

Abstract
Mutational activation of KRAS is a common oncogenic event in lung cancer and other epithelial cancer types. Efforts to develop therapies that counteract the oncogenic effects of mutant KRAS have been largely unsuccessful, and cancers driven by mutant KRAS remain among the most refractory to available treatments. Studies undertaken over the past decades have produced a wealth of information regarding the clinical relevance of KRAS mutations in lung cancer. Mutant Kras-driven mouse models of cancer, together with cellular and molecular studies, have provided a deeper appreciation for the complex functions of KRAS in tumorigenesis. However, a much more thorough understanding of these complexities is needed before clinically effective therapies targeting mutant KRAS-driven cancers can be achieved.
Keywords: KRAS, RAS, Oncogene, lung cancer
KRAS (chromosome 12p12.1) is a member of the canonical RAS family of genes that also includes HRAS (chromosome 11p15.5) and NRAS (chromosome 1p13.1). The importance of RAS in cancer pathogenesis was first recognized more than three decades ago when it was discovered that mutated versions of KRAS and HRAS were responsible for the transforming activities of sarcoma-inducing retroviruses in rats. We now know that somatic activating mutations in the cellular homologs of all three RAS family members occur in a wide spectrum of human cancers. These mutations predominantly occur at codons 12, 13, and 61, and result in constitutive activation of RAS. Overall, RAS mutations have been found in approximately 30% of all human cancers, with KRAS as the most commonly mutated family member[1].
The three RAS genes are highly conserved across different species and encode monomeric GTPases that cycle between active (GTP-bound) and inactive (GDP-bound) states in response to extracellular cues. Unlike HRAS and NRAS, KRAS undergoes alternative splicing, resulting in two proteins (KRAS4A and KRAS4B) that differ only at their carboxyl termini (Figure 1). RAS proteins are 188/189 amino acids in length, and the sequence of the first 165 amino acids is almost identical. This region contains highly conserved domains that are responsible for GTP binding and hydrolysis and functional interactions with regulators and downstream effectors (Figure 1). The hypervariable carboxyl domain is the most distinguishing feature among the RAS family members and contains sequences important for determining post-translational modification, including the terminal CAAX domain that is responsible for membrane targeting. The post-translational modification of RAS proteins is a complex multi-stage process that has been extensively reviewed elsewhere[2]. Briefly, all four RAS proteins are farnesylated, and with the exception of HRAS, they can also be geranylgeranylated. This modification is followed by proteolytic cleavage within the CAAX motif and carboxymethylation of the exposed cysteine residue. Finally, HRAS, NRAS, and KRAS4A undergo palmitoylation at cysteine residues located adjacent to the carboxyl end. While KRAS4B is not palmitoylated, it contains a polybasic lysine-rich sequence that enables association with the plasma membrane through electrostatic interactions. In this review, we provide a summary of the extensive body of knowledge of KRAS and the RAS gene family, emphasizing particular aspects of genetics and biology that are relevant to lung cancer.
Figure 1. Primary structures of RAS proteins.

The first 165 amino acids of RAS proteins are nearly identical and include motifs responsible for the binding and hydrolysis of GTP, binding of downstream effectors, and interactions with GAP and GEF. The hypervariable domain at the carboxy-terminus, including the CAAX motif, contains sequences important for the post-translational modification and membrane targeting of RAS proteins. The cysteine residue in the CAAX motif is a target for prenylation (i.e., farnesylation or geranylgeranylation). The cysteine residues (orange boxes) near the carboxy-termini of HRAS, NRAS, and KRAS4A are targets for palmitoylation. The poly-lysine track (green boxes) helps KRAS4B to associate with the membrane.
RAS Signaling
RAS signaling begins with the stimulation of a vast array of upstream receptors including receptor tyrosine kinases (RTKs) of which the epidermal growth factor receptor (EGFR) is perhaps most relevant to lung cancer. Adaptor proteins (e.g., Grb2) interact with the intracellular domain of activated EGFR and in turn recruit guanine nucleotide exchange factors (GEFs) such as Son of Sevenless (SOS) to the cellular membrane where they can associate with RAS to promote the exchange of GDP for GTP (Figure 2). RAS signaling is terminated upon the hydrolysis of GTP to GDP by the intrinsic GTPase activity of RAS through the interaction with GTPase-activating proteins (GAPs). Cancer-causing mutations in RAS drastically impair the GTPase activity, resulting in RAS proteins that are locked in the active GTP-bound conformation, regardless of the upstream signal.
Figure 2. The RAS signaling network.
