

Tumor cells of several cancers are known to constitutively express the coagulation initiating factor, tissue factor (TF). TF-mediated coagulation generates thrombin, platelet activation, and fibrin formation which altogether orchestrate cancer cell survival and proliferative pathways. Thrombin also induces activation of natural anticoagulant protein C. Activated protein C (APC) not only inhibits subsequent thrombin generation, but also induces cellular signaling through activation of protease activated receptor-1 (PAR1) [1,2]. Endothelial cell protein C receptor (EPCR) plays a key role in supporting APC-mediated cell signaling [1,2]. EPCR-APC-mediated cytoprotection and other cellular effects may accelerate cancer progression and metastasis. EPCR-dependent APC activation of PAR1 was shown to stimulate cell migration of breast cancer cells [3]. Recent studies have shown that EPCR-APC axis conferred a significant survival advantage to lung adenocarcinoma cells and favored their prometastatic activity [4]. Interestingly, our recent studies suggested that EPCR may also function as a negative regulator of cancer progression [5].
The present study was carried out to investigate the influence of EPCR on human breast cancer development. MDA-231t cells (tumor cells established from in vivo tumor developed by injection of MDA-MB-231 cells to a nude mouse) were stably transfected with a control vector (CV) or EPCR expression vector in pZeoSV plasmid vector. After 48 h of transfection, Zeocin (100 μg/ml) was added to the cells. After 3 weeks, stable transfectant colonies were isolated, expanded, and EPCR stable transfectants exhibiting similar TF activity as that of parental MDA-231t cells were selected for the present study. MDA-231t(+CV) and MDA-231t(+EPCR) cells expressed similar levels of TF antigen and activity (Fig. 1 panels A to C). MDA-231t(+CV) cells expressed very little EPCR, whereas EPCR expression levels in MDA-231t(+EPCR) cells was similar to that of HUVEC (Fig. 1A, 1B).
Fig. 1.
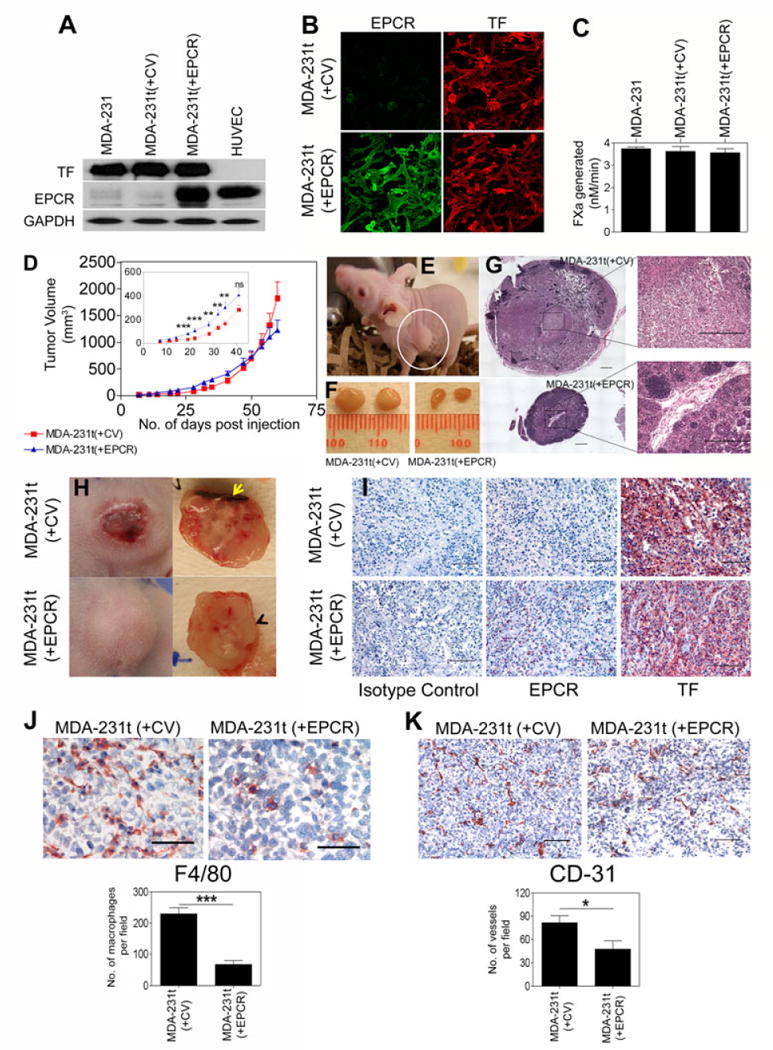
Influence of EPCR on tumor growth in a murine breast carcinoma model. TF and EPCR expression levels in MDA-231t cells stably transfected with a control or EPCR expression vector were analyzed by Western blot analysis (A) or immunofluorescence microscopy (B), and the cell surface TF activity was measured in a factor X activation assay (C). (D) MDA-231t(+CV) or MDA-231t(+EPCR) cells (106 cells/site) were injected into the mammary fat pad of nude mouse (BALB/c, NU/J, Stock number 002019, Jackson Laboratories, Maine) in 0.1 ml of Matrigel (BD Biosciences, diluted 1:4 in serum-free medium) and tumor growth was monitored for 60 days (n=9 mice/group, data shown is the cumulative data of two independent experiments). Tumor volumes were calculated by using formula (length × width1 × width2 × 0.5) by measuring 3 dimensions of the tumor with a digital caliper twice a week. (E) A swollen lymph node (white circle) in a mouse injected with MDA-231t(+CV) cells at day 60 following the injection of tumor cells. (F) A representative picture of lymph node size differences in mice injected with MDA-231t(+CV) or MDA-231t(+EPCR) cells. (G) Extensive infiltration of cells into