

Abstract
Discoidin domain receptor 2 (DDR2) is a receptor tyrosine kinase involved in a variety of cellular response pathways, including regulation of cell growth, proliferation and motility. Using a newly developed platform to identify the signaling pathway/molecular target of natural products, we identified a family of alkaloid natural products, discoipyrroles A–D (1–4), from Bacillus hunanensis that inhibit the DDR2 signaling pathway. The structure of 1–4, determined by detailed 2D NMR methods and confirmed by X-ray crystallographic analysis has an unusual 3H-benzo[ d]pyrrolo][1,3]oxazine-3,5-dione core. Discoipyrroles A–D potently inhibit DDR2 dependent migration of BR5 fibroblasts and show selective cytotoxicity to DDR2 mutant cell lung cancer cell lines (IC50 120–400 nM). Examination of the biosynthesis has led to the conclusion that the discoipyrroles are formed through a non-enzymatic process, leading to a one-pot total synthesis of 1.
INTRODUCTION
Discoidin domain receptors (DDR1 and DDR2) are cell-surface receptors, which are activated by binding collagen, which results in downstream signaling responses involved in regulation of cell growth, differentiation and motility.1 Prolonged stimulation of DDR2 has been associated with the upregulation of matrix metalloproteinase-1 expression. DDR2 also plays an important role in mediating fibroblast migration and proliferation by matrix metalloproteinase-2 dependent mechanisms.2 DDR2 has been implicated in obstructive diseases of blood vessels by mediation of collagen turnover in vascular smooth muscle cells.3
Recent reports have implicated DDR2 to play a role in a variety of cancers.4 Sequencing efforts by Meyerson and co-workers identified mutations in the DDR2 kinase gene in ~4.0% of human squamous cell lung cancer (SCC) and went on to establish that DDR2 mutant SCC cell lines were selectively killed by knockdown of DDR2 by RNAi or by treatment with the pan-kinase inhibitor dasatinib.5 This has been further validated by successful treatment of tumors established with DDR2 derived cell lines with dasatanib. In breast cancer, increased collagen deposits in the stroma leads to enhanced metastases. 6 In that context, DDR2 has been demonstrated to promotes breast cancer cell invasion and migration in vitro, and metastasis in vivo.7 With the emerging importance of this pathway, there are still a number of unknowns in the DDR2 signaling pathway and very few tools available to interrogate the system.
We have developed a natural products fraction library based on a collection of marine-derived bacteria.8 The library of 6500 fractions has been utilized in a number of target-specific and phenotypic HTS screens in the field of oncology. Screening for cytotoxicity against relevant human cancer cell lines continues to be a successful screening strategy, but is limited by a lack of mechanistic information.9 We have developed an assay that augments phenotypic screening, producing a FUnctional SIgnature-based ONtology (FUSION) map that links bioactive compounds to the molecular target or regulatory network they interact with in a mammalian cell (Figure 1a).10
Figure 1.

a) Overview of the FUSION assay to link natural products molecular target or pathway. HCT-116 cells were individually subjected to chemical and genetic perturbations and analyzed for their gene expression of a set of select genes. b) Hierarchical clustering of natural product fractions with discoidin domain receptor 2 (DDR2) based on gene expression of six genes. c) 2D-Matrigel cell migration assay with BR5 fibroblasts, platelet derived growth factor and a natural product fraction from B. hunanensis SNA-048 (fraction 7) at 1 μg/mL.
