

Abstract
Protein tyrosine phosphatase (PTP)–proline-, glutamate-, serine-, and threonine-rich sequence (PEST) is ubiquitously expressed and is a critical regulator of cell adhesion and migration. PTP-PEST activity can be regulated transcriptionally via gene deletion or mutation in several types of human cancers or via post-translational modifications, including phosphorylation, oxidation, and caspase-dependent cleavage. PTP-PEST interacts with and dephosphorylates cytoskeletal and focal adhesion-associated proteins. Dephosphorylation of PTP-PEST substrates regulates their enzymatic activities and/or their interaction with other proteins and plays an essential role in the tumor cell migration process.
Keywords: PTP-PEST, cell migration, metastasis
Protein tyrosine phosphorylation plays essential roles in many cellular physiological events, such as cell growth, division, differentiation, adhesion, motility, and death[1]. Aberrant regulation of protein phosphorylation can result in altered protein-protein interactions, protein instability, and abnormal protein activity, all of which can lead to many human diseases, including cancer[2],[3].
Protein tyrosine (Tyr) phosphorylation is a reversible, dynamic process that can be directly regulated by two types of antagonistic enzymes, protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs). PTKs phosphorylate proteins on tyrosine residues, a process that can be counteracted by PTP-mediated dephosphorylation. The level of protein tyrosine phosphorylation is a result of the equilibrium reached between the stringently controlled activities of PTKs and PTPs[4],[5].
The human genome encodes more than 100 PTPs[6]. On the basis of its structure and substrate specificity, the PTP superfamily consists of classical phospho-Tyr-specific PTPs, dual-specificity phosphatases that dephosphorylate both phospho-serine (Ser)/phospho-threonine (Thr) and phospho-Tyr residues in proteins, and low molecular weight PTPs. The classical phospho-Tyr-specific PTP group, which comprises approximately 40 PTPs, can be categorized into two subfamilies: transmembrane receptor-like proteins (RPTPs) and non-receptor PTPs[4],[7]. RPTPs are predominantly found at the plasma membrane, whereas non-receptor PTPs are localized to various intracellular compartments, including the cytosol, plasma membrane, and endoplasmic reticulum[8].
PTPs have similar core structures composed of a central parallel β-sheet with flanking α-helices containing a β-loop-α-loop that encompasses the PTP signature motif. Despite their conserved structural and catalytic properties for phosphate hydrolysis, PTPs have sufficient differences in their active site pockets and their immediate surrounding environment to ensure substrate specificity. Regulatory sequences that flank the catalytic domain control catalytic activity by interacting with the residues at the active sites or by controlling substrate specificity[7].
Overview of PTP-PEST
PTP-proline-, glutamate-, serine-, and threonine-rich sequence (PEST), which is encoded by the PTPN12 gene and was initially cloned in 1992, is a member of the non-receptor PTP subfamily[9],[10]. PTP-PEST contains 780 amino acids, has a molecular weight of 112 kDa, and is expressed ubiquitously in a wide variety of tissues and cell types. PTP-PEST is located mainly in the cytoplasm and is highly expressed in the hematopoietic system, particularly in the thymus, spleen, and liver, but it is also found in the brain and heart. PTP-PEST plays an essential function in early embryogenesis, and ablation of the PTPN12 gene leads to early embryonic lethality. Analysis of PTP-PEST–null mutant embryos revealed defects in the embryonic mesenchyme and subsequent defects in vascularization, liver development, and somatogenesis[11].
PTP-PEST consists of an NH2-terminal catalytic domain (amino acid residues 1-300) and a long COOH-terminal domain (amino acid residues 304-775) (Figure 1)[12],[13]. The sequence 229-IHCSAGCGRTG-239 in the N-terminal domain of PTP-PEST conforms to the highly conserved phosphatase signature motif (I/V)HCXAGXXR (S/T)G, and the cysteine residue within this motif is essential for its PTP activity. The large non-catalytic C-terminal region is rich in PEST sequences, which are found in many rapidly degrading proteins[14]. PTP-PEST, however, appears to be quite stable[15]. The main function of the non-catalytic segment of PTP-PEST is to mediate the interactions of PTP-PEST with its substrates and/or adaptor proteins[16].
Figure 1. Schematic structure of PTP-PEST.
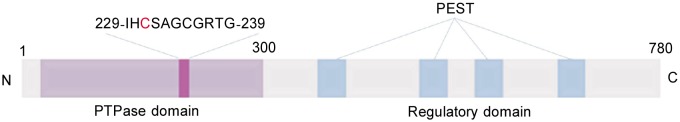
PTP-PEST consists of an NH2-terminal catalytic domain and a long COOH-terminal regulatory domain. The conserved phosphatase signature motif sequence in the catalytic domain (the essential cysteine residue is in red) is expanded. The regulatory domain contains several PEST-rich regions, which mediate interactions of PTP-PEST with its substrates and/or adaptor proteins.
PTP-PEST has important functions in cell spreading and migration[17]. Cell migration is a highly coordinated, dynamically, and precisely regulated multistep cyclical process involving the disassembly of focal adhesions at the leading edge of the cell, the polarization and protrusion of the leading edge followed by the formation and stabilization of cell-substrate adhesions, the contraction of the actin-based cytoskeleton to pull the cell body forward, and finally, the disassembly of adhesions at the rear region of the cell, resulting in the retraction of the trailing tail (Figure 2A)[18]–[20]. Successive waves of protein tyrosine phosphorylation and dephosphorylation, in which PTP-PEST plays an instrumental role, are essential for cell migration[17],[21].
Figure 2. PTP-PEST plays an instrumental role in cell migration.

A, cell migration is a highly coordinated, dynamic, and precisely regulated multistep cyclical process, which includes the disassembly of focal adhesions at the leading edge of the cell, the polarization and protrusion of the leading edge, the formation and stabilization of cell-substrate adhesions, the contraction of the cell body, the disassembly of adhesions at the rear of the cell, and the retraction of the trailing tail; B, PTP-PEST regulates multiple steps of the cell migration cycle, including membrane protrusion, tail retraction, and the dynamic regulation of focal adhesions via dephosphorylation of its associated proteins in the migration complex. Blunted arrows indicate dephosphorylation of substrate proteins by PTP-PEST.