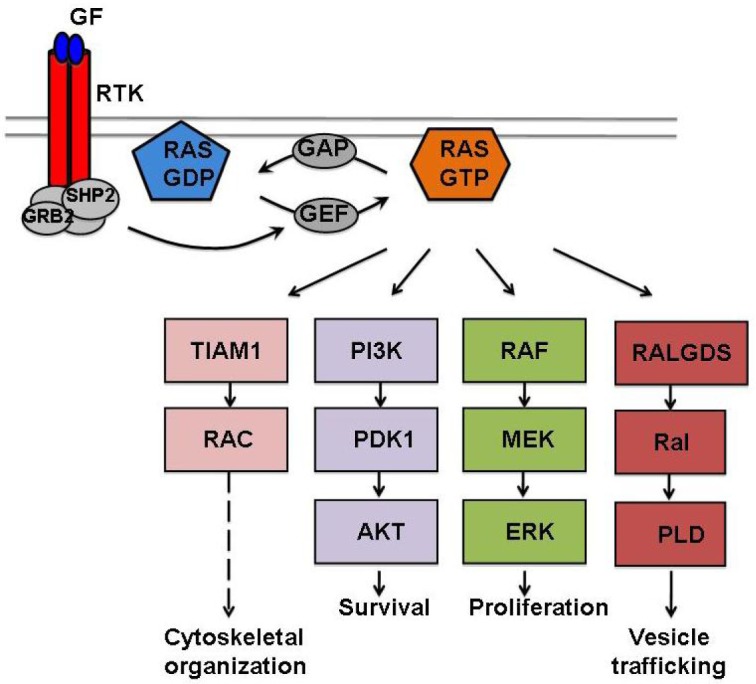
Activation of RTKs by growth factors (GFs) creates intracellular docking sites for adaptor proteins (e.g., GRB2 and SHP2) that recruit GEF to the membrane to interact with RAS and promote the exchange of GDP for GTP. In the active GTP-bound conformation, RAS engages and activates an array of downstream effector pathways to regulate many cellular responses. The RAS signaling is terminated upon hydrolysis of the bound GTP by the intrinsic GTPase activity of RAS with the help of GAP. Oncogenic mutations in RAS lock the proteins in a constitutively active state, resulting in the deregulation of many cellular functions that together contribute to the cancer phenotype.
In their active, GTP-bound conformations, the four RAS proteins engage and activate a large number of downstream signaling pathways to varying degrees. Signaling through these downstream pathways regulates diverse cellular responses, including proliferation, survival, and differentiation (Figure 2). The canonical RAS/RAF/MEK/ERK pathway controls cellular proliferation by modulating the levels of many cell cycle regulators and is frequently hyperactivated in cancers[3]. RAS also promotes cell survival by activating PI3K/PDK1/AKT signaling, a pathway that is also commonly deregulated in many cancer types[4]. RALGDS and RALGDS-like proteins and tumor invasion and metastasis-inducing protein 1 (TIAM1) can also be activated by RAS to control vesicle trafficking and cytoskeletal organization, respectively[5],[6]. Both RalGDS and Tiam1 have been shown to be required for Ras-dependent tumor formation in a mouse skin cancer model[7],[8]. Many of these downstream signaling pathways are involved in feedback regulation and crosstalk that together further contribute to the complexity of the RAS signaling network.
KRAS Mutations in Human Lung Cancer
Lung cancer is a common form of cancer in men and women, and it is responsible for the highest number of cancer-related deaths globally. Because of the high rate (>50%) of late diagnosis, the 5-year overall survival rate of patients with lung cancer has improved very little over the past 3 decades, hovering around the 13%–16% range[9]. Tobacco use is the most important risk factor for lung cancer, with ∼80% of cases arising in smokers.
KRAS is the most commonly mutated member of the RAS family in lung cancer, although HRAS and NRAS mutations have also been reported in a few cases[10]. KRAS mutations predominantly occur in lung adenocarcinomas, the most common histological subtype of non-small cell lung cancer (NSCLC), with frequencies ranging from 16% to 40% of the samples analyzed[10]–[13]. KRAS mutations have also been observed at a low frequency in squamous cell carcinoma (another subtype of NSCLC), but never in small cell lung cancer (SCLC)[10],[14]. Mutations predominantly occur at codon 12, occasionally at codon 13, and rarely at codon 61 of KRAS. Lung adenocarcinomas are usually associated with tobacco smoking[15], and KRAS mutations have been found to occur at a higher frequency in tumors in smokers compared to those in non-smokers[15]. In addition, mutations involving nucleotide transversions (i.e., G/C or G/T), which are known to be associated with exposure to tobacco smoke, are more common among smokers than non-smokers. Indeed, the two most common KRAS mutations in NSCLC, G12C (∼40%) and G12V (∼22%), arise from G/T transversions[16],[17].
