

Abstract
Impaired host defense after alcohol use is linked to altered cytokine production, however, acute and chronic alcohol differently modulate monocyte/macrophage activation. We hypothesized that in human monocytes, acute alcohol induces hyporesponsiveness to LPS, resulting in decreased TNF-α, whereas chronic alcohol increases TNF-α by sensitization to LPS. We found that acute alcohol increased IL-1R-associated kinase-monocyte (IRAK-M), a negative regulator of IRAK-1, in human monocytes. This was associated with decreased IκBα kinase activity, NFκB DNA binding, and NFκB-driven reporter activity after LPS stimulation. In contrast, chronic alcohol decreased IRAK-M expression but increased IRAK-1 and IKK kinase activities, NFκB DNA binding, and NFκB-reporter activity. Inhibition of IRAK-M in acute alcohol-exposed monocytes using small interfering RNA restored the LPS-induced TNF-α production whereas over-expression of IRAK-M in chronic alcohol macrophages prevented the increase in TNF-α production. Addition of inhibitors of alcohol metabolism did not alter LPS signaling and TNF-α production during chronic alcohol exposure. IRAK-1 activation induces MAPKs that play an important role in TNF-α induction. We determined that acute alcohol decreased but chronic alcohol increased activation of ERK in monocytes and ERK inhibitor, PD98059, prevented the chronic alcohol-induced increase in TNF-α. In summary, inhibition of LPS-induced NFκB and ERK activation by acute alcohol leads to hyporesponsiveness of monocytes to LPS due to increased IRAK-M. In contrast, chronic alcohol sensitizes monocytes to LPS through decreased IRAK-M expression and activation of NFκB and ERK kinases. Our data indicate that IRAK-M is a central player in the opposite regulation of LPS signaling by different lengths of alcohol exposure in monocytes.
Alcohol consumption has both beneficial and harmful effects based on the duration and extent of alcohol exposure. Although moderate alcohol use may have beneficial effects on the cardiovascular system, it also disrupts immune defenses against infections and increases susceptibility to bacterial pneumonia, hepatitis C, and HIV infections (1–3). In contrast, chronic alcohol drinking leads to organ injury and increased mortality (4, 5). The cellular mechanisms leading to these diverse effects of acute and chronic use are not clearly defined. Defects in innate immune cells such as monocytes and macrophages could contribute to alcohol-induced organ injury. Studies in animal and human models of acute and chronic alcohol consumption have shown evidence for the modulation of macrophage function (3, 6, 7).
Monocytes respond to endotoxin via recognition by the TLR4 complex resulting in production of inflammatory cytokines, particularly TNF-α. LPS binds to the CD14/TLR4 complex followed by activation of a cascade of down-stream signaling molecules including activation of IL-1R-associated kinase-1 (IRAK-1)3 that activates the IκBα kinase (IKK) complex and MAPK followed by dissociation of IκBα, nuclear translocation, and DNA binding of NFκB to the promoter region of target genes such as adhesion molecules and proinflammatory cytokines particularly, TNF-α, IL-1β, and IL-6 (8). Increased serum TNF-α has been observed in patients with alcoholic hepatitis (9). The critical role of TNF-α in alcoholic injury was elucidated by experiments that successfully used anti-TNF-α Abs to prevent liver injury in alcoholic hepatitis and alcohol-fed rats (10, 11).
Our earlier studies have shown that acute alcohol exposure is associated with LPS-induced anti-inflammatory effects in monocytic cells in humans both in vitro and in vivo (12–14). This is in contrast to the effects of chronic alcohol on inflammatory cytokine gene expression where TNF-α contributes to liver injury in humans and in mouse models (6, 15, 16). Hence, studying differences in regulation of LPS signaling by acute and chronic alcohol exposure is pertinent to the innate immune cell contribution to development and progression of alcoholic liver disease. In this study, we hypothesized that based on the length of exposure, acute and chronic alcohol renders monocytes hyporesponsive to subsequent LPS or sensitizes to LPS, respectively. To test our hypothesis, we developed a modified in vitro system to study the effects of acute and chronic alcohol exposure on LPS signaling events and TNF-α production in human monocytes (15). We report that acute alcohol increases IL-1R-associated kinase-monocyte (IRAK-M), a negative regulator of LPS signaling and leads to decreased downstream activation of the LPS signaling cascade culminating in impaired NFκB DNA binding and transactivation and, finally, decreased TNF-α production. In contrast, chronic alcohol switches the anti-inflammatory to a proinflammatory response by decreasing IRAK-M and increasing IRAK-1 and IKK kinase activation followed by enhanced NFκB DNA binding, transactivation, and ERK kinase activation and TNF-α production.
Materials and Methods
Cell culture and reagents
Monocytes from human peripheral blood were isolated by selective adherence from Ficoll-Hypaque-purified mononuclear cell preparations as previously described (14). Healthy individuals aged 18 to 60, females and males with no previous alcohol abuse history who consumed less than 6 drinks/week were recruited in the study. The study was reviewed and approved by the Institutional Committee for Protection of Human Subjects in Research.
RAW 264.7 macrophages were purchased from American Type Culture Collection and maintained in Dulbecco’s modified medium (Invitrogen Life Technologies) containing 10% FBS (HyClone). PD98059 and 4-methylpyrazole and cyanamide were purchased from Sigma-Aldrich. LPS (Echerichia coli strain 0111:B4) was from Difco and NFκB oligo synthesized by Oligos.
Alcohol exposure and cell stimulations
Monocytes or RAW 264.7 macrophages were stimulated with Escherichia coli-derived LPS (100 ng/ml), 25 mM ethanol, and the combination of LPS and ethanol at the times indicated in the figure legends. The 25 mM in vitro ethanol concentration approximates a 0.1g/dl blood alcohol level, which is achieved in vivo after a dose of moderate drink and is a little above the legal limit of blood alcohol concentration. Cell viability was not affected by ethanol or LPS treatment. Monocytes and/or macrophages were plated in 6-well plates and exposed to 25 mM alcohol over an extended period of up to 7 days. For short-term or acute alcohol exposure, cells were exposed to 25 mM alcohol for 24 h or less followed by LPS stimulation.
For prolonged alcohol exposure, cells exposed to 25 mM alcohol were placed in a Billups-Rothenburg chamber with twice the alcohol concentration in the bottom of the chamber to saturate the chamber and maintain the alcohol concentration in the wells for 7 days, as described earlier by other groups (15). The alcohol concentration in each well was measured using the Analox Alcohol Analyzer (Analox Instruments). The concentrations were maintained to 25 mM ± 4.5 over the 7-day period (mM EtOH: Day 1: 24 ± 0.1; Day 2: 21.5 ± 0.5; Day 3: 25 ± 0.1; Day 4: 22 ± 0.2; Day 5: 24 ± 0.7; Day 6: 22 ± 0.1; Day 7: 19.5 ± 0.5).
ELISA
TNF-α and IL-1β were estimated in cell-free supernatants using ELISA from BD Pharmingen.
Silencing of IRAK-M by small interfering RNA (siRNA)
For knock-down of IRAK-M by siRNA, Raw 264.7 macrophages were transfected with IRAK-M siRNA, and control siRNA (Applied Biosystems) using siPORT NeoFx transfection agent (Applied Biosystems) according to the manufacturer’s instructions. Transfection efficacy was determined by transfecting the cells with GAPDH siRNA. Forty-eight hours after transfection, macrophages were then exposed to 25 mM alcohol for 24 h followed by stimulation with LPS for 6 h. Total RNA extracts and whole cell lysates were analyzed for IRAK-M by real-time PCR and Western blotting to confirm IRAK-M knock-down and supernatants collected were analyzed for TNF-α by ELISA.