This methodology utilizes pattern matching of a gene-expression profile generated by exposing HCT-116 cells to a single siRNA, miRNA or natural product fraction. As the molecular target of an individual siRNA is known, the mechanism of action of a natural product fractions can be deduced by similar gene expression profiles (Figure S1, assay protocol in SI). Using the FUSION map approach we identified a natural product fraction from marine-derived Bacillus hunanensis strain SNA-048 that provided a functional signature identical to a siRNA knockout of DDR2 in HCT-116 cells (Figure 1b).10 Based on the functional signature, we showed fraction SNA-048-7 from B. hunanensis inhibits DDR2 dependent migration of BR5 fibroblasts through a 2D-Matrigel matrix at 1 μg/mL (Figure 1c).2a,11
Here we report the BR5 2D-Matrigel migration assay-guided isolation of discoipyrroles A-D (1–4) (Figure 2), a family of polycyclic alkaloids comprised of a 3H-benzo[d]pyrrolo][1,3]oxazine-3,5-dione core (for 1, 2 and 4) and adorned with aryl groups. The discoipyrroles are the first examples of a natural product containing this highly functionalized alkaloid core. In addition to 1–4, we have isolated hydroxysattabacin (5)12 and the related enol (6) (Figure 2), which provide insight into the biosynthetic origin of 1–4. The biosynthetic understanding ultimately led to a one-step total synthesis of 1. Furthermore, we report inhibition of DDR2 dependent migration of BR5 fibroblasts by discoipyrroles and further demonstrate their dose-dependent effect on post-translational modifications of DDR2. Evaluation of 1–4 against non-small cell lung cancer (NSCLC) cell-lines showed selective cytotoxicity toward DDR2 mutant cell-lines with IC50 values ~200 nM.
Figure 2.

Structures of 1 – 4 and biosynthetic precursors 5 and 6.
RESULTS AND DISCUSSION
Isolation and Structural Elucidation
B. hunanensis strain SNA-048 was isolated from a sediment sample collected at Galveston Bay, TX and selected using a humic acid based media. Fermentation and extraction were carried out using standard procedures.6 Purification of metabolites by sequential flash C18, Sephadex LH-20, and reversed phase C18 HPLC led to 1 (2.0 mg), 2 (0.8 mg), 3 (1.0 mg) and 4 (0.4 mg).
Discoipyrrole A (1) was obtained as a yellow solid and was determined to have a molecular formula of C27H23NO5, based on HR-ESIMS [M + H]+ m/z 442.1652 and interpretation of NMR data. The UV spectrum of 1 exhibited absorption bands at 396, 324, 263 and 214 nm, indicative of a highly conjugated system. The 1H NMR spectrum (Table S1, DMSO-d6) revealed signals indicative of di-substituted aromatic rings and a small ketide fragment, while the 13C NMR spectrum contained two downfield signals indicative of carbonyls. Because of the 11 quaternary carbons the structure elucidation of 1 proved a challenge. Detailed analysis of 1D and 2D NMR data allowed assignment of the simple substructures I–IV (Figure 3a). The full details of the structural elucidation can be found in the supporting information.
Figure 3.

Structure elucidation details of 1 – 4. a) Substructures of 1 (I–IV) with COSY, HMBC correlations. b) ORTEP drawing of crystal structure of (−)-1a. c) Key NMR correlations for 3.
Briefly, partial structures I (C-4 through C-8) and II (C-3, C-9 through C-12) were assigned as 1,4-disubstituted phenyl rings with an additional sp2 quaternary carbon at benzylic positions (C-4, C-3). The nature of the quaternary carbon substituent at the benzylic positions was not readily apparent from initial investigation of the NMR data. However, the difference in chemical shifts between H-6 and H-7 of ~ 0.3 ppm, provided evidence that C-4 was part of an olefin. This is predicated on considerable literature NMR data for p-hydroxycinnamates; where there is heteroatom substitution in the α or β position, the Δδ value is always ~ 0.3 ppm (Figure S2, S3). A similar Δδ of ~ 0.3 between H-10 and H-11 supports the structure of II.11 Partial structure III (C-17 through C-22) was readily determined to be an anthranilate moiety, while the aliphatic partial structure IV (C-13 through C-16) was established by COSY correlations and key HMBC correlations.