Many studies have suggested that the presence of KRAS mutations in NSCLC is associated with shortened survival and time to relapse[18]–[20], although some studies have reported a lack of association[21]. The specific type of KRAS mutation may also provide information on disease aggressiveness or drug sensitivity. For example, the G12D mutation in NSCLC has been associated with better prognosis compared to the G12V or G12R mutations[22]. In contrast, the G12D and G13D mutations in human colorectal cancer (CRC) have been associated with a decreased response to chemotherapy compared to other mutations at these codons[23]. Further research is needed to conclusively establish the relationship between KRAS mutation specificity and prognosis in NSCLC. In contrast, there is compelling data demonstrating the usefulness of KRAS status as a marker for predicting therapeutic response. Adjuvant treatment with cisplatin and vinorelbine has been found to increase the survival of NSCLC patients with wild-type (WT) KRAS but not those with KRAS mutations in their lung tumors[24],[25]. The presence of KRAS mutations is also associated with a lack of response to EGFR inhibitors[26],[27] not only for NSCLC but also for human CRC where KRAS is also frequently mutated[28]. Given that KRAS is downstream of EGFR, it seems intuitive that EGFR inhibition would have no impact on the activity of mutant KRAS. However, it is surprising that concurrent treatment of KRAS-mutant NSCLCs with erlotinib and chemotherapy resulted in shortened overall survival and progression-free survival compared with chemotherapy alone[26]. These observations underscore the complexity of KRAS biology and further emphasize the advantage of having molecular information available (e.g., KRAS mutational status) when deciding the appropriate course of treatment.
Insights from Mouse Models of Kras-driven NSCLC
The mouse is an invaluable in vivo model system that has been widely used to study the importance of genes and pathways in different physiologic and pathologic contexts. The extensive sequence homology together with the broad overlapping pattern of expression in mice and humans suggests a high degree of functional redundancy among the RAS family members. However, studies using mice have shown that knockout of the Kras locus results in embryonic lethality, whereas knockout of either Hras or Nras has no effect on embryonic development and welfare of adult mice [29]. Furthermore, genetic disruption involving exon 4A of Kras results in viable and healthy mice that express only Kras4B[30]. These observations indicate that Kras, namely Kras4B, is developmentally essential with unique functions that cannot be compensated by other Ras family members. Surprisingly, mice in which Hras is inserted into the Kras locus are viable despite completely lacking Kras proteins[31]. These data suggest that the role of Kras in development is not related to unique Kras protein functions, but rather involves regulatory properties that are specific to the Kras locus.
Several mouse models of mutant Kras-driven lung Carcinogenesis have been widely used to study the role of Kras in NSCLC in vivo. One model involves chemical carcinogen exposure, resulting in lung tumors that almost invariably harbor activating mutations at codon 12 or 61 of Kras[32]–[35]. A second involves a transgenic strategy in which the mutant Kras allele is incorporated into the genome at random locations and expression is induced by treating mice with doxycycline[36]. The third employs a knock-in strategy that involves targeting the endogenous Kras locus to generate a mutationally activated Kras allele that is maintained in a latent state by inclusion of a STOP cassette flanked by loxP elements[37],[38]. Removal of the STOP cassette, either by spontaneous recombination in the KrasLA2 model[37] or by intranasal administration of an adenovirus containing Cre recombinase in the KrasLSL−G12D model[38], results in mutant Kras expression and the development of lung tumors. These mouse models have demonstrated that mutational activation of Kras alone is sufficient for lung tumor development, suggesting that it is an early event during lung Carcinogenesis. The role of KRAS during the early stages of lung Carcinogenesis is further supported by studies in human NSCLC showing that KRAS mutations can be detected in precancerous lesions[39]. In addition, lung tumors induced by both carcinogen and genetic approaches stain positively for SP-C and negatively for CCA/CC10[33],[37],[38], suggesting that they derive from the same cell lineage as human lung adenocarcinomas. However, the majority of mouse lung tumors are adenomas[33],[37],[38], which are thought to be adenocarcinoma precursors. Progression from adenomas to adenocarcinomas in these mutant Kras-driven mouse models of NSCLC can be accelerated by loss of the p53 tumor suppressor gene[40]. Likewise, KRAS and P53 are mutated at similar frequencies in human lung adenocarcinomas and occur concurrently in many cases[11].
The Tumor Suppressor Function of Kras and Implications for Lung Cancer Susceptibility
While mutant Kras is potently oncogenic, studies in the mouse have elegantly demonstrated that WT Kras functions as a suppressor of this activity[41]. Mice with only one copy of Kras were found to be more susceptible to carcinogen-induced lung Carcinogenesis than mice with two copies of Kras[41]. These findings suggest that the remaining WT allele of Kras in mice of the latter genotype inhibits lung tumor development. This was further confirmed by in vitro studies in which ectopic expression of WT Kras attenuated the growth of cancer cells containing mutant Kras[41]. Analyses of lung tumors from mutant Kras-driven NSCLC models have shown that chromosomal duplication involving the Kras locus is the most common genetic event in these tumors that harbor Kras mutations[42],[43]. Studies of human lung adenocarcinomas have also shown frequent KRAS copy number gains and corresponding increases in RNA levels of KRAS[11]. In addition, loss of heterozygosity spanning the KRAS locus has been observed in human lung tumors that relate with KRAS mutation and preferentially target the WT KRAS allele[44]. The imbalance in favor of mutant KRAS in human and mouse lung tumors is consistent with the requirement of tumor cells to overcome the tumor suppressor effect of WT KRAS.