Cell migration is controlled by a complex signaling network consisting of integrins; protein kinases, such as the Src family kinases; focal adhesion associated-kinase (FAK) and proline-rich tyrosine kinase 2 (PYK2); small molecular weight G-proteins such as Rho, Rac, and Cdc42; guanine nucleotide exchange factors (GEFs) such as the Vav family members; and guanosine triphosphatase (GTPase)-activating proteins (GAPs) such as p190RhoGAP. In addition, adaptor molecules, such as paxillin, p130 Crk-associated protein (p130Cas), and Wiskott-Aldrich syndrome protein (WASP) and structural proteins such as tensin, talin, and actin are all involved in cell migration. Many studies have indicated that PTP-PEST, which binds to and regulates the tyrosine phosphorylation of its associated proteins in the migration complex, acts in multiple steps of the migration cycle, including membrane protrusion, tail retraction, and the dynamic regulation of focal adhesions[22]–[24]. PTP-PEST can exert both positive and negative effects on cell migration and other cellular activities via its substrates and associated molecules, as indicated in a subsequent section (Figure 2B)[4],[25].
PTP-PEST–dependent Dephosphorylation of Substrate Proteins in Cell Migration
Regulation of FAK, PYK2, p130Cas, and paxillin phosphorylation by PTP-PEST
Focal adhesions are the sites of contact between the extracellular matrix (ECM) and the cytoskeleton via the integrin family of transmembrane proteins and are major sites of dynamic tyrosine phosphorylation and dephosphorylation in cells[13]. Cell migration requires turnover of focal adhesions[26]. Either a lack or an excess of adhesions can inhibit migration.
More than 50 proteins are known to participate in cell-ECM adhesion[27]. PTP-PEST, along with FAK and Src, is required for coordinating the dephosphorylation and phosphorylation of focal adhesion proteins, which is necessary for promoting focal adhesion turnover and stimulating cell migration. As key components of focal adhesion, FAK, p130Cas, and paxillin become tyrosine-phosphorylated in response to the activation of the integrin signaling pathways. Once integrin is activated, FAK autophosphorylates at Y397, which triggers binding to the Src-homology-2 (SH2) domain of Src, leading to Src-dependent phosphorylation on multiple residues of FAK and an increase in FAK kinase activity. Activated FAK/Src may phosphorylate the adaptor protein p130Cas and the scaffold protein paxillin. Regulation of tyrosine phosphorylation of these proteins by PTP-PEST is involved in controlling the rate of focal adhesion turnover and cell migration[28].
PTP-PEST was first implicated in focal adhesion regulation when p130Cas was identified as its substrate using a substrate trapping approach[29]. Transient overexpression of PTP-PEST reduces the tyrosine phosphorylation of p130Cas stimulated by integrins and platelet-derived growth factor (PDGF) and retards cell motility[29]–[31]. PTP-PEST also interacts with and dephosphorylates paxillin[32]. The paxillin-binding site of PTP-PEST is located in its non-catalytic domain and resides close to but is independent of the p130Cas- binding site[32], whereas the LIM 3-4 motifs on the C terminus of paxillin are the binding regions for PTP-PEST[33]. It has been suggested that paxillin positively regulates cell migration, and this effect is inhibited by PTP-PEST, whereas reactive oxygen species (ROS) suppress PTP-PEST function[34]. In contrast, a report shows that the interaction between PTP-PEST and paxillin, which is required for paxillin dephosphorylation, is necessary for PTP-PEST stimulation of cell migration[35]. In addition, the paxillin LD4 motif binds to the paxillin kinase linker (PKL), which is another substrate of PTP-PEST. This PKL-paxillin interaction is required for PTP-PEST to inhibit cell spreading and promote cell migration[35].
Hic-5, a paralog of paxillin that shares all 11 exons of the paxillin gene, localizes to focal adhesions. The LIM 3 domain of Hic-5 binds to the second of the 5 proline-rich sequences in PTP-PEST[36],[37]. In addition to its focal adhesion localization, Hic-5 shuttles in and out of the nucleus via a redox-sensitive nuclear export signal. Hic-5 accumulates in the nucleus under oxidative conditions and participates in the transcription of the c-fos and p21 (Cip1) genes, and PTP-PEST inhibits the nuclear accumulation of Hic-5[37]. Leupaxin, a focal adhesion-associated adaptor protein in the paxillin extended family, associates with PTP-PEST to regulate migration of prostate cancer cells[38].
Studies of PTP-PEST–deficient fibroblasts have supported that PTP-PEST promotes cell migration. PTP-PEST–deficient fibroblasts exhibit elevated levels of tyrosine phosphorylation of p130Cas, paxillin, and FAK. These cells had more focal adhesions, spread faster on a fibronectin substrate, and displayed a strong defect in motility compared with their wild-type counterparts, thereby suggesting that PTP-PEST promotes cell migration[22],[39]. This assumption was further supported by the identification of FAK as a substrate of PTP-PEST[40]. The activation of epidermal growth factor receptor (EGFR), H-Ras, or K-Ras results in the dephosphorylation of the Y397 FAK autophosphorylation residue and the inhibition of FAK activity in several types of human cancer cells, leading to a reduction in the overall number of focal adhesions with a limited number of focal adhesions left at the lamellipodia, which are front leading edges of migrating cells[40],[41]. Ras-induced FAK Y397 dephosphorylation is mediated by a downstream protein Fgd1, a GEF that activates Cdc42, which in turn activates the PAK1-MEK-ERK signaling cascade. ERK primarily phosphorylates FAK at S910 and recruits PIN1 peptidyl-prolyl cis/trans isomerases and PTP-PEST, which primarily colocalize with FAK at the lamellipodia of migrating cells. PIN1 specifically recognizes phosphorylated serine or threonine in pS/TP peptide sequences[42]. The binding of PIN1 to phosphorylated FAK S910 and the prolyl isomerization of FAK cause PTP-PEST to interact with and dephosphorylate FAK Y397, leading to the dynamic turnover of focal adhesions in these regions. The inhibition of FAK that is mediated by this signal relay promotes Ras-induced cell migration, invasion, and metastasis[40],[41].