Based on 13C chemical shifts we could assign C-2 through C-4 as an α,β-unsaturated ketone, which in combination with I–IV, account for all of the carbon and hydrogens and 15 of the 17 double bond equivalents, suggesting 1 contained two additional rings. The distinct chemical shift of the C-1 quaternary carbon (δC 90.3) confirmed the presence of a hemiaminal functionality, which allowed us to propose the structure for 1 as depicted, containing a 3H-benzo[d]pyrrolo][1,3]oxazine-3,5-dione core. A zero [α]D was measured for 1, suggesting that the C1 quaternary stereocenter is racemic, as will be addressed later.
To verify the structure, a number of attempts to crystallize (±)-1 were made with no success. Ultimately, (±)-1 was separated via chiral-phase HPLC to give the individual enantiomers (Figure S6), and (−)-1 converted to the bis p-bromobenzoate derivative (−)-1a (p-bromobenzoyl chloride, Et3N, CH2Cl2) Crystallization of (−)-1a from 10:1 MeOH:CHCl3 resulted in yellow prisms, which gave an X-ray crystal structure (Figure 3b) with 0.84 Å resolution and a Flack parameter of 0.001 (6). Thus, we were able to confirm the NMR assignment of 1 and establish that (−)-1a has the 1R configuration.
The structures of the three remaining discoipyrroles 2–4 were assigned by NMR analysis and comparison to 1. Full details on the structure assignment can be found in Tables S2–S4 and the Supporting information. Of note, discoipyrrole C (3), lacks the C-17 to C-23 anthranilate moiety, instead is replaced by NH3. Acquisition of the 1H NMR of 3 in DMSO-d6 showed the exchangeable -NH (δH 7.96) of the C-1 hemiaminal, which gave HMBC correlations to C-1, C-2, C-3, C-4, C-5 and C-13, confirming the pyrrol-3-one core of 3 (Figure 3c). This provided further NMR evidence for the 3H-benzo[ d]pyrrolo][1,3]oxazine-3,5-dione core of 1.
Proposed biosynthesis and precursor-driven biosynthesis of 1 and analogs
A proposed biosynthetic pathway (Figure 4) accounts for the racemic nature of the discoipyrroles as well as the ability to incorporate different amines into the core structure. In the proposed pathway hydroxysattabacin (5), isolated from the non-polar partition of the crude extract, is oxidized to give enol 6, which was also isolated from B. hunanensis (Table S5). This is followed by an aldol reaction with p-hydroxybenzaldehyde and subsequent oxidation to give the tri-ketone 7. Following keto-enol tautomerization of 7, imine formation with anthranilic acid13 (or NH3 for 3) would give diketoenamine 8, which is followed by a non-enzymatic hemiaminal formation to give 9 as a racemate. We have isolated a small amount of 9 under stringent isolation conditions, utilizing only LH20 size exclusion chromatography, eliminating any trace acid from the purification. However, upon C18 chromatography 9 undergoes rapid dehydration-lactonization to give 1.
Figure 4.

Proposed biosynthetic pathway to 1.
Discoipyrrole B originates from incorporation of p-hydroxybenzaldehyde with sattabacin (lacking the phenoxyl group), while discoipyrrole D may arise from an elaborated anthranilate group or from nucleophilic attack of C-24 onto a suitably substituted indole analog. It should be noted that another reasonable biosynthetic pathway can be proposed that changes the order of events in a Mannich-type reaction, whereby anthranilic acid and p-hydroxybenzaldehyde first form an imine prior to nucleophilic attack by 6.14
Brief investigations of the biosynthetic hypotheses were carried out using a precursor-directed approach with various substituted benzaldehyde precursors, including p-nitrobenzaldehyde, p-bromobenzaldehyde and p-methoxybenzaldehyde. Separate additions of each precursor (1g.L−1) to A1 media and shake fermentation resulted only in oxidations to the corresponding benzoic acids, however, addition of p-hydroxybenzaldehyde-1-13C (1g.L−1) to solid agar A1 media resulted in production of 4-[13C]-discoipyrrole A (10), as analyzed by LC-MS and NMR (Scheme 1).
Scheme 1.

Precursor-directed biosynthesis of 13C labeled discoipyrrole analogs 10 and 12.
