

Abstract
BACKGROUND
‘Omics’ high-throughput analyses, including genomics, epigenomics, transcriptomics, proteomics and metabolomics, are widely applied in human endometrial studies. Analysis of endometrial transcriptome patterns in physiological and pathophysiological conditions has been to date the most commonly applied ‘omics’ technique in human endometrium. As the technologies improve, proteomics holds the next big promise for this field. The ‘omics’ technologies have undoubtedly advanced our knowledge of human endometrium in relation to fertility and different diseases. Nevertheless, the challenges arising from the vast amount of data generated and the broad variation of ‘omics’ profiling according to different environments and stimuli make it difficult to assess the validity, reproducibility and interpretation of such ‘omics’ data. With the expansion of ‘omics’ analyses in the study of the endometrium, there is a growing need to develop guidelines for the design of studies, and the analysis and interpretation of ‘omics’ data.
METHODS
Systematic review of the literature in PubMed, and references from relevant articles were investigated up to March 2013.
RESULTS
The current review aims to provide guidelines for future ‘omics’ studies on human endometrium, together with a summary of the status and trends, promise and shortcomings in the high-throughput technologies. In addition, the approaches presented here can be adapted to other areas of high-throughput ‘omics’ studies.
CONCLUSION
A highly rigorous approach to future studies, based on the guidelines provided here, is a prerequisite for obtaining data on biological systems which can be shared among researchers worldwide and will ultimately be of clinical benefit.
Keywords: endometrium, epigenomics, genomics, metabolomics, proteomics
Introduction
The completion of the Human Genome Project in 2000 (Venter et al., 2001) triggered a rapid development of several fields in molecular biology that together are described as ‘omics’, also known as the ‘omics revolution’. It was stated then that ‘with 35 000 genes and hundreds of thousands of protein states to identify, correlate and understand, it no longer suffices to rely on studies of one gene, gene product or process at a time. We have entered the ‘omic’ era in biology’ (Weinstein, 2001).
‘Omics’ refer to the application of high-throughput techniques which simultaneously examine changes in the genome (assessment of variability in DNA sequence in the genome, i.e. genomics), epigenome (epigenetic modifications of DNA, i.e. epigenomics), transcriptome (gene expression profiling, assessment of variability in composition and abundance of messenger RNA (mRNA) levels, i.e. transcriptomics), proteome (variability in composition and abundance of the proteins, i.e. proteomics) or metabolome (variability in composition and abundance of metabolites, i.e. metabolomics or metabonomics) in a biological sample. In addition to these well-established ‘omics’ fields, the ‘omics’ are ever expanding with new fields in biological data, such as exomics (exons in a genome, i.e. exome), lipidomics (collection of lipids, i.e. lipidome), secretomics (secreted protein, i.e. secretome), interactomics (interactome, or ‘systems biology’) and others (wikipedia.org; omics.org).
In reproductive medicine, Bellver et al. (2012) defined the ‘omics’ high-throughput analyses as ‘reproductomics’. At present, reproductomics applications are foreseen as: (i) genomics for whole-genome genetic screening in adults prior to conception, and diseases (male factor, implantation failure, recurrent abortion), comparative genome hybridization (CGH) analyses for PGD and preimplantation genetic screening, and in prenatal diagnosis (Wells and Levy, 2003; Rubio et al., 2007; Bellver et al., 2012; Treff, 2012); (ii) transcriptomics and proteomics to provide robust molecular tools for endometrial evaluation (Boomsma et al., 2009a, b; Diaz-Gimeno et al., 2011; Cheong et al., 2013; Diaz-Gimeno et al., 2013; Garrido-Gomez et al., 2013; Salamonsen et al., 2013), oocyte screening through cumulus cells investigation (Assou et al., 2010; Koks et al., 2010) and sperm selection (Seli et al., 2010; Altmäe and Salumets, 2011) and (iii) proteomics and metabolomics to provide future alternatives for non-invasive methods for embryo selection (Bellver et al., 2012) and for the diagnosis of endometriosis (Fassbender et al., 2012a, b; Fassbender et al., 2012a, b). Nevertheless, ‘omics’ technologies still require improved reproducibility and clinical predictive value based on large sample cohorts (thereby clinical applicability), before they can provide new diagnostic, prognostic and therapeutic tools.
The current review focuses on all ‘omics’ studies applied to human endometrium. While the ‘omics’ technologies have undoubtedly advanced our knowledge of human endometrium in health and different diseases, the challenges arising from the huge amount of data generated and broad variation of ‘omics’ profiling according to different environments and stimuli make it difficult to determine the use, validity, reproducibility and interpretation of ‘omics’ data. The current review provides guidelines for future ‘omics’ studies on human endometrium and summarizes the current status together with the promise and shortcomings of the high-throughput technologies.
‘Omics’ studies applied to human endometrium
The human endometrium is a dynamic tissue that undergoes cyclic growth, differentiation, desquamation and regeneration that are driven by the ovarian steroidal hormones oestrogen and progesterone as well as other hormones, cytokines and chemokines (Wilcox et al., 1999; Lessey, 2000; Salamonsen et al., 2009). The main function of the endometrium is to provide precisely timed support to enable embryo implantation and for further fetal growth and maturation. Although the endometrium is non-receptive to embryos for most of the menstrual cycle, it becomes receptive during a spatially and temporally restricted period in the secretory phase known as the ‘window of implantation’ (Harper, 1992; Giudice, 1999). Gaining insight into the complex mechanisms controlling changes within the endometrium is crucial to understand not only implantation but also gynaecological disorders, such as endometriosis, uterine fibroids or endometrial cancer, that can impact endometrial function leading to infertility or pregnancy loss.
From the first histologic dating methods (Noyes et al., 1950; Noyes et al., 1975) to the new ‘omics’ technologies, extensive efforts have been applied to understanding and characterizing receptive endometrium. Despite the common use of the traditional endometrial dating criteria, its accuracy, reproducibility and functional relevance have been questioned in randomized studies (Coutifaris et al., 2004; Murray et al., 2004), encouraging further research and application of new technologies to objectively diagnose endometrial receptivity, since reliable diagnostic markers are still lacking and the molecular mechanisms remain unclear (Brinsden et al., 2009; Lessey, 2011; Zhang et al., 2012). Pioneering studies have demonstrated that the application of high-throughput ‘omics’ technologies does hold the key to endometrial biomarker research (Boomsma et al., 2009a, b; Hannan et al., 2010; Diaz-Gimeno et al., 2011; Hannan et al., 2011a, b; Altmäe et al., 2012; Diaz-Gimeno et al., 2013; Garrido-Gomez et al., 2013). Nevertheless, further research is clearly warranted if a full understanding of the complex intercellular relationships is to be achieved. ‘Omics’ research in human endometrium is both complex and challenging, as the endometrium is regulated by cyclic hormones and autocrine/paracrine/juxtacrine factors, which when combined with the individual's genetic and environmental background, may result in different biological responses.
The past decade has witnessed an explosive growth in the number of the ‘omics’ studies involving the human endometrium (Figure 1). Analysis of the transcriptome pattern in endometrium in health and different disease states has been by far the most commonly applied ‘omics’ technique. However, as technologies improve, proteomics holds the next great promise for the studies in human endometrium.
Figure 1.
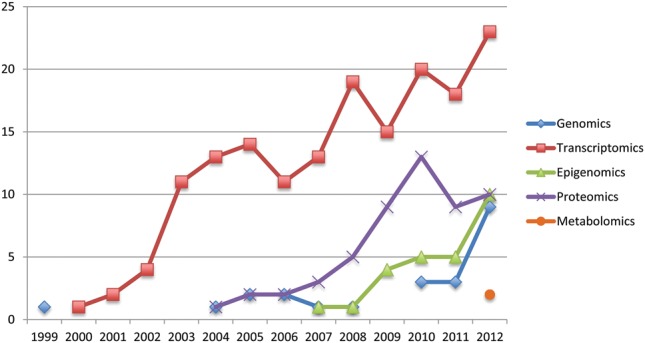
‘Omics’ publications in human endometrium studies. Y-axis indicates the number of studies, and X-axis denotes the year of publication. The systematic review of the literature in PubMed was conducted up to March 2013. The eligible studies were additionally identified using reference lists of review articles and other relevant studies. Abstracts from conference proceedings were also considered. No language or any other restrictions were applied. Search terms are presented in detail in Supplementary data, Table S1, and the search results in Supplementary data, Figure S1. In short, keywords ‘endometrium’, ‘endometriosis’ and ‘embryo implantation’ were one-by-one searched with each paired term. After excluding duplicates, a total of 2478 manuscripts were identified and following critical selection 269 manuscripts of ‘omics’ studies in human endometrium remained (including studies on normal endometrium, endometrial receptivity, implantation and implantation failure, endometriosis and endometrial cancer): 23 of genomics, 164 of transcriptomics, 26 of epigenomics, 54 of proteomics and 2 of metabolomics.
Genomics in human endometrium
Recent advances in genotyping technology together with detailed information of common gene variants (single nucleotide polymorphisms, SNPs) have led to a rapid development of genome-wide association studies (GWAS). GWAS are currently the most commonly used technique for searching for SNPs/loci associated with a trait of disease (Day and Loos, 2011). Genomics analyses in human endometrium are still limited; to date, four GWAS in endometriosis (Adachi et al., 2010; Uno et al., 2010; Painter et al., 2011; Nyholt et al., 2012) with summarizing tables of these findings (Rahmioglu et al., 2012; Burney, 2013) and one study in endometrial cancer patients (Ikeda et al., 2012) have been published. The inconsistency in the GWAS and association studies in general has been attributed to small sample sizes, proper controls and heterogeneity within populations (Guo, 2006; Sundqvist et al., 2013). The identification of true genetic association requires a large sample size and replication in different populations. Also a general problem in association studies is that papers with positive results may be published preferentially, thus masking the real situation (Altmäe et al., 2011). In the field of endometriosis, big advances in the search for risk loci have been achieved by two recent comprehensive and large-scale GWAS, where seven SNPs were identified and confirmed in different populations (Painter et al., 2011; Nyholt et al., 2012). These recent GWAS of >4600 cases and >9300 controls of Japanese and European ancestry demonstrated that many weakly associated SNPs represent true endometriosis risk loci that can be used for risk prediction and future targeted disease therapy across these different populations (Painter et al., 2011; Nyholt et al., 2012). No SNP-based microarray studies regarding endometrial receptivity or infertility have yet been published.
Although occurring less frequently than SNPs, copy number variations (CNVs) play an important role in genetic variation. CNVs located in the promoter regions of genes can influence gene expression levels and thereby contribute to the development of complex disease traits (Redon et al., 2006). Recent analysis of somatic copy number aberrations and CNVs in patients with endometriosis found no association with disease aetiology (Saare et al., 2012). As a small sample was analysed, the study results are rather preliminary and need to be confirmed in a larger study group. Previous GWAS focusing on genomic alterations in endometria of patients with endometriosis have shown various chromosomal alterations; however, only a few of these alterations have been observed in more than one study (Guo et al., 2004; Wu et al., 2006; Veiga-Castelli et al., 2010). These inconsistencies in findings highlight the need for studies that are powered sufficiently in order to detect (or rule out) the effects of interest, as has been neatly demonstrated on ∼15 000 women in a search for endometriosis risk loci (Painter et al., 2011; Nyholt et al., 2012).
Epigenomics in human endometrium
The cyclic changes in gene expression in the human endometrium during the menstrual cycle are believed, in part, to be under epigenetic control (Lessey, 2010; Munro et al., 2010; Guo, 2012). Several genes expressed by the endometrium have already been identified as being epigenetically regulated (Munro et al., 2010), including leukaemia inhibitory factor (LIF) (Uchida et al., 2005), glycodelin (Uchida et al., 2007), matrix metalloproteinases (Clark et al., 2007), E-cadherin (Rahnama et al., 2009), mucin I (Yamada et al., 2008) and others. Epigenetic modifications including DNA methylation, histone acetylation and RNA interference are involved in functional changes in endometrium associated with pregnancy (endometrial receptivity, decidualization) and can be altered in diseases affecting the endometrium (endometriosis, cancer, implantation failure) (see recent reviews Revel et al., 2011; Estella et al., 2012; Guo, 2012).
For epigenomics analysis of the endometrium, the most applied ‘omics’ platform so far has been microRNA (miRNA) arrays. miRNAs function as posttranscriptional regulators of gene expression and operate through RNA interference, either degrading or translationally repressing target mRNAs (Bhattacharyya et al., 2006; Carthew and Sontheimer, 2009). Array studies in the endometrium have demonstrated that aberrant miRNA expression is associated with benign gynaecological conditions, such as endometriosis (Burney et al., 2009; Ha, 2011; Zelenko et al., 2012), gynaecological malignancies (Gilabert-Estelles et al., 2012; Ramon et al., 2012; Torres et al., 2013) and fertility disorders (Li et al., 2011; Revel et al., 2011). The importance of miRNAs in human endometrial receptivity has also been highlighted (Kuokkanen et al., 2010; Li et al., 2011; Revel et al., 2011; Sha et al., 2011; Estella et al., 2012) and, in association with gene expression data, a subset of miRNA target genes, including LIF, that could serve as potential biomarkers for endometrial receptivity has been proposed (Altmäe et al., 2013). To date, only three studies have applied deep sequencing (RNA-seq, in which all of the RNA is sequenced) for identifying miRNA expression profiles in human endometrium related to infertility (Sha et al., 2011) and endometriosis (Creighton et al., 2010; Hawkins et al., 2011). Most recently, miRNAs identified as specific to endometrial exosomes have been described, providing a new paradigm for endometrial-embryo interactions (Ng et al., 2013) and maybe a new opportunity for biomarker discovery.
Methylation of DNA is another level of epigenetic control, which has important implications for diseases including endometriosis (Izawa et al., 2013). A recent whole-genome scanning of methylation status in >25 000 promoters using methylated DNA immunoprecipitation with hybridization to promoter microarrays demonstrated that the overall methylation profile was highly similar between the endometrium and the endometriotic lesions (Borghese et al., 2010).
Clearly, our current knowledge of the endometrial epigenome and its physiological and pathophysiological significance is somewhat limited. Assessing the impact of the epigenome on endometrial physiology and pathophysiology remains a challenge for future studies.