Overexpression of IRAK-M
For overexpression of IRAK-M, IRAK-M cDNA clone and control vector (pCMV6-XL-4) were purchased from Origene. Raw macrophages exposed to alcohol for 7 days, were transiently transfected with IRAK-M expression vector and control vector using Lipofectamine and plus reagents (Invitrogen) according to the manufacturer’s instructions. The transfected cells were either stimulated with LPS for 24 h to check the effect of overexpression or for 6 h to determine the production of TNF-α in IRAK-M overexpressed cells.
RNA analysis
RNA was extracted using the RNeasy Mini kit (Qiagen) according to the manufacturer’s instructions, and on-column DNase digestion was performed using the RNase-Free DNase Set (Qiagen). The RNA was quantified by spectrophotometric analysis and the quality of RNA was routinely checked by measurement of OD (260/280 and 260/230 ratio) and by gel electrophoresis. cDNA synthesis was performed by reverse transcription of RNA using the Reverse Transcription System (Promega). Real-time quantitative PCR was performed using the iCycler iQ Real-Time Detection System (Bio-Rad Laboratories) as described before (17). Human TNF-α primers were obtained from Maxim Biotech and human 18S primers (Forward 5′GTA ACC CGT TGA ACC CCA TT 3′; Reverse 5′ CCA TCC AAT CGG TAG TAG CG 3′) and IRAK-M primers (Forward 5′ AGA GCA GCT GTT CCT CCA AA 3′; Reverse 5′ AAT TGA GCG TGG ATT TGG TC 3′) were synthesized by IDT.
FACS analysis of surface markers
Monocytes were incubated with saturating concentrations of fluorochrome-conjugated mAbs at 4°C for 30 min, and then washed twice in PBS containing 2% FBS and fixed in 1% paraformaldehyde. Data were collected using a FACScan II flow cytometer (BD Biosciences), and data analysis was performed using FlowJo software (BD Biosciences). The fluorochrome-conjugated mAbs against human cell surface proteins CD14, TLR2, and TLR4 and their respective isotype controls were from Bio-Source International and BD Pharmingen.
Preparation of nuclear and cytoplasmic extracts and electrophoretic gel mobility shift assay
Nuclear and cytoplasmic extracts from cells with or without stimulation at 37°C were performed by the method of Schatzle et al. (18) as described earlier (19).
A consensus double-stranded NFκB oligonucleotide (5′AGTTGAG GGGACTTTCGC3′) was used and EMSA was performed (19).
Immunoblotting
Nuclear or cytoplasmic proteins (20 μg) were loaded onto each well, separated on 10% SDS-polyacrylamide gel and electroblotted onto nitro-cellulose membranes. Nonspecific binding was blocked by incubation of the membranes in TBS/1% nonfat dried milk/0.1% Tween 20 followed by Abs indicated in the figure legends. The Abs against phospho and total Erk, JNK, and p38 were purchased from Cell Signaling Technology and CYP2E1 and IRAK-M was from Chemicon. The rabbit Ab against IκBα was from Santa Cruz Biotechnology. β-actin and TATA-binding protein Abs were from Abcam. The Abs were detected using HRP-conjugated secondary Abs (Santa Cruz Biotechnology) and chemiluminescence assay reagents from Cell Signaling Technology.
NFκB reporter assay
The NFκB-dependent reporter plasmid p(κB)5-Luc, a gift from Dr. N. Mackman (Scripps Research Institute, La Jolla, CA) contains five tandem copies of the NFκB site from the human TNF-α gene. RAW264.7 macrophages either exposed to alcohol for 4 days or control macrophages were transfected with the reporter plasmids 5×NFκB-firefly luciferase and Renilla-Luciferase as control (Promega) using the transfection agent FuGENE 6 (Roche Applied Science) as reported earlier (20). After 24 h of incubation, control cells were treated with 25 mM alcohol in the presence or absence of LPS (100 ng/ml) or chronic alcohol exposed macrophages were exposed to LPS for 8 h and luciferase activity assessed with Dual Glo Luciferase Assay reagent (Promega) according to the manufacturer’s instructions. NFκB transcriptional activity as detected by firefly luciferase activity was normalized with the Renilla luciferase activity. The relative light units represent an average of triplicate samples.
Kinase assay
The IKK or IRAK-1 assay was performed as described previously (20). In brief, IKKβ and IRAK-1 from the cytoplasmic extract (500 μg) was precipitated with respective Abs in the lysis buffer (20mM Tris-HCl (pH 7.6), 150 mM NaCl, 1 mM EDTA, 1 mM DTT, 1% Triton X-100) followed by treatment with 50 μl of anti-rabbit IgG beads (eBioscience) and assayed in kinase assay buffer containing 20 mM HEPES (pH 7.6), 10 mM MgCl2, 1 mM DTT, 5 μCi [γ32P] ATP (NEN Life Science Products), 10 μM unlabeled ATP, and 0.5 μg of substrate (For IKKβ kinase: GST-IκBα (Santa Cruz Biotechnology); For IRAK-1 kinase: MBP (myelin basic protein) (Santa Cruz Biotechnology).
Statistical analysis
The Wilcoxon nonparametric analysis was used in the Statview (SAS Institute) program on a Mac G4 computer (Apple) to analyze TNF-α levels in human monocytes. For all other studies, the one-tailed t test was used. A p < 0.05 was considered significant in all studies.
Results
Opposite effects of acute and chronic alcohol exposure on TNF-α expression in human monocytes
Circulating monocytes play an important role in acute alcohol induced immunosuppression and chronic alcohol-related liver injury due to their response to increased endotoxin (LPS) during infections and in the serum of alcoholic hepatitis patients (9). In this study, we investigated the effects of acute (short-term) and chronic (prolonged) alcohol exposure of human monocytes using an in vitro system developed to maintain physiologically relevant (25 mM) alcohol concentrations over a period of 7 days in culture. Fig. 1A shows that acute alcohol exposure (1–2 days) decreases LPS-induced TNF-α production. However, extended exposure to alcohol for 4 –7 days (chronic) resulted in significantly increased LPS-induced TNF-α production in human monocytes. Changes in TNF-α protein levels correlated with TNF-α mRNA levels wherein initial alcohol exposure for 3 h to 1 day decreased LPS-induced TNF-α mRNA and 4 –7 days of alcohol exposure exhibited increased LPS-induced TNF-α mRNA (Fig. 1B) in human monocytes. These results indicate that alcohol exposure of monocytes induces hyporesponsiveness to LPS in the initial phase (acute) whereas prolonged (chronic) alcohol exposure sensitizes monocytes to LPS-induced cytokine production regulated at the transcriptional and translational level.
FIGURE 1.

Acute alcohol decreases but chronic alcohol treatment of human monocytes increases LPS-induced TNF-α production. A, Human monocytes were exposed to 25 mM alcohol (Et) from 1 to 7 days followed by LPS (100 ng/ml) treatment for 18 h and detection of TNF-α in the cell-free supernatants by ELISA. Mean values of TNF-α (pg/ml/106 cells) ± SE from a total of ten individuals are shown (compared with LPS; *, p < 0.001). B, LPS (100 ng/ml) treatment for 3 h and TNF-α mRNA determination by real-time PCR using specific TNF-α primers as described in Materials and Methods. The bar graph represents fold induction of mRNA ± SE (compared with LPS; *, p < 0.01; **, p < 0.001; n = 6).
