human tumors with acquired resistance to mTOR inhibitors for insight into additional resistance mechanisms. Such insight will provide new effective mTOR-targeted therapies for cancer patients.
MYC-dependent resistance to PI3K-mTOR-targeted therapy
In addition to activating PI3K-AKT and MAPK signaling, mTOR inhibition by rapamycin can also induce MYC phosphorylation and accumulation in colorectal cancer cells[21]. Functional investigations indicate that rapamycin-induced MYC phosphorylation is dependent on 3-phosphoinositide-dependent kinase 1 (PDK1) but independent of PI3K and AKT activity. We found that rapamycin-induced MYC activation is associated with the loss of PPP2R2B, which encodes for the B55β regulatory subunit of the serine/threonine protein phosphatase PP2A. Loss of PPP2R2B results in aberrant activation of PDK1, a master kinase often linked to AKT activation. We found that PDK1 inhibition by either gene knockdown or small-molecule kinase inhibitors markedly abolished MYC phosphorylation, leading to enhanced sensitivity to rapamycin in colon cancer cells, though it did not affect rapamycin-induced AKT phosphorylation. This suggests that mTOR inhibition may trigger a separate compensatory mechanism involving PDK1-MYC but not PI3K-AKT to attenuate rapamycin response. A role of MYC in mediating resistance to PI3K-mTOR inhibitors has also been reported in other models. For example, in a mouse model with established prostate cancer caused by either conditional deletion of PTEN or transgenic expression of MYC, tumors driven by MYC activation were highly resistant to NVP-BEZ235, a dual PI3K and mTORC1/2 inhibitor, compared with PTEN-deficient tumors[22]. Likewise, MYC amplification has been reported in PI3K-driven mammary tumors that recurred following treatment with GDC0941, a PI3K inhibitor[23],[24]. Further functional analysis indicates that MYC amplification contributed to this relapse and resistance through a PI3K pathway-independent manner[23]. These findings are also consistent with in vitro studies showing that MYC elevation is required to bypass pharmacologic inhibition of PI3K-mTOR with BEZ235 in breast cancer cells[24],[25]. These studies, along with our study, suggest that aberrant activation of MYC, either through increased phosphorylation or gene amplification, may contribute to acquired resistance to PI3K-mTOR-targeted therapy. Thus, combination therapies targeting both PI3K and MYC may be necessary to overcome resistance to PI3K-targeted therapy. Taken together, PI3K-mTOR inhibitors such as rapamycin, BEZ235, and GDC0941 induce either PI3K-dependent or MYC-dependent mechanisms, leading to acquired resistance to PI3K-mTOR-targeted therapy in cancer cells (Figure 1).
Figure 1. Potential mechanisms of resistance to PI3K-mTOR inhibitors in human cancer.
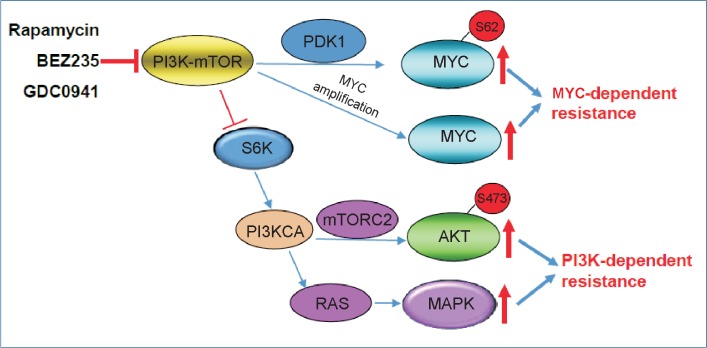
PI3K-mTOR inhibitors induce PI3K-dependent and/or MYC-dependent resistance mechanisms to PI3K-mTOR-targeted therapy. Targeting the PI3K-mTOR pathway causes MYC activation through PDK1-dependent MYC phosphorylation and MYC amplification, which is parallel to PIK3CA-dependent AKT and MAPK activation, attenuating therapeutic effect of PI3K-mTOR inhibitors. PI3K, phosphatidylinositide 3-kinase; mTOR, mammalian target of rapamycin; PDK1, 3-phosphoinositide-dependent kinase 1; PI3KCA, phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha; MAPK, mitogen-activated protein kinase.