Inbred strains of mice exhibit differential susceptibility to lung Carcinogenesis, and genetic crosses between different mouse strains have led to the identification of pulmonary adenoma susceptibility 1 (Pas1) as a major locus regulating predisposition to mutant Kras-driven lung cancer[45]. Of the genes located within Pas1, Kras emerged as the most likely candidate; however, a lack of coding sequence variants in Kras among the different strains of mice called into question the mechanisms through which Kras would regulate lung cancer susceptibility. However, mice that are susceptible to lung Carcinogenesis were found to express higher levels of Kras compared with mice classified as resistant[32]. We proposed that lung cancer susceptibility is regulated by the balance between the levels of mutant and WT Kras, taking into consideration the respective oncogenic and suppressor functions of these alleles[32]. Consistent with this notion, Kras mutations occur preferentially on the more highly expressed Kras allele inherited from the more susceptible parent[46]. In humans, a polymorphism in the 3′-untranslated region of KRAS, which results in increased KRAS expression via interference of binding by let-7 microRNA, is associated with an increased risk of developing NSCLC[47]. A number of additional genetic variants in the human KRAS gene have also been associated with the risk of developing lung adenocarcinomas[48],[49], and in some cases the susceptible allele is found to be expressed at relatively higher levels in normal lung tissues[48]. However, many of these associations were not reproducible in later studies[50],[51]. One possible explanation for the lack of consensus among human case-control studies may be the variations in frequency (16%–40%) of KRAS mutations in different cohorts of patients with NSCLC. Therefore, it may be necessary to consider the somatic events that occur in tumors (i.e., KRAS mutational status) in association studies to identify lung cancer susceptibility genes.
Isoform-specific Functions of Kras in Lung Carcinogenesis
As a result of alternative splicing, the human and mouse Kras loci encode two highly similar proteins, Kras4A and Kras4B, that are jointly affected by activating mutations commonly found in cancer (Figure 1). While Kras4B is ubiquitously expressed, albeit at varying levels across tissues, Kras4A expression is tissue-specific and not essential for embryonic development, suggesting that Kras4A has a minor role in Kras biology. However, we have shown that mice lacking Kras4A are highly resistant to carcinogen-induced lung tumor development[33]. Similar findings have been reported using a different mouse model that also lacks Kras4A[52]. These studies suggest that Kras4A is essential for lung Carcinogenesis. The requirement for Kras4A in Carcinogenesis is compatible with the observation that Kras4A is expressed in the lung and a number of other tissues from which arising tumors frequently harbor Kras mutations, namely the colon and the pancreas[53],[54]. In the lung, Kras4A is highly expressed in a subset of epithelial cells, which could potentially be the originating cells of NSCLC[33]. Studies of the cellular origin of NSCLC in the mouse have identified a number of candidates[55],[56], but their relationship to Kras4A remains to be determined. Nevertheless, the identification of Kras4A as an essential component of mutant Kras-driven lung tumors may have important implications for the design and development of KRAS-targeted therapeutics.
The Challenges and Future of Therapeutics for KRAS-mutant NSCLC
The development of KRAS-targeting cancer therapy has proven to be a challenging endeavor. Because cancer-causing mutations render KRAS oncogenic by impairing its GTPase activity, the KRAS oncoprotein has been generally deemed “undruggable” by conventional chemical approaches. In contrast, a number of drugs have been designed to inhibit the post-translational modification of the RAS proteins to prevent proper localization and function. However, farnesyl transferase inhibitors (FTIs) failed to inhibit KRAS due to alternative prenylation by geranylgeranyl transferase (GGTase)[57], and combined treatment with FTIs and the more recently developed GGTase inhibitors have displayed unacceptable levels of off-target effects and toxicity[58],[59]. An alternative strategy involving the use of RNA interference (RNAi) to deplete KRAS in cells has been shown to be effective in some human KRAS-mutant NSCLC cell lines[60], demonstrating that some tumors harboring KRAS mutations remain highly dependent on oncogenic KRAS for survival. While siRNA knockdown of gene expression is a promising strategy to treat such KRAS “addicted” tumors in the clinic, the lack of effective methods to deliver siRNA to tumors has precluded development of such therapeutics. However, recent advances in the use of nanoparticles for systemic siRNA delivery in humans may potentially help overcome this limitation[61].