Transcriptomics in human endometrium
Microarray-based gene expression technology that allows simultaneous monitoring of the expression of thousands of genes is the most widely used platform for transcriptome analysis. Our literature search (see Supplementary data, Table S1 and Figure S1) resulted in over 160 relevant manuscripts (61% of all identified ‘omics’ studies) (Figure 1). Endometrial transcriptomics has been applied to many aspects of endometrial physiology and pathophysiology, including the normal menstrual cycle (see reviews Horcajadas et al., 2004; White and Salamonsen, 2005; Giudice, 2006; Sherwin et al., 2006; Horcajadas et al., 2007; Aghajanova et al., 2008a, b; Bellver et al., 2012; Haouzi et al., 2012; Ruiz-Alonso et al., 2012), implantation and implantation failure (see reviews Toth et al., 2011; Koot et al., 2012), infertility including treatment protocols (see reviews Martinez-Conejero et al., 2007; Ruiz-Alonso et al., 2012), the impact of endometriosis (see reviews Matsuzaki, 2011; Fassbender et al., 2012a, b), endometrial cancer (see reviews Sherwin et al., 2006; Doll et al., 2008) and others (see reviews Horcajadas et al., 2007; Ruiz-Alonso et al., 2012; Garrido-Gomez et al., 2013). While any given study yields numerous candidate genes to explore, the number of genes, which have been identified in more than one study as potential biomarkers in endometrial physiology and pathophysiology, has remained somewhat small. This can be attributed to the differences in experimental design, timing and conditions of endometrial sampling, patient and control selection criteria, array platforms and annotation versions used, applied strategies for data processing and a lack of consistent standards for data presentation and deposition of complete data sets in public repositories (Horcajadas et al., 2007; Altmäe et al., 2010; Ruiz-Alonso et al., 2012; Ulbrich et al., 2013). Together, these constraints have made it nearly impossible to perform a meta-analysis of similar studies on specific stages of endometrial development (Ulbrich et al., 2013). Further, lack of uniformity in the validation of specific diseases such as endometriosis has limited the potential from this cumulative data to be realized.
Importantly, recent studies in which different cellular compartments of endometrium were analysed demonstrated cell type-specific gene expression profiles (Yanaihara et al., 2005; Evans et al., 2012a, b; Ulbrich et al., 2013). This was not surprising given the considerable changes in endometrial cellular composition with cycle stage. However, most transcriptome analyses have used biopsies of entire endometrial tissue containing all cell types: thus measured mRNA abundance reflects an average of all cell types present. In the two published ‘omics’ studies on human endometrium in which stromal and epithelial fractions were isolated by laser capture microdissection, distinct mRNA signatures were related to the day of the cycle (Yanaihara et al., 2005; Evans et al., 2012a, b). In addition, Evans et al., (2012a, b) compared two different microarray platforms, Affymetrix and Agilent, demonstrating concordance in their results. Clearly, one of the future tasks for transcriptome studies will be separate analyses of endometrial compartments, which will in turn provide a better understanding of endometrial physiology, the interactions between different cell types and their regulatory processes. However, the challenge is to obtain sufficient good quality RNA for expression analysis following microdissection. Recently published transcriptomes of endometrial cell constituents isolated by fluorescence-activated cell sorting (FACS) have also demonstrated cell-specific gene expression and identified multiple biological pathways and processes (Spitzer et al., 2012) and provided an alternative approach to laser dissection.
In addition to microarrays, there is the emerging alternative of RNA-seq, in which all RNAs are sequenced and most of the genes being expressed can be revealed. A comparison of the results derived from an Affymetrix microarray study and an RNA-seq study of bovine endometrium revealed a consistent overlap between the results but many more differentially expressed genes for the sequencing data (Ulbrich et al., 2013), as reported elsewhere (Malone and Oliver, 2011). RNA-seq technology detects more exons and alternative splicing events than microarray as it is entirely independent of prior knowledge. Microarrays routinely fail to pick up ∼25% of genes with low expression but such low-abundance transcripts are detected in RNA-seq reads (Werner, 2011). Nevertheless, while it is true that RNA-seq is independent of prior knowledge, the biological analysis of the data is not (Werner, 2011). RNA-seq analyses in human endometrium in health and diseased states are required.
The window of implantation is arguably the most relevant time to study gene expression profile as a way to establish biomarkers of a receptive endometrium (see reviews Bellver et al., 2012; Ruiz-Alonso et al., 2012; Garrido-Gomez et al., 2013). Based on earlier studies, Horcajadas et al. (2007) defined a list of 25 target genes for endometrial receptivity, including LIF, hyaluronan-binding protein 2, calpain, tissue factor pathway inhibitor 2, placental protein 14 and folate receptor. Subsequently, Dias-Gimeno et al. (2011) identified a set of 238 genes that are differentially expressed in the transition from the pre-receptive to the receptive state, creating a diagnostic tool named the endometrial receptivity array (ERA). The accuracy and consistency of this molecular test for defining endometrial cycle phases has been proved to be superior to classical histology methods (Horcajadas et al., 2007; Diaz-Gimeno et al., 2013). The clinical potential of the ERA for detecting the personalized window of implantation in patients with repetitive implantation failure, guiding their personalized embryo transfer as a novel therapeutic strategy has been demonstrated (Ruiz-Alonso et al., 2013). An ongoing randomized clinical trial should clarify the reliability and clinical efficacy of ERA in the general population. Interestingly, a recent study of biochemical markers in association with transcriptome expression analysis of separated stromal and glandular compartments and histological characterization, concluded that histology can provide an affordable, clinically applicable test for assessment of endometrial receptivity but not of implantation potential (Evans et al., 2012a, b). Further, such studies are clearly needed before this can replace the classical endometrial dating procedure (Noyes et al., 1950).
Another broadly studied area of transcriptomics is gene expression pattern analysis of eutopic endometrium compared with ectopic endometrium in women with and without endometriosis (Giudice et al., 2008). Multiple comparisons have been made, with primary endometriomas versus eutopic endometrium, revealing distinct transcriptomic differences and a variety of biological processes and signalling pathways unique to ectopic versus eutopic endometrium (Kao et al., 2003; Burney et al., 2007; Hansen and Eyster, 2010; Matsuzaki, 2011). Nevertheless, unique biomarkers for the pathophysiology and disease aetiology of endometriosis are still to be identified (Matsuzaki, 2011; Fassbender et al., 2013).
Proteomics in human endometrium
Proteomic research is currently considered a ‘hot topic’ and is increasingly being applied to human endometrium (Salamonsen et al., 2013). Any analysis of the full proteome is a challenging task as the proteome is large and of unknown complexity, being a result of alternative splicing of primary transcripts, the presence of sequence variation and epigenetic and post-translational modifications. This complexity is well reflected by the fact that although mRNA expression precedes protein translation, the correlation between a transcript level and the abundance of the corresponding protein product is poor (Fassbender et al., 2010; Ning et al., 2012), including in the endometrium (Stephens et al., 2010). Nevertheless, with the latest advances in mass spectrometry (MS) instrumentation, proteomics has emerged as a powerful tool for biomarker research.
Our literature search resulted in >50 manuscripts describing endometrial proteomic studies (21% of all identified ‘omics’ studies) (Figure 1). The most studied applications of proteomics include the search for biomarkers of the receptive endometrium (reviews Garrido-Gomez et al., 2010; Berlanga et al., 2011; Hannan et al., 2011a, b; Koot et al., 2012; Edgell et al., 2013; Upadhyay et al., 2013) and endometriosis (reviewed in Meehan et al., 2010; May et al., 2011; Fassbender et al., 2012a, b; Fassbender et al., 2013; Upadhyay et al., 2013). Edgell et al. (2013) presented very recently a list of nine validated proteins from endometrial tissue of relevance to endometrial receptivity, where membrane-associated progesterone receptor component 1 (PGRC1) and annexins (ANXA2 and ANXA4) together with others were validated. A recent systematic review of endometrial biomarkers of endometriosis assessing hormones, cytokines, proteomic factors and histological analysis of endometrial tissue concluded that none of the biomarkers alone or in a panel was ‘unequivocally clinically useful’ due to low numbers of subjects in discovery and replication studies, cycle- and stage dependence and low sensitivity and specificity (May et al., 2011).
Early proteomic studies focused on analysis of endometrial tissue which has the same limitations of changing cellular composition as those described for transcriptomics (reviewed in Salamonsen et al., 2013). Furthermore, the high abundance of structural proteins is also an issue as these can mask the lower abundance proteins during analysis. A more promising approach is the analysis of endometrial secretions, which can be accessed in uterine fluid (reviews Cheong et al., 2013; Edgell et al., 2013). Endometrial fluid collection is less invasive than endometrial biopsy. The fluid contains numerous secreted proteins associated with endometrial maturation and receptivity (Boomsma et al., 2009a, b Casado-Vela et al., 2009; Scotchie et al., 2009; Hannan et al., 2010; Hannan et al., 2011a, b). Furthermore, uterine secretions are less complex than tissue in terms of their protein repertoire and may provide a subset of biomarkers for functional endometrial disturbances. Although albumin and other plasma proteins make up some 90% of the total protein in uterine fluid, this can be removed (Hannan et al., 2009), enabling the lower abundance proteins to be more readily analysed. Uterine fluid can be harvested by either aspiration (provides ∼5 µl fluid for analysis) or lavage, which washes the endometrial surface, probably removing loosely attached proteins. A comparison of these two sampling methods demonstrated that although there were many similarities in protein profiles, the results differed somewhat between methodologies, both having advantages and disadvantages (Hannan et al., 2012). Therefore, consistency in the sampling technology is crucial to enable comparisons between data sets. Another potential non-invasive approach, namely analysis of the proteome of menstrual blood, has also been proposed for assessment of infertility and endometrial pathology (Yang et al., 2012).
Importantly, in the context of uterine fluid, is that many of the proteins proposed as biomarkers have been validated as produced locally by the endometrial epithelium following immunohistochemical staining of endometrial biopsies. Furthermore, their relative levels in uterine fluid represent changes in immunostaining intensity between the proliferative and secretory phases, and in mid-secretory phase in fertile versus infertile women (Hannan et al., 2010; Hannan et al., 2011a, b). In some cases, function within the embryo-maternal unit has also been defined (Hannan et al., 2011a, b), providing considerable confidence in these molecules as biomarkers.
Protein synthesis is the final result of gene expression (although not all mRNA expression leads to protein production) and is directly linked to the phenotype, holding high promise for biomarker discovery. However, post-translational modification including processing from latent to active forms, variable glycosylation and phosphorylation, are all common and require further examination. Such individual forms provide potential for unique endometrial markers.
The advent of new mass spectrometers, such as the LTQ Orbitrap Elite ETD, which is 100-fold more sensitive than earlier generation hardware (having an achievable ∼50 attomole peptide sensitivity), will lead to further developments in endometrial proteomics, as it will enable detection of the proteome in minute samples of protein. New techniques for pre-fractionation will further enhance sensitivity. Thus, the full potential of proteomics in endometrial research is yet to be achieved.
Metabolomics in human endometrium
The study of cellular metabolic products (metabolomics) as potential biomarkers lags behind the other ‘omics’ technologies discussed in this review. Metabolomics is the study of small, low-molecular weight products of metabolism (Nicholson and Lindon, 2008). Metabolomics reflects events well downstream of gene expression and is considered to be closer to the actual phenotype than genomics, transcriptomics or proteomics (Allen et al., 2003). The concentration of a specific metabolite is the cumulative effect of the activity of all enzymes involved in the synthesis and catabolism of a given compound, thereby having the potential to provide integrative information about tissue function within the larger context of the organism (Nielsen and Oliver, 2005). Although metabolomics should remain an integrated approach, its complexity leads to subdivisions, such as lipidomics and glycomics (Lagarde et al., 2003).
Two studies have been published on metabolomics in human endometrium, each novel in its focus (Vouk et al., 2012; Vilella et al., 2013). Vouk et al. (2012) provided the first report of a metabolomics approach to identification of biomarkers for the diagnosis of endometriosis from plasma (Vouk et al., 2012). Their data indicated that elevated levels of sphingomyelins and ether-phospholipids are associated with endometriosis, and eight lipids are presented as novel endometriosis-associated biomarkers (Vouk et al., 2012). Another recent study examined the lipidomics of human endometrium, demonstrating for the first time a significant increase of lipid levels in endometrial fluid at the window of implantation, which could provide a new tool for endometrial receptivity prediction (Vilella et al., 2013).
The high potential for metabolomics to unravel the genetics of human metabolism has been demonstrated (Illig et al., 2010); in contrast to most GWAS with clinically relevant end-points, most of the associations in metabolic traits were linked to genetic variants in genes with a matching metabolic function (Illig et al., 2010). If this is indeed the case, the use of ‘omics’ to understand the broad picture of the physiology needs to begin with an initial survey of the genomics of an individual/phenotype, allowing investigators to place in context any newly discovered metabolomic end-points.
Other ‘omics’ are awaiting elucidation in endometrial research, including microbiome in health and disease, glycome and exposome, to name a few. The integration of this information with the genome, epigenome, transcriptome, proteome and metabolome awaits a major effort of computational biology approaches and phenotypic assessment of patients with a variety of normal and abnormal presentations and hormonal milieu.
Study design in endometrial ‘omics’ studies
Study design is a crucial step in the general conduct of ‘omics’ studies. For the endometrium, the complex dynamic nature of the tissue response to the cyclic hormonal milieu makes the study design particularly important. The endometrium is composed of many cell types (epithelial, stromal fibroblasts or pre-decidual cells, leukocytes and cells of the vasculature), and there is considerable heterogeneity of cellular composition across the continuum of phases of the cycle. This is particularly the case for the epithelial compartments (luminal and glandular) and the numbers and subsets of leukocyte populations. This intrinsic variability needs to be considered in designing and analysing studies on endometrium (Salamonsen et al., 2009; Savaris and Giudice, 2009; Edgell et al., 2013). In addition, there could be inter-cycle variability; however, a recent study demonstrates consistency at the transcriptomic level between different biopsies from the same patient taken with an interval of 2 years (Diaz-Gimeno et al., 2013). Furthermore, pathological states of the endometrium must be considered, including the impact of structural (fibroids) or immune (endometriosis) alterations. Together with concurrent medication or exposures to environmental toxins as well as genetic susceptibility, all of these factors can alter ‘omics’ outcomes (Horcajadas et al., 2007; Savaris and Giudice, 2009). The importance of study design has previously been addressed in several reviews (White and Salamonsen, 2005; Horcajadas et al., 2007; Savaris and Giudice, 2009; Bellver et al., 2012; Ruiz-Alonso et al., 2012; Edgell et al., 2013) but we hereby briefly summarize the critical points.