Acute alcohol induces IRAK-M while chronic alcohol decreases IRAK-M to regulate down-stream LPS signaling
Various studies have explored the role of CD14 and TLR4, receptors of LPS, in chronic alcohol-induced sensitization to LPS-induced liver injury (21, 22) as well as in acute alcohol induced immune functions (4, 17). Because acute alcohol exposure of human monocytes reduces, whereas chronic alcohol exposure augments, LPS-induced TNF-α production, next we determined whether alcohol exposure of human monocytes for 1 day or 7 days had any effect on CD14 and TLR4 expression. Fig. 2A shows that acute (1 day) and chronic alcohol (7 days) exposure did not alter surface expression of CD14 and TLR4 as analyzed by flow cytometry, suggesting that acute and chronic alcohol mediated changes in TNF-α production is independent of surface receptor expression on human monocytes.
FIGURE 2.

Alcohol alters IRAK-M and IRAK-1 levels without any effect on CD14 and TLR4 expression. A, Human monocytes were either exposed to 25 mM alcohol (Et) for 1 day (acute) or 7 days (chronic), stained with CD14 and TLR4 Abs, and analyzed by flow cytometry. Iso-type IgG Abs were used as negative controls. B, Human monocytes were exposed to 25 mM alcohol for 7 days (Chr) followed by LPS (100 ng/ml) for 15 min. Immunoprecipitation (IP) of the cytoplasmic extracts with IRAK-1 Ab in lysis buffer was conducted as described in Materials and Methods. The kinase assay was performed with MBP (myelin basic protein) as substrate. The proteins were separated on SDS-PAGE and the gel depicts the 32P-phosphorylated MBP. Equal protein in the IP samples was determined by immunoblotting (WB) with IRAK-1 Abs. The bar graph below represents the cpm incorporated in the substrate as measured by scintillation counter. (compared with unst; *, p < 0.01; compared with LPS; **, p < 0.05; n = 4). C, Human monocytes were exposed to 25 mM alcohol (Et) for 1 day (Ac) or 7 days (Chr) followed by LPS (100 ng/ml) treatment for 6 h and IRAK-M mRNA determination by real-time PCR using specific IRAK-M primers as described in Materials and Methods. The bar graph represents fold induction of mRNA ± SE (compared with unst; *, p < 0.01; compared with LPS; **, p < 0.02; n = 4). D, Monocytes were exposed to 25 mM alcohol (Et) for 1 day (Ac) or 7 days (Chr) followed by LPS (100 ng/ml) treatment for 24 h and IRAK-M protein levels determined by immunoblotting using anti-IRAK-M Abs as described in Materials and Methods. Equal loading of protein is demonstrated using an internal control anti-β-actin Ab.
Engagement of the LPS receptor enhances activation of various adaptor proteins such as MyD88, IRAK-4, and IRAK-1 to the CD14-TLR4 complex (8). Subsequently, IRAK-1 becomes phosphorylated and then interacts with TRAF6 leading to activation of IKK kinase (23). Because we observed that acute and chronic alcohol exposure does not affect CD14 or TLR4 expression, we sought to determine whether any intracellular membrane-proximal signaling molecules were affected during alcohol exposure of human monocytes. In contrast to decreased LPS-induced IRAK-1 kinase activity by acute alcohol observed in our previous study (17), in this study we found that chronic alcohol augmented LPS-induced IRAK-1 kinase activity (Fig. 2B), indicating that acute and chronic alcohol exposure differently modulates IRAK-1 kinase activity.
IRAK-1 kinase activity is regulated by a negative regulator of LPS signaling, IRAK-M, expressed in monocytes and demonstrated to play a critical role in hyporesponsiveness and sensitization to LPS (24 –26). IRAK-M acts by inhibiting dissociation of MyD88 and IRAK-1 to turn off down-stream signaling (24). Thus, we hypothesized that acute and chronic alcohol differently regulates IRAK-M in human monocytes to influence IRAK-1 kinase activity. Fig. 2C shows that IRAK-M mRNA was induced after LPS stimulation for 6 h in human monocytes. Although LPS-induced IRAK-M mRNA was increased by acute alcohol, chronic alcohol significantly decreased IRAK-M mRNA at baseline and in response to LPS stimulation (Fig. 2C). IRAK-M protein expression followed a similar pattern wherein acute alcohol increased and chronic alcohol exposure decreased IRAK-M levels in the cytoplasm of human monocytes (Fig. 2D). Thus, it is likely that acute alcohol exposure up-regulates IRAK-M and thereby inhibits IRAK-1 kinase activity whereas chronic alcohol-mediated down-regulation of IRAK-M could permit persistent activation of IRAK-1 kinase.
To confirm the regulatory role of IRAK-M in acute and chronic alcohol-exposed monocytic cells, we either inhibited the IRAK-M using siRNA after acute alcohol exposure or overexpressed IRAK-M in chronic alcohol exposed macrophages. Fig. 3A shows that in acute alcohol-exposed macrophages with 70% knockdown of IRAK-M mRNA (upper panel) and protein (middle panel), significant restoration of LPS-induced TNF-α levels occurred. These data indicate that IRAK-M contributes to the reduction in LPS-induced TNF-α production during acute alcohol exposure. In contrast, efficient overexpression of IRAK-M indicated by western blot analysis (upper panel) in chronic alcohol exposed macrophages prevented up-regulation of LPS-induced TNF-α levels (Fig. 3B; lower panel) suggesting an important role for IRAK-M in increased proinflammatory responses during chronic alcohol exposure.
FIGURE 3.

IRAK-M expression regulates TNF-α during alcohol exposure. A, Raw macrophages (control cells) were transfected with control siRNA (scr) and IRAK-M-specific siRNA and after 48 h exposed to 25 mM alcohol followed by LPS for 6 h. Total RNA extracts were analyzed for IRAK-M mRNA by real time PCR and bar graph (upper panel) represented as percentage of IRAK-M gene expression (compared with control; *, p < 0.009). Whole cell lysates in the presence or absence of LPS for 6 h were analyzed for IRAK-M protein by Western blot analysis (middle panel) and supernatants analyzed for TNF-α production by ELISA (lower panel). Mean values of TNF-α (pg/ml/106 cells) ± SE from three experiments are shown (compared with control siRNA; #, p < 0.007; **, p < 0.01). B, Raw 264.7 macrophages were exposed to chronic alcohol for 7 days, transfected with either control vector or IRAK-M overexpression vector (Origene) for 36 h and treated without (inset graph) or with LPS for 6 h. Whole cell lysates were subject to Western blotting using anti- IRAK-M (upper panel) Ab and supernatents were analyzed for TNF-α by ELISA (lower panel). Mean values of TNF-α (pg/ml/106 cells) ± SE from a three experiments are shown (compared with vector control; *, p < 0.01; **, p < 0.001).
Acute and chronic alcohol exposure affect IKK activation and ERK MAP activation
Signals emanating from the IRAK-1 activation lead to activation of the inhibitor of κB kinase (IKK) complex that mediates phosphorylation of IκBα and activation of NFκB (23). Because we observed that acute and chronic alcohol regulates IRAK-1 activation inversely, we next evaluated whether the acute and chronic alcohol exposure of human monocytes regulates down-stream IKK kinase activity. Fig. 4 shows that while acute alcohol exposure decreased IKK-β kinase activity, chronic alcohol increased IKK-β kinase activity. These results are in agreement with decreased activation of proximal IRAK-1 in acute alcohol treated cells and enhanced IRAK-1 activation after chronic alcohol exposure in monocytes.
FIGURE 4.