PYK2 and FAK have a similar structural organization with a tyrosine kinase domain flanked by non-catalytic domains at both the N and C termini. Both the major autophosphorylation site of PYK2, Y402, and its activation loop tyrosine residues, Y579 and Y580, are targeted for dephosphorylation by PTP-PEST; as a result, the kinase activity of PYK2 is dramatically inhibited[16],[43]. The FERM domain of PYK2 binds to the pleckstrin homology domain of dynamin. The dephosphorylation of PYK2 by PTP-PEST, which requires the GTPase activity of dynamin, leads to the reorganization of the actin podosome belt/sealing zone (which contains highly dynamic, actin-rich structures and is important for cell attachment and for circumscribing the ruffled border membrane through which resorption occurs) and to a decrease in osteoclast bone-resorbing activity[44]. In addition, as demonstrated by conditional deletion of PTPN12 in mice, PTP-PEST dephosphorylates PYK2, which promotes the formation of T-cell homoaggregates that enhance T-cell activation, resulting in regulation of secondary T-cell responses, anergy prevention, and autoimmunity induction[45].
Regulation of Rho GTPase by PTP-PEST
In addition to directly regulating focal adhesion turnover, PTP-PEST regulates Rho GTPase for the coupling of membrane protrusion and tail retraction during cell migration. Migration-retarded PTP-PEST–deficient mouse fibroblasts have exaggerated membrane protrusions at the leading edge and long, unretracted tails in the rear that are mediated by enhanced Rac1 activity and decreased RhoA activity, respectively[46]. PTP-PEST inhibits Rac1 by dephosphorylating and reducing VAV2 activity while activating RhoA by dephosphorylating and inhibiting p190RhoGAP, which suggests that PTP-PEST–mediated RhoA activation and Rac1 suppression are required for tail retraction and membrane protrusion regulation, respectively. In the absence of PTP-PEST, excessive Rac1 activity combined with inhibited RhoA activity perturbs the balance between protrusion and retraction that is needed for migration[46]. In addition to inhibiting Rac1 by dephosphorylating and reducing VAV2, PTP-PEST inhibits Rac1 activity by interacting with paxillin. Phosphorylated paxillin binds to the adaptor protein Crkll, which, in turn, binds to an atypical Rac GEF complex, DOCK180/ELMO, and p130Cas to stimulate spreading by activating Rac1. PTP-PEST binds to the paxillin C terminus in focal adhesions and may suppress Rac activation by dephosphorylating p130Cas and inhibiting the activity of the Rac GEF DOCK180[35],[47],[48].
In contrast with its role in coupling protrusion and retraction during cell migration, PTP-PEST was detected in the adheren junctions of colon carcinoma cells and inhibited cell migration. Knockdown of PTP-PEST causes a disruption of cell-cell junctions, which is the result of a defect in junctional assembly accompanied by increased Rac1 activity and suppressed RhoA activity[49]. Overexpression of PTP-PEST in Chinese hamster ovary cells (CHOK1) impairs membrane protrusion by suppressing Rac1 activity, thereby inhibiting cell motility[24]; this finding contradicts those reported in studies of PTP-PEST–deficient mouse fibroblasts.
Regulation of Src, Abl, WASP, and filamin-A by PTP-PEST
Src activity is elevated in the majority of malignant and premalignant colon tumors. It has been shown that overexpression of PTP-PEST inactivates c-Src kinase by dephosphorylating a positive regulatory tyrosine, Y416, within the c-Src kinase domain to inhibit intestinal cell migration. In colorectal Carcinogenesis, down-regulation of PTP-PEST expression may promote cancer invasion and metastasis[50]. In contrast, a different report shows that PTP-PEST dephosphorylates c-Src at Y527 in osteoclasts to release the auto-inhibitory effect of Y527 phosphorylation and increase phosphorylation at Y418 in the catalytic domain. Activation of Src results in the phosphorylation of cortactin at Y421 and WASP at Y294; increased interaction of Src, cortactin, and Arp2 with WASP; and the promotion of sealing ring formation and bone-resorbing activity in osteoclasts[51]. PTP-PEST also interacts with C-terminal Src kinase (Csk), which phosphorylates Src Y527 and inhibits Src activation. This interaction involves the SH3 region of Csk and a proline-rich region outside the catalytic domain of PTP-PEST. It is not clear whether PTP-PEST regulates c-Src activation by modulating Csk activity[12].
The domain organization of Abl family members is similar to that of Src family members. These proteins have a tyrosine kinase domain that is preceded by both an SH2 domain and an SH3 domain[52]. c-Abl participates in membrane ruffling in response to growth factor stimulation[53] and antagonizes migration under some conditions[54]. c-Abl interacts with and phosphorylates proline-, serine-, and threonine-rich phosphatase-interacting protein (PSTPIP)[55], which is a substrate of PTP-PEST. PTP-PEST, which interacts directly via its carboxyterminal homology (CTH) domain with the coiled-coil domain of PSTPIP, dephosphorylates PSTPIP at Y344[56]. PSTPIP bridges PTP-PEST to c-Abl, resulting in the dephosphorylation and inhibition of c-Abl by PTP-PEST. Both c-Abl and PSTPIP are hyperphosphorylated in PTP-PEST–deficient cells, and platelet-derived growth factor (PDGF)-induced c-Abl kinase activation is prolonged in these cells[55].
In addition to acting as an adaptor to bridge the interaction between PTP-PEST and c-Abl, PSTPIP binds to WASP via its SH3 domain. The activation of the WASP proteins leads to actin nucleation and the formation of filopodia and lamellipodia, which are important events in the generation of cell polarity and directed cell migration[57]. PSTPIP serves as a scaffold protein between PTP-PEST and WASP and allows PTP-PEST to dephosphorylate WASP. PTP-PEST combines with PSTPIP to inhibit WASP-driven actin polymerization and synapse formation[56],[58].
PTP-PEST has been implicated in the regulation of cytokinesis, and PTP-PEST expression in HeLa cells results in the formation of multinucleated cells. Pro-teomic analyses revealed that the actin-binding protein filamin-A is a PTP-PEST substrate. The fourth proline-rich region of PTP-PEST was found to mediate filamin-A binding. A PTP-PEST mutant lacking both the fourth proline-rich region and filamin-A-binding ability failed to induce the multinucleated phenotype. Furthermore, depletion of filamin-A in HeLa cells was found to reduce the PTP-PEST–dependent multinucleated phenotype, thereby indicating an important role for the interaction between PTP-PEST and filamin-A in the control of cytokinesis in mammalian cells[12],[59]–[61].