First and foremost, researchers should be precise and consistent in defining the phenotype of the study population. Heterogeneity resulting from misclassifying participants decreases both sensitivity and power. The phenotypic heterogeneity across studies often makes it difficult to generalize study findings and replicate the results (Wu et al., 2013). While a clear phenotype definition could potentially reduce sample size, it will increase biological homogeneity and thus increase the statistical power (Gracie et al., 2011). For endometriosis, this problem of heterogeneity is an inherent problem. Half of women with this disease are infertile, while the other half are not. Many have pain that is unrelated to the extent of disease. Endometriosis may also not be diagnosed if it is symptom free. Once treated, the eutopic endometrial changes seen in the endometrium may revert to normal, while selection of patients ahead of laparoscopic diagnosis can be equally problematic. Endometriosis appears to present as mild and severe disease, with each having different genetic risk factors (Nyholt et al., 2012). Medications used to treat this disease may alter the progress and therefore the diagnostic biomarkers that ‘omics’ studies are attempting to establish. Further, the phenome of normal patients and those with endometrial disorders is important in interpreting the obtained data. Vigano et al. (2012) have pioneered this approach (Vigano et al., 2012), along with the World Endometriosis Research Foundation, which held a global consensus meeting on the endometriosis phenome (World Endometriosis Research Foundation: Endometriosis Phenome and Biobanking Harmonisation Project (EPHect), March 2013, http://endometriosisfoundation.org/ephect/). Although it is important to carefully define and phenotype cases with a trait, disease or susceptibility, it is as important to define adequately ‘normal’ controls (Savaris and Giudice, 2009; Edgell et al., 2013).
Other sources of biological variability can arise through differences in tissue composition of collected samples, phase or day of cycle, frequency of sample collection, medical history and others (Ulbrich et al., 2013). In endometrial tissue analysis, endometrial biopsy sampling using two different techniques, biopsy and curetting hysterectomy, resulted in identical results in transcriptome expression level (Talbi et al., 2006). A biopsy taken in 1 month may affect endometrial receptivity in the next biopsy (Barash et al., 2003). Nevertheless, the timing of the biopsy sampling is critical, as its composition varies considerably in a constantly changing endometrium (Giudice, 1999; Mirkin et al., 2005; Haouzi et al., 2009; Fassbender et al., 2010).
Histological dating of the endometrium is another critical aspect of studies on human endometrium. Although the classical histological dating by Noyes criteria has several limitations, it has remained the gold standard for establishing landmarks of endometrial development. Hormonal reference of the endometrium in natural cycles using urinary LH measurement or, even better, serum LH detection that has superior precision, is important (Evans et al., 2012a, b). In endometrial studies, the sample collection conditions, including the location and timing with respect to day of the cycle, should be as uniform as possible with adoption of standard operating procedures for tissue acquisition, processing, utilization, storage and distribution (Sheldon et al., 2011).
Race and ethnicity are also important. So far, ethnicity has been considered mainly in genetic/genomics studies but it applies to all ‘omics’ disciplines. Genetic variation may differ among racial/ethnic groups, which may influence the results at all ‘omics’ levels, a concept known as population stratification (see Figure 2 for the complexity of biological processes). This can lead to excess false-positive results and failure to detect true associations (Wu et al., 2013). Therefore, the studies should focus as far as possible on a single race/ethnic group or apply advanced analytic techniques for adjusting for ethnic genetic differences (Bryc et al., 2010). At the very least, these factors should be included in databases in the hopes that any differences may be identified in subsequent meta-analysis.
Figure 2.
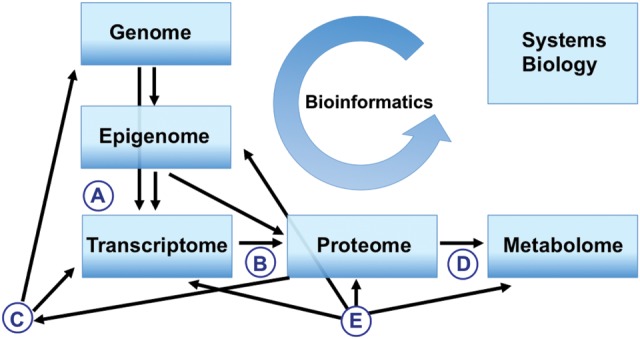
The complexity of ‘omics’ fields in biological processes that contribute to the study and understanding of biological systems. There are more than 25 000 genes in the human genome, encoding ∼100 000–200 000 transcripts and 1 million proteins, whereas there are as few as 2500–3000 metabolites that make up the human metabolome (Botros et al., 2008). The genome is essentially invariant among cells and tissues, while the epigenome has a low/moderate temporal variance and influences both transcriptome and proteome. The transcriptome has a high temporal variance and is translated into the proteome differentially in different tissues and physiological states, affecting the metabolome in a tissue-specific manner. This ‘simple’ model is modulated by multiple factors: (A) differential splicing that can be affected by the proteome; (B) post-translational modification of proteins; (C) transcription factor binding; (D) receptor ligand binding and (E) environmentally induced factors (adapted from Gracie et al. 2011; Bellver et al. 2012).
Sample size is critical to good study design, as discussed in detail in a prior review (Savaris and Giudice, 2009). Calculation of sample size demonstrates the power of the study, which takes into account the variance of individual measurements, the acceptable false-positive rate and the desired discriminatory power of the used platform (Savaris and Giudice, 2009). Researchers are strongly encouraged to maximize the sample size in their ‘omics’ studies. Collaboration between groups offers the optimal solution for this inherent problem in our field, as the sample collection is in general invasive and thus it can be difficult to motivate participants and clinicians to participate. Clear examples of adequately powered collaborative studies, where a large sample size was analysed and independent replicates performed, have resulted in strong candidate genes for endometriosis (Painter et al., 2011; Nyholt et al., 2012).
Although many significant results have been derived from ‘omics’ studies in the last decade, most would agree that many studies exhibit an unacceptably large degree of variability with low reproducibility. There is a need for better documentation and uniformity of the types of data collected. Groups of scientists have been working to establish standards of minimum information that must be collected and reported, to ensure the interpretability of the experimental results generated using ‘omics’ technologies (Brazma et al., 2001). These reporting standards include MIAME (Minimum Information about a Microarray Experiment), MIAPE (Minimum Information about a Proteomics Experiment), MIGS-MIMS (Minimum Information about a Genome/Metagenome Sequence), MIMIx (Minimum Information about a Molecular Interaction eXperiment), MINIMESS (Minimal Metagenome Sequence Analysis Standards), MINISEQE (Minimum Information about a high-throughput Nucleotide Sequencing Experiment) and CIMR (Core Information for Metabolomics Reporting), which have been discussed in detail in previous reports (Taylor et al., 2008; Chervitz et al., 2011). Furthermore, consideration of the complexity of human endometrial physiology in health and disease and additional documentation of the data collected are urgently required. Indeed, in a study evaluating the replication of data analyses in 18 articles on microarray-based gene expression profiling published in Nature Genetics in 2005–2006, only two analyses could be fully reproduced and six partially, while ten could not be reproduced (Ioannidis et al., 2009). The conclusion was that the main reason for lack of reproducibility was lack of access to data and discrepancies due to incomplete data annotation or specification of data processing and analysis. Therefore, more strict publication rules enforcing public data availability should be encouraged (Ioannidis et al., 2009). Table I highlights the points to consider for ‘omics’ studies on endometrium, and scientists in the field are encouraged to follow these guidelines when designing, conducting and reporting their projects.
Table I.
Points to consider for adequate study design and ‘good-reporting-practice’ in studies of human endometrium.
| Experimental design |
| Set the study hypothesis |
| Define study type (e.g. prospective, retrospective) |
| Precisely define phenotype of participants |
| Carefully select and describe controls |
| Calculate sample size and power |
| Provide adequate participant data (age, cycle characteristics, BMI, race/ethnicity, parity, obstetric and gynaecological history including family history of gynaecological complications/pathologies, hormonal profiles and other measured markers, medication including contraceptives) |
| Assess endometrial phase (histology, biomarkers) |
| Assess environmental exposure (tobacco, alcohol, drugs, nutritional status, socioeconomic status, education, psychological stress) |
| Identify risk factors and possible confounders |
| Design patient informed consent with the potential for possible international data sharing and complex integrated data analyses |
| Sample collection and preparation |
| Define and record sampling conditions (biopsy location, time) |
| Provide detailed protocol for sample processing and storage |
| Add biological duplicates for replication purposes (e.g. repeated sampling) |
| Avoid pooling of samples |
| Assess sample quality and quantity |
| Sample analysis |
| Provide detailed protocol for ‘omics’ technology to be applied |
| Consider technical duplicates |
| Define statistical methods, databases to be utilized for data analysis |
| Data validation |
| Validate results using alternative technologies (quantitative PCR, western blot, immunohistochemistry, in situ hybridization, etc.) |
| Data presentation |
| Upload raw ‘omics’ data and detailed sample/analysis data to public database (e.g. GEO, ArrayExpress) |
| Address limitations/strengths of the study |
Sample collection and processing
Sample collection for ‘omics’ technologies requires separate discussion of the considerations, obstacles and detailed protocols involved. For research in human endometrium, the biological sample collected is determined by the research question of interest, as a variety of sample types can be analysed (see Table II). Quality of the sample is determined at various steps in the process of collection and storage and is one of the most important factors in the overall success or failure of a study. Sampling, processing and preservation of study material should be carefully established and documented by standard operating procedures, a priori; the consistency in tissue handling should be closely followed (Huang et al., 2001; Spruessel et al., 2004; Shayeghi et al., 2005; Micke et al., 2006), to assure the highest-quality samples, representing as closely as possible the physiology of the original in vivo tissue.
Table II.
Sample processing for ‘omics’ studies in human endometrium (adapted from Gracie et al., 2011).
| ‘Omics’ platform | Sample | Considerations |
|---|---|---|
| Genomics (DNA) |
|
Stable at room temperature but best if refrigerated or frozen |
| Epigenomics (DNA, RNA) |
|
Require proper collection, meticulous processing and sample storage at −80°C to ensure sample integrity |
| Transcriptomics (RNA) |
|
Require appropriate collection, careful processing and storage at -80°C to ensure RNA integrity and quality for analysis |
| Proteomics (proteins) |
|
Require rapid sample preparation and preservation (±protease inhibitors) prior to storage at −80°C to prevent non-specific protein degradation |
| Metabolomics (metabolites) |
|
Require rapid metabolic ‘quenching’ (flash freezing or acid precipitation) to prevent degradation of metabolites |
Sample processing for different ‘omics’ techniques has been previously discussed in detail (White and Salamonsen, 2005; Horgan et al., 2009; Savaris and Giudice, 2009; Gunaratne et al., 2010; Robert, 2010; Pritchard et al., 2012; Slattery et al., 2012; Fassbender et al., 2013; Ulbrich et al., 2013). In terms of study design and sample collection, researchers are encouraged to take careful note of these guidelines. In Table II, we provide an outline of the ‘omics’ techniques and respective sample sources that can be utilized in endometrial studies.
Data processing and analysis
Adequate data analysis is always crucial for providing conclusive results, which can guide us to an appropriate interpretation of the data, in terms of both biological sense and clearly defined target clinical applications. Several excellent papers provide both protocols and instructions for data analysis for each ‘omic’ (see detailed references below, and general standards for ‘omic’ data description, exchange, terminology and experimental execution have also been described; Chervitz et al., 2011). The aim of these data standards is not only to improve reproducibility and to avoid discrepancies but also to determine a true clinical application. Thus, researchers are encouraged to read and follow such standards (Chervitz et al., 2011). In this section, an overview of the analysis and interpretation of ‘omics’ data provides some specific quality criteria details for each ‘omic’, with a special focus on human endometrium (see Table III for summary of data processing and analysis). The detailed procedures are thoroughly described in the cited literature.
Table III.
Data processing and analysis for ‘omics’ studies.
| Genomics | Epigenomics | Transcriptomics | Proteomics | Metabolomics | |
|---|---|---|---|---|---|
| Preprocessing/analysis | Calling; Standard deviation of the intensity ratios (DLR for CGH; MAPD for SNPs) | Background correction; quality control; cleaning and transformation | Background correction; quality control; cleaning and transformation | Extraction/derivation | Reproducible spectra |
| Data processing | Normalization (median; lowess) | Normalization (T-quantiles; VSN for miRNAs) | Normalization (quantiles for expression) | Alignment; baseline correction; peak deconvolution and identification; normalization; scaling | Chemometric software |
| Statistical analysis | Exploratory (PCA, clustering); parametric/non-parametric test; multiple hypothesis testing (FDR correction) | ||||
| Biological interpretation | Reference baseline library (for SNPs) | Gene enrichment functional analysis; gene targets (miRNAs); Gene networks; validation | Isoform and functional analysis; protein–protein interaction networks; validation | Metabolic networks; validation | |
CGH, comparative genome hybridization; DLR, derivative log ratio; FDR, false discovery rate; MAPD, median absolute pairwise difference; miRNA, microRNA; PCA, principal component analysis; SNPs, single nucleotide polymoprhisms; VSN, variance stabilizing normalization.
Genomics
For genomic microarrays, gains and losses of chromosomal regions can be detected after measuring the signal intensity ratio of labelled patient DNA hybridized to reference DNA with known genomic co-ordinates, the so-called array-based CGH (array CGH). Different types of genomic microarray technologies now exist (Brady and Vermeesch, 2012). Although SNP arrays were originally designed to detect common SNPs in GWAS, SNP platforms can also be used to ascertain the occurrence of CNVs. In SNP, arrays single channel signal intensities of patient DNA are obtained and compared with a reference dataset. On the other hand, CGH arrays are based on competitive hybridization of both patient and reference DNA samples (dual channel) to the same targets. In both cases, specialized software, either provided by the manufacturer or from other free or commercially available platforms, is used to obtain and assign the signal intensity data from the scanned array image to each target probe. For data analysis, different methods for normalization, segmentation and calling can be chosen, which may influence the final results (Pinto et al., 2011; Brady and Vermeesch, 2012). The reference baseline library used for SNP arrays is also important, with large in-house reference datasets improving the quality of the results.
Even though different somatic copy number aberrations (SCNAs) in endometriotic foci detected with CGH technology can be found in the literature, SNP genotyping arrays in human endometrial research are scarce. Recently, and as a good example of experimental design and appropriate technology and data analysis, no endometriosis specific de novo SCNAs, or regions of copy-neutral loss of heterozygosity (cn-LOH), were found in eutopic or ectopic endometrium (Saare et al., 2012). This array study applied the advantages of SNP targets, which enable the detection of cn-LOH from the common B-allele frequency measurement obtained with this technology. In addition, given the variability between calling algorithms, two different programs were used to minimize the number of false discoveries, providing robust results.
Epigenomics
The emerging science of epigenetics and genomics, coined epigenomics, is providing unique opportunities for the detection of heritable changes due to modifications in the DNA or chromatin that does not include alterations in DNA sequence. These modifications include DNA methylation and hydroxymethylation, chromatin remodelling, histone modifications (methylation, acetylation, ubiquitylation, phosphorylation and sumoylation) and gene regulation by non-coding RNAs (including miRNAs) (Weichenhan and Plass, 2013). The most studied epigenetic modifications at ‘omics’ level (miRNAs and DNA methylation) are also based on microarray platforms, although the substrates, pre-processing and data analysis depend on the modification to be studied (Callinan and Feinberg, 2006). A typical sequence for epigenomic array data analysis includes background correction, quality control (checking for positive and negative controls), data preprocessing (data cleaning and transformation), normalization and statistical analyses (exploratory and differential expression tests), together with appropriate further target validation (Deatherage et al., 2009), following similar quality criteria as described for other ‘omics’, but selecting specific algorithms, mainly related to the normalization method (Adriaens et al., 2012) depending on the platform used (Marabita et al., 2013).