Acute alcohol decreases whereas chronic alcohol increases LPS-induced IKK kinase activity in human monocytes. Human monocytes were exposed to alcohol (Et) for 1 day (Ac) or 7 days (Chr) followed by stimulation with LPS (100 ng/ml) for 15 min. IP of the cytoplasmic extracts with IKKβ Abs in lysis buffer was followed by kinase assays using the GST-IκBα as substrate as described in Materials and Methods. The proteins were separated on SDS-PAGE and the gels depict the 32P-phosphorylated GST-IκBα. Equal protein loading in the input samples was determined by immunoblotting with β-actin Ab. The bar graph represents the cpm incorporated in the substrate as measured by scintillation counter. (compared with unst; *, p < 0.05; compared with LPS; #, p < 0.05; **, p < 0.01; n = 3).
In addition to the IKK kinase activation, LPS-induced IRAK-1 activation triggers cascades of intracellular signaling events including the MAPK such as the ERK, the JNK, and the p38 kinase (27). In this study, we determined the effect of acute and chronic alcohol exposure on activation of MAPKs: ERK, JNK, and p38. Using Western blotting and phospho-specific MAPK Abs we observed that acute alcohol exposure decreased LPS-induced phospho-ERK kinase levels whereas chronic alcohol exposure significantly increased LPS-induced phospho-ERK kinase activity without any change in total ERK levels (Fig. 5, A and B). Furthermore, a minimal increase in LPS-induced phospho-p38 MAPK was noted in monocytes exposed to acute alcohol whereas chronic alcohol exposure did not have any effect on LPS-induced phospho-p38 MAPK (Fig. 5A). The JNK MAPK was not significantly affected by alcohol exposure in human monocytes. Importantly, inhibition of the ERK MAPK, using a specific inhibitor PD98059, in chronic alcohol exposed monocytes resulted in inhibition of chronic alcohol induced increase in LPS-induced TNF-α production (Fig. 5C) indicating an important role for ERK MAPK in inflammatory cytokine production after chronic alcohol exposure in human monocytes.
FIGURE 5.

Inhibition of MAPK-ERK activation by acute alcohol whereas chronic alcohol increases phospho-ERK levels in monocytes. Monocytes were exposed to 25 mM alcohol (Et) for 1 day (Ac) or 7 days (Chr) followed by LPS (100 ng/ml) treatment for 15 min. A, Total and phospho-ERK, phospho-p38 and phospho-JNK protein levels determined by immunoblotting using anti-specific phospho-antibodies as described in Materials and Methods. B, Mean density units ± SE of the phospho-ERK bands from a total of six individuals is shown. (compared with LPS; *, p < 0.05; #, p < 0.02, n = 6). C, Monocytes were exposed to 25 mM alcohol (Et) for 7 days (Chr) followed by incubation with ERK inhibitor (PD98059, 50 μM) for 4 h and stimulation with LPS (100 ng/ml) for 18 h. Cell-free supernatants were collected and assayed for TNF-α in an ELISA. The data are represented as mean ± SE (compared with LPS; **, p < 0.001; compared with Chr Et plus LPS; *, p < 0.02, n = 6).
Differential regulation of IκB-NFκB activation by acute vs chronic alcohol exposure in monocytes
NFκB activation is a major regulator of LPS-induced inflammatory cascade signaling. LPS induced IKK kinase activation culminates in rapid degradation of cytoplasmic IκBα (8). LPS stimulation of human monocytes revealed decreased IκBα in the cytoplasmic extracts suggesting that NFκB activation was accompanied by IκBα degradation (Fig. 6). As shown previously by our group (19, 20), our results in this study confirm that IκBα levels remained low in LPS plus acute alcohol exposed monocytes even though IKK kinase activity was reduced in these cells (Fig. 4). Interestingly, chronic alcohol exposure for 4 –7 days followed by LPS stimulation resulted in decreased IκBα levels in the cytoplasm, indicating that IKK kinase activation may induce IκBα degradation in chronic alcohol-exposed monocytes. These results suggest that while acute alcohol-induced IκBα degradation could be independent of IKK kinase, chronic alcohol-induced IκBα degradation is dependent on IKK kinase activity.
FIGURE 6.

Alcohol exposure, acute and chronic, promotes LPS-induced IκBα degradation in monocytes. Monocytes were exposed to alcohol for 1 (Ac), 4, and 7 days followed by simulation with LPS (100 ng/ml) for 60 min. Cytoplasmic extracts were prepared and immunoblotted with total IκBα Ab. The bar graph represents mean density units ± SE from a total of six individuals (compared with LPS; *, p < 0.02, n = 6).
LPS-induced IκBα degradation results in NFκB translocation, DNA binding, and transactivation of target genes such as TNF-α (8). When acute alcohol was combined with LPS, LPS-induced NFκB binding activity was significantly decreased in monocytes exposed to alcohol for 1 h (Fig. 7A). However, chronic alcohol exposure of monocytes (4 –7 days) resulted in a significant increase in LPS-induced NFκB activation (Fig. 6A). These results indicate that the opposite effects of acute and chronic alcohol on IRAK-1 and IKK kinase activation extend to differences in NFκB DNA binding activity in human monocytes.
FIGURE 7.

Opposite effects of acute and chronic alcohol exposure on NFκB binding activity is independent of alcohol metabolism in human monocytes. A, Monocytes were exposed to alcohol for 1 (Ac), 4, and 7 days followed by simulation with LPS (100 ng/ml) for 60 min. NFκB was detected in the nuclear extracts by EMSA using a 32P-labeled double-stranded NFκB oli-gonucleotide. The bar graph shows mean density ± SE of a total of six individuals (compared with LPS; *, p < 0.01). B, RAW 264.7 macrophages were exposed to alcohol (Et) for 1 (ac) or 4 days (chr) and then transiently transfected with NFκB reporter gene constructs carrying five tandem copies of the NFκB binding site in front of the firefly luciferase gene and the Renilla luciferase construct. At 24 h after transfection, cells were treated with LPS (100 ng/ml) for 8 h. Cells were then lysed to determine firefly luciferase activity and normalized to the Renilla luciferase activity. The bar graph represents fold activation of the luciferase gene as compared with the unstimulated control of a total of three experiments. (compared with LPS; *, p < 0.01; **, p < 0.02, n = 3). C, Monocytes were exposed to 25 mM alcohol (Et) for 1 to 7 days and CYP2E1 protein levels determined by immunoblotting using anti-CYP2E1 Abs as described in Materials and Methods. The bar graph represents mean density ± SE of a total of six individuals. D, Monocytes were exposed to alcohol for 7 days along with 4-methyl-prazole or cyanamide followed by stimulation with LPS (100 ng/ml) for 60 min. NFκB was detected in the nuclear extracts by EMSA using a 32P-labeled double-stranded NFκB oligonucleotide. The bar graph shows mean density ± SE of a total of six individuals (compared with unst; *, p < 0.001; compared with LPS; **, p < 0.001). E, Monocytes were exposed to 25 mM alcohol (Et) for 7 days (Chr) along with 4-methyl-prazole or cyanamide followed by stimulation with LPS (100 ng/ml) for 18 h. Cell-free supernatants were collected and assayed for TNF-α in an ELISA. The data are represented as mean ± SE (compared with LPS; *, p < 0.001, n = 6).