Regulation of Shc by PTP-PEST
PTP-PEST associates with the adaptor proteins Grb2[60] and Shc[62],[63], both of which participate in the growth factor signaling and motility. At least three genes, shcA, shcB, and shcC, are known to encode Shc proteins, which share an amino-terminal phosphor-tyrosine-binding (PTB) domain, a central proline/glycine-rich region (CH1), and a carboxy-terminal SH2 domain. ShcA exists in three isoforms in mammalian cells, p46Shc, p52Shc, and p66Shc, which differ only in the extent of their amino-terminal sequences and are produced via alternative splicing and differential use of translational initiation sites. The p52Shc/p66Shc proteins are associated with PTP-PEST via an NPxH (in which N is asparagine, P is proline, x is any residue, and H is histidine) motif in the non-catalytic domain of PTP-PEST[62],[63]. 12-O-tetradecanoylphorbol-13-acetate (TPA) induces the phosphorylation of S29 in p52Shc and S138 in p66Shc in a protein kinase C (PKC) activity-dependent manner. Phosphorylation of these residues and the presence of the intact PTB domain are essential for ShcA binding to PTP-PEST. Insulin also induces the association between ShcA and PTP-PEST. Phosphorylation of ShcA at S29 controls the ability of its PTB domain to bind to PTP-PEST, which is responsible for the Tyr dephosphorylation of ShcA and the inhibition of ShcA function in the induction of downstream signaling after insulin stimulation. Overexpression of a PTP-PEST binding-defective p52Shc S29A mutant, which increased Tyr phosphorylation, enhanced insulin-induced ERK activation[64].
Paradoxical Role of PTP-PEST in Tumor Cell Migration and Development
As illustrated in the regulation of PTP-PEST substrates, PTP-PEST has a paradoxical role in cell migration, which varies in cell line- and cellular signaling-dependent manners[22],[35],[39]. In addition, the reported functions of PTP-PEST in tumorigenesis are controversial. PTPN12 deletion was found in 13.8% of lung cancer cases[65]. Unlike normal breast tissue, in which PTP-PEST expression is consistent, 22.6% of breast cancer cases contain a deletion at the PTPN12 locus, and PTP-PEST protein expression is low in 37% of invasive breast cancers. In addition, low levels of PTP-PEST expression are more common in triple-negative breast cancer specimens, which do not express the genes for estrogen receptor, progesterone receptor, or Her2/neu. Previous studies have suggested that PTP-PEST suppresses triple-negative breast cancer cell transformation by interacting with and inhibiting multiple oncogenic tyrosine kinases, including EGFR and HER2[65],[66]. PTP-PEST may counter the activity of these receptor tyrosine kinases (RTKs) via dephosphorylation of the RTK substrates or the RTKs themselves to act as a tumor suppressor.
In contrast, active PTP-PEST mutants have been found in human tumor tissues. Three amino acid mutations at positions 1322, A573, and K709 were found in primary human breast and renal tumor samples. These mutations were also found in breast cancer cell lines and squamous carcinoma cell lines. Functional characterization of the PTP-PEST 1322 and A573 mutants revealed an enhancement of their in vitro phosphatase activity, although the catalytic activity of the K709 mutant was reduced. All three alterations are located between the proline-rich regions within the C-terminal portion of the regulatory domain of PTP-PEST. Thus, these mutants might affect intramolecular interactions or the overall 3-dimensional structure of the PTP-PEST protein, thereby affecting autoregulatory functions[67].
In line with the controversial results of studies of human tumor samples regarding the role of PTP-PEST in tumor development, analyses of cultured tumor cells have shown that both overexpression and genetic ablation of PTP-PEST cause profound inhibition of cell motility[22],[31]. These findings suggest that a precise balance of PTP-PEST function and dynamic regulation of PTP-PEST activity are required for regulating cell adhesion and maintaining an appropriate rate of focal adhesion turnover. Perturbation of phosphotyrosine homeostasis with either an excess or deficiency of PTP-PEST can interfere with focal adhesion turnover and retard cell movement. This hypothesis is consistent with a reported paradoxical role of the phosphorylation of FAK, a major PTP-PEST substrate, in tumor progression, suggesting a dynamic regulation of PTP-PEST and subsequent dynamic regulation of FAK phosphorylation during cell migration[26]. Activation of Ras by mutations or EGFR activation results in PTP-PEST–dependent FAK Y397 dephosphorylation and subsequent reduced focal adhesion and increased cell motility. In addition, Ras activation results in the colocalization of PTP-PEST and FAK in lamellipodia, suggesting a regulation of FAK by PTP-PEST in these regions[41]. However, when cells attach to fibronectin-containing matrices and integrins are activated, the integrin signaling (via unknown mechanisms) overrides the effects of Ras and EGFR on FAK, most likely by disrupting the interaction between FAK and PTP-PEST, and leads to FAK re-phosphorylation on Y397, which in turn promotes cell adhesion and spreading. However, once cells complete adhering to and spreading on ECM, FAK Tyr phosphorylation and activity are down-regulated again by Ras-induced and PTP-PEST-dependent dephosphorylation, leading to a reduction in the number of cell-ECM contacts[40],[41].
Thus, a model of Ras or EGFR activation-induced tumor cell migration could be as follows: activated Ras or EGFR has reduced overall FAK activity mediated by PTP-PEST–dependent dephosphorylation; as a result, the number of cell-ECM contacts is reduced, and cells become less adherent and more motile. At the same time, lamellipodial dynamics and focal adhesion turnover at the leading edges of cells, which are required for cell migration, are regulated by the dynamically integrated Ras- and integrin-induced signaling. During the formation of new lamellipodia driven by actin polymerization and stabilized by adherence to ECM, the integrin-FAK signaling overrides Ras-induced FAK Y397 dephosphorylation and facilitates the formation of new adhesions, which serve as traction sites for cell migration. However, these newly formed adhesions are quickly disassembled by Ras- and PTP-PEST–induced signaling, which regains dominance over signaling induced by integrin after adhesion formation. This disassembly of mature adhesions allows new adhesions, protrusions, and lamellipodia to form at the leading edge, thereby initiating another round of focal adhesion turnover[26],[40],[41].