Epigenomics also comprises the study of small and large non-coding RNAs. Studies on miRNA expression in both health and disease are widely carried out using microarray technology (Yin et al., 2008). While the general protocol for data analysis is similar to that for mRNA gene expression microarrays, the different nature of mRNA and miRNA expression experiments needs careful selection of the normalization method and quality assessment of data analysis (Sarkar et al., 2009). Such a miRNA analysis has been described for these small regulators in human endometrial receptivity (Altmäe et al., 2013).
Transcriptomics
Global gene expression analysis, functional genomics and transcriptomics are synonyms for describing and quantifying the set of mRNAs present in a given cell population or tissue at any point in time, and are mainly used to compare the global gene expression between different experimental or biomedical situations. Gene expression microarrays enable development of datasets that include the expression levels of all the genes of the genome just in one experiment. Once the raw data are obtained, preprocessing, normalization, statistical analysis for obtaining the differential gene expression between situations, multivariate data exploration and gene enrichment functional analyses are included in well-established protocols that have been described comprehensively (Cordero et al., 2007; Mocellin and Rossi, 2007; Weeraratna and Taub, 2007; Durinck, 2008; Suarez et al., 2009; Zhang et al., 2009). The need for an appropriate annotation of both the experimental approach and data analysis is a critical quality criterion for gene expression microarray data sharing, reanalysis and comparison. A particularly well-conducted initiative is the multidisciplinary EMERALD project that, based on MIAME standards, provides the quality metrics and even tools and platforms at any step of the microarray data, always aspiring to appropriate further model validations and practical clinical applications (Beisvag et al. 2011). Also, in terms of optimization and interpretation of gene expression data from different platforms, recent global gene expression analysis (Loven et al., 2012) has demonstrated that common assumptions (such as that cells produce similar levels of RNA per cell) led to erroneous interpretations which can be solved using the appropriate controls.
The transcriptomics of the human endometrium has been thoroughly reviewed (Ruiz-Alonso et al., 2012). This includes both the endometrial phase-specific transcriptomic gene profiles and common temporal gene expression patterns, also taking into account the necessary quality criteria. In addition, examples of endometrial transcriptomics as a diagnostic tool are provided.
Proteomics
The complexity of the proteome, and its functional interpretation, is one of the current challenges in biology (Cox and Mann, 2011; Matthiesen et al., 2011). Proteomic analyses are mainly based on high-throughput MS and protein array technologies and, as for other ‘omics’, the standards for data analysis, sharing and integration need to be carefully considered (Becnel and McKenna, 2012). Since protein array data analysis is adapted from gene expression microarrays (Sundaresh et al., 2006) the quality criteria described above are also valid for protein arrays. However, the uniquely quantitative nature of MS-based proteomics involves characteristic analysis with particular challenges, the most important being the peptide feature detection and quantification from the raw map (Cappadona et al., 2012). Rigorous methods for the assessment of quality of spectral data are also described (Cairns et al., 2008). Raw spectra processing methods and common data analysis strategies have been discussed recently (Matthiesen et al., 2011). Importantly, the ‘International Workshop on Proteomic Data Quality Metrics’ in 2010, identified and addressed issues regarding the development and use of open access proteomics data. The key principles underlying a framework for data quality assessment in MS data were enumerated and included both the need for an evolving list of comprehensive quality metrics and also standards accompanied by software analytics (Kinsinger et al., 2012). A revision of the bioinformatics analysis of qualitative and quantitative proteomic data has also been published (Kumar and Mann, 2009). To support the publication of MS studies, the proteomics identifications database repository includes a curation pipeline acting as a practical data deposition quality control (Csordas et al., 2012; Vizcaino et al., 2013).
Metabolomics
Metabolomics, the high-throughput identification of the general profile of metabolites in a system, shares with proteomics the use of MS methodology (Horgan et al., 2009). MS together with H nuclear magnetic resonance spectroscopy (NMR) is the most commonly applied techniques in metabolic profiling, with evaluation of different acquisition schemes generating reproducible spectra (high-quality NRM data) in different sample conditions. Data can be further assessed with automated processing (Aranibar et al., 2006). A comparison of the different metabolomics technologies and a detailed description of the spectral and statistical analysis tools in metabolic profiling studies can be found elsewhere (Wishart, 2010). Metabolites which are detected at different levels between control and disease samples are generally designated as biomarkers but they are not usually validated for practical clinical applications and their potential usefulness in the clinic needs careful consideration (Koulman et al., 2009).
Validation of biomarker sets
It is obvious that any set of biomarkers needs considerable validation to ensure that it differentiates between the physiologically or pathologically different groups of interest. Commonly, only very small sample sets are analysed and thus the risk of a type 1 error is significant. Independent validation of proposed markers in additional sets of clinical material is essential, preferably in a range of laboratories in different countries to allow for ethnic and environmental influences. To date, only a limited number of laboratories have published data on biomarkers in endometrium, most of these being in communities that are predominantly Caucasian. Multi-site collection and validation are essential to prove that any biomarker set is robust. Importantly, the patient groups tested need to be as uniform as possible; for example, in terms of exclusions (steroidal contraception, endometrial disorders not under study) or the stimulation protocols in an IVF cycle. As noted above, collaboration to achieve these requirements is essential. Other aspects of validation are discussed in detail in Edgell et al. (2013) and Dominguez et al. (2009).
Western blotting has been the method of choice for validation of individual proteins identified as potential biomarkers. However, accurate protein quantification by Western blotting presents a challenge, at least in part as it is dependent on the form of the protein recognized by the antibody selected for use. Recently, reliable MS-based methods to quantify proteins, known as selected reaction monitoring or multiple reaction monitoring, have become well established (Aebersold et al., 2013; Editorial, 2013) and are now easily developed for essentially any protein. Since this method outperforms Western blotting in terms of limit of detection, linear dynamic range, ability to multiplex and reproducibility, it is now clear that this must become the ‘gold standard’ for validation.
A recent development for validation of the cellular localization of any protein is matrix assisted laser desorption imaging mass spectrometry profiling which can establish molecular disease signatures in their histological context: essentially removing the need for immunohistochemistry with its dependence on antibodies. This has been applied to examination of mouse implantation sites (Burnum et al., 2008), for cervical cytology specimens (Schwamborn et al., 2011) and for serous ovarian cancer (Longuespee et al., 2013). It is likely also to prove useful in the context of validation of biomarkers in an endometrial setting.
Systems biology in integrative endometrial ‘omics’ studies
The integration of ‘omics’ techniques is called ‘systems biology’. This captures information from genomics, epigenomics, transcriptomics, proteomics, metabolomics, etc., and its combination with theoretical models for predicting the behaviour of a cell, tissue or organism (Figure 2).
The global analysis of the genome and the morphofunctional characterization of proteins and metabolites provide a vast amount of information with a very high potential to unravel the complex interaction of molecular networks underlying the function of any organism in health and disease. Thus, systems biology provides an integrative approach to understanding biology, entailing the functional analysis of the structure and dynamics of cells and focusing on complex interactions, rather than the characteristics of the isolated components of biological systems. The challenge of systems biology resides in the compilation of data derived from very different areas: biology, chemistry, statistics, physics, mathematics and computational engineering. As systems biology attempts to provide a comprehensive interpretation of all this knowledge, the high-throughput ‘omics’ platforms have to be integrated for the analysis, display and recording of information to guarantee compatibility and accessibility to these data sets (Chervitz et al., 2011).
Because systems biology approaches are focused on the global analysis of multiple interactions at different levels, the main strategy usually employs networks as a representation of interacting molecules. Modules are built around each discrete regulated function, with the interrelations among modules finally arising as complex networks. Thus, the process begins with a model based on sets of data, and conclusions are obtained when the experimental data and the model are juxtaposed (Weston and Hood, 2004).
From a pathophysiological point of view, the analysis of ‘-omics’ under a systems biology strategy could be widely used in gene finding, biomarker identification, normal endometrial physiology, endometrial disease classification, disease recurrence, drug discovery, therapy strategies and, in the last instance, predictive and preventive medicine. The first systems biology approach to the complex molecular network of the implantation process in humans has recently been described (Altmäe et al., 2012), in which embryonic and endometrial transcriptomic profiles were integrated with protein–protein interactions. The network included proteins, interaction modules and pathways that were activated within both the preimplanting blastocyst and the receptive endometrium and thus characterized the molecular network of the embryo-endometrium implantation interface (Figure 3). The methodology presented could inspire new analytical approaches to unravel complex networks in human endometrial physiology and pathophysiology. However, the authors acknowledge the limitation of microarray technology, with its focus on a static snapshot analysis of a dynamic process, and its unilateral analysis of either endometrium or in vitro cultured embryo. It is now well recognized that the endometrium is responsive to both the preimplantation and implanting embryo. In a very elegant in vivo study in women (Licht et al., 2001) demonstrated a clear responsiveness of the endometrium to infused hCG, which included changes in both vascular endothelial growth factor and LIF concentrations in the endometrium. This was further verified in primary endometrial epithelial cells, which responded to hCG by increased secretion of six cytokines/chemokines known to increase during the receptive phase and to be important for implantation (Paiva et al., 2011), indicating that embryo-derived factors can enhance endometrial epithelial receptivity. The decidualized endometrial stromal cells are also responsive to an implanting blastocyst, migrating around the embryo rather than simply being invaded by the embryo (Weimar et al., 2012), and have been proposed as ‘sensors’ for embryo quality (Teklenburg et al., 2010). While studies to assess such responsiveness using homologous co-cultures or perfusion studies are useful for analysing the specific components of the endometrium and their response to the embryo, all models are limited in their ability to represent the in vivo situation (Teklenburg and Macklon, 2009).
Figure 3.

High-confidence embryo-endometrium interaction network derived from protein–protein interaction data and literature curation. Node colour represents tissue-specific differential gene expression: blue, expressed in embryo; red, expressed in endometrium; grey, expressed in both tissues. The biggest interaction network highlights the importance of cell adhesion molecules, where integrins, collagens and laminins are present. The second largest interaction network represents proteins involved in cytokine–cytokine receptor interactions, where osteopontin, apolipoprotein D, leptin (LEP) and leukaemia inhibitory factor (LIF) pathways intertwine (from Altmäe et al., 2012; published with permission from Molecular Endocrinology).
Clearly the mass of data generated within ‘omics’ studies is far from being fully utilized. Over 1 million gene expression data sets are publicly available in repositories (Gene Expression Omnibus, GEO and ArrayExpress) but few scientists fully utilize this data to find new information (Baker, 2012); rather they use only a small set of the data to compare with their own findings (Baker, 2012). In human endometrium, only two studies apply a bioinformatic approach for ‘omics’ data mining (Tapia et al., 2011; Zhang et al., 2012). Zhang et al. (2012) analysed 45 microarrays from three independent studies of endometrial receptivity in the GEO database, and identified a series of potential biomarkers of endometrial receptivity. Tapia et al. (2011) used array data from seven different studies, comparing endometrial gene expression profiles from the proliferative versus mid-secretory phase, and from early secretory versus mid-secretory phase, and etected new transcription factors orchestrating human endometrial receptivity. These two studies open a promising new era for biomarker search in human endometrial physiology in health and disease. The challenge for the future will be analyse huge sets of data simultaneously, taking advantage of the existing data in publicly available databases for raising power, credibility and reliability of findings.
Conclusions and future perspectives
‘Omics’ high-throughput analyses have started to revolutionize our understanding of human endometrial physiology and pathophysiological conditions. Nevertheless, our understanding of the complex phenotypes of endometrial physiology, endometrial disorders and fertility complications remains incomplete, inconsistent and without strong clinical application.
This review has summarized the current status and trends in ‘omics’ technologies applied to human endometrium. While significant advances have been made in assessing endometrial receptivity and discovering potential biomarkers for endometriosis and receptive endometrium, many of the ‘omics’ studies have not been replicated and their practical value has been limited, without translation into clinical practice. In addition to sufficiently powered studies (i.e. larger sample size), there is a growing need for integrated approaches to investigate complex phenotypes across ‘omics’ categories. Most studies to date (i) have analysed a relatively small sample size and (ii) assessed a single level of ‘omics data in isolation, emphasising the need for highly powered studies and integrated ‘omics' approaches as a future path for research. For that, universal guidelines should be established so that data and sample collections can be merged, compared, validated and replicated.
We provide here a set of guidelines that we encourage are followed for conducting transparent and well-designed studies, and providing ‘good-reporting-practice’ in studies of the endometrium, as well as other biological systems. Central to these studies should be accurate phenotype definition, the choice and quality of the sample and adequate sample size. This necessitates collaboration among multiple research groups and the use of high-quality biological samples which have been collected and using consistent and precise approaches. This will provide international datasets that are transparent (Gracie et al., 2011) and will enable researchers to address unanswered questions and validate their results. We encourage scientists to create and join integrated databases and multicentre international networks, to advance the knowledge and potential biomarker search in health and disease conditions related to human endometrium. The World Endometriosis Research Foundation recent EPHect effort is a good example of phenotyping patients with an endometrial disorder. There are multiple other opportunities for ‘omics’ studies on endometrium in this regard.
Although the ‘omics’ technologies have high potential to deliver, there are still important technical limitations and constraints, including data analysis, that need further development. In research of human endometrium, one important technical consideration is the analysis of very small amounts of sample material. While there are a range of different commercial kits for sample material amplification, where linear amplification is assumed, it is well known that amplification creates errors (Vanneste et al., 2012). Furthermore, the trend towards analysis of endometrial compartments in isolation for understanding better the function of each compartment, requires prior sample amplification or FACS-isolated endometrial cell populations, or highly sensitive analytical tools such as are emerging for proteomic analyses. Moreover, technical advances are towards high-throughput single-cell analysis, which will better characterize cell populations and provide spatiotemporal resolution in systems biology (Mannello et al., 2012). However, next-generation single-cell analysis technology is still in its infancy, with a plethora of artefacts remaining (Prof. Thierry Voet, personal communication). Thus, it is important to be aware of and acknowledge the limitations of current technologies, while trusting that future developments will overcome these shortcomings.
