

Abstract
Recent advancements in molecular sequencing techniques have led to a surge in the number of phylogenetic studies that incorporate large amounts of genetic data. We test the assumption that analyzing large number of genes will lead to improvements in tree resolution and branch support using moths in the superfamily Bombycoidea, a group with some interfamilial relationships that have been difficult to resolve. Specifically, we use a next-gen data set that included 19 taxa and 938 genes (∼1.2M bp) to examine how codon position and saturation might influence resolution and node support among three key families. Maximum likelihood, parsimony, and species tree analysis using gene tree parsimony, on different nucleotide and amino acid data sets, resulted in largely congruent topologies with high bootstrap support compared with prior studies that included fewer loci. However, for a few shallow nodes, nucleotide and amino acid data provided high support for conflicting relationships. The third codon position was saturated and phylogenetic analysis of this position alone supported a completely different, potentially misleading sister group relationship. We used the program RADICAL to assess the number of genes needed to fix some of these difficult nodes. One such node originally needed a total of 850 genes but only required 250 when synonymous signal was removed. Our study shows that, in order to effectively use next-gen data to correctly resolve difficult phylogenetic relationships, it is necessary to assess the effects of synonymous substitutions and third codon positions.
Keywords: Bombycoidea, Lepidoptera, phylogeny, saturation, synonymous substitutions, transcriptome
Introduction
The revolution of next-generation sequencing methods has led to a surge in the number of phylogenetic studies incorporating an unprecedented number of genes (McCormack et al. 2013). Many methods have emerged as being very amenable for phylogenetic applications that target non-model taxa. These methods are aimed at sequencing specific gene regions by using polymerase chain reaction enrichment (e.g., Mamanova et al. 2010; Bybee et al. 2011), hybridization enrichment (e.g., Cronn et al. 2012; Faircloth et al. 2012; Lemmon et al. 2012), and transcriptomics (e.g., Hittinger et al. 2010; Nabholz et al. 2011; Oakley et al. 2012). Phylotranscriptomics, or the inclusion of transcriptomic data into phylogenetic studies, differs from other methods in that it is based on expressed mRNA sequences and does not require previous knowledge of specific gene regions (Cronn et al. 2012; McCormack et al. 2013). Phylotranscriptomics has been shown to be effective in analyzing and showing support for deep relationships within Pancrustacea (von Reumont et al. 2011; Oakley et al. 2012), Insecta (Simon et al. 2012), and Arthropoda (Meusemann et al. 2010). Transcriptomic data have further proven effective at estimating relationships for comparably younger divergences within Coleoptera (Hughes et al. 2006), Hymenoptera (Sharanowski et al. 2010), and between mosquito species (Hittinger et al. 2010).
Phylotranscriptomic studies that address arthropod relationships often utilize amino acid data sets (e.g., Meusemann et al. 2010; von Reumont et al. 2011; Oakley et al. 2012; Simon et al. 2012). Amino acid data matrices can eliminate the problem of saturated synonymous nucleotide substitutions and rate heterogeneity among codon positions but can also result in lower branch support than analyses based on nucleotide data alone (Regier et al. 2008b, 2010; Zwick et al. 2012). The negative effect of rate heterogeneity among codon positions on phylogenetic inference is well known (Kuhner and Felsenstein 1994; Sullivan 1996; Yang 1996a, 1996b; Cunningham et al. 1998; Buckley et al. 2001; Pagel and Meade 2004) and can affect small data sets based on a few genes (e.g., Sullivan 1996; Soltis et al. 2002), large data sets such as mitogenomic data (Song et al. 2010), and nuclear gene data sets (Regier et al. 2008b, 2010; Betancur-R. et al. 2013). Some strategies used to account for problems of saturated sites and rate heterogeneity in nucleotide data include removing third codon positions, degenerating synonymous substitutions (similar to RY coding), and using large amounts of genetic data to increase the signal-to-noise ratio (Regier et al. 2008b, 2010; Song et al. 2010; Zwick et al. 2012; Betancur-R. et al. 2013). Bentancur-R. et al. (2013) recently showed that a multi-gene concatenated data set (20 genes) can overcome systematic biases such as randomly distributed homoplasy and compositional heterogeneity. However, it is unknown whether saturation and rate heterogeneity among sites will have a significant negative effect on phylogeny estimation or whether the high amount of signal will compensate for these effects in large genomic nucleotide data sets.
Lepidoptera have become a premier model system for genetics with the completion of three recent genomes: the domesticated Bombyx silk moth (Xia et al. 2004), monarch butterfly (Zhan et al. 2011), and diamondback moth (You et al. 2013). The Lepidoptera Tree of Life initiative, has produced many impressive studies with up to 25 genes and extensive taxon sampling (e.g., Regier et al. 2008a, 2009, 2013; Cho et al. 2011; Zwick et al. 2011). Although relationships within superfamilies are generally well supported, several interfamilial relationships still remain uncertain.
Wild silk moths and relatives in the superfamily Bombycoidea are a diverse group of charismatic moths that include more than 4,700 species (van Nieukerken et al. 2011). Many species are model organisms, such as the tobacco hornworm (Manduca sexta) and domesticated silk moth (Bombyx mori). However, relationships among the three main bombycoid families, Bombycidae, Saturniidae, and Sphingidae, have been difficult to resolve. Studies that utilized mitochondrial (Kim et al. 2011; Huan-Na et al. 2012) and nuclear genes (e.g., Regier et al. 2008a, 2013; Zwick et al. 2011) as well as phylotranscriptomic studies on insects (Simon et al. 2012) and arthropods (Meusemann et al. 2010) that included a small number of bombycoid exemplars resulted in conflicting or poorly supported relationships among these families (fig. 1). We use next-gen transcriptomic data to test 1) whether transcriptomic data can conclusively estimate relationships among these three families, 2) the effect of saturated sites on phylogeny estimation, and 3) the relative phylogenetic signal from the first, second, and third codon positions. We test these hypotheses with phylogenetic analyses of concatenated nucleotide and amino acid data sets. We hypothesize that increasing the number of genes, as well as accounting for saturation, will provide greater resolution for a group whose interfamilial relationships are often characterized by short internal branch lengths.