Regulation of PTP-PEST
PTP-PEST plays an instrumental role in a variety of cellular functions, and its cellular activity is precisely regulated by several post-transcriptional modifications, including phosphorylation, oxidation, and proteolysis.
Phosphorylation of PTP-PEST
PTP-PEST can be regulated by phosphorylation, which affects its catalytic activity and access to substrates. The enzymatic activity of PTP-PEST can be down-regulated via the activation of PKC and cAMP-dependent protein kinase (PKA), which phosphorylate PTP-PEST at S435 in the non-catalytic domain and S39 in the catalytic domain. The phosphorylation of PTP-PEST at S435 has no direct effect on its enzyme activity, whereas the phosphorylation at S39 decreases its activity, possibly by reducing its affinity for substrates[5]. In contrast, protein phosphatase 1α (PP1α), which is a member of the serine/threonine-specific protein phosphatase family, binds to the non-catalytic domain of PTP-PEST and activates PTP via dephosphorylation of phospho-S39[68].
Mammalian sterile 20-like kinase 3 (MST3), a serine/threonine kinase, phosphorylates PTP-PEST and inhibits its tyrosine phosphatase activity, which correlates with enhanced phosphorylation of paxillin Y118 and Y31 and inhibited cell spreading and migration. These results suggest that MST3 inhibits cell migration in a fashion dependent on PTP-PEST–regulated paxillin dephosphory-lationt[69].
Consistent with that PTP-PEST can be regulated by phosphorylation, a recent study showed that the activation of ERK1/2 by Ras results in the phosphorylation of PTP-PEST at S571. The phosphorylation of S571, which by itself does not alter the catalytic activity of PTP-PEST toward phosphopeptide substrates, recruits PIN1 to bind to PTP-PEST. The conformational changes of phosphorylated PTP-PEST induced by PIN1, which may alter the structure of the protein-interacting domains adjacent to S571, increase the level of interaction between PTP-PEST and FAK, which leads to the dephosphorylation of FAK Y397 and promotes migration, invasion, and metastasis of v-H-Ras–transformed cells[70].
Oxidation of PTP-PEST
PTP-PEST can be regulated by reversible oxidation. PTP-mediated dephosphorylation requires a catalytically essential cysteine residue. This residue is highly sensitive to oxidation. Reversible oxidation is likely a physiologic mechanism of regulating PTP activity. One effect of ROS production is the oxidation of the cysteine residue within the signature motif of PTPs. This oxidation blocks the nucleophilic activity of PTPs and inactivates them in a reversible manner[71].
The cysteine residue in the catalytic domain of PTP-PEST was shown to be oxidized by ROS. Suppressing PTP-PEST activity by oxidation relieves its inhibitory effects on paxillin phosphorylation, which was proposed to be one of the mechanisms by which ROS promotes tumor cell migration and invasion[34]. The orphan adaptor TRAF4 was suggested to promote cell migration by regulating focal complexes for oxidative modification. TRAF4 binds to the NADPH oxidase subunit p47phox and the focal contact scaffold Hic-5, the latter of which is a substrate of PTP-PEST. An active mutant of TRAF4 activates the NADPH oxidase downstream from the Rho GTPases and PAK1 and oxidatively inactivates PTP-PEST. Active TRAF4 or knockdown of PTP-PEST initiates robust membrane ruffling. The knockdown of TRAF4 or Hic-5 protein expression or oxidant scavenging blocks cell migration[72].
Proteolysis of PTP-PEST
PTP-PEST can be regulated by proteolysis during apoptosis, which is a key process in cancer development and progression[73]. Previous studies suggested that PTP-PEST participates in tumor initiation by regulating apoptosis. PTP-PEST expression was shown to sensitize tumor cells to receptor-mediated apoptosis, whereas specific PTP-PEST cleavage, which generates PTP-PEST fragments with increased catalytic activity, was observed during apoptosis mediated by caspase-3-dependent cleavage of PTP-PEST at the 549-DSPD-552 motif in the non-catalytic domain. Given that PTP-PEST also acts as a scaffolding molecule connecting PSTPIP to a protein complex including paxillin, Shc, and Csk, the activation of caspase-3 disrupted the scaffolding properties of PTP-PEST and facilitated cellular detachment during apoptosis. Thus, PTP-PEST may participate in the amplification of apoptotic responses, likely via caspase-mediated changes in its scaffolding and enzymatic properties[74].
Concluding Remarks
In summary, PTP-PEST activity can be regulated at the transcriptional level by gene deletion or mutation or via post-translational modifications such as phosphorylation, oxidation, and caspase-dependent cleavage. Changes in the levels of PTP-PEST expression, activity, and interaction with substrates affect its Subcellular localization and substrate dephosphorylation, thereby regulating cellular activities such as cell adhesion, migration, and survival. Given the relatively high incidence of the genetic alteration of PTP-PEST in several types of human cancer, including breast and lung cancers, further studies are warranted to understand the dynamic regulation of PTP-PEST in tumor cell growth, migration, and metastasis so that novel targeted therapy against PTP-PEST can be rationally designed.
Acknowledgments
This work was supported by National Cancer Institute grants 2R01CA109035 (Z.L.) and CA16672 (Cancer Center Support Grant); research grant RP110252 (Z.L.) from the Cancer Prevention and Research Institute of Texas (CPRIT); American Cancer Society Research Scholar Award RSG-09-277-01-CSM (Z.L.); the James S. McDonnell Foundation 21st Century Science Initiative in Brain Cancer Research Award (220020318; Z.L.); and a Sister Institution Network Fund from The University of Texas MD Anderson Cancer Center (Z.L.).