Thus, the future of the endometrial ‘omics’ field lies in well designed, sufficiently powered studies together with the application of new-generation technologies, complex data analyses and integrated systems biology approaches. Provision of integrated databases and multicentre collaboration will enable new insights, and provide valid and reliable biomarkers. We are on the threshold of realizing the promise of the ‘omics’ technologies in endometrial research.
Supplementary data
Supplementary data are available at http://humupd.oxfordjournals.org/.
Authors' roles
Performed thorough literature search: S.A. Wrote the main body of the manuscript: S.A., F.J.E. Contribution to manuscript writing and editing: A.S.E., C.S., L.G., B.A.L., J.A.H., N.M., T.D.'H., C.C., B.C.F., L.A.S., A.S. Final editing: L.A.S.
Funding
This research was funded by Estonian Ministry of Education and Research (grant SF0180044s09); Enterprise Estonia (grant EU30020), EU-FP7 Eurostars Program (grant NOTED, EU41564) and EU-FP7 IAPP Project (grant SARM, EU324509); Marie Curie post-doctoral fellowship (FP7, no 329812, NutriOmics); Spanish Ministry of Education (Grant no. SB2010-0025); grant from Junta de Andalucía (BIO-302); the NHMRC of Australia (#1002028, #494802), The Monash IVF Education and Research Foundation and the Victorian Government's Operational Infrastructure Program;and the National Institutes of Health, Eunice Kennedy Shriver National Institute of Child Health and Human Development.
Conflict of Interest
None declared.
Supplementary Material
References
- Adachi S, Tajima A, Quan J, Haino K, Yoshihara K, Masuzaki H, Katabuchi H, Ikuma K, Suginami H, Nishida N, et al. Meta-analysis of genome-wide association scans for genetic susceptibility to endometriosis in Japanese population. J Hum Genet. 2010;55:816–821. doi: 10.1038/jhg.2010.118. [DOI] [PubMed] [Google Scholar]
- Adriaens ME, Jaillard M, Eijssen LM, Mayer CD, Evelo CT. An evaluation of two-channel ChIP-on-chip and DNA methylation microarray normalization strategies. BMC Genomics. 2012;13:42. doi: 10.1186/1471-2164-13-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aebersold R, Burlingame AL, Bradshaw RA. Western Blots vs. SRM Assays: Time to turn the tables? Mol Cell Proteomics. 2013 doi: 10.1074/mcp.E113.031658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aghajanova L, Hamilton AE, Giudice LC. Uterine receptivity to human embryonic implantation: histology, biomarkers, and transcriptomics. Semin Cell Dev Biol. 2008a;19:204–211. doi: 10.1016/j.semcdb.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aghajanova L, Simón C, Horcajadas JA. Are favorite molecules of endometrial receptivity still in favor? Expert Rev Obstet Gynecol. 2008b;3:487–501. [Google Scholar]
- Allen J, Davey HM, Broadhurst D, Heald JK, Rowland JJ, Oliver SG, Kell DB. High-throughput classification of yeast mutants for functional genomics using metabolic footprinting. Nat Biotechnol. 2003;21:692–696. doi: 10.1038/nbt823. [DOI] [PubMed] [Google Scholar]
- Altmäe S, Martinez-Conejero JA, Salumets A, Simon C, Horcajadas JA, Stavreus-Evers A. Endometrial gene expression analysis at the time of embryo implantation in women with unexplained infertility. Mol Hum Reprod. 2010;16:178–187. doi: 10.1093/molehr/gap102. [DOI] [PubMed] [Google Scholar]
- Altmäe S, Salumets A. A novel genomic diagnostic tool for sperm quality? Reprod Biomed Online. 2011 doi: 10.1016/j.rbmo.2010.12.009. [DOI] [PubMed] [Google Scholar]
- Altmäe S, Hovatta O, Stavreus-Evers A, Salumets A. Genetic predictors of controlled ovarian hyperstimulation: where do we stand today? Hum Reprod Update. 2011;17:813–828. doi: 10.1093/humupd/dmr034. [DOI] [PubMed] [Google Scholar]
- Altmäe S, Reimand J, Hovatta O, Zhang P, Kere J, Laisk T, Saare M, Peters M, Vilo J, Stavreus-Evers A, et al. Research resource: interactome of human embryo implantation: identification of gene expression pathways, regulation, and integrated regulatory networks. Mol Endocrinol. 2012;26:203–217. doi: 10.1210/me.2011-1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altmäe S, Martinez-Conejero JA, Esteban FJ, Ruiz-Alonso M, Stavreus-Evers A, Horcajadas JA, Salumets A. MicroRNAs miR-30b, miR-30d, and miR-494 Regulate Human Endometrial Receptivity. Reproduct Sci. 2013;20:308–317. doi: 10.1177/1933719112453507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranibar N, Ott KH, Roongta V, Mueller L. Metabolomic analysis using optimized NMR and statistical methods. Anal Biochem. 2006;355:62–70. doi: 10.1016/j.ab.2006.04.014. [DOI] [PubMed] [Google Scholar]
- Assou S, Haouzi D, De Vos J, Hamamah S. Human cumulus cells as biomarkers for embryo and pregnancy outcomes. Mol Hum Reproduct. 2010;16:531–538. doi: 10.1093/molehr/gaq032. [DOI] [PubMed] [Google Scholar]
- Baker M. Gene data to hit milestone. Nature. 2012;487:282–283. doi: 10.1038/487282a. [DOI] [PubMed] [Google Scholar]
- Barash A, Dekel N, Fieldust S, Segal I, Schechtman E, Granot I. Local injury to the endometrium doubles the incidence of successful pregnancies in patients undergoing in vitro fertilization. Fertil Steril. 2003;79:1317–1322. doi: 10.1016/s0015-0282(03)00345-5. [DOI] [PubMed] [Google Scholar]
- Becnel LB, McKenna NJ. Minireview: progress and challenges in proteomics data management, sharing, and integration. Mol Endocrinol. 2012;26:1660–1674. doi: 10.1210/me.2012-1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beisvag V, Kauffmann A, Malone J, Foy C, Salit M, Schimmel H, Bongcam-Rudloff E, Landegren U, Parkinson H, Huber W, et al. Contributions of the EMERALD project to assessing and improving microarray data quality. Biotechniques. 2011;50:27–31. doi: 10.2144/000113591. [DOI] [PubMed] [Google Scholar]
- Bellver J, Mundi M, Esteban FJ, Mosquera S, Horcajadas JA. ‘-OMICs’ technology and human reproduction: reproductomics. Expert Rev Obstet Gynecol. 2012;7:493–506. [Google Scholar]
- Berlanga O, Bradshaw HB, Vilella-Mitjana F, Garrido-Gomez T, Simon C. How endometrial secretomics can help in predicting implantation. Placenta. 2011;32(Suppl. 3):S271–275. doi: 10.1016/j.placenta.2011.06.002. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya SN, Habermacher R, Martine U, Closs EI, Filipowicz W. Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell. 2006;125:1111–1124. doi: 10.1016/j.cell.2006.04.031. [DOI] [PubMed] [Google Scholar]
- Boomsma CM, Kavelaars A, Eijkemans MJ, Amarouchi K, Teklenburg G, Gutknecht D, Fauser BJ, Heijnen CJ, Macklon NS. Cytokine profiling in endometrial secretions: a non-invasive window on endometrial receptivity. Reprod Biomed Online. 2009a;18:85–94. doi: 10.1016/s1472-6483(10)60429-4. [DOI] [PubMed] [Google Scholar]
- Boomsma CM, Kavelaars A, Eijkemans MJ, Lentjes EG, Fauser BC, Heijnen CJ, Macklon NS. Endometrial secretion analysis identifies a cytokine profile predictive of pregnancy in IVF. Hum Reprod. 2009b;24:1427–1435. doi: 10.1093/humrep/dep011. [DOI] [PubMed] [Google Scholar]
- Borghese B, Barbaux S, Mondon F, Santulli P, Pierre G, Vinci G, Chapron C, Vaiman D. Research resource: genome-wide profiling of methylated promoters in endometriosis reveals a subtelomeric location of hypermethylation. Mol Endocrinol. 2010;24:1872–1885. doi: 10.1210/me.2010-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botros L, Sakkas D, Seli E. Metabolomics and its application for non-invasive embryo assessment in IVF. Mol Hum Reprod. 2008;14:679–690. doi: 10.1093/molehr/gan066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady PD, Vermeesch JR. Genomic microarrays: a technology overview. Prenat Diagn. 2012;32:336–343. doi: 10.1002/pd.2933. [DOI] [PubMed] [Google Scholar]
- Brazma A, Hingamp P, Quackenbush J, Sherlock G, Spellman P, Stoeckert C, Aach J, Ansorge W, Ball CA, Causton HC, et al. Minimum information about a microarray experiment (MIAME)-toward standards for microarray data. Nat Genet. 2001;29:365–371. doi: 10.1038/ng1201-365. [DOI] [PubMed] [Google Scholar]
- Brinsden PR, Alam V, de Moustier B, Engrand P. Recombinant human leukemia inhibitory factor does not improve implantation and pregnancy outcomes after assisted reproductive techniques in women with recurrent unexplained implantation failure. Fertil Steril. 2009;91:1445–1447. doi: 10.1016/j.fertnstert.2008.06.047. [DOI] [PubMed] [Google Scholar]
- Bryc K, Auton A, Nelson MR, Oksenberg JR, Hauser SL, Williams S, Froment A, Bodo JM, Wambebe C, Tishkoff SA, et al. Genome-wide patterns of population structure and admixture in West Africans and African Americans. Proc Natl Acad SciUSA. 2010;107:786–791. doi: 10.1073/pnas.0909559107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burney RO. The genetics and biochemistry of endometriosis. Curr Opin Obstet Gynecol. 2013;25:280–286. doi: 10.1097/GCO.0b013e3283630d56. [DOI] [PubMed] [Google Scholar]
- Burney RO, Talbi S, Hamilton AE, Vo KC, Nyegaard M, Nezhat CR, Lessey BA, Giudice LC. Gene expression analysis of endometrium reveals progesterone resistance and candidate susceptibility genes in women with endometriosis. Endocrinology. 2007;148:3814–3826. doi: 10.1210/en.2006-1692. [DOI] [PubMed] [Google Scholar]
- Burney RO, Hamilton AE, Aghajanova L, Vo KC, Nezhat CN, Lessey BA, Giudice LC. MicroRNA expression profiling of eutopic secretory endometrium in women with versus without endometriosis. Mol Hum Reprod. 2009;15:625–631. doi: 10.1093/molehr/gap068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnum KE, Tranguch S, Mi D, Daikoku T, Dey SK, Caprioli RM. Imaging mass spectrometry reveals unique protein profiles during embryo implantation. Endocrinology. 2008;149:3274–3278. doi: 10.1210/en.2008-0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns DA, Perkins DN, Stanley AJ, Thompson D, Barrett JH, Selby PJ, Banks RE. Integrated multi-level quality control for proteomic profiling studies using mass spectrometry. BMC Bioinformatics. 2008;9:519. doi: 10.1186/1471-2105-9-519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callinan PA, Feinberg AP. The emerging science of epigenomics. Hum Mol Genet. 2006;15:R95–101. doi: 10.1093/hmg/ddl095. [DOI] [PubMed] [Google Scholar]
- Cappadona S, Baker PR, Cutillas PR, Heck AJ, van Breukelen B. Current challenges in software solutions for mass spectrometry-based quantitative proteomics. Amino Acids. 2012;43:1087–1108. doi: 10.1007/s00726-012-1289-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carthew RW, Sontheimer EJ. Origins and Mechanisms of miRNAs and siRNAs. Cell. 2009;136:642–655. doi: 10.1016/j.cell.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casado-Vela J, Rodriguez-Suarez E, Iloro I, Ametzazurra A, Alkorta N, Garcia-Velasco JA, Matorras R, Prieto B, Gonzalez S, Nagore D, et al. Comprehensive proteomic analysis of human endometrial fluid aspirate. J Proteome Res. 2009;8:4622–4632. doi: 10.1021/pr9004426. [DOI] [PubMed] [Google Scholar]
- Cheong Y, Boomsma C, Heijnen C, Macklon N. Uterine secretomics: a window on the maternal-embryo interface. Fertil Steril. 2013;99:1093–1099. doi: 10.1016/j.fertnstert.2013.01.144. [DOI] [PubMed] [Google Scholar]
- Chervitz SA, Deutsch EW, Field D, Parkinson H, Quackenbush J, Rocca-Serra P, Sansone SA, Stoeckert CJ, Jr., Taylor CF, Taylor R, et al. Data standards for Omics data: the basis of data sharing and reuse. Methods Mol Biol. 2011;719:31–69. doi: 10.1007/978-1-61779-027-0_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark IM, Swingler TE, Young DA. Acetylation in the regulation of metalloproteinase and tissue inhibitor of metalloproteinases gene expression. Front Biosci. 2007;12:528–535. doi: 10.2741/2079. [DOI] [PubMed] [Google Scholar]
- Cordero F, Botta M, Calogero RA. Microarray data analysis and mining approaches. Brief Funct Genomic Proteomic. 2007;6:265–281. doi: 10.1093/bfgp/elm034. [DOI] [PubMed] [Google Scholar]
- Coutifaris C, Myers ER, Guzick DS, Diamond MP, Carson SA, Legro RS, McGovern PG, Schlaff WD, Carr BR, Steinkampf MP, et al. Histological dating of timed endometrial biopsy tissue is not related to fertility status. Fertil Steril. 2004;82:1264–1272. doi: 10.1016/j.fertnstert.2004.03.069. [DOI] [PubMed] [Google Scholar]
- Cox J, Mann M. Quantitative, high-resolution proteomics for data-driven systems biology. Annu Rev Biochem. 2011;80:273–299. doi: 10.1146/annurev-biochem-061308-093216. [DOI] [PubMed] [Google Scholar]