Not surprisingly, this verified that C-4 through C-8 originates from p-hydroxybenzaldehyde. Similarly, we converted 5 to p-[13CH3]-methoxysattabacin (11) and added to A1 liquid media (0.250 g.L−1) and after 7 days of fermentation, observed production of a compound with a mass corresponding to 12-O-[13CH3]-methyl discoipyrrole A (12), which was verified by ESIMS and NMR (Table S6).
The foregoing precursor-directed experiments not only serve to validate our biosynthetic proposal, but as a means to generate structural diversity for optimization of biological activity. Therefore we decided to carry the precursor directed biosynthetic studies one step further based on the observation that the only enzymatic steps involved in our proposed biosynthesis of 1 involve oxidations of an α-hydroxy ketone and a benzylic alcohol. We hypothesized it would be possible to generate the entire discoipyrrole framework by providing the three substrates (5, p-hydroxybenzaldehyde, anthranilic acid) to cell-free “spent” A1 media (Figure 5a).
Figure 5.

a) Production of 1 from three biosynthetic precursors in “spent” media. b) SIM chromatograms, monitoring at m/z z 440 [M – H]−. for purified 1 from B. hunanensis strain SNA-048; B. aquamaris strain SNE-038; Spent media from B. aquamaris strain SNE-038 with 5, p-hydroxybenzaldehyde and anthranilic acid after 5 days at 37 °C. Shaded box indicates retention time of 1.
We wanted to use “spent” media because it contains a variety of metal ions and co-factors that are produced in the course of bacterial growth. Discoipyrrole producing strain B. hunanensis was avoided to preclude background production of 1. Instead, we chose media from cultures of Bacillus aquamaris strain SNE-038 which was devoid of discoipyrroles or 5 (LC-MS analysis, Figure 5b) and displayed, after a 7 day fermentation, a pH of 9.0, the same as spent media from B. hunanensis grown under the same conditions. After removal of the cells by centrifugation, the supernatant was heated to 90 °C for five minutes, and filtered through a 5KD filter to denature and/or remove enzymes. The resulting spent media was dosed with of 5 (10 mg.L−1), benzaldehyde (8 mg.L−1) and anthranilic acid ( 8 mg.L−1) at 37 °C and allowed to shake for 5 days. Gratifyingly, LC-MS analysis of the ethyl acetate extract revealed production of significant levels of 1 (Figure 5). This remarkable one-pot transformation involves the formation of one C-C bond, imine formation, hemiaminal formation, dehydration and two oxidations to generate 1. From our biosynthetic proposal it is clear that the last two steps, hemiaminal formation and dehydration, can be readily rationalized. The oxidation steps on the other hand require assistance from the media. The “spent” media does contain a number of metal species, including Fe, Co, Mn and Ni salts and are capable of promoting the transition-metal mediated benzylic oxidation.15 A second possibility is that the C-4 benzylic position is highly prone to air oxidation.
We further explored the possibility of carrying out a one-pot transformation of the three precursors to 1 in a variety of organic solvents, such as MeOH, MeOH/H2O and DMSO and at various pH ranges (2 – 11). Optimized conditions that lead to a 20% conversion to 1 were 1% trifluoroacetic acid in DMSO at 45 °C for 30 hours, but also produced a number of side-products, including 13 (Scheme 2). Similarly, utilizing p-hydroxybenzaldehyde-1-13C, 11 and anthranilic acid with 1% trifluoroacetic acid (TFA) in DMSO at 45 °C for 30 hours, we observed the production of 4-[13C]-12-O-[13CH3]-methyl discoipyrrole A (14), as characterized by MS and NMR (Table S7). It is likely that DMSO and O2 are participating in the two oxidation steps of the one-pot transformation, as when the reaction is carried out with deoxygenated DMSO under a N2 atmosphere, no transformation occurs.
Scheme 2.