Fig. 1.—
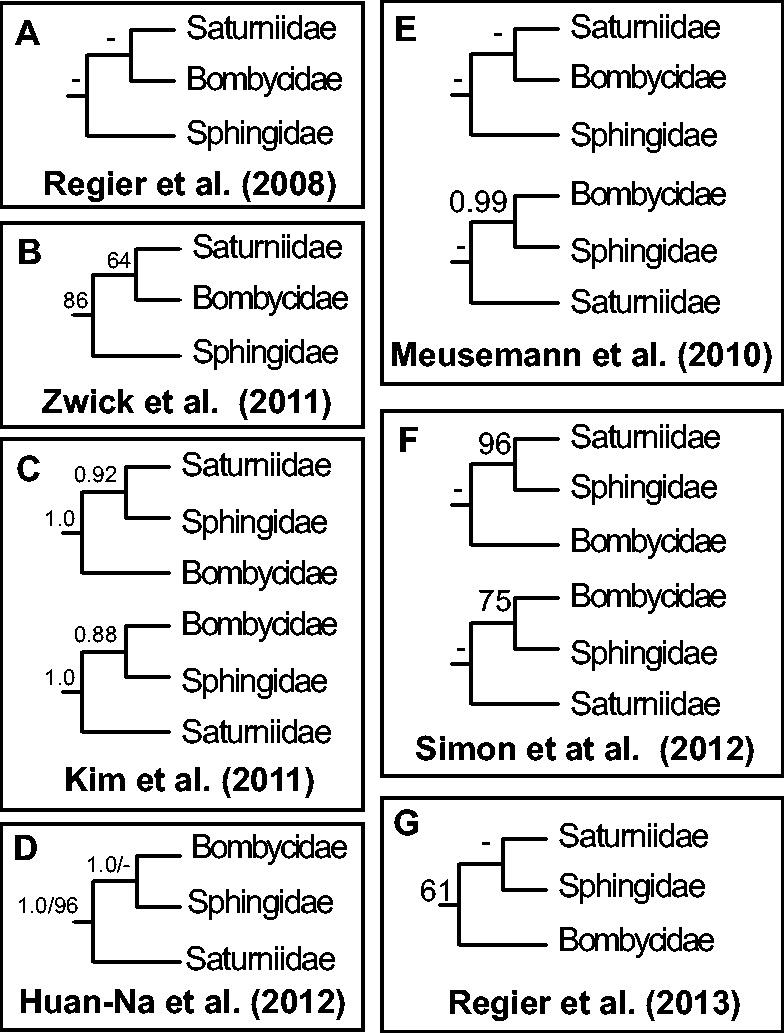
Published phylogenetic trees showing relationships among the families Bombycidae, Saturniidae, and Sphingidae. A dash (-) indicates branch support <60% bootstrap or <0.6 posterior probability. (A) Regier et al. (2008a): ML analysis of 5 protein-coding nuclear genes; (B) Zwick et al. (2011): ML analysis of 25 protein-coding nuclear genes; (C) Kim et al. (2011): Bayesian analysis of mitochondrial genome data, nt123 + rRNA (above) and nt12 + rRNA (below); (D) Huan-Na et al. (2012): 13 protein coding mitochondrial genes; Bayesian posterior probability before the slash and ML bootstraps after the slash; (E) Meusemann et al. (2010): 129 genes, ML tree (above) and Bayesian tree (below); (F) Simon et al. (2012): ML analysis of 335 genes (above) and 102 genes (below); (G) Regier et al. (2013): ML analysis of 19 protein-coding nuclear genes.
Materials and Methods
The general workflow from data collection to analysis is summarized in figure 2. Our taxon sampling included a total of 19 species, 14 ingroup and 5 outgroup. Ingroup taxa included seven species from Saturniidae, five from Sphingidae, and two from Bombycidae. Our outgroup sampling consisted of two species of Noctuidae, two species of Pyraloidea, and one butterfly. We collected available expressed sequence tag (EST) data from the B. mori (Xia et al. 2004) and M. sexta genomes (Agricultural Pest Genomics Resource Database, www.agripestbase.org, last accessed November 8, 2013) and from 12 taxa in the GenBank EST database (Benson et al. 2013). We generated six new transcriptomes, from two species of Saturniidae and four species of Sphingidae (supplementary table S1, Supplementary Material online). These transcriptomes were generated from purified RNA extracted with the SV Total RNA Isolation System (catalogue no. Z3100; Promega) from flash-frozen or RNAlater-preserved tissue. Purified RNA (5 µg) was sent to the University of Missouri DNA Core, where RNA quality check, RNA-Seq library construction, and Illumina HiSeq 2000 runs were performed as 100 bp SE reads with four samples per lane (one lane included two additional samples that were not included in this study). We used SOAPdenovo-Trans v1.01 for de novo assembly of the transcriptomes of each taxon using five k-mer values (13, 23, 33, 43, 63) following the additive multiple-k assembly method of Surget-Groba and Montoya-Burgos (2010). We combined redundant contigs from the multiple k-mer assembles with CD-HIT-EST (Li and Godzik 2006) and removed all sequences below 100 bp with FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/, last accessed November 4, 2013). To identify orthologous genes between transcriptomes and ESTs, we used HaMStR v8b (Ebersberger et al. 2009). HaMStR was implemented using the Insecta HMMER v3-2 core ortholog data (1,579 core orthologs) built upon the InParanoid transitive closure approach using proteomes from six primer taxa: Apis mellifera, B. mori, Capitella sp., Daphnia pulex, Ixodes scapularis, and Tribolium castaneum. The “-representative” option was implemented in HaMStR, and we used B. mori as the reference species for reciprocal Blast of candidate EST contigs.
Fig. 2.—
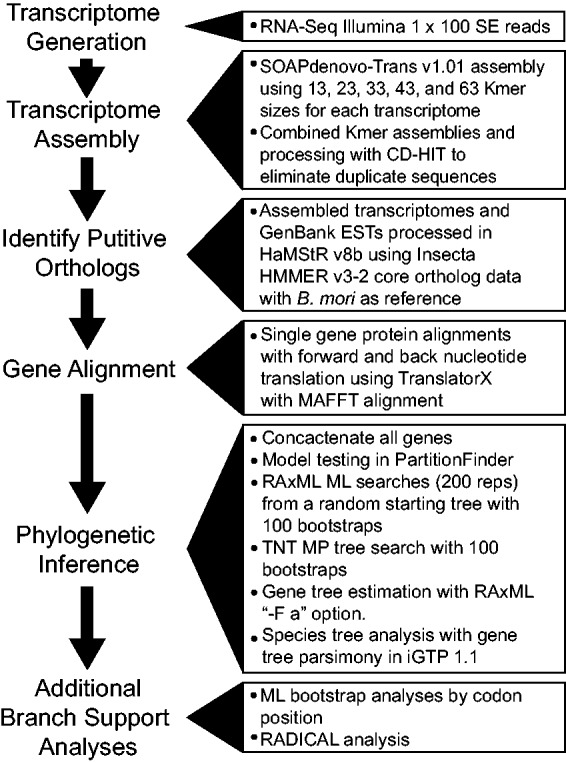
Diagram showing general workflow from data collection to analysis.
To reduce the amount of missing data in the final matrix, we included genes that were represented by at least half (10) of the taxa in this study. We translated the nucleotide data into amino acids in TranslatorX (Abascal et al. 2010) and aligned the data set using MAFFT v7.029b (Katoh and Standley 2013). The aligned amino acid sequence was then back-translated to nucleotide data in TranslatorX. Each gene alignment was visually inspected for misaligned regions, and if such regions were found, either the taxon was removed or the poorly aligned region was removed. Selected genes were concatenated using Geneious v5.5.8 (Biomatters 2013). We tested the concatenated alignment for nucleotide saturation at each codon position and created a saturation plot by nucleotide position in DAMBE v5.3.16 (Xia et al. 2003). The Akaike information criterion (AIC) score in PartitionFinder v1.0.1 (Lanfear et al. 2012) was used to select the best model for a concatenated amino acid alignment. Furthermore, we used the AIC score to choose the best model for the nucleotide alignment under three different partitioning strategies: 1) no partitions, 2) partitioned separately by codon position, and 3) two partitions, with the first and second codon position (nt1 and nt2) as one partition and the third codon position (nt3) as the other.