Strategies for Therapeutic Targeting of MYC
Because MYC activation may be an important mechanism underlying resistance to PI3K-mTOR inhibitors, developing an effective therapeutic strategy for targeting MYC may be necessary to overcome this resistance. The MYC oncoprotein is involved in many critical processes in malignant cells, including proliferation, growth, differentiation, and metabolism[26]. Its role in cancer stem cell initiation and maintenance and its association with tumor recurrence following treatment indicate that MYC induction following PI3K-mTOR inhibition may be a serious problem in the clinic. Although MYC has been validated as an important cancer target, direct inhibition of MYC has not yet been clinically successful. Thus, alternative strategies targeting MYC dependency have been proposed. These strategies include targeting MYC expression, synthetic lethal targeting of MYC activation, and interfering with key MYC target genes[27]–[29]. For example, a strategy directed towards targeting MYC transcription has been recently established. Bromodomain-containing protein 4 (BRD4) is a member of the bromodomain and extra terminal (BET) family of proteins that recently emerged as potent regulator of MYC transcription in different tumor types. Deregulation of BRD4 is reported in multiple myeloma, and inhibiting BRD4 with a small-molecule inhibitor significantly down-regulated MYC transcription, leading to apoptosis[27],[30]. These studies suggest that targeting druggable upstream effectors of MYC is feasible in selected cancers.
Aberrant activation of PDK1 and the positive regulation of PDK1 towards MYC signaling in our study suggest that targeting PDK1 may become a useful treatment strategy for MYC-driven tumors. Our data show that ectopic expression of PDK1 induces MYC signaling and cellular transformation (data not published); conversely, pharmacologic or genetic removal of PDK1 reduces MYC signaling and alleviates rapamycin resistance. This notion was further illustrated using a small-molecule PDK1 inhibitor, BX912, which abolished rapamycin-induced MYC phosphorylation and thus synergized with rapamycin in colon cancer cells. In conjunction with these findings, genetic ablation of PDK1 in human colorectal cancer was reported to markedly reduce proliferation and metastasis[31]. In addition, PDK1 inhibitor has been shown to be an effective anti-cancer agent in vitro and in vivo[32],[33] and is currently under clinical development. To overcome the resistance of mTOR inhibitors in targeted therapy, a combination approach involving PDK1 inhibition might be appropriate to overcome MYC activation and resistance to mTOR inhibitors. Furthermore, preclinical trials based on xenograft animal studies should be carried out to check the antitumor effects of combined treatment of PDK1 inhibitor and mTOR inhibitor. Overall, these studies will provide a potential strategy to overcome the resistance to mTOR inhibitors in clinical applications by targeting MYC signaling in cancer therapy.
References
- 1.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 2.Wong KK, Engelman JA, Cantley LC. Targeting the PI3K signaling pathway in cancer. Curr Opin Genet Dev. 2010;20:87–90. doi: 10.1016/j.gde.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Samuels Y, Wang Z, Bardelli A, et al. High frequency of mutations of the PICK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 4.Li J, Yen C, Liaw D, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 5.Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 6.Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6:729–734. doi: 10.1038/nrc1974. [DOI] [PubMed] [Google Scholar]
- 7.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 8.Dowling RJ, Topisirovic I, Alain T, et al. mTORC1-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science. 2010;328:1172–1176. doi: 10.1126/science.1187532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guertin DA, Sabatini DM. The pharmacology of mTOR inhibition. Sci Signal. 2009;2:pe24. doi: 10.1126/scisignal.267pe24. [DOI] [PubMed] [Google Scholar]