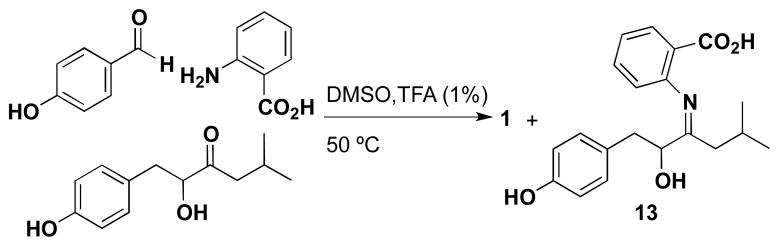
Synthesis of 1 in organic solvent.
We monitored the oxidation of 5 to 6 by NMR and LC-MS in DMSO-d6 and 1% deuterated TFA and could observe a 3:1 ratio of 5 to 6 after 6 hours (Figure S7). In addition to 6, we observed conversion of 5 to p-hydroxybenzaldehyde (~5%), which presumably derives from tautomerization of 6 to give the quinone, followed by nucleophilic addition of H2O and a subsequent retro-aldol (Figure S8).
The combination of the “spent” media induced formation and the DMSO, TFA promoted formation of 1 offer complimentary routes for generation of discoipyrrole analogs for further evaluation.
Inhibition of DDR2 signaling by 1 – 4
Based on our initial prediction, we evaluated the ability of 1–4 to inhibit BR5 fibroblast migration in a 2D-Matrigel matrix at 1 μM, with 1 displaying the most potent inhibition at 95% followed by 2 and 4 with 85% inhibition and only modest inhibition by 3 at 50% (Figures 6a, Figure S9). All four compounds exhibited no cytotoxicity against BR5 fibroblasts up to 10 μM, ruling out the possibility that migration inhibition was caused by reduced cell viability (Figure 6a). As a positive control in these assays, we measured the migration inhibition ability of the pan-kinase inhibitor dasatinib, which has been shown to be an inhibitor of the kinase domain of DDR2. As expected, dasatinib inhibited cell migration at 0.1 μM, however was modestly cytotoxic to the BR5 fibroblasts at this concentration.16
Figure 6.
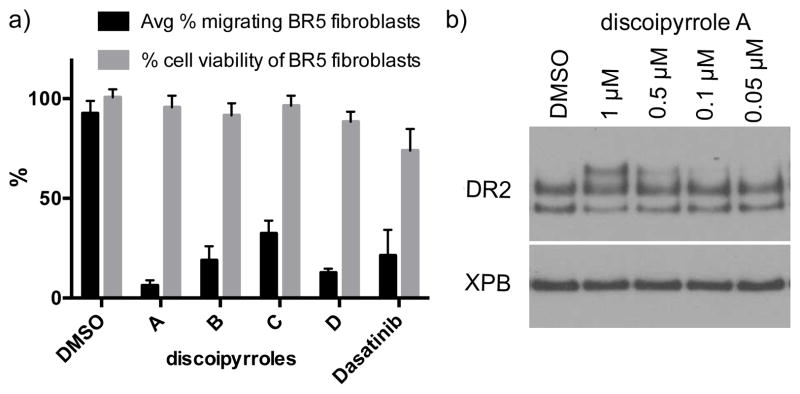
a) Quantification of migration inhibition in a BR5 2D-Matrigel fibroblast migration assay with 1 μM of 1 – 4 + platelet derived growth factor, DMSO as a negative control and 0.1 μM dasatinib (0.1 μM) as a positive control. To ensure activity was not due to cell death, cell viability of BR5 fibroblasts was measured by Cell Titer-glo® at the same concentration. b) Dose-dependent effect of 1 on DDR2 in BR5 fibroblasts.