We estimated our phylogeny with two different approaches: a standard concatenation approach and a species tree analysis using gene tree parsimony. For phylogeny estimation using concatenation, we ran maximum likelihood (ML) analyses in RAxML (Stamatakis 2006) and maximum parsimony (MP) analyses in TNT (Goloboff et al. 2008). For ML analysis, we created six separate data matrices consisting of 19 taxa for all genes: 1) the full nucleotide data matrix consisting of all nucleotide positions (nt123), 2) a matrix consisting of only the first and second codon positions (nt12), 3) a matrix consisting of only the third codon position (nt3), 4) a data set that removed all synonymous signal, thus leaving only non-synonymous changes at all coding positions (degen1), 5) a data set consisting of only nt3 from the degen1 matrix (degen1-nt3), and 6) an amino acid matrix (AA). In RAxML, we estimated ML trees for each of these six concatenated matrices. We conducted 200 tree searches starting from a random topology and also implemented the “–F a” option for a combined ML tree search with 100 bootstrap replicates. The concatenated matrices were partitioned following the optimal partitioning strategy as identified in PartitionFinder. The degen1 data sets were created with the degen v1.4 perl script from Zwick et al. (2012), which degenerates nucleotides to IUPAC ambiguity codes at all sites that have synonymous substitutions. A benefit of using degen1 over nt12 is that degen1 removes all synonymous signal while keeping non-synonymous substitutions from all nucleotide positions, whereas nt12 retains synonymous signal at nt1 and completely removes all signal from nt3 (Zwick et al. 2012). The CAT model of Lartillot and Philippe (2004) implemented in PhyloBayes (Lartillot et al. 2009) has also been shown to effectively deal with saturated sites and synonoymous signal at nt3. However, because PhyloBayes cannot analyze large data sets without jackknifing the data into smaller data sets (Delsuc et al. 2008), we chose not to use this program.
Parsimony analyses were conducted in TNT (Goloboff et al. 2008). We used the 19 taxon, nt123 data set and conducted a new technology search including three rounds of tree fusing, ten rounds of drifting, and tree ratcheting for 100 random addition replicates (xmult = rss, fuse 2, drift 3, ratchet 10, replications 100). One hundred parsimony bootstrap replicates were estimated in TNT with a new technology search including three rounds of tree fusing, ten rounds of drift, and tree ratcheting for ten random addition replicates (xmult = rss, fuse 3, drift 10, ratchet 10, replications 10). We also conducted 100 ML and MP bootstrap analyses using methods described above for each codon position to determine how each codon position supported different regions of the tree.
To examine the effect of concatenation on our resulting topology, we used the program Random Addition Concatenation Analysis (RADICAL) (Narechania et al. 2012). RADICAL produces multiple random concatenation paths by sequentially concatenating genes arbitrarily, without replacement, starting with a single gene and ending with all genes in the data set. RADICAL analysis examines how many genes are necessary to resolve a node. RADICAL produces two statistics for each node: 1) a fixation/degradation point, which is estimated as the number of genes needed to fix a node or ensure a node no longer appears in any concatenation path, and a area under the curve (AUC) value which represents the percent of the total number of trees from the RADICAL analysis that have that particular node. The RADICAL curve provides a visual representation of the emergent phylogenetic support of a node by plotting the normalized average consensus fork index (CFI) (Colless 1980) of all concatenation paths at each concatenation point by the number of genes at these points. RADICAL analyses were conducted on six data matrices (nt123, nt12, nt3, degen1, degen1-nt3, and AA) with ten random concatenation paths using the “–step 25” option, which adds 25 genes to each step in the concatenation path using RAxML for tree construction. We deleted five taxa (Antheraea mylitta, A. pernyi, B. mandarina, Lonomia obliqua, and Ostrinia nubilalis) to limit the amount of missing data and reduce the possible effects of missing data on smaller concatenation points in the analysis. We chose a step of 25 to reduce the number of total trees estimated, thereby significantly reducing computational time needed to complete the analysis. Applying a RADICAL approach to different matrices allows us to examine the phylogenetic contribution of each codon position, the effect of saturated sites on phylogenetic analysis, and the amount of agreement/disagreement for nodes among data matrices.
To account for individual gene history, we estimated species trees using gene tree topologies. Individual gene trees for all 938 genes were estimated in RAxML using the –F a option with 100 bootstraps on nucleotide alignments including all codon positions. Species trees were estimated with gene tree parsimony implemented in iGTP1.1 (Chaudhary et al. 2010) using 50 replicate searches that minimize duplications (MD), minimize duplications and losses (MDL), or minimize deep coalescence (MDC). To estimate confidence in our estimated species tree, we created 100 bootstrap pseudoreplicate gene tree sets by sampling our gene trees with replacement (Felsenstein 1985) and analyzed them in iGTP under the “minimize duplications and losses” model.
Results
The six newly assembled transcriptomes had an average of 202,444 contigs that were above 100 bp and an average N50 of 605 (supplementary table S1, Supplementary Material online). The assembled transcriptomes are deposited at the Dryad data depository (http://datadryad.org, last accessed November 8, 2013) (dryad accession doi:10.5061/dryad.r5cq0). The Illumina data used to assemble the transcriptomes have been submitted to the GenBank Sequence Read Archive database with accession numbers SRR1002974 (Actias luna, BioSample SAMN02364143), SRR1002985 (Ceratomia undulosa, BioSample SAMN02364254), SRR1002983 (Enyo lugubris, BioSample SAMN02364253), SRR1002987 (Hemaris diffinis, BioSample SAMN02364259), SRR1002986 (Darapsa myron, BioSample SAMN02364260), and SRR1002994 (Attacus atlas, BioSample SAMN02364261) under the BioProject PRJNA221547. HaMStR identified an average of 1,436 genes from these transcriptomes as putative homologs to B. mori. We reduced the number of loci to 938 so that included genes were represented by at least half the number of taxa in the data set (10 taxa). These new transcriptomes had an average of 932 genes (supplementary table S2, Supplementary Material online). We deposited our putative homologs as a concatenated nucleotide and amino acids nexus files to the Dryad data depository (http://datadryad.org, last accessed November 8, 2013) (dryad accession doi:10.5061/dryad.r5cq0). EST libraries excluding B. mori had 17–870 genes with an average of 412 (supplementary table S2, Supplementary Material online). Our visual inspection of each gene found no obvious misalignment of nucleotides, and therefore no data were trimmed from our alignments. The resulting concatenated matrix consisted of 1,210,419 bp with 62% missing data and 64% gene coverage for 19 taxa. The data set used in the RADICAL analysis consisted of 14 taxa, 49% missing data, and 85% gene coverage. Xia et al.’s (2003) test for saturation reveled that the first and second codon positions (nt1 and nt2) were not saturated (both P ≤ 0), whereas nt3 was substantially saturated (P ≥ 0.3774). Saturation plots of model-corrected genetic distance by codon position plotted against transitions and transversions also indicate that nt3 was partially saturated (supplementary fig. S1, Supplementary Material online). Degenerating synonymous signal significantly altered the proportion of variable sites at each codon position (supplementary fig. S2, Supplementary Material online). Examination of changes in the number of variable sites from nt3 in the nt123 and degen1 matrices indicates that many (>200,000, 86.6%) of these variable sites were associated with synonymous amino acid substitutions (supplementary fig. S2, Supplementary Material online). Partitioning by codon position was the best partitioning scheme for the nt123 data set, as indicated by the AIC score in PartitionFinder, and this model was used for all ML analyses that included all three codon positions. For the amino acid alignment, the best AIC score was JTT + F + GAMMA. Due to the large number of analyses, we summarize the analyses performed, models, and partitions in table 1.