the lymph node of mice injected with MDA-231t(+CV) and not MDA-231t(+EPCR) cells. Images of multiple fields obtained using 10× lens were digitally stitched together to obtain a full view (left side, bar represents 500 μm); one of the fields, marked by square, was shown at its original 10× magnification (Nikon Eclipse Ti microscope with software NIS Elements BR3.2). (H) Tumors developed in mice injected with MDA-231t(+CV) were inflamed and necrotic. Yellow arrow mark points the necrotic region. Tumor tissue sections were stained for TF and EPCR (I) (please note that although it was not clearly visual from the small size image, a small fraction of tumor cells from MDA-231t(+EPCR) tumors stained positive for EPCR while most of the tumor cells were negative for EPCR. In case of MDA-231t(+CV) tumors, none of the tumor cells stained positive for EPCR), F4/80 antigen (mouse macrophage marker; J), or CD31 (endothelial cell marker; K) and immunostained sections were viewed using 20× (I and K, bar represents 100 μm) or 40× lens (J, bar represents 50 μm). Number of macrophages and microvessels per field was obtained by counting 8 to 12 randomly chosen fields in 3 tumors in each experimental group (at 20× magnification). Statistical significance between the groups was calculated by Mann-Whitney test; *, P < 0.05, ** P <0.01, and *** P <0.001.
MDA-231t(+CV) or MDA-231t(+EPCR) cells were injected into the mammary fat pad (m.f.p) of nude mice, and the growth of tumor in m.f.p. was monitored for 2 months. As shown in Fig. 1D (in set), tumor growth rate is statistically significantly higher in mice injected with MDA-231t(+EPCR) cells compared to MDA-231t(+CV) cells until 40 days following tumor cell implantation. However, in the last two weeks, tumors derived from MDA231t(+EPCR) cells grew less rapidly than tumors originating from MDA-231t(+CV) cells. At the end of 60 days, the tumor volume of MDA-231t(+EPCR) cell-derived tumors was about 30% lower than that of MDA-231t(+CV) cell-derived tumors (Fig. 1D). Although this difference did not reach statistical significance, it was substantial and consistent. At the time of euthanasia (day 60), the mice bearing MDA-231t(+CV) cell-derived tumors appeared to be lethargic, and developed swollen lymph nodes (Fig. 1E and 1F), whereas mice bearing MDA-231t(+EPCR) cell-derived tumors exhibited no outward sickness and did not develop any swollen lymph nodes (Fig. 1F). Histological examination of lymph node sections showed extensive infiltration of cells into this region in mice injected with MDA-231t(+CV) cells and not in mice injected with MDA-231(+EPCR) cells (Fig. 1G). The skin over the tumors of the mice injected with MDA-231t(+CV) cells turned blood red and looked different from that of the tumors generated by MDA-231(+EPCR) cells, starting around 30 to 35 days following tumor cell inoculation. At the time of sacrifice (60 days), all tumors developed in mice injected with MDA-231t(+CV) cells were highly inflamed and necrotic, most of which developed hematogenous ulcers at the top skin of tumors (Fig. 1H). Some necrotic tumors collapsed and had leaky liquid centers. None of the tumors in mice bearing MDA-231t(+EPCR) cells showed necrotic ulcerations.