Closer investigations of the direct effect of 1 on DDR2 was carried out with human BR-5 fibroblast cells in culture with indicated concentrations of 1 (DMSO control) for 18 hours. Whole cell lysate was collected, separated by electrophoresis through an SDS-PAGE gel, transferred to a PVDF membrane, and detected by anti-DDR2 antibody. In DMSO-treated cells, DDR2 protein ran as a two bands of approximately 100 kDa apparent size. Treatment with the initial active fraction SNA-048-7 as well as 1 and 2 resulted in appearance of a third DDR2-reactive band of approximately 130 kDa apparent size, indicating modification of the DDR2 protein (Figure 6b). The nature of the modification is unknown at this time, but is consistent with glycosylation, based on electrophoretic mobility.17 Further work is necessary to elucidate the full mechanistic details for inhibition and to identify the molecular target.
As part of our UTSW panel of non-small cell lung cancer (NSCLC) lines, we have identified lines HCC366 as harboring a DDR2 mutation. Measurement of cytotoxicity of 1 against HCC366 gave an IC50 = 120 nM, while the cytotoxicity against the DDR2 wild-type NSCLC cell-line A549 had a decrease in cytotoxicity with an IC50 = 10 μM. The IC50 values for 2 and 4 are similar to 1 (0.190 μM and 0.275 μM against HCC366, respectively; 8.8 μM and 13.4 μM against A549, respectively), while the IC50 values for 3 are significantly reduced (0.7 μM against HCC366 and 19.6 μM against A549, also see Table S8). As a positive control, dasatinib had IC50 values of 170 nM against HCC366 and and 9.8 μM against A549, consistent with previous reports.5,18
The only other natural product that has been shown to inhibit DDR2 activity is actinomycin D. Although actinomycin D has been shown to possess potent DNA intercalating activity (< 10 nM), when cells are treated with actinomycin D for short periods (< 8 hours) there is significant inhibition of DDR2 activation by collagen.19 The mechanistic details suggest actinomycin D disrupts the ability of collagen to bind to DDR2, but does not interrupt DDR2 localization in the cell membrane.
CONCLUSIONS
Through the use of an unbiased screening strategy using the recently developed FUSION platform, we were able to identify inhibitors of the DDR2 pathway. DDR2 has emerged as an important target in cancer therapy, particularly in a significant percentage of squamous cell carcinomas, such as non-small cell lung cancers. The discovery of the discoipyrroles represents a second class of natural products that inhibit the DDR2 signaling pathway.
In addition to the structural novelty and biological activity, the non-enzymatic production of the discoipyrrole family of compounds is an example of a growing number of natural products that involve non-enzymatic steps in their production. Other examples of de novo non-enzymatic natural products synthesis include jadomycin (DNA alkylator)20, elansolid (antibiotic) 21 and the ammosamides (quinone reductase 2 inhibitors, myosin modifiers).22 In these examples, the non-enzymatic reaction pathway has been exploited to generate analogs with enhanced biological activity. Non-enzymatic multi-step synthesis of analogs of 1 provide us an opportunity to explore and discover analogs with enhanced potency and pharmacokinetic attributes.
Supplementary Material
Acknowledgments
The authors thank S. Wang, B. Posner and members of the UTSW High-Throughput Screening Core Facility. This work was funded by grants from the US National Institutes of Health (R01 CA149833 and RC2 CA148225) and the Welch Foundation (I- 1689) J.B.M. is the Chilton/Bell Scholar in Biochemistry. M.B.P. was supported by a Komen for the Cure Postdoctoral Fellowship.