Table 1.
Summary of the Number of Taxa, % of Gene Coverage, Analyses Performed, Models, and Partitions for Each Data Set
| Data Set | Number of Taxa | % Gene Coverage | Analysis | Models | Partitions |
|---|---|---|---|---|---|
| nt123 | 19 | 64 | MP | — | — |
| 19 | 64 | ML | GTR-GAMMA | 3: nt1, 2, 3 | |
| 19 | 64 | ML (GT) | GTR-GAMMA | by gene | |
| 19 | 64 | iGTP (GT) | MDC, MDL, MD | — | |
| 14 | 85 | ML (R) | GTR-GAMMA | — | |
| nt12 | 19 | 64 | MP | — | — |
| 19 | 64 | ML | GTR-GAMMA | 2: nt1, 2 | |
| 14 | 85 | ML (R) | GTR-GAMMA | — | |
| nt3 | 19 | 64 | MP | — | — |
| 19 | 64 | ML | GTR-GAMMA | — | |
| 14 | 85 | ML (R) | GTR-GAMMA | — | |
| Degen1 | 19 | 64 | MP | — | — |
| 19 | 64 | ML | GTR-GAMMA | 3: nt1, 2, 3 | |
| 14 | 85 | ML (R) | GTR-GAMMA | — | |
| Degen1-nt3 | 19 | 64 | MP | — | — |
| 19 | 64 | ML | GTR-GAMMA | — | |
| 14 | 85 | ML (R) | GTR-GAMMA | — | |
| Amino acid | 19 | 64 | ML | GAMMA JTT+F | — |
| 14 | 85 | ML (R) | GAMMA JTT+F | — |
Note.—MP analyses were conducted in TNT and ML analyses were conducted in RAxML unless otherwise noted. (GT) indicates a gene-tree analysis and (R) indicates a RADICAL analysis.
Optimal phylogenetic trees estimated from nt123 with ML (fig. 3), MP (supplementary fig. S3A, Supplementary Material online), and gene tree–species tree estimation (supplementary fig. S3B–C, Supplementary Material online) were nearly identical. ML topologies from nt123 (fig. 3) and AA (supplementary fig. S4E, Supplementary Material online) differed from parsimony and gene tree–species tree estimation (supplementary fig. S3, Supplementary Material online) in that the two pyraloid outgroups were monophyletic in the ML analyses. The placement of A. luna differed among methods, but this taxon was placed confidently (100% bootstrap) within the Saturniidae in every analysis, corroborating previous morphological and molecular hypotheses (e.g., Minet 1991, 1994; Regier et al. 2002, 2008a).
Fig. 3.—

ML tree estimated from 938 genes in RAxML with bootstrap values placed on each branch. AA, amino acid; iGTP, gene tree parsimony; ML, maximum likelihood; MP, parsimony. Codon positions: first = nt1, second = nt2, third = nt3.
The MDC iGTP analysis that minimizes deep coalescence placed B. mori as sister to Plodia interpunctella, which appears misplaced, as these taxa belong in different superfamilies and no previous phylogenetic analysis has indicated that these taxa are closely related. The placement of P. interpunctella was the only difference between this tree and other species tree methods. ML analyses of nt123 (fig. 3), nt12 (supplementary fig. S4A, Supplementary Material online), degen1 (supplementary fig. S3B, Supplementary Material online), and AA (supplementary fig. S4E, Supplementary Material online) only differed in a few terminal relationships within the Saturniidae and Sphingidae. The ML nt123 and AA analyses differed in the placement of two sphingid taxa (nt123 = H. diffinis + D. myron; AA = H. diffinis + E. lugubris). The nt3 analysis resulted in a topology (supplementary fig. S4D, Supplementary Material online) that strongly conflicted with the nt123 topology. The nt3 tree supported the Bombycidae + Saturniidae (91% bootstrap) with a basal Sphingidae (95% bootstrap). However, all other analyses provided very strong support for Saturniidae + Sphingidae (>97% bootstrap for all trees resulting from the nt123, nt12, degen1, and AA data sets including species tree methods) with a basal Bombycidae that had >97% bootstrap support for species tree methods.
Our estimated relationships (fig. 3) are consistent with the recent Lepidoptera phylogeny of Regier et al. (2013) and provide stronger branch support among Bombycidae, Saturniidae, and Sphingidae. Estimated relationships among saturniid species are also consistent with previous phylogenetic studies of Bombycoidea (Regier et al. 2005; Zwick et al. 2011). Relationships within Sphingidae differ from a recent hawkmoth phylogeny (Kawahara et al. 2009), which placed Hemaris basal to all Macroglossinae (including Darapsa and Enyo). However, these relationships were not strongly supported in Kawahara et al.’s study. In this study, the relationship D. myron + H. diffinis is supported by the third codon position alone.
Bootstrap support for several key nodes differed significantly among trees generated from each codon position. For Saturniidae + Sphingidae (fig. 4A, Node 2), nt1 and nt2 provided strong support, whereas no support came from nt3; nt3 provided strong support for a different relationship (fig. 3 and supplementary fig. S4C, Supplementary Material online). The RADICAL analysis on nt123 indicates that estimating this node is especially challenging (fig. 4A, Node 2). The number of genes needed for all 10 concatenation paths to fix this node was 850, despite having a 73% AUC value (73% of trees estimated in RADICAL have sphingid + saturniid sister group relationship) (fig. 4A and C). The AA, degen1, and nt12 data sets fixed this node at 175, 150, and 225 genes, respectively (fig 4A and C). When comparing the tree generated from nt3 and degen1-nt3 (supplementary fig. S4C and D, Supplementary Material online) and examining the RADICAL traces (fig. 4C), it is clear that the conflicting signal lies in the saturated synonymous substitutions at nt3. Another node that appears to be difficult to estimate with nt123 is Node 4 (Act. luna and A. assamensis, fig. 4A and E). This node received no support from nt3, and the tree estimated from nt3 alone places Act. luna basal to the two saturniid clades (supplementary fig. S4C, Supplementary Material online). The AA, degen1, and nt12 data sets resolved this node with fewer loci, but required >375 genes. The nt3 and nt3_degen1 data sets both had difficulty estimating this node and both reached degradation points (fig. 4E). The relationship between D. myron and H. diffinis (fig. 4A and D, Node 3) is interesting in that the nt123 fixes this relationship at 600 genes. The amino acid, nt12, and degen1 data sets have degradation points at which beyond 850, 850, and 938 genes, respectively, the node is not found in any of the trees in the RADICAL analysis. The AUC value for this node in AA, degen1, and nt12 analyses are ≤34% (fig. 4A, Node 3). The signal supporting this node comes from synonymous third codon substitutions, as nt12, degen1, AA, and degen1-nt3 analyses do not provide support to this node (fig. 4E).