A great deal of effort has been placed on developing inhibitors of effector pathways downstream of RAS with the understanding that inhibition of these pathways could counteract the activity of oncogenic RAS. The RAF/MEK/ERK pathway was the first RAS effector pathway identified and the best characterized. Activating mutations in BRAF have been identified in different human cancers, including lung cancer, but generally never together with RAS mutations[62]. BRAF-selective inhibitors that effectively block proliferation of BRAF-mutant cell lines have been developed but are surprisingly ineffective against RAS-mutant cells[63]–[65]. In fact, these inhibitors paradoxically potentiate RAF/MEK/ERK signaling in RAS-mutant cells[63]–[65], and may cause severe adverse effects when given to patients with RAS-mutant cancers. Indeed, a subset of patients with melanoma treated with BRAF inhibitors developed cutaneous squamous cell carcinomas that contained RAS mutations[66]. The mechanistic explanation for the paradoxical activation of RAF/MEK/ERK in RAS-mutant cells treated with BRAF inhibitors involves CRAF activation[63]–[65], suggesting that CRAF-selective inhibitors could potentially be effective against cancers driven by mutant RAS. In support of this notion, studies in the mouse have shown that CRaf, rather than BRaf, is essential for the development of mutant Kras-driven NSCLC[67].
Recently, there has been growing interest in exploiting the concept of synthetic lethality to target KRAS-mutant cancer cells[68]. This approach involves the selective killing of KRAS-mutant cancer cells through inhibition of a second protein. Studies in the mouse identified a synthetic lethal interaction between mutant Kras and Cdk4, where Cdk4 ablation caused lung cells expressing mutant Kras to undergo senescence and prevented tumor growth[69]. A number of RNAi-based synthetic lethal screens in cell lines have identified many potential synthetic lethal therapeutic targets that preferentially cause death of KRAS-mutant cells[70]–[72]. Some of these targets, including CDK4, STK33, TBK1, and PLK1, encode protein kinases and thus may be tractable to inhibition by selective small molecular inhibitors, such as those that have demonstrated success against mutant EGFR and BRAF.
Conclusions
Much progress has been made over the years in our understanding of RAS genes and the critical role they play in tumorigenesis, yet we have been unable to exploit this knowledge to significantly improve the outcome of cancers driven by mutant KRAS. While recognizing that NSCLC and CRC with KRAS mutations are not likely to be responsive to EGFR inhibitors is an important step forward in improving patient treatment, there remains a pressing need for the development of effective KRAS-directed cancer therapies. Although efforts to develop KRAS-targeting therapies have so far been met with disappointment, we have gained important insights into the complex biochemistry of KRAS proteins and KRAS signaling networks. At the same time, in vivo studies in mice have and will continue to make important contributions to our understanding of the underlying biology of KRAS proteins and their roles in cancer. Going forward, it will be critical to continue interrogating the role of KRAS in cancer through mouse and molecular studies and to bridge this knowledge with clinical studies to facilitate the development of truly effective therapies against mutant KRAS-driven cancers.
Acknowledgments
P.M.K.W. is supported by the NIH Training Grant T32 GM007175 and a National Science Foundation Graduate Research Fellowship. M.D.T. was supported by a UCSF Academic Senate Research Grant and the Nan Tucker McEvoy Research Fund in Thoracic Oncology.