Footnotes
ASSOCIATED CONTENT
General procedures, bioassay protocols, purification proceures, data tables and NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.a) Vogel W, Gish GD, Alves F, Pawson T. Mol Cell. 1997;1:13–23. doi: 10.1016/s1097-2765(00)80003-9. [DOI] [PubMed] [Google Scholar]; (b) Shrivastava A, Radziejewski C, Campbell E, Kovac L, McGlynn M, Ryan TE. Mol Cell. 1997;1:25–34. doi: 10.1016/s1097-2765(00)80004-0. [DOI] [PubMed] [Google Scholar]
- 2.a) Olaso E, Labrador JP, Wang L, Ikeda K, Eng FJ, Klein R, Lovett DH, Lin HC, Friedman SL. J Biol Chem. 2002;277:3606–3613. doi: 10.1074/jbc.M107571200. [DOI] [PubMed] [Google Scholar]; (b) Leitinger B. J Biol Chem. 2003;278:16761–16769. doi: 10.1074/jbc.M301370200. [DOI] [PubMed] [Google Scholar]
- 3.Xu L, Servais J, Polur I, Kim D, Lee PL, Chung K, Li Y. Arthritis Rheum. 2010;62:2736–2744. doi: 10.1002/art.27582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ford CE, Lau SK, Zhu CQ, Andersson T, Tsao MS, Vogel WF. Br J Cancer. 2007;96:808–814. doi: 10.1038/sj.bjc.6603614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hammerman PS, et al. Cancer Disc. 2011;1:78–89. doi: 10.1158/2159-8274.CD-11-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kauppila S, Stenback F, Risteli J, Jukkola A, Risteli L. J Pathol. 1998;186:262–268. doi: 10.1002/(SICI)1096-9896(1998110)186:3<262::AID-PATH191>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 7.Zhang K, Corsa CA, Ponik SM, Prior JL, Piwnica-Worms D, Eliceiri KW, Keely PJ, Longmore GD. Nat Cell Biol. 2013;15:677–687. doi: 10.1038/ncb2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Hu Y, Legako AG, Espindola A-PDM, MacMillan JB. J Org Chem. 2012;77:3401–3307. doi: 10.1021/jo300197z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Pan E, Jamison M, Yousufuddin M, MacMillan JB. Org Lett. 2012;14:2390–2393. doi: 10.1021/ol300806e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hu Y, MacMillan JB. 2011;13:6580–6583. doi: 10.1021/ol202944g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hu Y, Espindola APDM, Stewart NA, Wei S, Posner BA, MacMillan JB. Bioorg Med Chem. 2011;19:5183–5189. doi: 10.1016/j.bmc.2011.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Hu Y, Wang K, MacMillan JB. Org Lett. 2013;15:390–393. doi: 10.1021/ol303376c. [DOI] [PubMed] [Google Scholar]
- 9.Garrett MD, Walton MI, McDonald E, Judon I, Workman P. Prog Cell Cycle Res. 2003;5:145–158. [PubMed] [Google Scholar]
- 10.a) Potts MB, Kim HY, Fisher KW, Hu Y, Carrasco Y, Bulut GB, Ou Y-H, Herrera-Herrera ML, Cubillos F, Mendiratta S, Xiao G, Hofree M, Ideker T, Xie Y, Huang LJ-S, Lewis RE, MacMillan JB, White MA. Cell. doi: 10.1126/scisignal.2004657. in review. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet J-P, Subramanian A, Ross KN, Reich M, Hieronymus H, Wei G, Armstrong SA, Haggarty SJ, Clemons PA, Wei R, Carr SA, Lander ES, Golub TR. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- 11.a) Menezes GC, Miron-Mendoza M, Ho CH, Jiang H, Grinnell F. Exp Cell Res. 2008;314:3081–3091. doi: 10.1016/j.yexcr.2008.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Grinnell F, Rocha LB, Iucu C, Rhee S, Jiang H. Exp Cell Res. 2006;312:86–94. doi: 10.1016/j.yexcr.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 12.Lampis G, Deidda D, Maullu C, Madeddu MA, Pompei R, Monachie F, Satta G. J Antibiotics. 1995;48:967–972. doi: 10.7164/antibiotics.48.967. [DOI] [PubMed] [Google Scholar]