Fig. 4.—

The effect of nucleotide position and synonymous signal on phylogeny. (A) ML tree with RADICAL results from six matrices (nt123, nt12, degen1, nt3, degen1-nt3, and AA). Values are shown on each branch. Fixation/degradation points are shown to the left of the central bar, and the AUC score is shown to the right of the bar. Degradation is indicated with asterisks. (B–E) RADICAL curves for Nodes 1 through 4, with the average CFI of the 10 concatenation paths on the y axis and the number of concatenated genes on the x axis. (B) Node 1, Bombycidae + Saturniidae + Sphingidae. (C) Node 2, Saturniidae + Sphingidae. (D) Node 3, Darapsa myron + Hemaris diffinis. (E) Node 4, Actias luna + Antheraea assamensis.
Discussion
We tested whether phylotranscriptomics can resolve relationships among three major bombycoid moth families on a large data set of >900 genes. Increasing the number of loci can provide high branch support for relationships that have been challenging to estimate with fewer genes. Relationships among these three families are robust and there is general agreement among trees generated from ML (nt123 and AA) and MP analyses on concatenated data sets (fig. 1 and supplementary figs. S3A and S4E, Supplementary Material online). Resulting topologies are also in general congruence with species tree analysis using gene tree parsimony (supplementary fig. S3, Supplementary Material online). Comparison of bootstrap support from nucleotide and amino acid analyses did not show an increase in branch support when using nucleotide data, unlike similar comparisons in smaller arthropod data sets (Regier et al. 2008b; Zwick et al. 2012). Furthermore, the ML analysis of the amino acid data set failed to resolve a shallow relationship that received a majority of support from the third codon position (fig. 4, Node 4).
Our analyses support the common assumption that large concatenated next-gen datasets can compensate for some saturation/homoplasy and rate heterogeneity among sites. However, the RADICAL analysis clearly shows that saturated sites at nt3 can have detrimental effects on the estimation of Saturniidae + Sphingidae (Node 2, fig. 4A) when less than 850 genes are used to estimate phylogeny. By removing nt3 altogether, degenerating synonymous changes, or analyzing data as amino acids, this node can reach fixation with fewer genes (Node 2, fig. 4A). One clear indication of the effect of saturation is the ML topology, estimated from nt3 alone, which resulted in a strongly supported (91% bootstrap) sister group relationship of Bombycidae + Saturniidae. In contrast, analyses from degen1-nt3 alone resolved a strongly supported (100% bootstrap) sister group relationship of Saturniidae +Sphingidae, consistent with our other analyses.
Past studies have shown that phylogenetic signal from nt1 and nt2 conflicts with nt3 especially for rapidly evolving coding chloroplast genes (e.g., Chaw et al. 2005; Goremykin et al. 2009; Wu et al. 2013) and mitochondrial genomes (e.g., Nakatani et al. 2011; Talavera and Vila 2011; Song et al. 2012). Studies that utilize nuclear protein coding genes for lepidopteran phylogenetics have also documented conflicts in signal from nt1 and nt2 with nt3 codon positions (e.g., Regier et al. 2009, 2013; Cho et al. 2011; Kawahara et al. 2011; Zwick et al. 2011; Sohn et al. 2013). Although degen1, nt12, and AA analyses resolved the same deep relationships as nt123, they failed to resolve or provide strong support for at least one shallow divergence (fig. 4, node 3), a result which is largely consistent with studies based on a fewer number of genes (e.g., Regier et al. 2008b, 2009; Cho et al. 2011; Kawahara et al. 2011; Zwick et al. 2011). Synonymous substitutions at nt3 that are saturated at deeper nodes in the tree might be critical in estimating shallow nodes. For instance, the sister group relationship of D. myron + H. diffinis received high support in the nt123 analysis, but further examination by codon position reveals that support for this relationship is mostly from nt3. The RADICAL analysis of degen1, nt12, AA, and degen1-nt3 shows high degradation points and a low AUC for this node, suggesting that increasing the number of concatenated genes in these data sets leads to greater difficulty in estimating this node. Therefore, if the nt123 topology represents the true tree, third codon positions are necessary to estimate this node, and multiple analyses that partition the data set and model synonymous and non-synonymous changes differently are required to effectively estimate shallow and deep nodes. However, if the true relationship is not D. myron + H. diffinis, then saturated sites at nt3 are strongly driving the estimation of an incorrect relationship in the nt123 analysis. Because taxon sampling is limited in our study, adding more taxa might provide a better estimate of relationships and more insight about the true relationships among these taxa.
Differences between ML (which uses models that can account for saturation) and MP (which does not account for saturation) can indicate areas of the tree affected by saturation at nt3. For example, there are several instances where ML bootstraps are high and parsimony bootstraps are low in the nt123 trees. There are also several nodes with high ML bootstraps and >50% parsimony bootstraps estimated from nt3 alone (fig. 3). Given the instances that differ between ML and MP, it appears that partitioning by each codon position has effectively accounted for some degree of saturation at nt3. In our nt123 RADICAL analysis, each of the ten paths includes the node supporting Saturniidae + Sphingidae (Node 2) for 725 concatenated genes and only four replicates above 725 genes do not include this node. This result might be attributed to the current version of RADICAL, which does not allow partition by codon position along the concatenation path. Presumably, if each matrix along the concatenation path were partitioned by codon, this node would fix much faster due to the model’s ability to account for the saturated sites at nt3.
The most recent mitochondrial genome analysis that included bombycoids was based on amino acid alignments of 13 coding genes (Huan-Na et al. 2012). These authors found high posterior probability but no ML bootstrap support for Bombycidae + Sphingidae (fig. 1D). We presume that the difference between their topology and ours could be due to the fact that they were using mitochondrial data, which can result in different topologies because mitogenomes evolve differently than nuclear genes and functionally represent a single locus (Palumbi and Baker 1994). Alternatively, different results could be achieved because their study was based on a smaller number of nucleotides, which might not have captured enough information to estimate relationships among these groups. In a separate study based on mitochondrial genomes and rRNA, Kim et al. (2011) found high support for two different topologies (fig. 1B) depending on whether nt3 was included or different partitioning strategies were implemented. Although Kim et al. (2011) were unable to conclude which topology was the best estimate for bombycoid families, they showed that partitions can strongly affect the resulting topology. The several previous multi-gene studies also failed to resolve relationships among these three families, which is consistent with our results that it might take up to 150 genes (for nt12) and up to 850 genes (for nt123) to solidify the relationship among these three families. The latest study from the LepTree initiative (Regier et al. 2013), based on 19 genes, recovered the same bombycoid relationships as ours when accounting for saturated sites, but their relationships were not strongly supported (fig. 1F). One reason these studies might have failed to strongly support relationships among these families is that the split between these families might have taken place during a rapid radiation event. Many of these previous studies show some characteristics attributable to a rapid radiation, such as short branch lengths and low bootstrap support. Rapid radiation events have been shown to blur phylogenetic relationships and are a problem not only within Lepidoptera but also across Holometabola (Whitfield and Kjer 2008; Trautwein et al. 2012).