References
- 1.Lu Z, Hunter T. Degradation of activated protein kinases by ubiquitination. Annu Rev Biochem. 2009;78:435–475. doi: 10.1146/annurev.biochem.013008.092711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tonks NK, Neel BG. Combinatorial control of the specificity of protein tyrosine phosphatases. Curr Opin Cell Biol. 2001;13:182–195. doi: 10.1016/s0955-0674(00)00196-4. [DOI] [PubMed] [Google Scholar]
- 3.Hunter T. Tyrosine phosphorylation: thirty years and counting. Curr Opin Cell Biol. 2009;21:140–146. doi: 10.1016/j.ceb.2009.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol. 2006;7:833–846. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- 5.Garton AJ, Tonks NK. PTP-PEST: a protein tyrosine phosphatase regulated by serine phosphorylation. EMBO J. 1994;13:3763–3771. doi: 10.1002/j.1460-2075.1994.tb06687.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alonso A, Sasin J, Bottini N, et al. Protein tyrosine phosphatases in the human genome. Cell. 2004;117:699–711. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 7.Wang WQ, Sun JP, Zhang ZY. An overview of the protein tyrosine phosphatase superfamily. Curr Top Med Chem. 2003;3:739–748. doi: 10.2174/1568026033452302. [DOI] [PubMed] [Google Scholar]
- 8.Stoker AW. Protein tyrosine phosphatases and signalling. J Endocrinol. 2005;185:19–33. doi: 10.1677/joe.1.06069. [DOI] [PubMed] [Google Scholar]
- 9.Takekawa M, Itoh F, Hinoda Y, et al. Cloning and characterization of a human cDNA encoding a novel putative cytoplasmic protein-tyrosine-phosphatase. Biochem Biophys Res Commun. 1992;189:1223–1230. doi: 10.1016/0006-291x(92)92335-u. [DOI] [PubMed] [Google Scholar]
- 10.Yang Q, Co D, Sommercorn J, et al. Cloning and expression of PTP-PEST. A novel, human, nontransmembrane protein tyrosine phosphatase. J Biol Chem. 1993;268:6622–6628. [PubMed] [Google Scholar]
- 11.Sirois J, Cote JF, Charest A, et al. Essential function of PTP-PEST during mouse embryonic vascularization, mesenchyme formation, neurogenesis and early liver development. Mech Dev. 2006;123:869–880. doi: 10.1016/j.mod.2006.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davidson D, Cloutier JF, Gregorieff A, et al. Inhibitory tyrosine protein kinase p50csk is associated with protein-tyrosine phosphatase PTP-PEST in hemopoietic and non-hemopoietic cells. J Biol Chem. 1997;272:23455–23462. doi: 10.1074/jbc.272.37.23455. [DOI] [PubMed] [Google Scholar]
- 13.Shen Y, Schneider G, Cloutier JF, et al. Direct association of protein-tyrosine phosphatase PTP-PEST with paxillin. J Biol Chem. 1998;273:6474–6481. doi: 10.1074/jbc.273.11.6474. [DOI] [PubMed] [Google Scholar]
- 14.Rogers SW, Rechsteiner MC. Microinjection studies on selective protein degradation: relationships between stability, structure, and location. Biomed Biochim Acta. 1986;45:1611–1618. [PubMed] [Google Scholar]
- 15.Charest A, Wagner J, Muise ES, et al. Structure of the murine mPTP-PEST gene: genomic organization and chromosomal mapping. Genomics. 1995;28:501–507. doi: 10.1006/geno.1995.1181. [DOI] [PubMed] [Google Scholar]
- 16.Davidson D, Veillette A. PTP-PEST, a scaffold protein tyrosine phosphatase, negatively regulates lymphocyte activation by targeting a unique set of substrates. EMBO J. 2001;20:3414–3426. doi: 10.1093/emboj/20.13.3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ostman A, Hellberg C, Bohmer FD. Protein-tyrosine phosphatases and cancer. Nat Rev Cancer. 2006;6:307–320. doi: 10.1038/nrc1837. [DOI] [PubMed] [Google Scholar]
- 18.Frame MC, Fincham VJ, Carragher NO, et al. v-Src's hold over actin and cell adhesions. Nat Rev Mol Cell Biol. 2002;3:233–245. doi: 10.1038/nrm779. [DOI] [PubMed] [Google Scholar]
- 19.Larsen M, Tremblay ML, Yamada KM. Phosphatases in cell-matrix adhesion and migration. Nat Rev Mol Cell Biol. 2003;4:700–711. doi: 10.1038/nrm1199. [DOI] [PubMed] [Google Scholar]
- 20.Ridley AJ, Schwartz MA, Burridge K, et al. Cell migration: integrating signals from front to back. Science. 2003;302:1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- 21.Burridge K, Wennerberg K. Rho and Rac take center stage. Cell. 2004;116:167–179. doi: 10.1016/s0092-8674(04)00003-0. [DOI] [PubMed] [Google Scholar]
- 22.Angers-Loustau A, Cote JF, Charest A, et al. Protein tyrosine phosphatase-PEST regulates focal adhesion disassembly, migration, and cytokinesis in fibroblasts. J Cell Biol. 1999;144:1019–1031. doi: 10.1083/jcb.144.5.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Angers-Loustau A, Cote JF, Tremblay ML. Roles of protein tyrosine phosphatases in cell migration and adhesion. Biochem Cell Biol. 1999;77:493–505. [PubMed] [Google Scholar]
- 24.Sastry SK, Lyons PD, Schaller MD, et al. PTP-PEST controls motility through regulation of Rac1. J Cell Sci. 2002;115:4305–4316. doi: 10.1242/jcs.00105. [DOI] [PubMed] [Google Scholar]
- 25.Julien SG, Dube N, Hardy S, et al. Inside the human cancer tyrosine phosphatome. Nat Rev Cancer. 2011;11:35–49. doi: 10.1038/nrc2980. [DOI] [PubMed] [Google Scholar]
- 26.Zheng Y, Lu Z. Paradoxical roles of FAK in tumor cell migration and metastasis. Cell Cycle. 2009;8:3474–3479. doi: 10.4161/cc.8.21.9846. [DOI] [PubMed] [Google Scholar]
- 27.Zamir E, Geiger B. Molecular complexity and dynamics of cell-matrix adhesions. J Cell Sci. 2001;114:3583–3590. doi: 10.1242/jcs.114.20.3583. [DOI] [PubMed] [Google Scholar]
- 28.Guo W, Giancotti FG. Integrin signalling during tumour progression. Nat Rev Mol Cell Biol. 2004;5:816–826. doi: 10.1038/nrm1490. [DOI] [PubMed] [Google Scholar]