Interestingly, analysis of tumor tissue sections for EPCR and TF expression showed that a majority of tumor cells stained negative for EPCR irrespective of whether MDA-231t(+CV) or MDA-231t(+EPCR) cells were used for implantation (Fig. 1I). In both cases, tumor cells stained intensively positive for TF. Analysis of tumor tissue sections for macrophage infiltration and angiogenesis by staining them for F4/80 antigen and CD31, respectively, showed significant reduction in macrophage infiltration (Fig. 1J) and microvessel density (Fig. 1K) in tumors derived from MDA-231t(+EPCR) cells compared to tumors derived from MDA-231t(+CV) cells. It may be pertinent to note here that tissue sections analyzed for tumors derived from both MDA-231t(+CV) and MDA-231t(+EPCR) cells represent the actively growing regions of the tumor.
During the preparation of this manuscript, Schaffner et al. [6] reported that EPCR-expressing cells, selected from expansion of EPCR+ cancer stem cell-like population from MDA-MB-231 mfp cells, had markedly increased tumor cell-initiating activity compared to EPCR− cells. Although the experimental approach and MDA-MB-231 cells used for implantation in the present study and the recently published study [6] differed, the results obtained from both the studies are similar to some extent. Schaffner et al. [6] observed that MDA-MB-231 mfp cells exhibit two distinct populations, EPCR+ cells with moderate levels of TF and EPCR negative cells with high levels of TF expression, and cell sorting was used to select EPCR+ and EPCR− cells for their experiments. Although varied levels of EPCR and TF expression were also found in our MDA-MB-231t cell population, we did not find two clearly distinctive populations of cells (see Fig. 1C). Most of the cells expressed very low levels of EPCR and high levels of TF. Therefore, we genetically engineered them to express EPCR to obtain EPCR+ cells. As reported by Schaffner et al. [6], EPCR expression in breast cancer cells increased the tumor cell growth potential, although not drastically, but in a statistically significant fashion. However, we found that in the later stages of tumor progression, the differences in tumor growth between tumors derived from EPCR+ or EPCRlow cells vanished. In fact, at the end of experimental period, tumor volume in mice injected with EPCR+ cells was 30% lower than in mice injected with EPCRlow cells. It is difficult to predict whether the earlier study [6] could have found similar results if their observation was not terminated when the tumor size reached less than 1 cm3. It is interesting to note that, as found in the earlier study [6], irrespective of the EPCR status in tumor cells that were inoculated, the majority of outgrown tumor cells were EPCR negative, which indicates a conversion from EPCR+ cells to EPCR− cells in the tumor microenvironment. Here, it is important to point out that our observation on loss of EPCR expression in tumor tissues derived from EPCR+ tumor cells is not due to a lack of sensitivity to detect EPCR in tumor tissues by immunohistochemistry method. Loss of EPCR in tumors derived from EPCR+ cells is also confirmed by immunoblot analysis of tumor tissue extracts (data not shown). At present, it is unknown at which stage of tumor growth the EPCR expression was lost and the underlying mechanism for it.