References
- 1.Bos JL. Ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–4689. [PubMed] [Google Scholar]
- 2.Konstantinopoulos PA, Karamouzis MV, Papavassiliou AG. Post-translational modifications and regulation of the RAS superfamily of GTPases as anticancer targets. Nat Rev Drug Discov. 2007;6:541–555. doi: 10.1038/nrd2221. [DOI] [PubMed] [Google Scholar]
- 3.Seger R, Krebs EG. The MAPK signaling cascade. FASEB J. 1995;9:726–735. [PubMed] [Google Scholar]
- 4.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 5.Wolthuis RM, Bos JL. Ras caught in another affair: the exchange factors for Ral. Curr Opin Genet Dev. 1999;9:112–117. doi: 10.1016/s0959-437x(99)80016-1. [DOI] [PubMed] [Google Scholar]
- 6.Lambert JM, Lambert QT, Reuther GW, et al. Tiam1 mediates Ras activation of Rac by a PI (3)K-independent mechanism. Nat Cell Biol. 2002;4:621–625. doi: 10.1038/ncb833. [DOI] [PubMed] [Google Scholar]
- 7.Gonzalez-Garcia A, Pritchard CA, Paterson HF, et al. RalGDS is required for tumor formation in a model of skin Carcinogenesis. Cancer Cell. 2005;7:219–226. doi: 10.1016/j.ccr.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 8.Malliri A, van der Kammen RA, Clark K, et al. Mice deficient in the Rac activator Tiam1 are resistant to Ras-induced skin tumours. Nature. 2002;417:867–871. doi: 10.1038/nature00848. [DOI] [PubMed] [Google Scholar]
- 9.Jemal A, Siegel R, Xu J, et al. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 10.Suzuki Y, Orita M, Shiraishi M, et al. Detection of ras gene mutations in human lung cancers by single-strand conformation polymorphism analysis of polymerase chain reaction products. Oncogene. 1990;5:1037–1043. [PubMed] [Google Scholar]
- 11.Ding L, Getz G, Wheeler DA, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Graziano SL, Gamble GP, Newman NB, et al. Prognostic significance of K-ras codon 12 mutations in patients with resected stage I and II non-small-cell lung cancer. J Clin Oncol. 1999;17:668–675. doi: 10.1200/JCO.1999.17.2.668. [DOI] [PubMed] [Google Scholar]
- 13.Nelson MA, Wymer J, Clements N., Jr Detection of K-ras gene mutations in non-neoplastic lung tissue and lung cancers. Cancer Lett. 1996;103:115–121. doi: 10.1016/0304-3835(96)04202-4. [DOI] [PubMed] [Google Scholar]
- 14.Mitsudomi T, Viallet J, Mulshine JL, et al. Mutations of ras genes distinguish a subset of non-small-cell lung cancer cell lines from small-cell lung cancer cell lines. Oncogene. 1991;6:1353–1362. [PubMed] [Google Scholar]
- 15.Ahrendt SA, Decker PA, Alawi EA, et al. Cigarette smoking is strongly associated with mutation of the K-ras gene in patients with primary adenocarcinoma of the lung. Cancer. 2001;92:1525–1230. doi: 10.1002/1097-0142(20010915)92:6<1525::aid-cncr1478>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 16.Forbes SA, Bindal N, Bamford S, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011;39:D945–D950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garassino MC, Marabese M, Rusconi P, et al. Different types of K-Ras mutations could affect drug sensitivity and tumour behaviour in non-small-cell lung cancer. Ann Oncol. 2011;22:235–237. doi: 10.1093/annonc/mdq680. [DOI] [PubMed] [Google Scholar]
- 18.Gautschi O, Huegli B, Ziegler A, et al. Origin and prognostic value of circulating KRAS mutations in lung cancer patients. Cancer Lett. 2007;254:265–273. doi: 10.1016/j.canlet.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 19.Mascaux C, Iannino N, Martin B, et al. The role of RAS Oncogene in survival of patients with lung cancer: a systematic review of the literature with meta-analysis. Br J Cancer. 2005;92:131–139. doi: 10.1038/sj.bjc.6602258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sasaki H, Okuda K, Kawano O, et al. Nras and Kras mutation in Japanese lung cancer patients: genotyping analysis using LightCycler. Oncol Rep. 2007;18:623–628. [PubMed] [Google Scholar]
- 21.Camps C, Sirera R, Bremnes R, et al. Is there a prognostic role of K-ras point mutations in the serum of patients with advanced non-small cell lung cancer? Lung Cancer. 2005;50:339–346. doi: 10.1016/j.lungcan.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 22.Keohavong P, DeMichele MA, Melacrinos AC, et al. Detection of K-ras mutations in lung carcinomas: relationship to prognosis. Clin Cancer Res. 1996;2:411–418. [PubMed] [Google Scholar]
- 23.Gnanasampanthan G, Elsaleh H, McCaul K, et al. Ki-ras mutation type and the survival benefit from adjuvant chemotherapy in Dukes' C colorectal cancer. J Pathol. 2001;195:543–548. doi: 10.1002/path.990. [DOI] [PubMed] [Google Scholar]
- 24.Tsao MS, Aviel-Ronen S, Ding K, et al. Prognostic and predictive importance of p53 and RAS for adjuvant chemotherapy in non small-cell lung cancer. J Clin Oncol. 2007;25:5240–5247. doi: 10.1200/JCO.2007.12.6953. [DOI] [PubMed] [Google Scholar]