Missing data have been shown to adversely affect phylogenetic results under certain conditions (Wiens 2003; Lemmon et al. 2009; Wiens and Morrill 2011). The bombycoid transcriptomes we generated for this data set had 23% missing data and 99% gene coverage. This indicates that many of the genes from our transcriptomes are not full-length compared with the complete genes from the Bombyx genome. The majority of missing data in our matrix came from the two pyraloid outgroups and taxa that were included from EST data. Our results (as few as 17 genes) echo the results of other phylotranscriptomic studies (e.g., Oakley et al. 2012), which indicate that taxa with limited amounts of data can be placed with confidence. We recovered 84% gene coverage across the matrix used for the RADICAL analyses after eliminating just five taxa that have the fewest number of genes. Taxa with high gene numbers were generally distributed across our phylogeny, and exclusion of taxa with limited data did not change the resulting phylogeny. Therefore, it is unlikely that missing data had a significant impact on our results.
The RADICAL analyses revealed that the difficulty in recovering relationships among the families Bombycidae, Saturniidae, and Sphingidae might be largely attributed to saturated synonymous changes from nt3. Accounting for synonymous substitutions or excluding nt3 reduces the difficulty in estimating these relationships, but it might also remove signal needed to resolve shallow divergences. Recent phylotranscriptomic studies have demonstrated that support values generally increase with the use of large amounts of next-gen data (e.g., Meusemann et al. 2010; von Reumont et al. 2011; Oakley et al. 2012; Simon et al. 2012; Wheat and Wahlberg 2013), and our results also indicate that large amounts of data can resolve many difficult nodes and overcome some of the issues faced by traditional analyses. Our study clearly demonstrates that transcriptomic data sets should examine the effects of including, excluding, and degenerating synonymous substitutions when estimating phylogeny. Careful analyses of data are critical, especially at a time when more next-gen data are becoming available for inclusion into phylogenetic studies.
Supplementary Material
Supplementary figures S1–S4 and tables S1 and S2 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The authors thank Lei Xiao for preparing RNA extracts of the six transcriptomes. The authors acknowledge the University of Florida HPC Cluster for providing computational resources and support that contributed to the research results. Finally, they thank H. Bracken-Grissom, David Plotkin, and two anonymous reviewers for comments that significantly improved this manuscript. This work was partially supported by the National Science Foundation (IOS-1121739 to A.Y.K.).
Literature Cited
- Abascal F, Zardoya R, Telford MJ. TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 2010;38(Suppl 2):W7–W13. doi: 10.1093/nar/gkq291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson DA, et al. GenBank. Nucleic Acids Res. 2013;41(1):36–42. doi: 10.1093/nar/gks1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betancur-R R, Li C, Munroe TA, Ballesteros JA, Ortí G. Addressing gene tree discordance and non-stationarity to resolve a multi-locus phylogeny of the flatfishes (Teleostei: Pleuronectiformes) Syst Biol. 2013;62(5):763–785. doi: 10.1093/sysbio/syt039. [DOI] [PubMed] [Google Scholar]
- Biomatters. Geneious v5.5.8. 2013 [cited 2013 Nov 8]. Available from: http://www.geneious.com. [Google Scholar]
- Buckley TR, Simon C, Chambers GK. Exploring among-site rate variation models in a maximum likelihood framework using empirical data: the effects of model assumptions on estimates of topology, branch lengths, and bootstrap support. Syst Biol. 2001;50:67–86. [PubMed] [Google Scholar]
- Bybee SM, et al. Directed next generation sequencing for phylogenetics: an example using Decapoda (Crustacea) Zool Anz. 2011;250(4):497–506. [Google Scholar]
- Chaudhary R, Bansal MS, Wehe A, Fernández-Baca D, Eulenstein O. iGTP: a software package for large-scale gene tree parsimony analysis. BMC Bioinformatics. 2010;11:574. doi: 10.1186/1471-2105-11-574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaw SM, Walters TW, Chang CC, Hu SH, Chen SH. A phylogeny of cycads (Cycadales) inferred from chloroplast matK gene, trnK intron, and nuclear rDNA ITS region. Mol Phylogenet Evol. 2005;37(1):214–234. doi: 10.1016/j.ympev.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Cho S, et al. Can deliberately incomplete gene sample augmentation improve a phylogeny estimate for the advanced moths and butterflies (Hexapoda: Lepidoptera)? Syst Biol. 2011;60(6):782–796. doi: 10.1093/sysbio/syr079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colless DH. Congruence between morphometric and allozyme data for menidia species: a reappraisal. Syst Zool. 1980;29(3):288–299. [Google Scholar]
- Cronn R, et al. Targeted enrichment strategies for next-generation plant biology. Am J Bot. 2012;99(2):291–311. doi: 10.3732/ajb.1100356. [DOI] [PubMed] [Google Scholar]
- Cunningham CW, Zhu H, Hillis DM. Best-fit maximum-likelihood models for phylogenetic inference: empirical tests with known phylogenies. Evolution. 1998;52(4):978–987. doi: 10.1111/j.1558-5646.1998.tb01827.x. [DOI] [PubMed] [Google Scholar]
- Delsuc F, Tsagkogeorga G, Lartillot N, Philippe H. Additional molecular support for the new chordate phylogeny. Genesis. 2008;46(11):592–604. doi: 10.1002/dvg.20450. [DOI] [PubMed] [Google Scholar]
- Ebersberger I, Strauss S, von Haeseler A. HaMStR: profile hidden Markov model based search for orthologs in ESTs. BMC Evol Biol. 2009;9(1):157. doi: 10.1186/1471-2148-9-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faircloth BC, et al. Ultraconserved elements anchor thousands of genetic markers spanning multiple evolutionary timescales. Syst Biol. 2012;61(5):717–726. doi: 10.1093/sysbio/sys004. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- Goloboff P, Farris J, Nixon K. TNT: a free program for phylogenetic. Cladistics. 2008;24:774–786. [Google Scholar]
- Goremykin VV, Viola R, Hellwig FH. Removal of noisy characters from chloroplast genome-scale data suggests revision of phylogenetic llacements of Amborella and Ceratophyllum. J Mol Evol. 2009;68(3):197–204. doi: 10.1007/s00239-009-9206-9. [DOI] [PubMed] [Google Scholar]