- 13.Dalgliesh CE, Knox WE, Neuberger E. Nature. 1951;168:20–22. doi: 10.1038/168020a0. [DOI] [PubMed] [Google Scholar]
- 14.Arend M, Westermann B, Risch N. Angew Chem Int Ed. 1998;37:1044–1070. doi: 10.1002/(SICI)1521-3773(19980504)37:8<1044::AID-ANIE1044>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 15.a) Moore CM, Helmann JD. Curr Opin Microbiol. 2005;8:188–195. doi: 10.1016/j.mib.2005.02.007. [DOI] [PubMed] [Google Scholar]; (b) Imlay JA. Annu Rev Microbiol. 2003;57:395–418. doi: 10.1146/annurev.micro.57.030502.090938. [DOI] [PubMed] [Google Scholar]
- 16.Shor AC, Keschman EA, Lee FY, Muro-Cacho C, Letson GD, Trent JC, Pledger WJ, Jove R. Cancer Res. 2007;67:2800–2808. doi: 10.1158/0008-5472.CAN-06-3469. [DOI] [PubMed] [Google Scholar]
- 17.a) Konitsiotis AD, Raynal N, Bihan D, Hohenester E, Farndale RW, Leitinger B. J Biol Chem. 2008;283:6861–6868. doi: 10.1074/jbc.M709290200. [DOI] [PubMed] [Google Scholar]; (b) Ali BR, Xu H, Akawi NA, John A, Karuvantevida NS, Langer R, Al-Gazali L, Leitinger B. Human Mol Genet. 2010;19:2239–2250. doi: 10.1093/hmg/ddq103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnson FM, Saigal B, Talpaz M, Donato NJ. Clin Cancer Res. 2005;11:6924–6932. doi: 10.1158/1078-0432.CCR-05-0757. [DOI] [PubMed] [Google Scholar]
- 19.Siddiqui K, Kim GW, Lee DH, Shin HR, Yang EG, Lee NT, Yang BS. Biol Parm Bull. 2009;32:136–141. doi: 10.1248/bpb.32.136. [DOI] [PubMed] [Google Scholar]
- 20.a) Rix U, Zheng J, Remsing Rix LL, Greenwell L, Yang K, Rohr J. J Am Chem Soc. 2004;126:4496–4497. doi: 10.1021/ja031724o. [DOI] [PubMed] [Google Scholar]; (b) Cottreau KM, Spencer C, Wentzell JR, Graham CL, Borissow CN, Jakeman DL, McFarland SA. Org Lett. 2010;12:1172–1175. doi: 10.1021/ol902907r. [DOI] [PubMed] [Google Scholar]; (c) Dupuis SN, Robertson AW, Veinot T, Monro SMA, Douglas SE, Syvitski RT, Goralski KB, McFarland SA, Jakeman DL. Chem Sci. 2012;3:1640–1644. [Google Scholar]
- 21.a) Steinmetz H, Gerth K, Jansen R, Schläger N, Dehn R, Reinecke S, Kirschning A, Müller R. Angew Chem Int Ed. 2011;50:532–536. doi: 10.1002/anie.201005226. [DOI] [PubMed] [Google Scholar]; (b) Jansen R, Gerth K, Steinmetz H, Reinecke S, Kessler W, Kirschning A, Müller R. Chem Eur J. 2011;17:7739–7744. doi: 10.1002/chem.201100457. [DOI] [PubMed] [Google Scholar]; (c) Dehn R, Katsuyama Y, Weber A, Gerth K, Jansen R, Steinmetz H, Hofle G, Müller R, Kirschning A. Angew Chem Int Ed. 2011;50:3882–3887. doi: 10.1002/anie.201006880. [DOI] [PubMed] [Google Scholar]; (d) Steinmetz H, Zander W, Shushni MAM, Jansen R, Gerth K, Dehn R, Dräger G, Kirschning A, Müller R. ChemBioChem. 2012;13:1813–1817. doi: 10.1002/cbic.201200228. [DOI] [PubMed] [Google Scholar]
- 22.a) Hughes CC, MacMillan JB, Gaudencio SP, Jensen PR, Fenical W. 2009;48:725–727. doi: 10.1002/anie.200804890. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hughes CC, MacMillan JB, Gaudencio SP, Fenical W, LaClair JJ. Angew Chemie Int Ed. 2009;48:728–732. doi: 10.1002/anie.200804107. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Pan E, Oswald N, Life JM, Posner BA, MacMillan JB. Chem Sci. 2013;4:482–488. doi: 10.1039/C2SC21442C. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.