- 25.Winton T, Livingston R, Johnson D, et al. Vinorelbine plus cisplatin vs. observation in resected non-small-cell lung cancer. N Engl J Med. 2005;352:2589–2597. doi: 10.1056/NEJMoa043623. [DOI] [PubMed] [Google Scholar]
- 26.Eberhard DA, Johnson BE, Amler LC, et al. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol. 2005;23:5900–5909. doi: 10.1200/JCO.2005.02.857. [DOI] [PubMed] [Google Scholar]
- 27.Pao W, Wang TY, Riely GJ, et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005;2:e17. doi: 10.1371/journal.pmed.0020017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tol J, Koopman M, Cats A, et al. Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer. N Engl J Med. 2009;360:563–572. doi: 10.1056/NEJMoa0808268. [DOI] [PubMed] [Google Scholar]
- 29.Johnson L, Greenbaum D, Cichowski K, et al. K-ras is an essential gene in the mouse with partial functional overlap with N-ras. Genes Dev. 1997;11:2468–2481. doi: 10.1101/gad.11.19.2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Plowman SJ, Williamson DJ, O'Sullivan MJ, et al. While K-ras is essential for mouse development, expression of the K-ras 4A splice variant is dispensable. Mol Cell Biol. 2003;23:9245–9250. doi: 10.1128/MCB.23.24.9245-9250.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Potenza N, Vecchione C, Notte A, et al. Replacement of K-Ras with H-Ras supports normal embryonic development despite inducing cardiovascular pathology in adult mice. EMBO Rep. 2005;6:432–437. doi: 10.1038/sj.embor.7400397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.To MD, Perez-Losada J, Mao JH, et al. A functional switch from lung cancer resistance to susceptibility at the Pas1 locus in Kras2LA2 mice. Nat Genet. 2006;38:926–930. doi: 10.1038/ng1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.To MD, Wong CE, Karnezis AN, et al. Kras regulatory elements and exon 4A determine mutation specificity in lung cancer. Nat Genet. 2008;40:1240–1244. doi: 10.1038/ng.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.You M, Candrian U, Maronpot RR, et al. Activation of the Ki-ras protooncogene in spontaneously occurring and chemically induced lung tumors of the strain A mouse. Proc Natl Acad Sci USA. 1989;86:3070–3074. doi: 10.1073/pnas.86.9.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.You M, Wang Y, Lineen AM, et al. Mutagenesis of the K-ras protooncogene in mouse lung tumors induced by N-ethyl-N-nitrosourea or N-nitrosodiethylamine. Carcinogenesis. 1992;13:1583–1586. doi: 10.1093/carcin/13.9.1583. [DOI] [PubMed] [Google Scholar]
- 36.Fisher GH, Wellen SL, Klimstra D, et al. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev. 2001;15:3249–3262. doi: 10.1101/gad.947701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnson L, Mercer K, Greenbaum D, et al. Somatic activation of the K-ras Oncogene causes early onset lung cancer in mice. Nature. 2001;410:1111–1116. doi: 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- 38.Jackson EL, Willis N, Mercer K, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yokota J, Kohno T. Molecular footprints of human lung cancer progression. Cancer Sci. 2004;95:197–204. doi: 10.1111/j.1349-7006.2004.tb02203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jackson EL, Olive KP, Tuveson DA, et al. The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res. 2005;65:10280–10288. doi: 10.1158/0008-5472.CAN-05-2193. [DOI] [PubMed] [Google Scholar]
- 41.Zhang Z, Wang Y, Vikis HG, et al. Wildtype Kras2 can inhibit lung Carcinogenesis in mice. Nat Genet. 2001;29:25–33. doi: 10.1038/ng721. [DOI] [PubMed] [Google Scholar]
- 42.Sweet-Cordero A, Tseng GC, You H, et al. Comparison of gene expression and DNA copy number changes in a murine model of lung cancer. Genes Chromosomes Cancer. 2006;45:338–348. doi: 10.1002/gcc.20296. [DOI] [PubMed] [Google Scholar]
- 43.To MD, Quigley DA, Mao JH, et al. Progressive genomic instability in the FVB/Kras (LA2) mouse model of lung cancer. Mol Cancer Res. 2011;9:1339–1345. doi: 10.1158/1541-7786.MCR-11-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li J, Zhang Z, Dai Z, et al. LOH of chromosome 12p correlates with Kras2 mutation in non-small cell lung cancer. Oncogene. 2003;22:1243–1245. doi: 10.1038/sj.onc.1206192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gariboldi M, Manenti G, Canzian F, et al. A major susceptibility locus to murine lung Carcinogenesis maps on chromosome 6. Nat Genet. 1993;3:132–136. doi: 10.1038/ng0293-132. [DOI] [PubMed] [Google Scholar]
- 46.You M, Wang Y, Stoner G, et al. Parental bias of Ki-ras oncogenes detected in lung tumors from mouse hybrids. Proc Natl Acad Sci USA. 1992;89:5804–5808. doi: 10.1073/pnas.89.13.5804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chin LJ, Ratner E, Leng S, et al. A SNP in a let-7 microRNA complementary site in the KRAS 3′ untranslated region increases non–small cell lung cancer risk. Cancer Res. 2008;68:8535–8540. doi: 10.1158/0008-5472.CAN-08-2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kohno T, Kunitoh H, Suzuki K, et al. Association of KRAS polymorphisms with risk for lung adenocarcinoma accompanied by atypical adenomatous hyperplasias. Carcinogenesis. 2008;29:957–963. doi: 10.1093/carcin/bgn048. [DOI] [PubMed] [Google Scholar]