Transcription of proinflammatory cytokine genes is controlled by binding of specific transcription factors in the promoter of target genes. NFκB, a pivotal transcription factor binds in the promoter region of the TNF-α gene and initiates transcription (18). Because we found modulation of NFκB binding activity, we wanted to evaluate whether alcohol affects NFκB promoter-driven transcriptional activity. Transient transfection of the NFκB-driven luciferase reporter in control RAW 264.7 cells or cells exposed to alcohol for 4 days, a time-point showing enhanced NFκB binding in human monocytes, was chosen. Fig. 7B shows that LPS stimulation significantly induced NFκB-mediated luciferase reporter activity. Furthermore, as reported earlier (20), exposure of 25 mM alcohol for up to 24 h or 1 day followed by LPS resulted in a significant reduction of the NFκB-mediated luciferase reporter activity. However, transfection of chronic alcohol-exposed RAW 264.7 cells showed a significant increase in LPS-induced NFκB-mediated luciferase activity (Fig. 7B). These results propose that acute and chronic alcohol exposure modulates NFκB binding activity as well as transactivation suggesting a pivotal role for NFκB in regulating TNF-α expression during alcohol exposure.
Alcohol metabolizing enzymes such as CYP2E1 play an important role in oxidative stress related liver injury (29) and is directly involved in production of reactive oxygen species. Human monocyte-derived macrophages induce CYP2E1 in response to alcohol treatment at a level lower than the liver cells but similar to other extrahepatic tissues (30). Reactive oxygen species generation due to CYP2E1 has been shown to directly affect NFκB activation, and this could influence inflammatory cytokine gene expression (31). Because chronic alcohol induces NFκB activation, using 4-methyl pyrazole, a pharmacological inhibitor of CYP2E1 and cyanamide, an aldehyde dehydrogenase inhibitor we determined whether alcohol metabolism was involved in this effect. Fig. 7C shows that chronic alcohol exposure over a period of 7 days moderately induced CYP2E1 in human monocytes. Inhibition of alcohol metabolizing enzymes using 4-methyl pyrazole and cyanamide however, did not affect chronic alcohol-induced NFκB DNA binding activity (Fig. 7D) and LPS-induced TNF-α levels (Fig. 7E), suggesting that although chronic alcohol induces CYP2E1 in human monocytes, the effect of alcohol on NFκB regulation is CYP2E1 independent.
Discussion
Studies on acute alcohol-induced immunosuppression leading to increased susceptibility to infections and chronic alcohol-mediated liver disease have indicated a key role for innate immune cells. In vivo studies in mouse models have shown that while chronic alcohol increases sensitivity of macrophages to LPS (7), acute alcohol in human peripheral blood monocytes decreases LPS-mediated proinflammatory cytokines (14). The precise intracellular signaling molecules involved in reversing the acute immunosuppressive effects of alcohol to a hyperinflammatory state are not well understood. In this study, for the first time using human peripheral blood monocytes, we demonstrate the signaling molecules involved in acute alcohol-induced hyporesponsiveness to LPS and chronic alcohol-mediated sensitivity of monocytes to subsequent LPS stimulation. Our results show that the primary target of alcohol is IRAK-M, a negative modulator of LPS signaling, that is involved in the opposite effects of acute and chronic alcohol on the LPS-induced signaling pathway (Fig. 8). Acute alcohol exposure of monocytes appears to induce hyporesponsiveness to LPS by increasing IRAK-M levels. However, chronic alcohol exposure decreases IRAK-M levels resulting in sensitization to subsequent LPS stimulation. IRAK-M thus serves as a molecular switch to convert an acute alcohol-induced anti-inflammatory phenotype to chronic alcohol-induced hyperinflammatory phenotype. Our results revealed that mechanisms involved in acute alcohol-induced LPS hyporesponsiveness involve decreased IRAK-1 and IKK kinase activity, decreased MAPK-ERK activation, and reduced NFκB activation independent of IκBα degradation. In contrast, reduced IRAK-M after chronic alcohol exposure of monocytes was associated with increased IRAK-1 and IKK kinase activity, increased MAPK-ERK activity, and IκBα-dependent NFκB activity (Fig. 7). These effects of acute or chronic alcohol exposure were found to be independent of CD14 and TLR4 surface expression, indicating that alcohol exclusively alters intracellular signaling molecules to exert its effect on proinflammatory cytokine expression.
FIGURE 8.
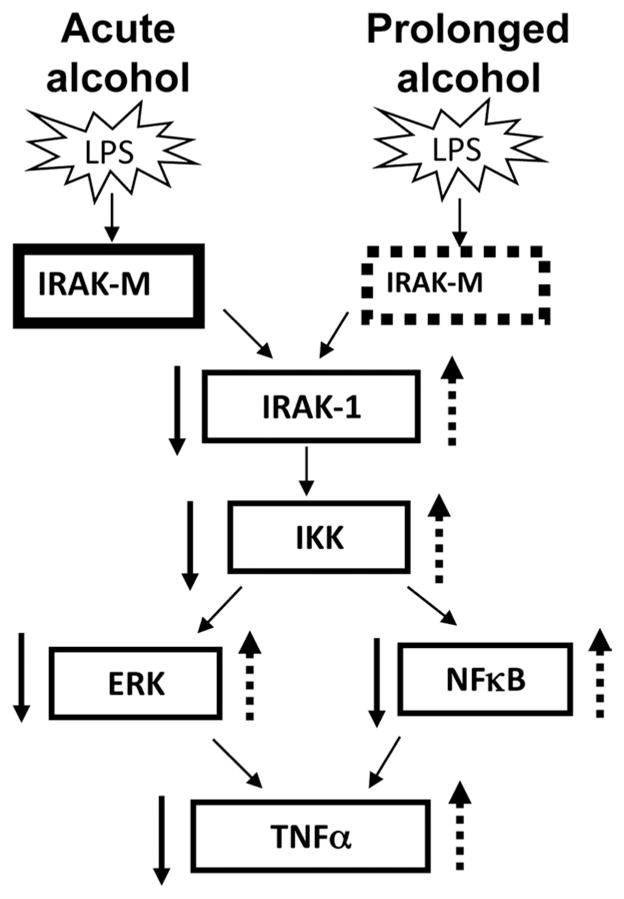
Alcohol, based on the length of exposure, alters IRAK-M to differently regulate LPS signaling in human monocytes. Acute alcohol exposure induces whereas chronic alcohol decreases IRAK-M in human monocytes to either reduce or augment activity of down-stream LPS signaling molecules IRAK-1, IKK, ERK, and NFκB, and finally TNF-α production, respectively.
The influence of acute and chronic alcohol exposure on innate immune cells from peripheral blood monocytes to tissue macrophages originating from spleen, lungs, and liver can be diverse. Earlier studies have shown that sterilization of the gut with antibiotics abrogated the in vivo effects of acute alcohol on Kupffer cells, indicating that circulating endotoxin played a key role in the effects of alcohol (32). Furthermore, in vivo acute alcohol exposure of murine Kupffer cells show decreased responsiveness to LPS, and this has been related to decreased IRAK expression, reduced NFκB activity (33), and decreased intracellular Ca2+ concentration (32). Subsequent studies also revealed that acute in vivo ethanol exposure of Kupffer cells increased CD14 expression through a mechanism dependent on AP-1 activation (34). In addition, splenic and alveolar macrophages exposed to a single dose of in vivo alcohol followed by ex vivo stimulation of LPS showed a significant decrease in proinflammatory responses and impaired phagocytic function, likely due to inhibition of pattern recognition receptors and down-stream MAPK (36). In our studies, acute alcohol did not affect surface expression of LPS receptors, CD14, and TLR4, whereas LPS-mediated signaling was altered in peripheral blood human monocytes.