- Hittinger CT, Johnston M, Tossberg JT, Rokas A. Leveraging skewed transcript abundance by RNA-Seq to increase the genomic depth of the tree of life. Proc Nat Acad Sci U S A. 2010;107(4):1476–1481. doi: 10.1073/pnas.0910449107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huan-Na C, Yu-Zhou D, Bao-Ping Z. Characterization of the complete mitochondrial genomes of Cnaphalocrocis medinalis and Chilo suppressalis (Lepidoptera: Pyralidae) Int J Biol Sci. 2012;8(4):561–579. doi: 10.7150/ijbs.3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes J, et al. Dense taxonomic EST sampling and its applications for molecular systematics of the Coleoptera (beetles) Mol Biol Evol. 2006;23(2):268–278. doi: 10.1093/molbev/msj041. [DOI] [PubMed] [Google Scholar]
- Katoh K, Kuma K, Toh H, Miyata T. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005;33(2):511–518. doi: 10.1093/nar/gki198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara AY, Mignault AA, Regier JC, Kitching IJ, Mitter C. Phylogeny and biogeography of hawkmoths (Lepidoptera: Sphingidae): evidence from five nuclear genes. PLoS One. 2009;4(5):e5719. doi: 10.1371/journal.pone.0005719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara AV, et al. Increased gene sampling strengthens support for higher-level groups within leaf-mining moths and relatives (Lepidoptera: Gracillariidae) BMC Evol Biol. 2011;11:182. doi: 10.1186/1471-2148-11-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MJ, Kang AR, Jeong HC, Kim K-G, Kim I. Reconstructing intraordinal relationships in Lepidoptera using mitochondrial genome data with the description of two newly sequenced lycaenids, Spindasis takanonis and Protantigius superans (Lepidoptera: Lycaenidae) Mol Phylogenet Evol. 2011;61(2):436–445. doi: 10.1016/j.ympev.2011.07.013. [DOI] [PubMed] [Google Scholar]
- Kuhner MK, Felsenstein J. A simulation comparison of phylogeny algorithms under equal and unequal evolutionary rates. Mol Biol Evol. 1994;11(3):459–468. doi: 10.1093/oxfordjournals.molbev.a040126. [DOI] [PubMed] [Google Scholar]
- Lanfear R, Calcott B, Ho S, Guindon S. PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol. 2012;29(6):1695–1701. doi: 10.1093/molbev/mss020. [DOI] [PubMed] [Google Scholar]
- Lartillot N, Lepage T, Blanquart S. PhyloBayes 3: a Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics. 2009;25(17):2286–2288. doi: 10.1093/bioinformatics/btp368. [DOI] [PubMed] [Google Scholar]
- Lartillot N, Philippe H. A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol Biol Evol. 2004;21(6):1095–1109. doi: 10.1093/molbev/msh112. [DOI] [PubMed] [Google Scholar]
- Lemmon AR, Brown JM, Stanger-Hall K, Lemmon EM. The effect of ambiguous data on phylogenetic estimates obtained by maximum likelihood and Bayesian inference. Syst Biol. 2009;58(1):130–145. doi: 10.1093/sysbio/syp017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon AR, Emme S, Lemmon EM. Anchored hybrid enrichment for massively high-throughput phylogenomics. Syst Biol. 2012;61(5):727–744. doi: 10.1093/sysbio/sys049. [DOI] [PubMed] [Google Scholar]
- Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22(13):1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- Mamanova L, et al. Target-enrichment strategies for next-generation sequencing. Nat Methods. 2010;7(2):111–118. doi: 10.1038/nmeth.1419. [DOI] [PubMed] [Google Scholar]
- McCormack JE, Hird SM, Zellmer AJ, Carstens BC, Brumfield RT. Applications of next-generation sequencing to phylogeography and phylogenetics. Mol Phylogenet Evol. 2013;66(2):526–538. doi: 10.1016/j.ympev.2011.12.007. [DOI] [PubMed] [Google Scholar]
- Meusemann K, et al. A phylogenomic approach to resolve the arthropod tree of life. Mol Biol Evol. 2010;27(11):2451–2464. doi: 10.1093/molbev/msq130. [DOI] [PubMed] [Google Scholar]
- Minet J. Tentative reconstruction of the ditrysian phylogeny (Lepidoptera: Glossata) Entomol Scand. 1991;22:69–95. [Google Scholar]
- Minet J. The Bombycoidea: phylogeny and higher classification (Lepidoptera: Glossata) Entomol Scand. 1994;25:63–88. [Google Scholar]
- Nabholz B, Künstner A, Wang R, Jarvis ED, Ellegren H. Dynamic evolution of base composition: causes and consequences in avian phylogenomics. Mol Biol Evol. 2011;28(8):2197–2210. doi: 10.1093/molbev/msr047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatani M, Miya M, Mabuchi K, Saitoh K, Nishida M. Evolutionary history of Otophysi (Teleostei), a major clade of the modern freshwater fishes: Pangaean origin and Mesozoic radiation. BMC Evol Biol. 2011;11(1):177. doi: 10.1186/1471-2148-11-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narechania A, et al. Random addition concatenation analysis: a novel approach to the exploration of phylogenomic signal reveals strong agreement between core and shell genomic partitions in the Cyanobacteria. Genome Biol Evol. 2012;4(1):30–43. doi: 10.1093/gbe/evr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley TH, Wolfe JM, Lindgren AR, Zaharoff AK. Phylotranscriptomics to bring the understudied into the fold: monophyletic Ostracoda, fossil placement and pancrustacean phylogeny. Mol Biol Evol. 2012;30(1):215–233. doi: 10.1093/molbev/mss216. [DOI] [PubMed] [Google Scholar]
- Pagel M, Meade A. A phylogenetic mixture model for detecting pattern-heterogeneity in gene sequence of character-state data. Syst Biol. 2004;53:571–581. doi: 10.1080/10635150490468675. [DOI] [PubMed] [Google Scholar]
- Palumbi SR, Baker CS. Contrasting population structure from nuclear intron sequences and mtDNA of humpback whales. Mol Biol Evol. 1994;11(3):426–435. doi: 10.1093/oxfordjournals.molbev.a040115. [DOI] [PubMed] [Google Scholar]
- Regier JC, Mitter C, Peigler RS, Friedlander TP. Monophyly, composition, and relationships within Saturniinae (Lepidoptera: Saturniidae): evidence from two nuclear genes. Insect Syst Evol. 2002;33(1):9–21. [Google Scholar]
- Regier JC, Paukstadt U, Paukstadt LH, Mitter C, Peigler RS. Phylogenetics of eggshell morphogenesis in Antheraea (Lepidoptera: Saturniidae): unique origin and repeated reduction of the aeropyle crown. Syst Biol. 2005;54(2):254–267. doi: 10.1080/10635150590923281. [DOI] [PubMed] [Google Scholar]
- Regier JC, et al. Phylogenetic relationships of wild silkmoths (Lepidoptera: Saturniidae) inferred from four protein-coding nuclear genes. Syst Entomol. 2008a;33(2):219–228. [Google Scholar]
- Regier JC, et al. Resolving arthropod phylogeny: exploring phylogenetic signal within 41 kb of protein-coding nuclear gene sequence. Syst Biol. 2008b;57(6):920–938. doi: 10.1080/10635150802570791. [DOI] [PubMed] [Google Scholar]