- 29.Garton AJ, Flint AJ, Tonks NK. Identification of p130(cas) as a substrate for the cytosolic protein tyrosine phosphatase PTP-PEST. Mol Cell Biol. 1996;16:6408–6418. doi: 10.1128/mcb.16.11.6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garton AJ, Burnham MR, Bouton AH, et al. Association of PTP-PEST with the SH3 domain of p130cas; a novel mechanism of protein tyrosine phosphatase substrate recognition. Oncogene. 1997;15:877–885. doi: 10.1038/sj.onc.1201279. [DOI] [PubMed] [Google Scholar]
- 31.Garton AJ, Tonks NK. Regulation of fibroblast motility by the protein tyrosine phosphatase PTP-PEST. J Biol Chem. 1999;274:3811–3818. doi: 10.1074/jbc.274.6.3811. [DOI] [PubMed] [Google Scholar]
- 32.Shen Y, Lyons P, Cooley M, et al. The noncatalytic domain of protein-tyrosine phosphatase-PEST targets paxillin for dephosphorylation in vivo. J Biol Chem. 2000;275:1405–1413. doi: 10.1074/jbc.275.2.1405. [DOI] [PubMed] [Google Scholar]
- 33.Brown MC, Turner CE. Paxillin: adapting to change. Physiol Rev. 2004;84:1315–1339. doi: 10.1152/physrev.00002.2004. [DOI] [PubMed] [Google Scholar]
- 34.Wu WS, Wu JR, Hu CT. Signal cross talks for sustained mapk activation and cell migration: the potential role of reactive oxygen species. Cancer Metastasis Rev. 2008;27:303–314. doi: 10.1007/s10555-008-9112-4. [DOI] [PubMed] [Google Scholar]
- 35.Jamieson JS, Tumbarello DA, Halle M, et al. Paxillin is essential for PTP-PEST–dependent regulation of cell spreading and motility: a role for paxillin kinase linker. J Cell Sci. 2005;118:5835–5847. doi: 10.1242/jcs.02693. [DOI] [PubMed] [Google Scholar]
- 36.Nishiya N, Iwabuchi Y, Shibanuma M, et al. Hic-5, a paxillin homologue, binds to the protein-tyrosine phosphatase PEST (PTP-PEST) through its LIM 3 domain. J Biol Chem. 1999;274:9847–9853. doi: 10.1074/jbc.274.14.9847. [DOI] [PubMed] [Google Scholar]
- 37.Shibanuma M, Mori K, Kim-Kaneyama JR, et al. Involvement of FAK and PTP-PEST in the regulation of redox-sensitive nuclear-cytoplasmic shuttling of a LIM protein, Hic-5. Antioxid Redox Signal. 2005;7:335–347. doi: 10.1089/ars.2005.7.335. [DOI] [PubMed] [Google Scholar]
- 38.Sahu SN, Nunez S, Bai G, et al. Interaction of Pyk2 and PTP-PEST with leupaxin in prostate cancer cells. Am J Physiol Cell Physiol. 2007;292:C2288–2296. doi: 10.1152/ajpcell.00503.2006. [DOI] [PubMed] [Google Scholar]
- 39.Cote JF, Charest A, Wagner J, et al. Combination of gene targeting and substrate trapping to identify substrates of protein tyrosine phosphatases using PTP-PEST as a model. Biochemistry. 1998;37:13128–13137. doi: 10.1021/bi981259l. [DOI] [PubMed] [Google Scholar]
- 40.Lu Z, Jiang G, Blume-Jensen P, et al. Epidermal growth factor-induced tumor cell invasion and metastasis initiated by dephosphorylation and downregulation of focal adhesion kinase. Mol Cell Biol. 2001;21:4016–4031. doi: 10.1128/MCB.21.12.4016-4031.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zheng Y, Xia Y, Hawke D, et al. FAK phosphorylation by ERK primes ras-induced tyrosine dephosphorylation of FAK mediated by PIN1 and PTP-PEST. Mol Cell. 2009;35:11–25. doi: 10.1016/j.molcel.2009.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu KP, Zhou XZ. The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat Rev Mol Cell Biol. 2007;8:904–916. doi: 10.1038/nrm2261. [DOI] [PubMed] [Google Scholar]
- 43.Lyons PD, Dunty JM, Schaefer EM, et al. Inhibition of the catalytic activity of cell adhesion kinase beta by protein-tyrosine phosphatase-PEST–mediated dephosphorylation. J Biol Chem. 2001;276:24422–24431. doi: 10.1074/jbc.M011080200. [DOI] [PubMed] [Google Scholar]
- 44.Eleniste PP, Du L, Shivanna M, et al. Dynamin and PTP-PEST cooperatively regulate Pyk2 dephosphorylation in osteoclasts. Int J Biochem Cell Biol. 2012;44:790–800. doi: 10.1016/j.biocel.2012.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Davidson D, Shi X, Zhong MC, et al. The phosphatase PTP-PEST promotes secondary T cell responses by dephos-phorylating the protein tyrosine kinase Pyk2. Immunity. 2010;33:167–180. doi: 10.1016/j.immuni.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 46.Sastry SK, Rajfur Z, Liu BP, et al. PTP-PEST couples membrane protrusion and tail retraction via VAV2 and p190RhoGAP. J Biol Chem. 2006;281:11627–11636. doi: 10.1074/jbc.M600897200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Valles AM, Beuvin M, Boyer B. Activation of Rac1 by paxillin-Crk-DOCK180 signaling complex is antagonized by Rap1 in migrating NBT-II cells. J Biol Chem. 2004;279:44490–44496. doi: 10.1074/jbc.M405144200. [DOI] [PubMed] [Google Scholar]
- 48.Feller SM. Crk family adaptors—signalling complex formation and biological roles. Oncogene. 2001;20:6348–6371. doi: 10.1038/sj.onc.1204779. [DOI] [PubMed] [Google Scholar]
- 49.Espejo R, Rengifo-Cam W, Schaller MD, et al. PTP-PEST controls motility, adherens junction assembly, and Rho GTPase activity in colon cancer cells. Am J Physiol Cell Physiol. 2010;299:C454–463. doi: 10.1152/ajpcell.00148.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mathew S, George SP, Wang Y, et al. Potential molecular mechanism for c-Src kinase–mediated regulation of intestinal cell migration. J Biol Chem. 2008;283:22709–22722. doi: 10.1074/jbc.M801319200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chellaiah MA, Schaller MD. Activation of Src kinase by protein-tyrosine phosphatase-PEST in osteoclasts: comparative analysis of the effects of bisphosphonate and protein-tyrosine phosphatase inhibitor on Src activation in vitro. J Cell Physiol. 2009;220:382–393. doi: 10.1002/jcp.21777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hantschel O, Superti-Furga G. Regulation of the c-Abl and Bcr-Abl tyrosine kinases. Nat Rev Mol Cell Biol. 2004;5:33–44. doi: 10.1038/nrm1280. [DOI] [PubMed] [Google Scholar]