In addition to down-regulation of LPS receptors, negative regulators of LPS signaling play an important role in LPS hyporesponsiveness (37). Negative regulators such as IRAK-M, SOCS-1, A20, and Tollip affect LPS signaling at different levels (37) and are important in LPS hyporesponsiveness and sensitization. Although IRAK-M and SOCS-1 directly inhibit LPS-induced IRAK-1 kinase activity, Tollip affects IL-1 receptor-associated IRAK-1 kinase and A20 directly inhibits IKK kinase and NFκB activation (37). Recent studies have shown that up-regulation of IRAK-M is essential for hyporesponsiveness to a second endotoxin stimulus in Kupffer cells (26). In this study, we show that acute exposure of human monocytes to alcohol for up to 1 day results in increased IRAK-M mRNA and protein levels. Previous studies from our laboratory have shown that acute in vivo and in vitro alcohol induces SOCS-1 expression to affect proinflammatory cytokine signaling (38). Increased IRAK-M levels were accompanied by decreased LPS-induced IRAK-1 (17) and IKK kinase activity and decreased NFκB binding along with IκBα degradation. Acute alcohol exposure not only decreased NFκB DNA binding but also reduced its transactivation potential. This is in agreement with previous data showing that acute alcohol promotes DNA binding of the NF-κB p50/p50 homodimer that has minimal transactivational capacity (39). NFκB dimers are released from their complex with IκB proteins followed by their translocation to the nucleus. Although acute alcohol reduced NFκB binding activity with ongoing IκBα degradation, IRAK-1 and IKK kinase activity were suppressed by acute alcohol in monocytes. Our earlier results also show that acute alcohol exposure affects NFκB p65 phosphorylation via modulation of IKK kinase to result in altered NFκB binding and TNF-α expression (20) in human monocytes. Conflicting reports show either a down-regulation or no change in ERK, JNK, and p38 MAPK activity during LPS hyporesponsiveness in macrophages (40, 41). In our experiments, acute alcohol selectively reduced ERK MAPK levels with no significant effect on JNK and p38 MAPK. Our results overall indicate that, similar to the in vivo effects of acute alcohol on Kupffer cells (32), short-term alcohol exposure induces an “LPS hyporesponsive” state in human blood monocytes resulting in suppressed TNF-α production. Additionally, acute alcohol exposure could lead to LPS hyporesponsiveness in human peripheral blood monocytes limiting inflammation and favoring an increased susceptibility to infections similar to that described in septic shock patients (42).
In contrast to acute alcohol, chronic alcohol exposure of human monocytes induced sensitization to LPS resulting in increased TNF-α expression. Exposure of monocytes to alcohol from 4 to 7 days shows a gradual increase in NFκB activation as well as TNF-α production. These results are consistent with the hypothesis that prolonged alcohol sensitizes to LPS as has been proposed in alcoholic liver disease wherein alcohol-induced increase in gut-derived endotoxin results in increased proinflammatory cytokine induction (43). Several studies have shown that Kupffer cells treated with chronic alcohol in vitro or in vivo were highly sensitive to LPS and showed increased TNF-α production (44). Chronic alcohol-induced sensitization in Kupffer cells to LPS has been attributed to increased IRAK kinase expression and activity and in part due to increased CD14 expression (33). Our studies demonstrated that chronic alcohol exposure of human monocytes decreases IRAK-M expression and increases IRAK-1 kinase activity without changes in CD14 and TLR4 expression. This is consistent with studies where monocytes from patients with advanced cirrhosis showed that up-regulation of TNF-α production was due to increased IRAK kinase, increased NFκB activity, and less IκBα protein levels, a lack of IRAK-M induction, and decreased TLR4 expression (45). Previous studies in monocytes have shown that IFN-γ and granulocyte/monocyte CSF can prevent or reverse decreased responses to LPS by inhibition of IRAK kinase degradation and promoting its association to MyD88 without any effect on IRAK kinase activity (46). Considering that chronic alcohol exposure of human monocytes resulted in increased IRAK-1 kinase activity and up-regulation of IKK kinase activity leading to augmentation of LPS-induced NFκB binding and TNF-α expression in our experiments, it cannot be ruled out that chronic alcohol increases IFN-γ production by circulating T and NK cells in vivo to influence this hyperactivity of monocytes and increased inflammation. This possibility awaits further investigation. However, in our experiments, chronic alcohol alone exhibited inhibition of IRAK-M at the mRNA and protein level in monocytes even in the absence of other cell types. Recent studies show that using a human monocytic cell line Mono Mac 6 cells, chronic alcohol increased activation of the TNF-α promoter and its production and this was dependent on activation of the TRIF-IRF3 pathway (47). Whereas chronic alcohol-induced NFκB activation observed in our studies could occur via the MyD88-IRAK-1 pathway, a role for its activation via the TRIF-IRF3-TBK-1 pathway cannot be ruled out.
Unlike monocytes and Kupffer cells, chronic alcohol ingestion decreased TNF-α production (48) in alveolar macrophages that was related to oxidative stress, decreased glutathione availability, and decreased GM-CSF receptors (35). Because GM-CSF has been shown to result in loss of TLR tolerance, it is tempting to speculate that the persistent TNF-α hyporesponsiveness of alveolar macrophages after chronic alcohol use could be related to the decreased GM-CSFR signaling (35).
Although the role of MAPK regulation in LPS hyporesponsiveness of monocytes and macrophages have been established (38, 39), there are no studies indicating a role for ERK, JNK, and p38 MAPK in increased sensitization to LPS. In this study, we show that prior chronic alcohol exposure for 7 days selectively increases subsequent LPS-induced ERK MAPK activation that contributes to the increased LPS-induced TNF-α production after chronic alcohol treatment. This was suggested by the observation that a specific ERK kinase inhibitor could prevent the chronic alcohol-induced increase in TNF-α production. Similar to chronic alcohol-exposed Kupffer cells (49), these studies indicate an important role for ERK MAPK kinases in addition to increased NFκB activity in chronic alcohol exposed human monocytes. We also found that chronic alcohol exposure does not affect p38 and JNK MAPK. It appears that reduced IRAK-M plays a pivotal role in down-stream activation of IRAK-1 kinase and subsequent activation of IKK and ERK-MAPK in chronic alcohol induced TNF-α production in human monocytes. The precise mechanisms of IRAK-M reduction by chronic alcohol exposure of human monocytes will be explored in future studies.
Although alcohol itself has direct effects on cell function and signaling molecules, products of its metabolism have also been implicated in immunomodulatory effects (29, 50). In our experiments, CYP2E1, a major metabolizing enzyme of alcohol in macrophages was slightly up-regulated by chronic and not by acute alcohol treatment. Interestingly, alterations in NFκB binding activity and TNF-α production were independent of CYP2E1 activation, suggesting that the modulatory effects of alcohol on LPS signaling were direct and not due to its metabolites.
In summary, our studies indicate that alcohol-mediated regulation of IRAK-M, a negative regulator of LPS signaling expressed in monocytes, may be responsible for the switch from a LPS hyporesponsiveness after acute alcohol exposure to reprogramming to increased TNF-α gene expression after chronic alcohol exposure. Thus, it is evident from these studies that the length of alcohol exposure and the resulting immune phenotype of peripheral blood monocytes could set the stage for increased susceptibility to infections or contribution to alcoholic liver injury.
Footnotes
This work was supported by Public Health Service Grant No. AA011576 (to G.S.) and AA008577 (to G.S.) from the National Institute of Alcohol Abuse and Alcoholism and by the resources of University of Massachusetts Medical School Center for AIDS Research (Grant 5P30 AI42845) and the Diabetes Endocrinology Research Center (PHS Grant DK32520) and its contents are solely the responsibility of the authors and do not necessarily represent the views of the National Institute of Alcohol Abuse and Alcoholism.