- 49.Manenti G, De Gregorio L, Pilotti S, et al. Association of chromosome 12p genetic polymorphisms with lung adenocarcinoma risk and prognosis. Carcinogenesis. 1997;18:1917–1920. doi: 10.1093/carcin/18.10.1917. [DOI] [PubMed] [Google Scholar]
- 50.Dragani TA, Hirohashi S, Juji T, et al. Population-based mapping of pulmonary adenoma susceptibility 1 locus. Cancer Res. 2000;60:5017–5020. [PubMed] [Google Scholar]
- 51.Manenti G, Nomoto T, De Gregorio L, et al. Predisposition to lung tumorigenesis. Toxicol Lett. 2000;112–113:257–263. doi: 10.1016/s0378-4274(99)00232-5. [DOI] [PubMed] [Google Scholar]
- 52.Patek CE, Arends MJ, Wallace WA, et al. Mutationally activated K-ras 4A and 4B both mediate lung Carcinogenesis. Exp Cell Res. 2008;314:1105–1114. doi: 10.1016/j.yexcr.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 53.Pells S, Divjak M, Romanowski P, et al. Developmentally-regulated expression of murine K-ras isoforms. Oncogene. 1997;15:1781–1786. doi: 10.1038/sj.onc.1201354. [DOI] [PubMed] [Google Scholar]
- 54.Wang Y, You M, Wang Y. Alternative splicing of the K-ras gene in mouse tissues and cell lines. Exp Lung Res. 2001;27:255–267. doi: 10.1080/019021401300054028. [DOI] [PubMed] [Google Scholar]
- 55.Kim CF, Jackson EL, Woolfenden AE, et al. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 2005;121:823–835. doi: 10.1016/j.cell.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 56.Xu X, Rock JR, Lu Y, et al. Evidence for type II cells as cells of origin of K-Ras-induced distal lung adenocarcinoma. Proc Natl Acad Sci USA. 2012;109:4910–4915. doi: 10.1073/pnas.1112499109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fiordalisi JJ, Johnson RL, 2nd, Weinbaum CA, et al. High affinity for farnesyltransferase and alternative prenylation contribute individually to K-Ras4B resistance to farnesyltransferase inhibitors. J Biol Chem. 2003;278:41718–41727. doi: 10.1074/jbc.M305733200. [DOI] [PubMed] [Google Scholar]
- 58.Lobell RB, Omer CA, Abrams MT, et al. Evaluation of farnesyl: protein transferase and geranylgeranyl:protein transferase inhibitor combinations in preclinical models. Cancer Res. 2001;61:8758–8768. [PubMed] [Google Scholar]
- 59.Lobell RB, Liu D, Buser CA, et al. Preclinical and clinical pharmacodynamic assessment of L-778,123, a dual inhibitor of farnesyl:protein transferase and geranylgeranyl:protein transferase type-I. Mol Cancer Ther. 2002;1:747–758. [PubMed] [Google Scholar]
- 60.Singh A, Greninger P, Rhodes D, et al. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell. 2009;15:489–500. doi: 10.1016/j.ccr.2009.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Davis ME, Zuckerman JE, Choi CH, et al. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature. 2010;464:1067–1070. doi: 10.1038/nature08956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 63.Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431–435. doi: 10.1038/nature08833. [DOI] [PubMed] [Google Scholar]
- 64.Heidorn SJ, Milagre C, Whittaker S, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209–221. doi: 10.1016/j.cell.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Poulikakos PI, Zhang C, Bollag G, et al. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with Wildtype BRAF. Nature. 2010;464:427–430. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366:207–215. doi: 10.1056/NEJMoa1105358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Blasco RB, Francoz S, Santamaria D, et al. c-Raf, but not B-Raf, is essential for development of K-Ras oncogene-driven non-small cell lung carcinoma. Cancer Cell. 2011;19:652–663. doi: 10.1016/j.ccr.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kaelin WG., Jr The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005;5:689–698. doi: 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- 69.Puyol M, Martin A, Dubus P, et al. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell. 2010;18:63–73. doi: 10.1016/j.ccr.2010.05.025. [DOI] [PubMed] [Google Scholar]
- 70.Barbie DA, Tamayo P, Boehm JS, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–112. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Luo J, Emanuele MJ, Li D, et al. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras Oncogene. Cell. 2009;137:835–848. doi: 10.1016/j.cell.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Scholl C, Frohling S, Dunn IF, et al. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell. 2009;137:821–834. doi: 10.1016/j.cell.2009.03.017. [DOI] [PubMed] [Google Scholar]