A notable finding of the present study is that while we observed solid and liquefaction necrosis in tumors originated from EPCRlow cells, no necrosis was found in tumors originated from EPCR+ cells. Necrosis is a common feature of aggressive breast cancer and has been associated with a poor prognosis [7]. Tumor necrosis is the direct result of chronic ischemia caused by vascular collapse when the rate of tumor cell growth exceeds that of angiogenesis [8]. Necrotic liquefaction occurs when the cellular structures are broken down by proteolytic enzymes released from ruptured lysosomes of tumor cells and/or similar enzymes released by infiltrating inflammatory cells [9]. Although it appears to be counterintuitive at first, necrosis resulting from chronic ischemia is associated with increased angiogenesis [8]. Prolonged hypoxic conditions were known to increase tumor progression and angiogenesis, and to promote metastatic potential [10]. Therefore, EPCR expression in breast cancer cells, despite having initial growth advantage, may limit cancer progression at an advanced stage.
Acknowledgments
The study was partly supported by a research seed grant from contributions made by local community to the research at our institute and NIH grant HL107483 (LVMR).
Footnotes
Addendum
S. Keshava performed all experiments and analyzed the data. H. Kothari performed immunofluorescence experiments. L.V.M. Rao participated in study design and wrote the manuscript. U. Pendurthi designed the study and wrote the manuscript.
Disclosure of Conflict of Interests
None
References
- 1.Mosnier LO, Zlokovic BV, Griffin JH. The cytoprotective protein C pathway. Blood. 2007;109:3161–72. doi: 10.1182/blood-2006-09-003004. [DOI] [PubMed] [Google Scholar]
- 2.Rezaie AR. Regulation of the protein C anticoagulant and antiinflammatory pathways. Curr Med Chem. 2010;17:2059–69. doi: 10.2174/092986710791233706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beaulieu LM, Church FC. Activated protein C promotes breast cancer cell migration through interactions with EPCR and PAR-1. Exp Cell Res. 2007;313:677–87. doi: 10.1016/j.yexcr.2006.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anton I, Molina E, Luis-Ravelo D, Zandueta C, Valencia K, Ormazabal C, Martinez-Canarias S, Perurena N, Pajares MJ, Agorreta J, Montuenga LM, Segura V, Wistuba II, De Las RJ, Hermida J, Lecanda F. Receptor of activated protein C promotes metastasis and correlates with clinical outcome in lung adenocarcinoma. Am J Respir Crit Care Med. 2012;186:96–105. doi: 10.1164/rccm.201110-1826OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keshava S, Sahoo S, Tucker TA, Idell S, Rao LV, Pendurthi UR. Endothelial cell protein C receptor opposes mesothelioma growth driven by tissue factor. Cancer Res. 2013;73:3963–73. doi: 10.1158/0008-5472.CAN-12-1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schaffner F, Yokota N, Carneiro-Lobo T, Kitano M, Schaffer M, Anderson GM, Mueller BM, Esmon CT, Ruf W. Endothelial protein C receptor function in murine and human breast cancer development. PLoS One. 2013;8:e61071. doi: 10.1371/journal.pone.0061071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Richards CH, Mohammed Z, Qayyum T, Horgan PG, McMillan DC. The prognostic value of histological tumor necrosis in solid organ malignant disease: a systematic review. Future Oncol. 2011;7:1223–35. doi: 10.2217/fon.11.99. [DOI] [PubMed] [Google Scholar]
- 8.Leek RD, Landers RJ, Harris AL, Lewis CE. Necrosis correlates with high vascular density and focal macrophage infiltration in invasive carcinoma of the breast. Br J Cancer. 1999;79:991–5. doi: 10.1038/sj.bjc.6690158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Woolf N. Cell, Tissue and Disease. London: Bailliere Tindall; 1986. Cell and tissue death; pp. 25–30. [Google Scholar]
- 10.Liao D, Johnson RS. Hypoxia: a key regulator of angiogenesis in cancer. Cancer Metastasis Rev. 2007;26:281–90. doi: 10.1007/s10555-007-9066-y. [DOI] [PubMed] [Google Scholar]