- Creighton CJ, Benham AL, Zhu H, Khan MF, Reid JG, Nagaraja AK, Fountain MD, Dziadek O, Han D, Ma L, et al. Discovery of novel microRNAs in female reproductive tract using next generation sequencing. PLoS ONE. 2010;5:e9637. doi: 10.1371/journal.pone.0009637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordas A, Ovelleiro D, Wang R, Foster JM, Rios D, Vizcaino JA, Hermjakob H. PRIDE: quality control in a proteomics data repository. Database (Oxford) 2012;2012:bas004. doi: 10.1093/database/bas004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day FR, Loos RJ. Developments in obesity genetics in the era of genome-wide association studies. J Nutrigenet Nutrigenomics. 2011;4:222–238. doi: 10.1159/000332158. [DOI] [PubMed] [Google Scholar]
- Deatherage DE, Potter D, Yan PS, Huang TH, Lin S. Methylation analysis by microarray. Methods Mol Biol. 2009;556:117–139. doi: 10.1007/978-1-60327-192-9_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Gimeno P, Horcajadas JA, Martinez-Conejero JA, Esteban FJ, Alama P, Pellicer A, Simon C. A genomic diagnostic tool for human endometrial receptivity based on the transcriptomic signature. Fertil Steril. 2011;95:50–60. doi: 10.1016/j.fertnstert.2010.04.063. 60 e51–15. [DOI] [PubMed] [Google Scholar]
- Diaz-Gimeno P, Ruiz-Alonso M, Blesa D, Bosch N, Martinez-Conejero JA, Alama P, Garrido N, Pellicer A, Simon C. The accuracy and reproducibility of the endometrial receptivity array is superior to histology as a diagnostic method for endometrial receptivity. Fertil Steril. 2013;99:508–517. doi: 10.1016/j.fertnstert.2012.09.046. [DOI] [PubMed] [Google Scholar]
- Doll A, Abal M, Rigau M, Monge M, Gonzalez M, Demajo S, Colas E, Llaurado M, Alazzouzi H, Planaguma J, et al. Novel molecular profiles of endometrial cancer-new light through old windows. J Steroid Biochem Mol Biol. 2008;108:221–229. doi: 10.1016/j.jsbmb.2007.09.020. [DOI] [PubMed] [Google Scholar]
- Dominguez F, Garrido-Gomez T, Lopez JA, Camafeita E, Quinonero A, Pellicer A, Simon C. Proteomic analysis of the human receptive versus non-receptive endometrium using differential in-gel electrophoresis and MALDI-MS unveils stathmin 1 and annexin A2 as differentially regulated. Hum Reprod. 2009;24:2607–2617. doi: 10.1093/humrep/dep230. [DOI] [PubMed] [Google Scholar]
- Durinck S. Pre-processing of microarray data and analysis of differential expression. Methods Mol Biol. 2008;452:89–110. doi: 10.1007/978-1-60327-159-2_4. [DOI] [PubMed] [Google Scholar]
- Edgell TA, Rombauts LJF, Salamonsen LA. Assessing receptivity in the endometrium: the need for a rapid, non-invasive test. Reprod Biomed Online. 2013 doi: 10.1016/j.rbmo.2013.05.014. in press. [DOI] [PubMed] [Google Scholar]
- Editorial Method of the Year 2012. Nat Methods. 2013;10:1. doi: 10.1038/nmeth.2329. [DOI] [PubMed] [Google Scholar]
- Estella C, Herrer I, Moreno-Moya JM, Quinonero A, Martinez S, Pellicer A, Simon C. miRNA signature and Dicer requirement during human endometrial stromal decidualization in vitro. PLoS ONE. 2012;7:e41080. doi: 10.1371/journal.pone.0041080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans GE, Martinez-Conejero JA, Phillipson GT, Simon C, McNoe LA, Sykes PH, Horcajadas JA, Lam EY, Print CG, Sin IL, et al. Gene and protein expression signature of endometrial glandular and stromal compartments during the window of implantation. Fertil Steril. 2012a;97:1365–1373. doi: 10.1016/j.fertnstert.2012.03.007. e1361–1362. [DOI] [PubMed] [Google Scholar]
- Evans GE, Phillipson GT, Sin IL, Frampton CM, Kirker JA, Bigby SM, Evans JJ. Gene expression confirms a potentially receptive endometrium identified by histology in fertile women. Hum Reprod. 2012b;27:2747–2755. doi: 10.1093/humrep/des233. [DOI] [PubMed] [Google Scholar]
- Fassbender A, Simsa P, Kyama CM, Waelkens E, Mihalyi A, Meuleman C, Gevaert O, Van de Plas R, de Moor B, D'Hooghe TM. TRIzol treatment of secretory phase endometrium allows combined proteomic and mRNA microarray analysis of the same sample in women with and without endometriosis. Reprod Biol Endocrinol. 2010;8:123. doi: 10.1186/1477-7827-8-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassbender A, Verbeeck N, Bornigen D, Kyama CM, Bokor A, Vodolazkaia A, Peeraer K, Tomassetti C, Meuleman C, Gevaert O, et al. Combined mRNA microarray and proteomic analysis of eutopic endometrium of women with and without endometriosis. Hum Reprod. 2012a;27:2020–2029. doi: 10.1093/humrep/des127. [DOI] [PubMed] [Google Scholar]
- Fassbender A, Waelkens E, Verbeeck N, Kyama CM, Bokor A, Vodolazkaia A, Van de Plas R, Meuleman C, Peeraer K, Tomassetti C, et al. Proteomics analysis of plasma for early diagnosis of endometriosis. Obstet Gynecol. 2012b;119:276–285. doi: 10.1097/AOG.0b013e31823fda8d. [DOI] [PubMed] [Google Scholar]
- Fassbender A, Vodolazkaia A, Saunders P, Lebovic D, Waelkens E, De Moor B, D'Hooghe T. Biomarkers of endometriosis. Fertil Steril. 2013;99:1135–1145. doi: 10.1016/j.fertnstert.2013.01.097. [DOI] [PubMed] [Google Scholar]
- Garrido-Gomez T, Dominguez F, Simon C. Proteomics of embryonic implantation. Handb Exp Pharmacol. 2010:67–78. doi: 10.1007/978-3-642-02062-9_5. [DOI] [PubMed] [Google Scholar]
- Garrido-Gomez T, Ruiz-Alonso M, Blesa D, Diaz-Gimeno P, Vilella F, Simon C. Profiling the gene signature of endometrial receptivity: clinical results. Fertil Steril. 2013;99:1078–1085. doi: 10.1016/j.fertnstert.2012.12.005. [DOI] [PubMed] [Google Scholar]
- Gilabert-Estelles J, Braza-Boils A, Ramon LA, Zorio E, Medina P, Espana F, Estelles A. Role of microRNAs in gynecological pathology. Curr Med Chem. 2012;19:2406–2413. doi: 10.2174/092986712800269362. [DOI] [PubMed] [Google Scholar]
- Giudice LC. Potential biochemical markers of uterine receptivity. Hum Reprod. 1999;14(Suppl. 2):3–16. doi: 10.1093/humrep/14.suppl_2.3. [DOI] [PubMed] [Google Scholar]
- Giudice LC. Application of functional genomics to primate endometrium: insights into biological processes. Reprod Biol Endocrinol. 2006;4(Suppl. 1):S4. doi: 10.1186/1477-7827-4-S1-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giudice LC, Talbi S, Hamilton AE, Lessey BA. The Endometrium. London: Informa; 2008. Transcriptomics. [Google Scholar]
- Gracie S, Pennell C, Ekman-Ordeberg G, Lye S, McManaman J, Williams S, Palmer L, Kelley M, Menon R, Gravett M. An integrated systems biology approach to the study of preterm birth using ‘-omic’ technology – a guideline for research. BMC Pregnancy Childbirth. 2011;11:71. doi: 10.1186/1471-2393-11-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunaratne PH, Creighton CJ, Watson M, Tennakoon JB. Large-scale integration of MicroRNA and gene expression data for identification of enriched microRNA-mRNA associations in biological systems. Methods Mol Biol. 2010;667:297–315. doi: 10.1007/978-1-60761-811-9_20. [DOI] [PubMed] [Google Scholar]
- Guo SW. The association of endometriosis risk and genetic polymorphisms involving dioxin detoxification enzymes: a systematic review. Eur J Obstet Gynecol Reprod Biol. 2006;124:134–143. doi: 10.1016/j.ejogrb.2005.10.002. [DOI] [PubMed] [Google Scholar]
- Guo SW. The endometrial epigenome and its response to steroid hormones. Mol Cell Endocrinol. 2012;358:185–196. doi: 10.1016/j.mce.2011.10.025. [DOI] [PubMed] [Google Scholar]
- Guo SW, Wu Y, Strawn E, Basir Z, Wang Y, Halverson G, Montgomery K, Kajdacsy-Balla A. Genomic alterations in the endometrium may be a proximate cause for endometriosis. Eur J Obstet Gynecol Reprod Biol. 2004;116:89–99. doi: 10.1016/j.ejogrb.2004.02.004. [DOI] [PubMed] [Google Scholar]
- Ha TY. MicroRNAs in human diseases: from lung, liver and kidney diseases to infectious disease, sickle cell disease and endometrium disease. Immune Netw. 2011;11:309–323. doi: 10.4110/in.2011.11.6.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannan NJ, Stoikos CJ, Stephens AN, Salamonsen LA. Depletion of high-abundance serum proteins from human uterine lavages enhances detection of lower-abundance proteins. J Proteome Res. 2009;8:1099–1103. doi: 10.1021/pr800811y. [DOI] [PubMed] [Google Scholar]
- Hannan NJ, Stephens AN, Rainczuk A, Hincks C, Rombauts LJ, Salamonsen LA. 2D-DiGE analysis of the human endometrial secretome reveals differences between receptive and nonreceptive states in fertile and infertile women. J Proteome Res. 2010;9:6256–6264. doi: 10.1021/pr1004828. [DOI] [PubMed] [Google Scholar]
- Hannan NJ, Evans J, Salamonsen LA. Alternate roles for immune regulators: establishing endometrial receptivity for implantation. Expert Rev Clin Immunol. 2011a;7:789–802. doi: 10.1586/eci.11.65. [DOI] [PubMed] [Google Scholar]
- Hannan NJ, Paiva P, Meehan KL, Rombauts LJ, Gardner DK, Salamonsen LA. Analysis of fertility-related soluble mediators in human uterine fluid identifies VEGF as a key regulator of embryo implantation. Endocrinology. 2011b;152:4948–4956. doi: 10.1210/en.2011-1248. [DOI] [PubMed] [Google Scholar]
- Hannan NJ, Nie G, Rainzcuk A, Rombauts LJ, Salamonsen LA. Uterine lavage or aspirate: which view of the intrauterine environment? Reprod Sci. 2012;19:1125–1132. doi: 10.1177/1933719112443879. [DOI] [PubMed] [Google Scholar]
- Hansen KA, Eyster KM. Genetics and genomics of endometriosis. Clin Obstet Gynecol. 2010;53:403–412. doi: 10.1097/GRF.0b013e3181db7ca1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haouzi D, Mahmoud K, Fourar M, Bendhaou K, Dechaud H, De Vos J, Reme T, Dewailly D, Hamamah S. Identification of new biomarkers of human endometrial receptivity in the natural cycle. Hum Reprod. 2009;24:198–205. doi: 10.1093/humrep/den360. [DOI] [PubMed] [Google Scholar]
- Haouzi D, Dechaud H, Assou S, De Vos J, Hamamah S. Insights into human endometrial receptivity from transcriptomic and proteomic data. Reprod Biomed Online. 2012;24:23–34. doi: 10.1016/j.rbmo.2011.09.009. [DOI] [PubMed] [Google Scholar]
- Harper MJ. The implantation window. Baillieres Clin Obstet Gynaecol. 1992;6:351–371. doi: 10.1016/s0950-3552(05)80092-6. [DOI] [PubMed] [Google Scholar]
- Hawkins SM, Creighton CJ, Han DY, Zariff A, Anderson ML, Gunaratne PH, Matzuk MM. Functional microRNA involved in endometriosis. Mol Endocrinol. 2011;25:821–832. doi: 10.1210/me.2010-0371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horcajadas JA, Riesewijk A, Dominguez F, Cervero A, Pellicer A, Simon C. Determinants of endometrial receptivity. Ann N Y Acad Sci. 2004;1034:166–175. doi: 10.1196/annals.1335.019. [DOI] [PubMed] [Google Scholar]
- Horcajadas JA, Pellicer A, Simon C. Wide genomic analysis of human endometrial receptivity: new times, new opportunities. Hum Reprod Update. 2007;13:77–86. doi: 10.1093/humupd/dml046. [DOI] [PubMed] [Google Scholar]
- Horgan RP, Clancy OH, Myers JE, Baker PN. An overview of proteomic and metabolomic technologies and their application to pregnancy research. BJOG. 2009;116:173–181. doi: 10.1111/j.1471-0528.2008.01997.x. [DOI] [PubMed] [Google Scholar]
- Huang J, Qi R, Quackenbush J, Dauway E, Lazaridis E, Yeatman T. Effects of ischemia on gene expression. J Surg Res. 2001;99:222–227. doi: 10.1006/jsre.2001.6195. [DOI] [PubMed] [Google Scholar]
- Ikeda Y, Oda K, Nakagawa S, Murayama-Hosokawa S, Yamamoto S, Ishikawa S, Wang L, Takazawa Y, Maeda D, Wada-Hiraike O, et al. Genome-wide single nucleotide polymorphism arrays as a diagnostic tool in patients with synchronous endometrial and ovarian cancer. Int J Gynecol Cancer. 2012;22:725–731. doi: 10.1097/IGC.0b013e31824c6ea6. [DOI] [PubMed] [Google Scholar]
- Illig T, Gieger C, Zhai G, Romisch-Margl W, Wang-Sattler R, Prehn C, Altmaier E, Kastenmuller G, Kato BS, Mewes HW, et al. A genome-wide perspective of genetic variation in human metabolism. Nat Genet. 2010;42:137–141. doi: 10.1038/ng.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannidis JP, Allison DB, Ball CA, Coulibaly I, Cui X, Culhane AC, Falchi M, Furlanello C, Game L, Jurman G, et al. Repeatability of published microarray gene expression analyses. Nat Genet. 2009;41:149–155. doi: 10.1038/ng.295. [DOI] [PubMed] [Google Scholar]
- Izawa M, Taniguchi F, Terakawa N, Harada T. Epigenetic aberration of gene expression in endometriosis. Front Biosci (Elite Ed) 2013;E5:900–910. doi: 10.2741/e669. [DOI] [PubMed] [Google Scholar]
- Kao LC, Germeyer A, Tulac S, Lobo S, Yang JP, Taylor RN, Osteen K, Lessey BA, Giudice LC. Expression profiling of endometrium from women with endometriosis reveals candidate genes for disease-based implantation failure and infertility. Endocrinology. 2003;144:2870–2881. doi: 10.1210/en.2003-0043. [DOI] [PubMed] [Google Scholar]
- Kinsinger CR, Apffel J, Baker M, Bian X, Borchers CH, Bradshaw R, Brusniak MY, Chan DW, Deutsch EW, Domon B, et al. Recommendations for mass spectrometry data quality metrics for open access data (corollary to the Amsterdam Principles) J Proteome Res. 2012;11:1412–1419. doi: 10.1021/pr201071t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koks S, Velthut A, Sarapik A, Altmae S, Reinmaa E, Schalkwyk LC, Fernandes C, Lad HV, Soomets U, Jaakma U, et al. The differential transcriptome and ontology profiles of floating and cumulus granulosa cells in stimulated human antral follicles. Mol Hum Reprod. 2010;16:229–240. doi: 10.1093/molehr/gap103. [DOI] [PubMed] [Google Scholar]