Abbreviations used in this paper: IRAK-1, IL-1R-associated kinase 1; IKK, IκBα kinase; siRNA, small interfering RNA.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Nelson S, Kolls JK. Alcohol, host defense and society. Nat Rev Immunol. 2002;2:205–209. doi: 10.1038/nri744. [DOI] [PubMed] [Google Scholar]
- 2.Szabo G. Monocytes, alcohol use, and altered immunity. Alcoholism: Clin Exp Res. 1998;22:217s–220s. doi: 10.1097/00000374-199805001-00002. [DOI] [PubMed] [Google Scholar]
- 3.Cook RT. Alcohol abuse, alcoholism, and damage to the immune system: a review. Alcoholism: Clin Exp Res. 1998;22:1927–1942. [PubMed] [Google Scholar]
- 4.McClain C, Barve S, Deaciuc I, Kugelmas M, Hill D. Cytokines in alcoholic liver disease. Semin Liver Disease. 1999;19:205–219. doi: 10.1055/s-2007-1007110. [DOI] [PubMed] [Google Scholar]
- 5.Nelson S, Bagby GJ, Bainton G, Warren WR. The effects of acute and chronic alcoholism on tumor necrosis factor and inflammatory response. J Infect Dis. 1989;1989:422– 429. doi: 10.1093/infdis/160.3.422. [DOI] [PubMed] [Google Scholar]
- 6.Nagy LE. Recent insights into the role of the innate immune system in the development of alcoholic liver disease. Exp Biol Med. 2003;228:882– 890. doi: 10.1177/153537020322800803. [DOI] [PubMed] [Google Scholar]
- 7.Thakur V, McMullen MR, Pritchard MT, Nagy LE. Regulation of macrophage activation in alcoholic liver disease. J Gastroenterol Hepatol. 2007;22:S53–S56. doi: 10.1111/j.1440-1746.2006.04650.x. [DOI] [PubMed] [Google Scholar]
- 8.Guha M, Mackman N. LPS indcution of gene expression in human monocytes. Cell Signal. 2001;13:85–94. doi: 10.1016/s0898-6568(00)00149-2. [DOI] [PubMed] [Google Scholar]
- 9.McClain CJ, Cohen DA. Increased tumor necrosis factor production by monocytes in alcoholic patients. Hepatology. 1989;9:349–351. doi: 10.1002/hep.1840090302. [DOI] [PubMed] [Google Scholar]
- 10.Tilg H, Jalan R, Kaser A, Davies NA, Offner FA, Hodges SJ, Ludwiczek O, Shwacross D, Zoller H, Alisa A, et al. Anti-tumor necrosis factor-α monoclonal antibody therapy in severe alcoholic hepatitis. J Hepatol. 2003;38:419– 425. doi: 10.1016/s0168-8278(02)00442-7. [DOI] [PubMed] [Google Scholar]
- 11.Iimuro Y, Gallucci RM, Luster MI, Kono H, Thurman RG. Antibodies to TNFα attenuate hepatic necrosis and inflammation caused by chronic exposure to ethanol in the rat. Hepatology. 1997;26:1530–1537. doi: 10.1002/hep.510260621. [DOI] [PubMed] [Google Scholar]
- 12.Verma BK, Fogarasi M, Szabo G. Down-regulation of TNFα activity by acute ethanol treatment in human peripheral blood monocytes. J Clin Immunol. 1993;13:8–22. doi: 10.1007/BF00920631. [DOI] [PubMed] [Google Scholar]
- 13.Szabo G, Chavan S, Mandrekar P, Catalano D. Acute alcohol consumption attenuates chemokine induction in response to ex vivo stimulation. J Clin Immunol. 1999;19:67–76. doi: 10.1023/a:1020518703050. [DOI] [PubMed] [Google Scholar]
- 14.Szabo G, Mandrekar P, Newman L, Catalano D. Regulation of human monocyte functions by acute ethanol treatment: decreased TNFα, IL-1ß and elevated IL-10, and TGFα production. Alcoholism: Clin Exp Res. 1996;14:900–907. doi: 10.1111/j.1530-0277.1996.tb05269.x. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Z, Bagby GJ, Stoltz D, Oliver P, Schwarzenberger PO, Kolls JK. Prolonged ethanol treatment enhances lippolysaccharide/phorbol myristate acetate induced tumor necrosis factor-α production in human monocytic cells. Alcoholism: Clin Exp Res. 2001;25:444– 449. [PubMed] [Google Scholar]
- 16.Yin M, Wheeler M, Kono H, Thurman RG. Essential role for TNF in alcohol-induced liver inury in mice. Gastroenterology. 1999;117:942–952. doi: 10.1016/s0016-5085(99)70354-9. [DOI] [PubMed] [Google Scholar]
- 17.Oak S, Mandrekar P, Catalano D, Kodys K, Szabo G. TLR2- and TLR4-mediated signals determine attenuation or augmentation of inflammation by acute alcohol in monocytes. J Immunol. 2006;176:7628–7635. doi: 10.4049/jimmunol.176.12.7628. [DOI] [PubMed] [Google Scholar]
- 18.Schatzle JD, Kralova J, Bose HR., Jr Avian IκBα is transcriptionally induced by c-Rel and v-Rel with different kinetics. J Virol. 1995;69:5383–5390. doi: 10.1128/jvi.69.9.5383-5390.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mandrekar P, Catalano D, Szabo G. Inhibition of LPS-mediated NF-κB activation by ethanol in human monocytes. Int Immunol. 1999;11:1781–1790. doi: 10.1093/intimm/11.11.1781. [DOI] [PubMed] [Google Scholar]
- 20.Mandrekar P, Jeliazkova V, Catalano D, Szabo G. Acute alcohol exposure exerts anti-inflammatory effects by inhibiting IκB kinase activity and p65 phosphorylation in monocytes. J Immunol. 2007;178:7686–7693. doi: 10.4049/jimmunol.178.12.7686. [DOI] [PubMed] [Google Scholar]
- 21.Uesugi T, Froh M, Arteel GE, Bradford B, Thurman RG. Toll-like receptor 4 is invovled in the mechanimsms of early alcohol-induced liver injury in mice. Hepatology. 2001;34:101–108. doi: 10.1053/jhep.2001.25350. [DOI] [PubMed] [Google Scholar]
- 22.Yin M, Bradford B, Wheeler M, Uesugi T, Froh M, Goyert S, Thurman RG. Reduced early alcohol-induced liver injury is CD14-deficient mice. J Immunol. 2001;166:4737– 4742. doi: 10.4049/jimmunol.166.7.4737. [DOI] [PubMed] [Google Scholar]
- 23.Janssens S, Beyaert R. Functional diversity and regulation of different interleukin-1 receptor-associated kinase (IRAK) famiky members. Mol Cell. 2003;11:293–302. doi: 10.1016/s1097-2765(03)00053-4. [DOI] [PubMed] [Google Scholar]
- 24.Kobayashi KS, Hernandez LD, Galan JE, Janeway CA, Jr, Medzhitov R, Flavell RA. IRAK-M is a negative regulator of toll-like receptor signaling. Cell. 2002;110:191–202. doi: 10.1016/s0092-8674(02)00827-9. [DOI] [PubMed] [Google Scholar]
- 25.van’tVeer C, vandenPangaart PS, vanZoelen MA, deKruif M, Birjmohun RS, Stroes ES, deVos AF, vanderPoll T. Induction of IRAK-M is associated with lippolysaccharide tolerance in a human endotoxemia model. J Immunol. 2007;179:7110–7120. doi: 10.4049/jimmunol.179.10.7110. [DOI] [PubMed] [Google Scholar]
- 26.Lui ZJ, Yan LN, Li XH, Xu FL, Chen XF, You HB, Gong JP. Up-regulation of IRAK-M is essential for endotoxin tolerance induced by a low dose of lipopolysaccharide in Kupffer cells. J Surg Res. 2008;150:34–39. doi: 10.1016/j.jss.2007.12.759. [DOI] [PubMed] [Google Scholar]