- 53.Plattner R, Kadlec L, DeMali KA, et al. c-Abl is activated by growth factors and Src family kinases and has a role in the cellular response to PDGF. Genes Dev. 1999;13:2400–2411. doi: 10.1101/gad.13.18.2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kain KH, Klemke RL. Inhibition of cell migration by Abl family tyrosine kinases through uncoupling of Crk-CAS complexes. J Biol Chem. 2001;276:16185–16192. doi: 10.1074/jbc.M100095200. [DOI] [PubMed] [Google Scholar]
- 55.Cong F, Spencer S, Cote JF, et al. Cytoskeletal protein PSTPIP1 directs the PEST-type protein tyrosine phosphatase to the c-Abl kinase to mediate Abl dephosphorylation. Mol Cell. 2000;6:1413–1423. doi: 10.1016/s1097-2765(00)00138-6. [DOI] [PubMed] [Google Scholar]
- 56.Cote JF, Chung PL, Theberge JF, et al. PSTPIP is a substrate of PTP-PEST and serves as a scaffold guiding PTP-PEST toward a specific dephosphorylation of WASP. J Biol Chem. 2002;277:2973–2986. doi: 10.1074/jbc.M106428200. [DOI] [PubMed] [Google Scholar]
- 57.Rohatgi R, Ma L, Miki H, et al. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell. 1999;97:221–231. doi: 10.1016/s0092-8674(00)80732-1. [DOI] [PubMed] [Google Scholar]
- 58.Badour K, Zhang J, Shi F, et al. Fyn and PTP-PEST–mediated regulation of Wiskott-Aldrich syndrome protein (WASP) tyrosine phosphorylation is required for coupling T cell antigen receptor engagement to WASP effector function and T cell activation. J Exp Med. 2004;199:99–112. doi: 10.1084/jem.20030976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cloutier JF, Veillette A. Association of inhibitory tyrosine protein kinase p50csk with protein tyrosine phosphatase PEP in T cells and other hemopoietic cells. EMBO J. 1996;15:4909–4918. [PMC free article] [PubMed] [Google Scholar]
- 60.Charest A, Wagner J, Kwan M, et al. Coupling of the murine protein tyrosine phosphatase PEST to the epidermal growth factor (EGF) receptor through a Src homology 3 (SH3) domain-mediated association with Grb2. Oncogene. 1997;14:1643–1651. doi: 10.1038/sj.onc.1201008. [DOI] [PubMed] [Google Scholar]
- 61.Playford MP, Lyons PD, Sastry SK, et al. Identification of a filamin docking site on PTP-PEST. J Biol Chem. 2006;281:34104–34112. doi: 10.1074/jbc.M606277200. [DOI] [PubMed] [Google Scholar]
- 62.Habib T, Herrera R, Decker SJ. Activators of protein kinase C stimulate association of Shc and the PEST tyrosine phosphatase. J Biol Chem. 1994;269:25243–25246. [PubMed] [Google Scholar]
- 63.Charest A, Wagner J, Jacob S, et al. Phosphotyrosine-independent binding of SHC to the NPLH sequence of murine protein-tyrosine phosphatase-PEST. Evidence for extended phosphotyrosine binding/phosphotyrosine interaction domain recognition specificity. J Biol Chem. 1996;271:8424–8429. doi: 10.1074/jbc.271.14.8424. [DOI] [PubMed] [Google Scholar]
- 64.Faisal A, el-Shemerly M, Hess D, et al. Serine/threonine phosphorylation of ShcA. Regulation of protein-tyrosine phosphatase-pest binding and involvement in insulin signaling. J Biol Chem. 2002;277:30144–30152. doi: 10.1074/jbc.M203229200. [DOI] [PubMed] [Google Scholar]
- 65.Sun T, Aceto N, Meerbrey KL, et al. Activation of multiple proto-oncogenic tyrosine kinases in breast cancer via loss of the PTPN12 phosphatase. Cell. 2011;144:703–718. doi: 10.1016/j.cell.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 67.Streit S, Ruhe JE, Knyazev P, et al. PTP-PEST phosphatase variations in human cancer. Cancer Genet Cytogenet. 2006;170:48–53. doi: 10.1016/j.cancergencyto.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 68.Nakamura K, Palmer HE, Ozawa T, et al. Protein phosphatase 1alpha associates with protein tyrosine phosphatase-PEST inducing dephosphorylation of phospho-serine 39. J Biochem. 2010;147:493–500. doi: 10.1093/jb/mvp191. [DOI] [PubMed] [Google Scholar]
- 69.Lu TJ, Lai WY, Huang CY, et al. Inhibition of cell migration by autophosphorylated mammalian sterile 20-like kinase 3 (MST3) involves paxillin and protein-tyrosine phosphatase-PEST. J Biol Chem. 2006;281:38405–38417. doi: 10.1074/jbc.M605035200. [DOI] [PubMed] [Google Scholar]
- 70.Zheng Y, Yang W, Xia Y, et al. Ras-induced and extracellular signal-regulated kinase 1 and 2 phosphorylation-dependent isomerization of protein tyrosine phosphatase (PTP)-PEST by PIN1 promotes FAK dephosphorylation by PTP-PEST. Mol Cell Biol. 2011;31:4258–4269. doi: 10.1128/MCB.05547-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tonks NK. Redox redux: revisiting PTPs and the control of cell signaling. Cell. 2005;121:667–670. doi: 10.1016/j.cell.2005.05.016. [DOI] [PubMed] [Google Scholar]
- 72.Wu RF, Xu YC, Ma Z, et al. Subcellular targeting of oxidants during endothelial cell migration. J Cell Biol. 2005;171:893–904. doi: 10.1083/jcb.200507004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 74.Halle M, Liu YC, Hardy S, et al. Caspase-3 regulates catalytic activity and scaffolding functions of the protein tyrosine phosphatase PEST, a novel modulator of the apoptotic response. Mol Cell Biol. 2007;27:1172–1190. doi: 10.1128/MCB.02462-05. [DOI] [PMC free article] [PubMed] [Google Scholar]