- 10.Sarbassov DD, Ali SM, Sengupta S, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 11.Carracedo A, Baselga J, Pandolfi PP. Deconstructing feedback-signaling networks to improve anticancer therapy with mTORC1 inhibitors. Cell Cycle. 2008;7:3805–3809. doi: 10.4161/cc.7.24.7244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carracedo A, Ma L, Teruya-Feldstein J, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118:3065–3074. doi: 10.1172/JCI34739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O'Reilly KE, Rojo F, She QB, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–1508. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fan QW, Knight ZA, Goldenberg DD, et al. A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell. 2006;9:341–349. doi: 10.1016/j.ccr.2006.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maira SM, Stauffer F, Brueggen J, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7:1851–1863. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- 16.Janes MR, Limon JJ, So L, et al. Effective and selective targeting of leukemia cells using a TORC1/2 kinase inhibitor. Nat Med. 2010;16:205–213. doi: 10.1038/nm.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sarbassov DD, Guertin DA, Ali SM, et al. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 18.Hsieh AC, Costa M, Zollo O, et al. Genetic dissection of the oncogenic mTOR pathway reveals druggable addiction to translational control via 4EBP-EIF4E. Cancer Cell. 2010;17:249–261. doi: 10.1016/j.ccr.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Serra V, Markman B, Scaltriti M, et al. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res. 2008;68:8022–8030. doi: 10.1158/0008-5472.CAN-08-1385. [DOI] [PubMed] [Google Scholar]
- 20.Thomas GV, Tran C, Mellinghoff IK, et al. Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat Med. 2006;12:122–127. doi: 10.1038/nm1337. [DOI] [PubMed] [Google Scholar]
- 21.Tan J, Lee PL, Li Z, et al. B55beta-associated PP2A complex controls PDK1-directed myc signaling and modulates rapamycin sensitivity in colorectal cancer. Cancer Cell. 2010;18:459–471. doi: 10.1016/j.ccr.2010.10.021. [DOI] [PubMed] [Google Scholar]
- 22.Carver BS, Chapinski C, Wongvipat J, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. 2011;19:575–586. doi: 10.1016/j.ccr.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu P, Cheng H, Santiago S, et al. Oncogenic PIK3CA-driven mammary tumors frequently recur via PI3K pathway-dependent and PI3K pathway-independent mechanisms. Nat Med. 2011;17:1116–1120. doi: 10.1038/nm.2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ilic N, Utermark T, Widlund HR, et al. PI3K-targeted therapy can be evaded by gene amplification along the MYC-eukaryotic translation initiation factor 4E (eIF4E) axis. Proc Natl Acad Sci U S A. 2011;108:E699–708. doi: 10.1073/pnas.1108237108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muellner MK, Uras IZ, Gapp BV, et al. A chemical-genetic screen reveals a mechanism of resistance to PI3K inhibitors in cancer. Nat Chem Biol. 2011;7:787–793. doi: 10.1038/nchembio.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dang CV. Myc on the path to cancer. Cell. 2012;149:22–35. doi: 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Delmore JE, Issa GC, Lemieux ME, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kessler JD, Kahle KT, Sun T, et al. A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science. 2012;335:348–353. doi: 10.1126/science.1212728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kress TR, Cannell IG, Brenkman AB, et al. The MK5/PRAK kinase and Myc form a negative feedback loop that is disrupted during colorectal tumorigenesis. Mol Cell. 2011;41:445–457. doi: 10.1016/j.molcel.2011.01.023. [DOI] [PubMed] [Google Scholar]
- 30.Zuber J, Shi J, Wang E, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524–528. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ericson K, Gan C, Cheong I, et al. Genetic inactivation of AKT1, AKT2, and PDPK1 in human colorectal cancer cells clarifies their roles in tumor growth regulation. Proc Natl Acad Sci U S A. 2010;107:2598–2603. doi: 10.1073/pnas.0914018107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peifer C, Alessi DR. Small-molecule inhibitors of PDK1. ChemMedChem. 2008;3:1810–1838. doi: 10.1002/cmdc.200800195. [DOI] [PubMed] [Google Scholar]
- 33.Maurer M, Su T, Saal LH, et al. 3-phosphoinositide-dependent kinase 1 potentiates upstream lesions on the phosphatidylinositol 3-kinase pathway in breast carcinoma. Cancer Res. 2009;69:6299–6306. doi: 10.1158/0008-5472.CAN-09-0820. [DOI] [PMC free article] [PubMed] [Google Scholar]