- Koot YE, Teklenburg G, Salker MS, Brosens JJ, Macklon NS. Molecular aspects of implantation failure. Biochim Biophys Acta. 2012;1822:1943–1950. doi: 10.1016/j.bbadis.2012.05.017. [DOI] [PubMed] [Google Scholar]
- Koulman A, Lane GA, Harrison SJ, Volmer DA. From differentiating metabolites to biomarkers. Anal Bioanal Chem. 2009;394:663–670. doi: 10.1007/s00216-009-2690-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar C, Mann M. Bioinformatics analysis of mass spectrometry-based proteomics data sets. FEBS Lett. 2009;583:1703–1712. doi: 10.1016/j.febslet.2009.03.035. [DOI] [PubMed] [Google Scholar]
- Kuokkanen S, Chen B, Ojalvo L, Benard L, Santoro N, Pollard JW. Genomic profiling of microRNAs and messenger RNAs reveals hormonal regulation in microRNA expression in human endometrium. Biol Reprod. 2010;82:791–801. doi: 10.1095/biolreprod.109.081059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagarde M, Geloen A, Record M, Vance D, Spener F. Lipidomics is emerging. Biochim Biophys Acta. 2003;1634:61. doi: 10.1016/j.bbalip.2003.11.002. [DOI] [PubMed] [Google Scholar]
- Lessey BA. The role of the endometrium during embryo implantation. Hum Reprod. 2000;15(Suppl. 6):39–50. [PubMed] [Google Scholar]
- Lessey BA. Fine tuning of endometrial function by estrogen and progesterone through microRNAs. Biol Reprod. 2010;82:653–655. doi: 10.1095/biolreprod.110.083667. [DOI] [PubMed] [Google Scholar]
- Lessey BA. Assessment of endometrial receptivity. Fertil Steril. 2011;96:522–529. doi: 10.1016/j.fertnstert.2011.07.1095. [DOI] [PubMed] [Google Scholar]
- Li R, Qiao J, Wang L, Li L, Zhen X, Liu P, Zheng X. MicroRNA array and microarray evaluation of endometrial receptivity in patients with high serum progesterone levels on the day of hCG administration. Reprod Biol Endocrinol. 2011;9:29. doi: 10.1186/1477-7827-9-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licht P, Russu V, Lehmeyer S, Wildt L. Molecular aspects of direct LH/hCG effects on human endometrium – lessons from intrauterine microdialysis in the human female in vivo. Reprod Biol. 2001;1:10–19. [PubMed] [Google Scholar]
- Longuespee R, Gagnon H, Boyon C, Strupat K, Dauly C, Kerdraon O, Ighodaro A, Desmons A, Dupuis J, Wisztorski M, et al. Proteomic analyses of serous and endometrioid epithelial ovarian cancers - cases studies - molecular insights of a possible histological etiology of serous ovarian cancer. Proteomics Clin Appl. 2013;7:337–354. doi: 10.1002/prca.201200079. [DOI] [PubMed] [Google Scholar]
- Loven J, Orlando DA, Sigova AA, Lin CY, Rahl PB, Burge CB, Levens DL, Lee TI, Young RA. Revisiting global gene expression analysis. Cell. 2012;151:476–482. doi: 10.1016/j.cell.2012.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone JH, Oliver B. Microarrays, deep sequencing and the true measure of the transcriptome. BMC Biol. 2011;9:34. doi: 10.1186/1741-7007-9-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannello F, Ligi D, Magnani M. Deciphering the single-cell omic: innovative application for translational medicine. Expert Rev Proteomics. 2012;9:635–648. doi: 10.1586/epr.12.61. [DOI] [PubMed] [Google Scholar]
- Marabita F, Almgren M, Lindholm M, Ruhrmann S, Fagerstrom-Billai F, Jagodic M, Sundberg CJ, Ekstrom TJ, Teschendorff AE, Tegner J, et al. An evaluation of analysis pipelines for DNA methylation profiling using the Illumina HumanMethylation450 BeadChip platform. Epigenetics. 2013;8:333–346. doi: 10.4161/epi.24008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Conejero JA, Simon C, Pellicer A, Horcajadas JA. Is ovarian stimulation detrimental to the endometrium? Reprod Biomed Online. 2007;15:45–50. doi: 10.1016/s1472-6483(10)60690-6. [DOI] [PubMed] [Google Scholar]
- Matsuzaki S. DNA microarray analysis in endometriosis for development of more effective targeted therapies. Front Biosci (Elite Ed) 2011;3:1139–1153. doi: 10.2741/e317. [DOI] [PubMed] [Google Scholar]
- Matthiesen R, Azevedo L, Amorim A, Carvalho AS. Discussion on common data analysis strategies used in MS-based proteomics. Proteomics. 2011;11:604–619. doi: 10.1002/pmic.201000404. [DOI] [PubMed] [Google Scholar]
- May KE, Villar J, Kirtley S, Kennedy SH, Becker CM. Endometrial alterations in endometriosis: a systematic review of putative biomarkers. Hum Reprod Update. 2011;17:637–653. doi: 10.1093/humupd/dmr013. [DOI] [PubMed] [Google Scholar]
- Meehan KL, Rainczuk A, Salamonsen LA, Stephens AN. Proteomics and the search for biomarkers of female reproductive diseases. Reproduction. 2010;140:505–519. doi: 10.1530/REP-10-0226. [DOI] [PubMed] [Google Scholar]
- Micke P, Ohshima M, Tahmasebpoor S, Ren ZP, Ostman A, Ponten F, Botling J. Biobanking of fresh frozen tissue: RNA is stable in nonfixed surgical specimens. Lab Invest. 2006;86:202–211. doi: 10.1038/labinvest.3700372. [DOI] [PubMed] [Google Scholar]
- Mirkin S, Arslan M, Churikov D, Corica A, Diaz JI, Williams S, Bocca S, Oehninger S. In search of candidate genes critically expressed in the human endometrium during the window of implantation. Hum Reprod. 2005;20:2104–2117. doi: 10.1093/humrep/dei051. [DOI] [PubMed] [Google Scholar]
- Mocellin S, Rossi CR. Principles of gene microarray data analysis. Adv Exp Med Biol. 2007;593:19–30. doi: 10.1007/978-0-387-39978-2_3. [DOI] [PubMed] [Google Scholar]
- Munro SK, Farquhar CM, Mitchell MD, Ponnampalam AP. Epigenetic regulation of endometrium during the menstrual cycle. Mol Hum Reprod. 2010;16:297–310. doi: 10.1093/molehr/gaq010. [DOI] [PubMed] [Google Scholar]
- Murray MJ, Meyer WR, Zaino RJ, Lessey BA, Novotny DB, Ireland K, Zeng D, Fritz MA. A critical analysis of the accuracy, reproducibility, and clinical utility of histologic endometrial dating in fertile women. Fertil Steril. 2004;81:1333–1343. doi: 10.1016/j.fertnstert.2003.11.030. [DOI] [PubMed] [Google Scholar]
- Ng YH, Rome S, Jalabert A, Forterre A, Singh H, Hincks CL, Salamonsen LA. Endometrial exosomes/microvesicles in the uterine microenvironment: a new paradigm for embryo-endometrial cross talk at implantation. PLoS ONE. 2013;8:e58502. doi: 10.1371/journal.pone.0058502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson JK, Lindon JC. Systems biology: Metabonomics. Nature. 2008;455:1054–1056. doi: 10.1038/4551054a. [DOI] [PubMed] [Google Scholar]
- Nielsen J, Oliver S. The next wave in metabolome analysis. Trends Biotechnol. 2005;23:544–546. doi: 10.1016/j.tibtech.2005.08.005. [DOI] [PubMed] [Google Scholar]
- Ning K, Fermin D, Nesvizhskii AI. Comparative analysis of different label-free mass spectrometry based protein abundance estimates and their correlation with RNA-Seq gene expression data. J Proteome Res. 2012;11:2261–2271. doi: 10.1021/pr201052x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noyes RW, Hertig AT, Rock J. Dating the endometrial biopsy. Fertil Steril. 1950;1:3–25. doi: 10.1016/j.fertnstert.2019.08.079. [DOI] [PubMed] [Google Scholar]
- Noyes RW, Hertig AT, Rock J. Dating the endometrial biopsy. Am J Obstet Gynecol. 1975;122:262–263. doi: 10.1016/s0002-9378(16)33500-1. [DOI] [PubMed] [Google Scholar]
- Nyholt DR, Low SK, Anderson CA, Painter JN, Uno S, Morris AP, MacGregor S, Gordon SD, Henders AK, Martin NG, et al. Genome-wide association meta-analysis identifies new endometriosis risk loci. Nat Genet. 2012;44:1355–1359. doi: 10.1038/ng.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painter JN, Anderson CA, Nyholt DR, Macgregor S, Lin J, Lee SH, Lambert A, Zhao ZZ, Roseman F, Guo Q, et al. Genome-wide association study identifies a locus at 7p15.2 associated with endometriosis. Nat Genet. 2011;43:51–54. doi: 10.1038/ng.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paiva P, Hannan NJ, Hincks C, Meehan KL, Pruysers E, Dimitriadis E, Salamonsen LA. Human chorionic gonadotrophin regulates FGF2 and other cytokines produced by human endometrial epithelial cells, providing a mechanism for enhancing endometrial receptivity. Hum Reprod. 2011;26:1153–1162. doi: 10.1093/humrep/der027. [DOI] [PubMed] [Google Scholar]
- Pinto D, Darvishi K, Shi X, Rajan D, Rigler D, Fitzgerald T, Lionel AC, Thiruvahindrapuram B, Macdonald JR, Mills R, et al. Comprehensive assessment of array-based platforms and calling algorithms for detection of copy number variants. Nature Biotechnol. 2011;29:512–520. doi: 10.1038/nbt.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard CC, Cheng HH, Tewari M. MicroRNA profiling: approaches and considerations. Nat Rev Genet. 2012;13:358–369. doi: 10.1038/nrg3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahmioglu N, Missmer SA, Montgomery GW, Zondervan KT. Insights into Assessing the Genetics of Endometriosis. Curr Obstet Gynecol Rep. 2012;1:124–137. doi: 10.1007/s13669-012-0016-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahnama F, Thompson B, Steiner M, Shafiei F, Lobie PE, Mitchell MD. Epigenetic regulation of E-cadherin controls endometrial receptivity. Endocrinology. 2009;150:1466–1472. doi: 10.1210/en.2008-1142. [DOI] [PubMed] [Google Scholar]
- Ramon LA, Braza-Boils A, Gilabert J, Chirivella M, Espana F, Estelles A, Gilabert-Estelles J. microRNAs related to angiogenesis are dysregulated in endometrioid endometrial cancer. Hum Reprod. 2012;27:3036–3045. doi: 10.1093/humrep/des292. [DOI] [PubMed] [Google Scholar]
- Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revel A, Achache H, Stevens J, Smith Y, Reich R. MicroRNAs are associated with human embryo implantation defects. Hum Reprod. 2011;26:2830–2840. doi: 10.1093/humrep/der255. [DOI] [PubMed] [Google Scholar]
- Robert C. Microarray analysis of gene expression during early development: a cautionary overview. Reproduction. 2010;140:787–801. doi: 10.1530/REP-10-0191. [DOI] [PubMed] [Google Scholar]
- Rubio C, Rodrigo L, Mercader A, Mateu E, Buendia P, Pehlivan T, Viloria T, De los Santos MJ, Simon C, Remohi J, et al. Impact of chromosomal abnormalities on preimplantation embryo development. Prenat Diagn. 2007;27:748–756. doi: 10.1002/pd.1773. [DOI] [PubMed] [Google Scholar]
- Ruiz-Alonso M, Blesa D, Simon C. The genomics of the human endometrium. Biochim Biophys Acta. 2012;1822:1931–1942. doi: 10.1016/j.bbadis.2012.05.004. [DOI] [PubMed] [Google Scholar]
- Ruiz-Alonso M, Blesa D, Diaz-Gimeno P, Vilella F, Pellicer A, Simon C. The endometrial receptivity array as diagnosis and personalized embryo transfer as treatment for patients with receptive implantation failure. Fertil Steril. 2013;100:818–824. doi: 10.1016/j.fertnstert.2013.05.004. [DOI] [PubMed] [Google Scholar]
- Saare M, Soritsa D, Vaidla K, Palta P, Remm M, Laan M, Karro H, Soritsa A, Salumets A, D'Hooghe T, et al. No evidence of somatic DNA copy number alterations in eutopic and ectopic endometrial tissue in endometriosis. Hum Reprod. 2012;27:1857–1864. doi: 10.1093/humrep/des125. [DOI] [PubMed] [Google Scholar]
- Salamonsen LA, Nie G, Hannan NJ, Dimitriadis E. Society for Reproductive Biology Founders’ Lecture 2009. Preparing fertile soil: the importance of endometrial receptivity. Reprod Fertil Dev. 2009;21:923–934. doi: 10.1071/RD09145. [DOI] [PubMed] [Google Scholar]
- Salamonsen LA, Edgell T, Rombauts LJ, Stephens AN, Robertson DM, Rainczuk A, Nie G, Hannan NJ. Proteomics of the human endometrium and uterine fluid: a pathway to biomarker discovery. Fertil Steril. 2013;99:1086–1092. doi: 10.1016/j.fertnstert.2012.09.013. [DOI] [PubMed] [Google Scholar]
- Sarkar D, Parkin R, Wyman S, Bendoraite A, Sather C, Delrow J, Godwin AK, Drescher C, Huber W, Gentleman R, et al. Quality assessment and data analysis for microRNA expression arrays. Nucleic Acids Res. 2009;37:e17. doi: 10.1093/nar/gkn932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savaris RF, Giudice LC. Genomics analysis: endometrium. Methods Mol Biol. 2009;590:91–113. doi: 10.1007/978-1-60327-378-7_6. [DOI] [PubMed] [Google Scholar]
- Schwamborn K, Krieg RC, Uhlig S, Ikenberg H, Wellmann A. MALDI imaging as a specific diagnostic tool for routine cervical cytology specimens. Int J Mol Med. 2011;27:417–421. doi: 10.3892/ijmm.2010.587. [DOI] [PubMed] [Google Scholar]
- Scotchie JG, Fritz MA, Mocanu M, Lessey BA, Young SL. Proteomic analysis of the luteal endometrial secretome. Reprod Sci. 2009;16:883–893. doi: 10.1177/1933719109337165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seli E, Robert C, Sirard MA. OMICS in assisted reproduction: possibilities and pitfalls. Mol Hum Reprod. 2010;16:513–530. doi: 10.1093/molehr/gaq041. [DOI] [PubMed] [Google Scholar]