- 27.Cuschieri J, Maeir RV. Mitogen-activated protein kinase (MAPK) Crit Care Med. 2005;33:S417–S419. doi: 10.1097/01.ccm.0000191714.39495.a6. [DOI] [PubMed] [Google Scholar]
- 28.Collart MA, Bauerle P, Vassali P. Regulation of tumor necrosis factor α transcription in macrophages: involvement of four NF-κB motifs and constitutive and inducible form of NF-kB. Mol Cell Biol. 1990;10:1478–1506. doi: 10.1128/mcb.10.4.1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu Y, Cederbaum AI. CY2E1 and oxidative liver injury by alcohol. Free Radical Biol Med. 2008;44:723–738. doi: 10.1016/j.freeradbiomed.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hutson JL, Wickramasinghe SN. Expression of CYP2E1 by human monocyte-derived macrophages. J Pathol. 1999;188:197–200. doi: 10.1002/(SICI)1096-9896(199906)188:2<197::AID-PATH295>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 31.Asehnoune K, Strassheim D, Mitra S, Kim JY, Abraham E. Involvement of reactive oxygen species in toll-like receptor 4-dependent activation of NF-κB. J Immunol. 2004;172:2522–2529. doi: 10.4049/jimmunol.172.4.2522. [DOI] [PubMed] [Google Scholar]
- 32.Enomoto N, Ikejima K, Bradford B, Rivera C, Kono H, Brenner DA, Thurman RG. Alcohol causes both tolerance and sensitization of rat Kupffer cells via mechanisms dependent on endotoxin. Gastroenterology. 1998;115:443– 451. doi: 10.1016/s0016-5085(98)70211-2. [DOI] [PubMed] [Google Scholar]
- 33.Yamashina S, Wheeler MD, Rusyn I, Ikejima K, Sato N, Thurman RG. Tolerance and sensitization to endotoxin in Kupffer cells caused by acute ethanol involve interleukin-1 receptor-associated kinase. Biochem Biophys Res Comm. 2000;277:686– 690. doi: 10.1006/bbrc.2000.3738. [DOI] [PubMed] [Google Scholar]
- 34.Wheeler MD, Thurman RG. Up-regulation of CD14 in liver caused by acute ethanol involves oxidant-dependent AP-1 pathway. J Biol Chem. 2003;278:8435– 8441. doi: 10.1074/jbc.M212076200. [DOI] [PubMed] [Google Scholar]
- 35.Joshi PC, Guidot DM. The alcoholic lung: epidemiology, pathophysiology and potential therapies. Am J Physiol. 2007;292:L813–L823. doi: 10.1152/ajplung.00348.2006. [DOI] [PubMed] [Google Scholar]
- 36.Karavitis J, Murdoch EL, Gomez CR, Ramirez L, Kovacs EJ. Acute ethanol exposure attenuates pattern recognition receptor activated macrophage functions. J Interferon Cytokine Res. 2008;28:413– 422. doi: 10.1089/jir.2007.0111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kobayashi KS, Flavell RA. Shielding the double-edged sword:negative regulation of the innate immune system. J Leukocyte Biol. 2004;75:428– 433. doi: 10.1189/jlb.0703321. [DOI] [PubMed] [Google Scholar]
- 38.Norkina O, Dolganiuc A, Catalano D, Kodys K, Mandrekar P, Syed A, Efros M, Szabo G. Acute alcohol intake induces SOCS1 and SOCS3 and inhibits cytokine-induced STAT1 and STAT3 signaling in human monocytes. Alcohol Clin Exp Res. 2008;132:1565–1573. doi: 10.1111/j.1530-0277.2008.00726.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mandrekar P, Catalano D, Szabo G. Alcohol-induced regulation of nuclear regulatory factor-κB in human monocytes. Alcoholism: Clin Exp Res. 1997;21:988–994. [PubMed] [Google Scholar]
- 40.Kraatz J, Clair L, Rodriguez JL, West MA. Macrophage TNF secretion in endotoxin tolerance: role of SAPK, p38 and MAPK. J Surg Res. 1999;83:158–164. doi: 10.1006/jsre.1999.5587. [DOI] [PubMed] [Google Scholar]
- 41.Heagy W, Hansen C, Neiman K, Rodriguez JL, West MA. Impaired mitogen-activated protein kinase activation and altered cytokine secretion in endotoxin-tolerant human monocytes. J Trauma. 2000;49:806– 814. doi: 10.1097/00005373-200011000-00003. [DOI] [PubMed] [Google Scholar]
- 42.Wilson CS, Seatter SC, Rodriguez JL, Bellingham J, Clair L, West MA. In vivo endotoxin tolerance: impaired LPS-stimulated TNF release of monocytes from patients with sepsis, but not SIRS. J Surg Res. 1997;69:101–106. doi: 10.1006/jsre.1997.5040. [DOI] [PubMed] [Google Scholar]
- 43.Thurman RG., II Alcoholic liver injury involves activation of Kupffer cells by endotoxin. Am J Physiol. 1998;275:G605–G611. doi: 10.1152/ajpgi.1998.275.4.G605. [DOI] [PubMed] [Google Scholar]
- 44.Enomoto N, Schemmer P, Ikejima K, Tahei Y, Sato N, Brenner DA, Thurman RG. Long-term alcohol exposure changes sensitivity of rat Kupffer cells to lipopolysaccharide. Alcoholism: Clin Exp Res. 2001;25:1360–1367. [PubMed] [Google Scholar]
- 45.Tazi KA, Quioc JJ, Saada V, Bezeaud A, Lebrec D, Moreau R. Upregulation of TNF-α production signaling pathways in monocytes from patients with advanced cirrhosis: possible role of Akt and IRAK-M. J Hepatol. 2006;45:280–289. doi: 10.1016/j.jhep.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 46.Adib-Conquy M, Cavaillon JM. Gamma interferon and granulocyte/monocyte colony-stimulating factor prevent endotoxin tolerance in human monocytes by promoting interleukin-1 receptor-associated kinase expression and its association to MyD88 and not by modulating TLR4 expression. J Biol Chem. 2002;277:27927–27934. doi: 10.1074/jbc.M200705200. [DOI] [PubMed] [Google Scholar]
- 47.Zhao XJ, Dong Q, Bindas J, Piganelli JD, Magill A, Kolls JK. TRIF and IRF3 binding to the TNF promoter results in macrophage TNF dysregulation and steatosis induced by chronic ethanol. J Immunol. 2008;181:3049–3056. doi: 10.4049/jimmunol.181.5.3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Omidvari K, Casey R, Nelson S, Olariu R, Shellito JE. Alveolar macrophage release of tumor necrosis factor-α in chronic alcoholics without liver disease. Alcoholism: Clin Exp Res. 1998;22:567–572. doi: 10.1111/j.1530-0277.1998.tb04294.x. [DOI] [PubMed] [Google Scholar]
- 49.Kishore R, Hill J, McMuller M, Frenkel J, Nagy LE. ERK1/2 and Egr-1 contribute to increased TNF production in rat Kupffer cells after chronic alcohol feeding. Am J Physiol. 2002;282:G6–G15. doi: 10.1152/ajpgi.00328.2001. [DOI] [PubMed] [Google Scholar]
- 50.Vidali M, Stewart SF, Albano E. Interplay between oxidative stress and immunity in the progression of alcohol-mediated liver injury. Trends Mol Med. 2008;14:63–71. doi: 10.1016/j.molmed.2007.12.005. [DOI] [PubMed] [Google Scholar]