- Regier JC, et al. Toward reconstructing the evolution of advanced moths and butterflies (Lepidoptera: Ditrysia): an initial molecular study. BMC Evol Biol. 2009;9(1):280. doi: 10.1186/1471-2148-9-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regier JC, et al. Arthropod relationships revealed by phylogenomic analysis of nuclear protein-coding sequences. Nature. 2010;463(7284):1079–1083. doi: 10.1038/nature08742. [DOI] [PubMed] [Google Scholar]
- Regier JC, et al. A large-scale, higher-level, molecular phylogenetic study of the insect order Lepidoptera (moths and butterflies) PLoS One. 2013;8(3):e58568. doi: 10.1371/journal.pone.0058568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharanowski BJ, et al. Expressed sequence tags reveal Proctotrupomorpha (minus Chalcidoidea) as sister to Aculeata (Hymenoptera: Insecta) Mol Phylogenet Evol. 2010;57(1):101–112. doi: 10.1016/j.ympev.2010.07.006. [DOI] [PubMed] [Google Scholar]
- Simon S, Narechania A, DeSalle R, Hadrys H. Insect phylogenomics: exploring the source of incongruence using new transcriptomic data. Genome Biol Evol. 2012;4(12):1295–1309. doi: 10.1093/gbe/evs104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn J-C, et al. A molecular phylogeny for Yponomeutoidea (Insecta, Lepidoptera, Ditrysia) and its implications for classification, biogeography and the evolution of host plant use. PLoS One. 2013;8(1):e55066. doi: 10.1371/journal.pone.0055066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltis PS, Soltis DE, Savolainen V, Crane PR, Barraclough TG. Rate heterogeneity among lineages of tracheophytes: integration of molecular and fossil data and evidence for molecular living fossils. Proc Nat Acad Sci U S A. 2002;99(7):4430–4435. doi: 10.1073/pnas.032087199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song N, Liang A-P, Bu C-P. A Molecular phylogeny of Hemiptera inferred from mitochondrial genome sequences. PLoS One. 2012;7(11):e48778. doi: 10.1371/journal.pone.0048778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H, Sheffield NC, Cameron SL, Miller KB, Whiting MF. When phylogenetic assumptions are violated: base compositional heterogeneity and among-site rate variation in beetle mitochondrial phylogenomics. Syst Entomol. 2010;35(3):429–448. [Google Scholar]
- Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22(21):2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- Sullivan J. Combining data with different distributions of among-site rate variation. Syst Biol. 1996;45(3):375–380. [Google Scholar]
- Surget-Groba Y, Montoya-Burgos JI. Optimization of de novo transcriptome assembly from next-generation sequencing data. Genome Res. 2010;20(10):1432–1440. doi: 10.1101/gr.103846.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talavera G, Vila R. What is the phylogenetic signal limit from mitogenomes? The reconciliation between mitochondrial and nuclear data in the Insecta class phylogeny. BMC Evol Biol. 2011;11(1):315. doi: 10.1186/1471-2148-11-315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trautwein MD, Wiegmann BM, Beutel R, Kjer KM, Yeates DK. Advances in insect phylogeny at the dawn of the postgenomic era. Annu Rev Entomol. 2012;57(1):449–468. doi: 10.1146/annurev-ento-120710-100538. [DOI] [PubMed] [Google Scholar]
- van Nieukerken EJ, et al. Order Lepidoptera Linnaeus, 1758. In: Zhang Z-Q, editor. Animal biodiversity: an outline of higher-level classification and survey of taxonomic richness. 2011. Zootaxa 3148:212–221. [DOI] [PubMed] [Google Scholar]
- von Reumont BM, et al. Pancrustacean phylogeny in the light of new phylogenomic data: support for Remipedia as the possible sister group of Hexapoda. Mol Biol Evol. 2011;29:1031–1045. doi: 10.1093/molbev/msr270. [DOI] [PubMed] [Google Scholar]
- Wheat CW, Wahlberg N. Phylogenomic insights into the cambrian explosion, the colonization of land and the evolution of flight in Arthropoda. Syst Biol. 2013;62(1):93–109. doi: 10.1093/sysbio/sys074. [DOI] [PubMed] [Google Scholar]
- Whitfield JB, Kjer KM. Ancient rapid radiations of insects: challenges for phylogenetic analysis. Annu Rev Entomol. 2008;53:449–472. doi: 10.1146/annurev.ento.53.103106.093304. [DOI] [PubMed] [Google Scholar]
- Wiens JJ. Missing data, incomplete taxa, and phylogenetic accuracy. Syst Biol. 2003;52(4):528–538. doi: 10.1080/10635150390218330. [DOI] [PubMed] [Google Scholar]
- Wiens JJ, Morrill MC. Missing data in phylogenetic analysis: reconciling results from simulations and empirical data. Syst Biol. 2011;60(5):719–731. doi: 10.1093/sysbio/syr025. [DOI] [PubMed] [Google Scholar]
- Wu C-S, Chaw S-M, Huang Y-Y. Chloroplast phylogenomics indicates that Ginkgo biloba is sister to cycads. Genome Biol Evol. 2013;5(1):243–254. doi: 10.1093/gbe/evt001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X, Xie Z, Salemi M, Chen L, Wang Y. An index of substitution saturation and its application. Mol Phylogenet Evol. 2003;26(1):1–7. doi: 10.1016/s1055-7903(02)00326-3. [DOI] [PubMed] [Google Scholar]
- Xia Q, et al. A draft sequence for the genome of the domesticated silkworm (Bombyx mori) Science. 2004;306(5703):1937–1940. doi: 10.1126/science.1102210. [DOI] [PubMed] [Google Scholar]
- Yang Z. Among-site rate variation and its impact on phylogenetic analyses. Trends Ecol Evol. 1996a;11(9):367–372. doi: 10.1016/0169-5347(96)10041-0. [DOI] [PubMed] [Google Scholar]
- Yang Z. Maximum-likelihood models for combined analyses of multiple sequence data. J Mol Evol. 1996b;42(5):587–96. doi: 10.1007/BF02352289. [DOI] [PubMed] [Google Scholar]
- You M, et al. A heterozygous moth genome provides insights into herbivory and detoxification. Nat Genet. 2013;45(2):220–225. doi: 10.1038/ng.2524. [DOI] [PubMed] [Google Scholar]
- Zhan S, Merlin C, Boore JL, Reppert SM. The monarch butterfly genome yields insights into long-distance migration. Cell. 2011;147(5):1171–1185. doi: 10.1016/j.cell.2011.09.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwick A, Regier JC, Mitter C, Cummings MP. Increased gene sampling yields robust support for higher-level clades within Bombycoidea (Lepidoptera) Syst Entomol. 2011;36(1):31–43. [Google Scholar]
- Zwick A, Regier JC, Zwickl DJ. Resolving discrepancy between nucleotides and amino acids in deep-level arthropod phylogenomics: differentiating serine codons in 21-amino-acid models. PLoS One. 2012;7(11):e47450. doi: 10.1371/journal.pone.0047450. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.