- Sha AG, Liu JL, Jiang XM, Ren JZ, Ma CH, Lei W, Su RW, Yang ZM. Genome-wide identification of micro-ribonucleic acids associated with human endometrial receptivity in natural and stimulated cycles by deep sequencing. Fertil Steril. 2011;96:150–155. doi: 10.1016/j.fertnstert.2011.04.072. e155. [DOI] [PubMed] [Google Scholar]
- Shayeghi M, Latunde-Dada GO, Oakhill JS, Laftah AH, Takeuchi K, Halliday N, Khan Y, Warley A, McCann FE, Hider RC, et al. Identification of an intestinal heme transporter. Cell. 2005;122:789–801. doi: 10.1016/j.cell.2005.06.025. [DOI] [PubMed] [Google Scholar]
- Sheldon E, Vo KC, McIntire RA, Aghajanova L, Zelenko Z, Irwin JC, Giudice LC. Biobanking human endometrial tissue and blood specimens: standard operating procedure and importance to reproductive biology research and diagnostic development. Fertil Steril. 2011;95:2120–2122. doi: 10.1016/j.fertnstert.2011.01.164. 2122, e2121–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwin R, Catalano R, Sharkey A. Large-scale gene expression studies of the endometrium: what have we learnt? Reproduction. 2006;132:1–10. doi: 10.1530/rep.1.00355. [DOI] [PubMed] [Google Scholar]
- Slattery M, Ankisetty S, Corrales J, Marsh-Hunkin KE, Gochfeld DJ, Willett KL, Rimoldi JM. Marine proteomics: a critical assessment of an emerging technology. J Nat Prod. 2012;75:1833–1877. doi: 10.1021/np300366a. [DOI] [PubMed] [Google Scholar]
- Spitzer TL, Rojas A, Zelenko Z, Aghajanova L, Erikson DW, Barragan F, Meyer M, Tamaresis JS, Hamilton AE, Irwin JC, et al. Perivascular human endometrial mesenchymal stem cells express pathways relevant to self-renewal, lineage specification, and functional phenotype. Biol Reprod. 2012;86:58. doi: 10.1095/biolreprod.111.095885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruessel A, Steimann G, Jung M, Lee SA, Carr T, Fentz AK, Spangenberg J, Zornig C, Juhl HH, David KA. Tissue ischemia time affects gene and protein expression patterns within minutes following surgical tumor excision. Biotechniques. 2004;36:1030–1037. doi: 10.2144/04366RR04. [DOI] [PubMed] [Google Scholar]
- Stephens AN, Hannan NJ, Rainczuk A, Meehan KL, Chen J, Nicholls PK, Rombauts LJ, Stanton PG, Robertson DM, Salamonsen LA. Post-translational modifications and protein-specific isoforms in endometriosis revealed by 2D DIGE. J Proteome Res. 2010;9:2438–2449. doi: 10.1021/pr901131p. [DOI] [PubMed] [Google Scholar]
- Suarez E, Burguete A, McLachlan GJ. Microarray data analysis for differential expression: a tutorial. P R Health Sci J. 2009;28:89–104. [PubMed] [Google Scholar]
- Sundaresh S, Doolan DL, Hirst S, Mu Y, Unal B, Davies DH, Felgner PL, Baldi P. Identification of humoral immune responses in protein microarrays using DNA microarray data analysis techniques. Bioinformatics. 2006;22:1760–1766. doi: 10.1093/bioinformatics/btl162. [DOI] [PubMed] [Google Scholar]
- Sundqvist J, Xu H, Vodolazkaia A, Fassbender A, Kyama C, Bokor A, Gemzell-Danielsson K, D'Hooghe TM, Falconer H. Replication of endometriosis-associated single-nucleotide polymorphisms from genome-wide association studies in a Caucasian population. Hum Reprod. 2013;28:835–839. doi: 10.1093/humrep/des457. [DOI] [PubMed] [Google Scholar]
- Talbi S, Hamilton AE, Vo KC, Tulac S, Overgaard MT, Dosiou C, Le Shay N, Nezhat CN, Kempson R, Lessey BA, et al. Molecular phenotyping of human endometrium distinguishes menstrual cycle phases and underlying biological processes in normo-ovulatory women. Endocrinology. 2006;147:1097–1121. doi: 10.1210/en.2005-1076. [DOI] [PubMed] [Google Scholar]
- Tapia A, Vilos C, Marin JC, Croxatto HB, Devoto L. Bioinformatic detection of E47, E2F1 and SREBP1 transcription factors as potential regulators of genes associated to acquisition of endometrial receptivity. Reprod Biol Endocrinol. 2011;9:14. doi: 10.1186/1477-7827-9-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor CF, Field D, Sansone SA, Aerts J, Apweiler R, Ashburner M, Ball CA, Binz PA, Bogue M, Booth T, et al. Promoting coherent minimum reporting guidelines for biological and biomedical investigations: the MIBBI project. Nat Biotechnol. 2008;26:889–896. doi: 10.1038/nbt.1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teklenburg G, Macklon NS. Review: in vitro models for the study of early human embryo-endometrium interactions. Reprod Sci. 2009;16:811–818. doi: 10.1177/1933719109334966. [DOI] [PubMed] [Google Scholar]
- Teklenburg G, Salker M, Molokhia M, Lavery S, Trew G, Aojanepong T, Mardon HJ, Lokugamage AU, Rai R, Landles C, et al. Natural selection of human embryos: decidualizing endometrial stromal cells serve as sensors of embryo quality upon implantation. PLoS ONE. 2010;5:e10258. doi: 10.1371/journal.pone.0010258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres A, Torres K, Pesci A, Ceccaroni M, Paszkowski T, Cassandrini P, Zamboni G, Maciejewski R. Diagnostic and prognostic significance of miRNA signatures in tissues and plasma of endometrioid endometrial carcinoma patients. Int J Cancer. 2013;132:1633–1645. doi: 10.1002/ijc.27840. [DOI] [PubMed] [Google Scholar]
- Toth B, Wurfel W, Germeyer A, Hirv K, Makrigiannakis A, Strowitzki T. Disorders of implantation–are there diagnostic and therapeutic options? J Reprod Immunol. 2011;90:117–123. doi: 10.1016/j.jri.2011.05.002. [DOI] [PubMed] [Google Scholar]
- Treff NR. Genome-wide analysis of human preimplantation aneuploidy. Semin Reprod Med. 2012;30:283–288. doi: 10.1055/s-0032-1313907. [DOI] [PubMed] [Google Scholar]
- Uchida H, Maruyama T, Nagashima T, Asada H, Yoshimura Y. Histone deacetylase inhibitors induce differentiation of human endometrial adenocarcinoma cells through up-regulation of glycodelin. Endocrinology. 2005;146:5365–5373. doi: 10.1210/en.2005-0359. [DOI] [PubMed] [Google Scholar]
- Uchida H, Maruyama T, Ohta K, Ono M, Arase T, Kagami M, Oda H, Kajitani T, Asada H, Yoshimura Y. Histone deacetylase inhibitor-induced glycodelin enhances the initial step of implantation. Hum Reprod. 2007;22:2615–2622. doi: 10.1093/humrep/dem263. [DOI] [PubMed] [Google Scholar]
- Ulbrich SE, Groebner AE, Bauersachs S. Transcriptional profiling to address molecular determinants of endometrial receptivity – lessons from studies in livestock species. Methods. 2013;59:108–115. doi: 10.1016/j.ymeth.2012.10.013. [DOI] [PubMed] [Google Scholar]
- Uno S, Zembutsu H, Hirasawa A, Takahashi A, Kubo M, Akahane T, Aoki D, Kamatani N, Hirata K, Nakamura Y. A genome-wide association study identifies genetic variants in the CDKN2BAS locus associated with endometriosis in Japanese. Nat Genet. 2010;42:707–710. doi: 10.1038/ng.612. [DOI] [PubMed] [Google Scholar]
- Upadhyay RD, Balasinor NH, Kumar AV, Sachdeva G, Parte P, Dumasia K. Proteomics in reproductive biology: beacon for unraveling the molecular complexities. Biochim Biophys Acta. 2013;1834:8–15. doi: 10.1016/j.bbapap.2012.10.004. [DOI] [PubMed] [Google Scholar]
- Vanneste E, Bittman L, Van der Aa N, Voet T, Vermeesch JR. New array approaches to explore single cells genomes. Front Genet. 2012;3:44. doi: 10.3389/fgene.2012.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiga-Castelli LC, Silva JC, Meola J, Ferriani RA, Yoshimoto M, Santos SA, Squire JA, Martelli L. Genomic alterations detected by comparative genomic hybridization in ovarian endometriomas. Braz J Med Biol Res. 2010;43:799–805. doi: 10.1590/s0100-879x2010007500072. [DOI] [PubMed] [Google Scholar]
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- Vigano P, Somigliana E, Panina P, Rabellotti E, Vercellini P, Candiani M. Principles of phenomics in endometriosis. Hum Reprod Update. 2012;18:248–259. doi: 10.1093/humupd/dms001. [DOI] [PubMed] [Google Scholar]
- Vilella F, Ramirez LB, Simon C. Lipidomics as an emerging tool to predict endometrial receptivity. Fertil Steril. 2013 doi: 10.1016/j.fertnstert.2012.12.026. [DOI] [PubMed] [Google Scholar]
- Vizcaino JA, Cote RG, Csordas A, Dianes JA, Fabregat A, Foster JM, Griss J, Alpi E, Birim M, Contell J, et al. The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 2013;41:D1063–D1069. doi: 10.1093/nar/gks1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vouk K, Hevir N, Ribic-Pucelj M, Haarpaintner G, Scherb H, Osredkar J, Moller G, Prehn C, Rizner TL, Adamski J. Discovery of phosphatidylcholines and sphingomyelins as biomarkers for ovarian endometriosis. Hum Reprod. 2012;27:2955–2965. doi: 10.1093/humrep/des152. [DOI] [PubMed] [Google Scholar]
- Weeraratna AT, Taub DD. Microarray data analysis: an overview of design, methodology, and analysis. Methods Mol Biol. 2007;377:1–16. doi: 10.1007/978-1-59745-390-5_1. [DOI] [PubMed] [Google Scholar]
- Weichenhan D, Plass C. The evolving epigenome. Human Mol Genet. 2013 doi: 10.1093/hmg/ddt348. in press. [DOI] [PubMed] [Google Scholar]
- Weimar CH, Kavelaars A, Brosens JJ, Gellersen B, de Vreeden-Elbertse JM, Heijnen CJ, Macklon NS. Endometrial stromal cells of women with recurrent miscarriage fail to discriminate between high- and low-quality human embryos. PLoS ONE. 2012;7:e41424. doi: 10.1371/journal.pone.0041424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein JN. Searching for pharmacogenomic markers: the synergy between omic and hypothesis-driven research. Dis Markers. 2001;17:77–88. doi: 10.1155/2001/435746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells D, Levy B. Cytogenetics in reproductive medicine: the contribution of comparative genomic hybridization (CGH) BioEssays. 2003;25:289–300. doi: 10.1002/bies.10232. [DOI] [PubMed] [Google Scholar]
- Werner T. Next generation sequencing allows deeper analysis and understanding of genomes and transcriptomes including aspects to fertility. Reprod Fertil Dev. 2011;23:75–80. doi: 10.1071/RD10247. [DOI] [PubMed] [Google Scholar]
- Weston AD, Hood L. Systems biology, proteomics, and the future of health care: toward predictive, preventative, and personalized medicine. J Proteome Res. 2004;3:179–196. doi: 10.1021/pr0499693. [DOI] [PubMed] [Google Scholar]
- White CA, Salamonsen LA. A guide to issues in microarray analysis: application to endometrial biology. Reproduction. 2005;130:1–13. doi: 10.1530/rep.1.00685. [DOI] [PubMed] [Google Scholar]
- Wilcox AJ, Baird DD, Weinberg CR. Time of implantation of the conceptus and loss of pregnancy. N Engl J Med. 1999;340:1796–1799. doi: 10.1056/NEJM199906103402304. [DOI] [PubMed] [Google Scholar]
- Wishart DS. Computational approaches to metabolomics. Methods Mol Biol. 2010;593:283–313. doi: 10.1007/978-1-60327-194-3_14. [DOI] [PubMed] [Google Scholar]
- Wu Y, Strawn E, Basir Z, Wang Y, Halverson G, Jailwala P, Guo SW. Genomic alterations in ectopic and eutopic endometria of women with endometriosis. Gynecol Obstet Invest. 2006;62:148–159. doi: 10.1159/000093130. [DOI] [PubMed] [Google Scholar]
- Wu JM, Ward RM, Allen-Brady KL, Edwards TL, Norton PA, Hartmann KE, Hauser ER, Edwards DR. Phenotyping clinical disorders: lessons learned from pelvic organ prolapse. Am J Obstet Gynecol. 2013;208:360–365. doi: 10.1016/j.ajog.2012.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada N, Nishida Y, Tsutsumida H, Hamada T, Goto M, Higashi M, Nomoto M, Yonezawa S. MUC1 expression is regulated by DNA methylation and histone H3 lysine 9 modification in cancer cells. Cancer Res. 2008;68:2708–2716. doi: 10.1158/0008-5472.CAN-07-6844. [DOI] [PubMed] [Google Scholar]
- Yanaihara A, Otsuka Y, Iwasaki S, Aida T, Tachikawa T, Irie T, Okai T. Differences in gene expression in the proliferative human endometrium. Fertil Steril. 2005;83(Suppl. 1):1206–1215. doi: 10.1016/j.fertnstert.2004.11.032. [DOI] [PubMed] [Google Scholar]
- Yang H, Zhou B, Prinz M, Siegel D. Proteomic analysis of menstrual blood. Mol Cell Proteomics. 2012;11:1024–1035. doi: 10.1074/mcp.M112.018390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin JQ, Zhao RC, Morris KV. Profiling microRNA expression with microarrays. Trends Biotechnol. 2008;26:70–76. doi: 10.1016/j.tibtech.2007.11.007. [DOI] [PubMed] [Google Scholar]
- Zelenko Z, Aghajanova L, Irwin JC, Giudice LC. Nuclear receptor, coregulator signaling, and chromatin remodeling pathways suggest involvement of the epigenome in the steroid hormone response of endometrium and abnormalities in endometriosis. Reprod Sci. 2012;19:152–162. doi: 10.1177/1933719111415546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Szustakowski J, Schinke M. Bioinformatics analysis of microarray data. Methods Mol Biol. 2009;573:259–284. doi: 10.1007/978-1-60761-247-6_15. [DOI] [PubMed] [Google Scholar]
- Zhang D, Sun C, Ma C, Dai H, Zhang W. Data mining of spatial-temporal expression of genes in the human endometrium during the window of implantation. Reprod Sci. 2012;19:1085–1098. doi: 10.1177/1933719112442248. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.